
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps†
S.
Van den Bosch‡
a,
W.
Schutyser‡
a,
R.
Vanholme
bc,
T.
Driessen
a,
S.-F.
Koelewijn
a,
T.
Renders
a,
B.
De Meester
bc,
W. J. J.
Huijgen
d,
W.
Dehaen
e,
C. M.
Courtin
f,
B.
Lagrain
a,
W.
Boerjan
bc and
B. F.
Sels
*a
aCenter for Surface Chemistry and Catalysis, KU Leuven, Kasteelpark Arenberg 23, 3001 Heverlee, Belgium. E-mail: bert.sels@biw.kuleuven.be
bDepartment of Plant Systems Biology, VIB, Technologiepark 927, 9052 Ghent, Belgium
cDepartment of Plant Biotechnology and Bioinformatics, Ghent University, Technologiepark 927, 9052 Ghent, Belgium
dEnergy Research Centre of the Netherlands (ECN), Biomass & Energy Efficiency, Westerduinweg 3, 1755 LE, Petten, The Netherlands
eMolecular Design and Synthesis, KU Leuven, Celestijnenlaan 200F, 3001 Heverlee, Belgium
fCenter for Food and Microbial Technology, KU Leuven, Kasteelpark Arenberg 22, 3001 Heverlee, Belgium
First published on 20th April 2015
Abstract
A catalytic lignocellulose biorefinery process is presented, valorizing both polysaccharide and lignin components into a handful of chemicals. To that end, birch sawdust is efficiently delignified through simultaneous solvolysis and catalytic hydrogenolysis in the presence of a Ru on carbon catalyst (Ru/C) in methanol under a H2 atmosphere at elevated temperature, resulting in a carbohydrate pulp and a lignin oil. The lignin oil yields above 50% of phenolic monomers (mainly 4-n-propylguaiacol and 4-n-propylsyringol) and about 20% of a set of phenolic dimers, relative to the original lignin content, next to phenolic oligomers. The structural features of the lignin monomers, dimers and oligomers were identified by a combination of GC/MS, GPC and 2D HSQC NMR techniques, showing interesting functionalities for forthcoming polymer applications. The effect of several key parameters like temperature, reaction time, wood particle size, reactor loading, catalyst reusability and the influence of solvent and gas were examined in view of the phenolic product yield, the degree of delignification and the sugar retention as a first assessment of the techno-economic feasibility of this biorefinery process. The separated carbohydrate pulp contains up to 92% of the initial polysaccharides, with a nearly quantitative retention of cellulose. Pulp valorization was demonstrated by its chemocatalytic conversion to sugar polyols, showing the multiple use of Ru/C, initially applied in the hydrogenolysis process. Various lignocellulosic substrates, including genetically modified lines of Arabidopsis thaliana, were finally processed in the hydrogenolytic biorefinery, indicating lignocellulose rich in syringyl-type lignin, as found in hardwoods, as the ideal feedstock for the production of chemicals.
Broader contextThere is a growing consensus that lignin valorization is essential for the environmental sustainability and economics of a lignocellulosic biorefinery. The thermal conversion of lignocellulose to renewable gas and bio-oils has been heavily researched and their benefits and challenges for industrial implementation have become clear now. However, to preserve Nature's highly functionalized materials, milder treatments are required, fractionating lignocellulose into its main components, namely carbohydrates and lignin. Inspired by old delignification processes, many initiatives have been presented to remove lignin, while producing pure hemicellulose and cellulose products. Initially the chemical occurrence of disassembled lignin and its use for the production of chemicals were not a primary concern, but because of the importance of lignin valorization, it is currently one of the foremost challenges of new biorefinery strategies. This work promotes a lignin-first biorefinery approach, converting lignin into useful chemicals during fractionation, while keeping the pulp fraction available for further processing. |
Introduction
Research on the novel ‘biorefinery’ concept has lately received a lot of attention as a sustainable alternative for the current petrochemical industry. Renewable biomass, instead of fossil resources, is herein used to produce heat, power, fuels, chemicals and materials.1–11 Lignocellulose, a sustainable and highly abundant source of biomass, is typically presented as a promising feedstock.2,5 Since its three main components, cellulose, hemicellulose and lignin, are located in the cell wall as a complex rigid matrix, thermal and solvolytic processing is required before the selective conversion towards value-added products is possible.2,3Numerous lignocellulose conversion efforts have been made, often preferring integrated biorefinery strategies with use of the entire plant because of feasibility reasons. A well-known example is the gasification of lignocellulose to syngas, ultimately generating electricity/heat or leading to the production of chemicals like alkanes, methanol and H2.12 The production of bio-oils via pyrolysis or liquefaction is another option, typically yielding unstable bio-oils, containing hundreds of different oxygenates. These bio-oils can be upgraded catalytically before being used as biofuels.13–17 Through fast-hydropyrolysis, the production of a high-quality liquid fuel (C4–C8) can be achieved using a single procedure, combining pyrolysis with subsequent catalytic hydrodeoxygenation in the gas phase.18,19 Also, liquefaction in supercritical methanol in the presence of a Cu catalyst was recently demonstrated, resulting in a combustible liquid of complex composition.20,21
One may argue if forthcoming biorefineries should merely focus on the strong defunctionalization of the highly functionalized bio-based (macro)molecules or whether a milder and more selective conversion of Nature's precious resources to a handful of value-added chemicals is a better research focus. Exploiting the original chemical structure and functionality, hence preserving a high atom efficiency, is probably the best and most encouraging strategy for the creation of value, if techno-economically applicable.10
Other biorefinery approaches therefore encourage a prior fractionation of the lignocellulose matrix into its different components (i.e. carbohydrates, lignin,…), thereby reducing the complexity of downstream separation and conversion processes. Most lignocellulosic fractionations involve the removal of lignin (delignification), often accompanied by a major part of hemicellulose, to yield a rather pure cellulose substrate. Some of these methods are industrially applied in paper mills or will be used in the production of next generation biofuels like bioethanol as well as biofuel precursors like bio-derived naphtha.2,3,18,19,22–26 Two intriguing fractionation methods were recently introduced that perform a complete solubilisation of the lignocellulose substrate. A mechanocatalytic approach was demonstrated, converting lignocellulose into water soluble oligosaccharides and lignin fragments.27 Further processing can result in various products like sugars,28 furfurals29 or γ-valerolactone,30 next to a lignin precipitate. Interestingly, the sugar processing towards γ-valerolactone has been achieved in a continuous flow mode.30 The second method is based on the promoting effect of γ-valerolactone on the acid-catalyzed saccharification of lignocellulose, enabling very high yields of soluble carbohydrates and a water insoluble lignin fraction.31 However, for all aforementioned fractionation methods, the chemical structure of the obtained lignin precipitates is inevitably degraded to some extent, when compared to that of the original ‘protolignin’.25,26,32–36 Even under relatively mild conditions, such as those used in typical organosolv fractionations, the lignin structure is altered.37–40 Such alterations, amplified in the presence of acid or base, are the result of several side-reactions like the breaking of readily cleavable ether linkages and the formation of new stable C–C bonds.35–39,41 Besides lignin's recalcitrant behaviour, it also shows a species-specific distribution of bonds and building blocks, further complicating a governable conversion process to a handful of valuable products. Lignin recovery and its subsequent valorization to chemicals have never been of primary concern. Instead, lignin side streams are typically burned for energy recuperation or used in low-value material applications.42,43
However, since lignin constitutes the largest direct source of renewable aromatic/phenolic compounds on the Earth, the conversion opportunities towards aromatics but also other chemicals should not be underestimated.22,32,36,44,45 With the emergence of next-generation biofuels, a huge amount of lignin is expected to enter the market and with that, an increased awareness of lignin's potential.26,46,47 Recent reports have also predicted the essential role of lignin valorization in the economics of lignocellulosic biorefining.22,26 Finding efficient processing routes to convert lignin into valuable products, while maintaining a maximum valorization of the carbohydrate pulp, may thus be regarded essential to strongly expand the economic feasibility and environmental sustainability of the lignocellulosic biorefinery. Numerous efforts have recently led to great progress in the conversion of various types of lignin streams, e.g. originating from pulp- and paper industries and organosolv processes, to valuable chemicals.32,45,48–57
In our view, a forthcoming biorefinery should deal with the unfavorable fractionation side-effects so as to allow for processing lignin in its most reactive and workable form. Milder solvolytic fractionation conditions, currently under investigation using less acidic or basic compounds, are a valid option to improve the potential valorization of the resulting lignin fraction.3,31,37 However, perhaps the most promising strategy is a fractionation process including catalytic hydrogenolysis in the liquid phase, starting from raw unfractionated lignocellulose. In contrast to previous methods, typically forming a condensed lignin polymer fraction, the thermal and solvolytic disassembly of lignin (delignification) is here immediately followed by the reductive stabilization of lignin's most reactive intermediates like olefins and carbonyls into a handful of soluble and stable low-molecular-weight phenolic products. This fractionation strategy can be denominated as a ‘lignin-first’ biorefinery, as the valorization of lignin to chemicals is performed before carbohydrate processing. Though being conveyed in the old literature,58–61 its integration in a contemporary biorefinery was only recently discussed by a handful of research groups.62–67 Interestingly, high lignin monomer yields, ranging from 10 to 54%, have thus far been reported.62–65,67 For instance, Li et al. presented a Ni-W2C/AC catalytic system in water that not only depolymerized lignin, but also converted the carbohydrate fraction into C2–C3 diols.65 However, the presence of all products in the same liquid phase might ultimately complicate product separation, while the integrated carbohydrate conversion reduces the versatility of carbohydrate processing towards other chemicals or materials. Using Ni on carbon as a catalyst and methanol as both solvent and hydrogen donor, Song et al. showed the selective hydrogenolysis of protolignin to 4-n-propylguaiacol and 4-n-propylsyringol.63 On the other hand, Galkin et al. obtained high yields of 4-(prop-1-enyl) guaiacol and 4-(prop-1-enyl) syringol using Pd on carbon in a water–ethanol solvent system, with formic acid from wood acting as the hydrogen source.64 Both systems elegantly avoid the use of an external H2-source, while yielding a solid carbohydrate pulp. Although these studies enable high phenolic monomer yields, other aspects like the degree of delignification, the carbohydrate retention in the pulp or further processing opportunities of the pulp are not studied. Rinaldi et al. proposed RANEY® Ni in an isopropanol–water solvent mixture, with isopropanol as the hydrogen donor. High degrees of delignification and carbohydrate retention in the pulp as well as the enzymatic processing of the pulp were demonstrated. The results however also showed the complexity of the low-molecular weight lignin product mixture.66 Recently Abu-Omar et al. presented a selective hydrogenolysis of protolignin with ZnII modified Pd nanoparticles on carbon with external H2, focusing on the lignin monomers and the enzymatic conversion of the retained pulp.67
This paper presents a lignocellulosic fractionation process that results in a high degree of delignification, a selective conversion of lignin towards a handful of useful chemicals and a maximal sugar retention, obtaining a carbohydrate pulp that is applicable for a myriad of downstream processes (Scheme 1). Mainly because of sugar solubilization issues, which lower the polysaccharide retention, but also due to the expected process and product separation issues later on, it was decided to avoid the additional use of water. Instead, lignin is disassembled from the lignocellulose matrix in condensed methanol at elevated temperature. Meanwhile, the lignin fragments are selectively depolymerized in the presence of a commercial Ru/C catalyst preferably under a H2 atmosphere. The hydrogenolysis reaction results in the formation of methoxyphenolic monomers, structurally related dimers and short oligomers, which together form a ‘lignin oil’. Whereas in our hands other alcohols, like ethanol, and metal catalysts, like Ni, are applicable as well, the combination of methanol and Ru/C showed minor methanation and thus a loss of solvent and H2. In addition, methanol is a relatively cheap solvent and is easily recoverable from both product fractions. Moreover, demethoxylation of the lignin-derived products has been demonstrated to provide bio-derived methanol,17,32,68 thus nicely exemplifying the integrated nature of the proposed biorefinery.
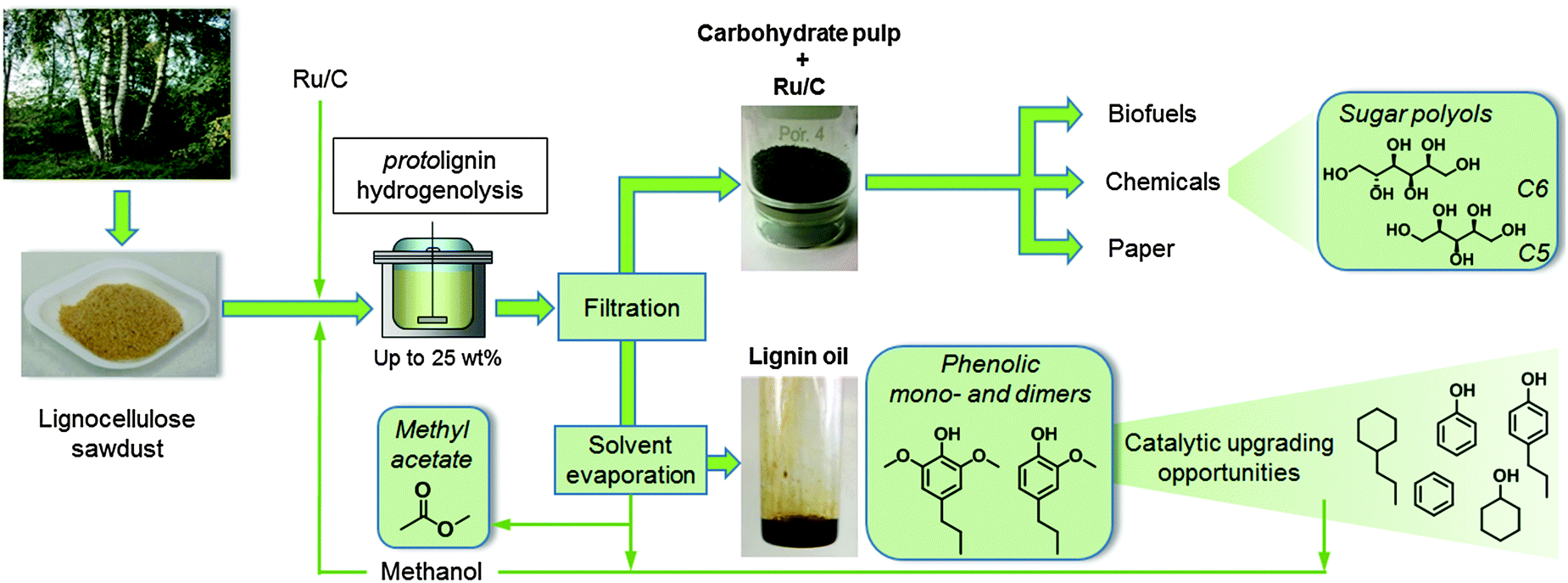 | ||
| Scheme 1 Schematic representation of the proposed integrated biorefinery process. A hydrogenolysis reaction was executed on lignocellulose sawdust in the presence of Ru on carbon (Ru/C) in methanol under H2 pressure at elevated temperature. The lignocellulose substrate was fractionated into a solid carbohydrate pulp (containing cellulose, hemicellulose and the solid Ru/C catalyst) and a depolymerized lignin fraction that was solubilized in the methanol solution. The carbohydrate pulp was easily separated by filtration while after solvent recovery from the liquid phase, a brown lignin oil was obtained. The lignin oil consists of a select set of methoxyphenol mono- and dimers, which can be upgraded further into a myriad of downstream products, including possibilities to produce methanol. The recovered solvent fraction also contains methyl acetate, produced by the transesterification of hemicellulose acetyl groups. Catalytic reductive splitting converts the pulp into bio-derived sugar polyols, also demonstrating the multiple use of the Ru/C catalyst. | ||
Various biomass feedstocks, including several wood and grass types, but also genetically modified lines of Arabidopsis thaliana,69–71 were examined to investigate the impact of different lignin compositions on the product yield. Irrespective of plant species, we noted that the lignin-derived product yield strongly depends on the protolignin monomer composition. Lignin rich in S-units showed the highest degree of delignification as well as the highest monomer yield, suggesting the preferred use of hardwood substrates such as poplar and birch in the proposed lignin-first biorefinery. The remaining solid fraction, primarily composed of Ru/C and the polysaccharides, cellulose and hemicellulose, may be valorized into paper, biofuels and chemicals. Here, the amenability of the carbohydrate pulp towards chemocatalytic conversion is successfully illustrated by its conversion to a mixture of sugar polyols. Earlier reported bifunctional acid-redox catalysis was applied here,72–77 to demonstrate the reusability of the Ru/C catalyst, originally used in the first hydrogenolysis step (Scheme 1).
Experimental section
For a list of all used chemicals and materials as well as a more complete description of the experimental procedures, the reader is kindly referred to the ESI.†In a typical catalytic hydrogenolysis experiment, 2 g of birch sawdust (Betula pendula from Ecobois, Ghent), 0.3 g of Ru/C and 40 mL of methanol were loaded into a 100 mL stainless steel batch reactor (Parr Instruments Co.). The reactor was sealed, flushed with N2 and pressurized with 3 MPa H2 at room temperature (RT). The mixture was stirred at 700 rpm and the temperature was increased to 523 K (∼10 K min−1) at which the pressure reached ∼12 MPa (∼6.5 MPa at 473 K) and the reaction was started. After reaction, the autoclave was cooled in water and depressurized at RT.
To analyze the lignin monomers, a weighed amount of external standard (2-isopropylphenol) was added and mixed in the reactor. The reactor content was filtered and a sample of the filtrate was used for GC analysis. To analyze the dimers, a derivatization step, via trimethylsilylation with N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA), was introduced to increase their volatility before GC analysis.78–80 GC/MS was used to identify the phenolic mono- and dimers, while gel permeation chromatography (GPC) and 2D HSQC NMR were applied for qualitative analyses of the lignin oil. To determine the degree of delignification, the raw filtered methanol product mixture was evaporated and a brown ‘lignin oil’ was obtained. The lignin oil was subjected to threefold liquid–liquid extraction using dichloromethane (DCM) and water to separate the soluble lignin- and sugar-derived products. Finally the DCM-extracted phase was dried to obtain the ‘DCM lignin oil’. The weight of the DCM lignin oil is then used to determine the degree of delignification (based on Klason lignin weight). A corrected value was added in the results, to account for the expected presence of birch extractives in the DCM lignin oil. The sugar retention was based on the amount of sugars in the lignocellulose substrates and in the carbohydrate pulp after hydrogenolysis, using a standard total sugar procedure, adapted with hydrolysis conditions for cellulose-rich materials.81–83
The chemocatalytic conversion of the carbohydrate pulp was demonstrated in a hydrolytic hydrogenation experiment. The carbohydrate pulp (∼1.4 g including 0.3 g Ru/C) was mixed with tungstosilicic acid (0.5 g) and water (50 mL) in a 100 mL stainless steel batch reactor (Parr Instruments Co.). The reactor was sealed, flushed with N2 and subsequently pressurized with 5 MPa H2 at RT. The mixture was stirred at 700 rpm and the temperature was increased to 463 K (∼13 K min−1), at which the pressure reached ∼7 MPa and the reaction was started. After reaction, the autoclave was rapidly cooled in water and depressurized at RT. A sample of the reaction product was taken and centrifuged. External standard (myo-inositol) was added to the supernatant and dried under vacuum, after which it was derivatized via trimethylsilylation and analyzed by GC.
Results and discussion
Catalytic delignification of birch wood
Initial catalytic reactions were executed using birch sawdust as a benchmark hardwood substrate and Ru/C as the solid redox catalyst typically at 3 MPa H2 (RT) and 523 K (Table 1). All reactions in Table 1 show 4-n-propylguaiacol (PG) and 4-n-propylsyringol (PS) as main compounds, with a PS/PG ratio of around three, being in close agreement with the syringyl/guaiacyl ratio of birch wood lignin.84 Besides PG and PS, other monomers like 4-n-propanolguaiacol, 4-n-propanolsyringol and 4-ethylsyringol were identified (see Fig. 1a). A more detailed monomer distribution for all experiments in Table 1 is presented in the ESI,† Table S1. Next to the lignin monomer yields, also the dimer yields, the degree of delignification and the retention of sugars in the carbohydrate pulp, as defined in the Experimental section and the ESI,† are listed in Table 1.| Entry | t (h) | Birch (g mL−1) | Phenolic product yieldsj (C%) | Delignificationk (wt%) | Sugar retentionl (C%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| PG + PSg | Total monomers | Dimers | C6 | C5 | Total | ||||
| a Reaction conditions: 2 g of birch sawdust (particle size 0.25–0.50 mm; composition: 19.5 wt% lignin, 2.5 wt% extractives, 39.3 wt% C6 sugars, and 20.7 wt% C5 sugars), 0.3 g of 5% Ru/C, 40 mL of methanol, 523 K and 3 MPa H2 at RT (∼12 MPa at 523 K). b 40 mL of water as the solvent, no delignification value due to the complete dissolution of lignocellulose. c Reaction without catalyst. d Reaction temperature 473 K (∼6.5 MPa). e 1 MPa H2 at RT. f Atmospheric pressure of N2 at RT. g Reuse of the catalyst (0.3 g) after liquid–liquid (methanol/decane) separation of Ru/C and the carbohydrate fraction. h 10 g of birch sawdust in 40 mL of methanol and 1 g of 5% Ru/C. i Reaction performed in a 600 mL batch reactor with 60 g of birch sawdust in 240 mL of methanol, and 6 g of 5% Ru/C. j Yields are carbon-based, assuming a birch protolignin carbon content of 64 wt% (ESI). Primary products are 4-n-propylguaiacol (PG) and 4-n-propylsyringol (PS), PS/PG ratios vary around 3, and values in parentheses refer to the selectivity of both products based on the total phenolic monomer yield. k Based on the weight of the dichloromethane (DCM) extracted fraction, specified in the text as ‘DCM lignin oil’, and the Klason lignin weight. These values slightly overestimate the real delignification degree due to the concomitant removal of other extractives. Values in parentheses are corrected for the weight of these birch extractives. l Based on the amount of carbon in the sugar fractions of birch sawdust and the produced carbohydrate pulps (ESI). | |||||||||
| 1 | 6 | 0.05 | 41 (79) | 52 | 16 | 92 (79) | 95 | 47 | 78 |
| 2b | 6 | 0.05 | 17 (70) | 25 | 11 | — | <1 | <1 | 1 |
| 3c | 6 | 0.05 | 0.9 (12) | 8 | 9 | 95 (82) | 86 | 68 | 79 |
| 4d | 6 | 0.05 | 33 (77) | 43 | 16 | 78 (65) | 97 | 84 | 92 |
| 5 | 3 | 0.05 | 42 (84) | 50 | 18 | 93 (80) | 95 | 56 | 81 |
| 6 | 0.5 | 0.05 | 33 (84) | 39 | 18 | 81 (68) | 96 | 67 | 86 |
| 7e | 3 | 0.05 | 47 (92) | 51 | 14 | 98 (85) | 94 | 63 | 83 |
| 8f | 3 | 0.05 | 35 (87) | 40 | 17 | 92 (79) | 99 | 65 | 87 |
| 9g | 3 | 0.05 | 30 (62) | 48 | 15 | 92 (79) | 93 | 83 | 90 |
| 10h | 3 | 0.25 | 44 (87) | 50 | 14 | 94 (81) | 90 | 52 | 77 |
| 11i | 3 | 0.25 | 44 (89) | 49 | 15 | 92 (79) | 92 | 55 | 79 |
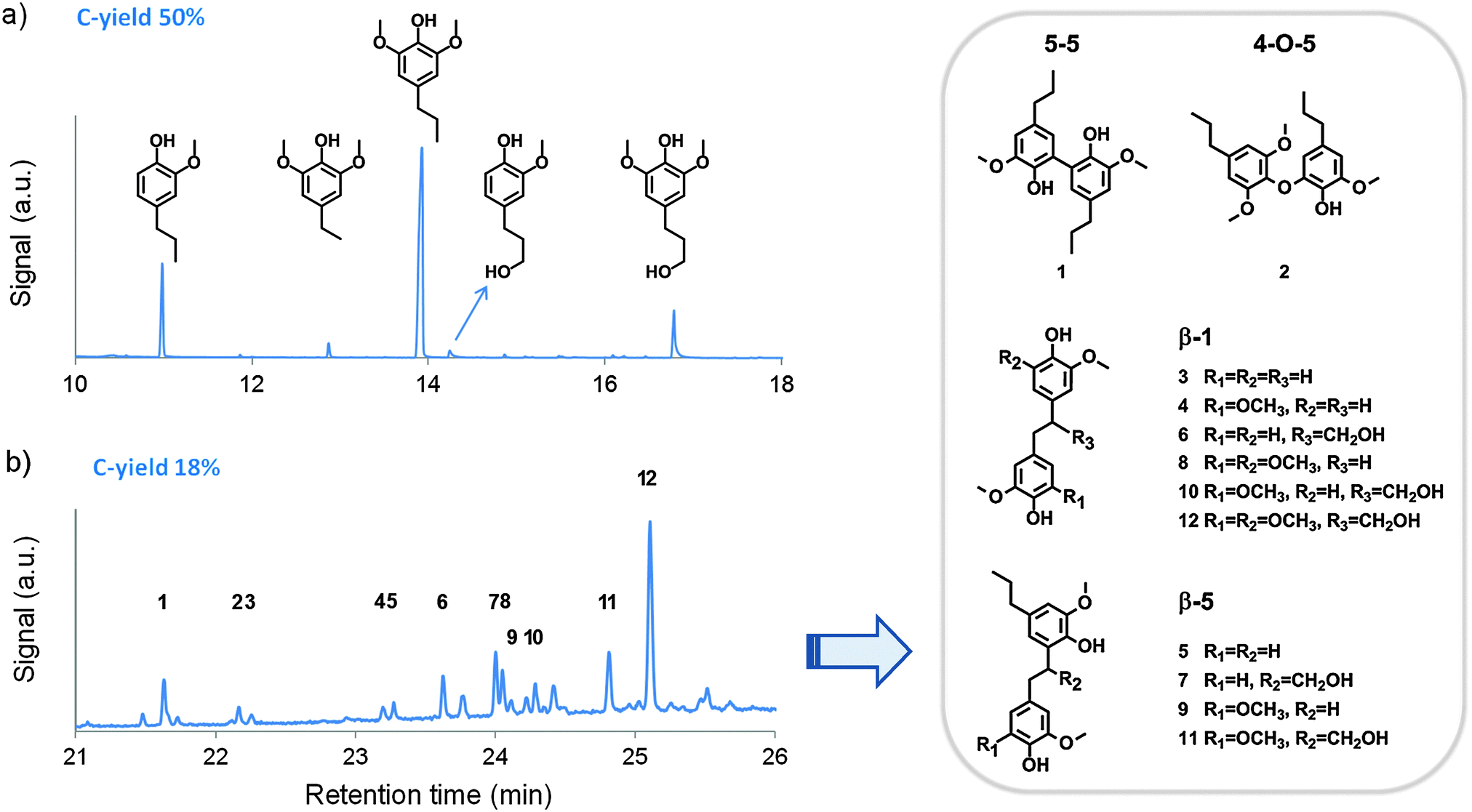 | ||
| Fig. 1 Gas-chromatograms and peak identification of (a) the lignin monomer fraction (left to right: 4-n-propylguaiacol, 4-ethylsyringol, 4-n-propylsyringol, 4-n-propanolguaiacol and 4-n-propanolsyringol) and (b) the trimethylsilylated dimer fraction after birch hydrogenolysis (reaction conditions from entry 5, Table 1). C-yield represents the carbon yield, as defined in the ESI.† | ||
Entries 1 and 2 compare the results for reactions in methanol and water, respectively, as commonly used solvents in lignocellulose pretreatment and lignin valorization. With methanol, more than 90% of lignin was solubilized, yielding 52% phenolic monomers with a selectivity of 79% towards PG and PS. Next to monomers, a phenolic dimer yield of 16% was obtained, totaling a well-defined lignin product yield of almost 70% to phenolic mono- and dimers. The nature of the dimer structures is discussed below. Furthermore, a total carbohydrate pulp retention as high as 78% was obtained in methanol, the retention of C6 sugars being almost quantitative as opposed to a 47% retention for C5 sugars. The higher retention of C6 sugars compared to C5 sugars in the pulp is due to a better protection of glucose (C6) in the crystalline cellulose structure, while C5 sugars like xylose in the amorphous hemicellulose are more prone to solvolysis. The released C5 sugars mainly appear in methanol as the corresponding methyl sugars, which may be useful in the detergent and soap industries, or could be hydrolyzed readily into the C5 sugar. Hence, with birch sawdust, the primary sugar product in methanol was methylated xylose, corresponding to 33% of the initial carbon in hemicellulose. In addition, the acetyl groups in hemicellulose, representing about 3 to 4 wt% of birch,85 were entirely removed and appeared in the methanol phase as methyl acetate, an interesting bio-derived solvent86 and precursor for chemicals like acetic anhydride and vinyl acetate.87–89 Separation of methyl acetate and methanol is common practice in industry.89 In comparison with methanol, the use of water in entry 2 resulted in a lower phenolic monomer yield of 25% and a complete dissolution of the carbohydrate fraction (no pulp remaining). The carbohydrates mainly appeared as water soluble polyols. For the envisioned biorefinery concept, water is thus not a suitable solvent.
Next, the essential role of Ru/C is demonstrated in entry 3. Without catalyst a phenolic monomer yield of only 8% and a dimer yield of 9% were obtained when using the conditions from entry 1. The much higher monomer yield with Ru/C is likely due to an efficient hydrogenolysis of most of the ether-bonds between phenolic units, combined with a reductive stabilization of reactive intermediates. This prevents repolymerization reactions leading to new stable C–C bonds within the lignin structural network. The product spectrum of the uncatalyzed reaction indeed shifts towards phenolic compounds with unsaturated C3-chains (ESI,† Table S1). Lowering of the reaction temperature to 473 K (entry 4) resulted in a higher retention of the C5 sugars (here 84%), corresponding to a total pulp retention of 92%, with only a small decrease in the phenolic monomer yield as well as in the delignification efficiency. A similar trend is observed by lowering the contact time at 523 K (entries 5 and 6), leading to a monomeric phenol yield of 50% after 3 h and 39% after 0.5 h, along with a C6/C5 sugar retention in the pulp of 95/56 and 96/67, respectively. With a shorter reaction time, the selectivity to PG and PS within the monomeric fraction increased slightly to 84%. The gas chromatogram in Fig. 1a illustrates the monomer distribution for entry 5. Moreover, gas analysis, showing low amounts of CO and methane (ESI,† Table S2), reveals a minor loss of carbon in the gas phase, indicating a high stability of the solvent, methanol, in the presented biorefinery process.
To conclude, various parameters, determining the severity of the reaction conditions, need to be well-balanced to optimize the phenolic monomer yield, the product selectivity and the degree of delignification as well as the carbohydrate pulp retention. Based on the aforementioned results, the reaction conditions from entry 5 were used for the following experiments.
Several potential constraints were additionally tested to anticipate the technical and economic feasibility of an industrial implementation. Interestingly, a reaction executed at reduced H2 pressure (1 MPa at RT) showed a similar catalytic performance (entry 7). Here, birch wood is efficiently delignified, yielding 51% of phenolic monomers with up to 92% of PG and PS, while the retention of C6 sugars is nearly complete and 63% for the C5 sugars. The use of N2 at atmospheric pressure, thus implying methanol or lignocellulose itself as a reducing source, also proved to be possible, in agreement with previous reports.63,64,90 However, in our hands the phenolic monomer yield was considerably lower (entry 8) than when executed under a H2 atmosphere. Crucial for the viability of the biorefinery is also the reusability of the Ru/C catalyst. Ru/C was separated from the carbohydrate pulp by a liquid–liquid extraction as described in the ESI.† In entry 9, the recycled catalyst showed a phenolic monomer yield of 48%, very similar to the 50% obtained with a fresh catalyst. A shift in selectivity towards more propanolsyringol and propanolguaiacol as well as a higher C5 sugar retention of 83% was observed. Next, the substrate to solvent ratio was increased from 5 wt% up to 25 wt%, which corresponds to the highest values reported in typical organosolv pretreatments,3 forming a paste of 10 g of birch sawdust wetted with 40 mL of methanol. The use of such a highly concentrated feed resulted in a similarly high sugar retention in the pulp as well as a high degree of delignification, yielding 50% of phenolic monomers, corresponding to 25 g L−1 (entry 10). The concentrated reaction was then repeated in a 600 mL batch reactor, using 60 g of birch sawdust in 240 mL of methanol (entry 11). Nearly the same results were obtained at this enlarged scale, which is a good sign for our planned future pilot scale experiments. Finally, the influence of the wood particle diameter was examined in an attempt to reduce the cost by avoiding fine-milling. Hence, a larger birch fraction, retained by a 1.5 mm sieve with an irregular shape and a broad average size, was tested (ESI,† Fig. S1). No undesirable impact on the pulp retention, delignification and phenolic monomer yield (and selectivity) was observed (ESI,† Table S1). Overall, the above experiments provided a promise (high wood loading, reuse of the catalyst, realistic particle size and low H2 pressure) towards the industrial feasibility of this catalytic biorefinery process. The experiments indicate that the most favorable conditions to process birch wood were the ones used in entry 7, as these result in a high lignin product yield, while leaving the sugars essentially unaltered for further processing.
Chemical composition of the lignin oil
Liquid–liquid extraction of the raw lignin oil with DCM and water was applied to remove the soluble sugar-derived products prior to a detailed analysis of the chemical composition. Next to the earlier discussed phenolic monomers, the isolated birch ‘DCM lignin oil’ (see Experimental section) also contains a set of dimers and some small oligomers. This can be derived from the GPC chromatogram in Fig. 2a (blue line), which shows two major signals, at circa 200 and 450 g mol−1 (based on polystyrene standards), suggesting a successful depolymerization mainly towards monomers and dimers. In an effort to elucidate their chemical structure, both GC/MS as well as NMR analyses were conducted on the DCM lignin oil.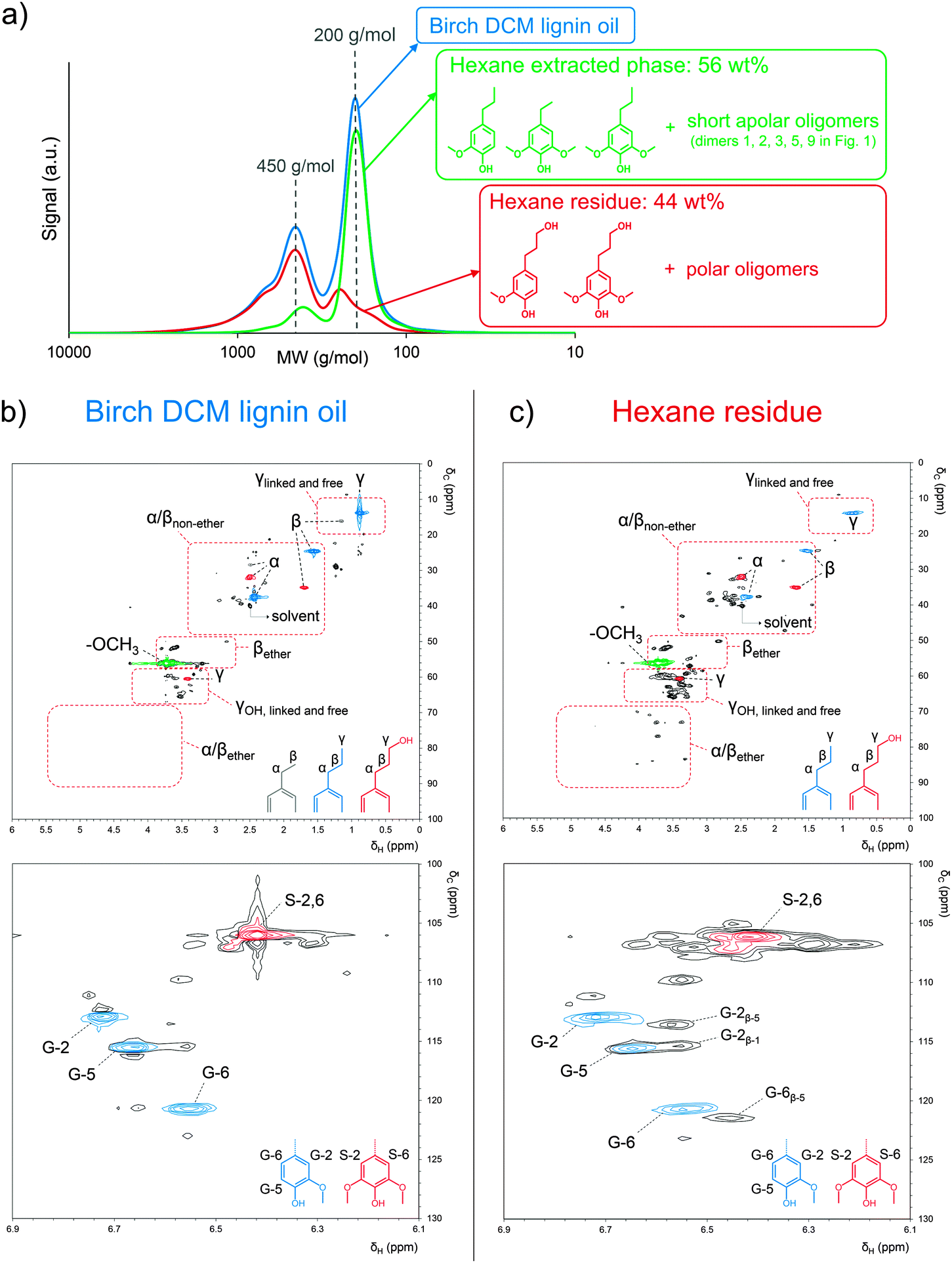 | ||
| Fig. 2 Characterization of the lignin product mixture by (a) gel permeation chromatography of the birch DCM lignin oil (calibration with polystyrene standards), the hexane extracted phase and the hexane retained phase (hexane residue) combined with 2D HSQC NMR analysis of (b) the DCM lignin oil and (c) the hexane residue. | ||
Before GC/MS analysis, the DCM lignin oil was first derivatized via trimethylsilylation to improve the volatility of the dimers. The identification of these dimers was supported by literature78–80 and the results are presented in the chromatographic analysis in Fig. 1b. The first observation is the absence of ether bonds in the present dimer fraction, except for a minor signal at 22.2 min, representing compound 2 with a relatively stable 4-O-5 ether bond.91,92 This suggests a nearly complete hydrogenolysis of the ether bonds, present in the original protolignin structure. Taking into account the ether function density of a typical birch lignin and assuming that most C–C bonds are not broken under the applied conditions, one can estimate that the previously determined monomer yield of about 50% is close to the expected theoretical maximum monomer yield of birch wood, as discussed in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45,62–64
45,62–64
Within the identified dimer fraction, the largest part of C–C linkages is represented by β-1 bonds, followed by β-5 and to a lesser extent 5–5 bonds, as illustrated in the structures of compounds 1 to 12 in Fig. 1b. These interunit linkages also represent the most important C–C bonds in birch lignin.32,85 Although β–β linkages are also common in birch lignin, no dimers with this bond were identified in the product mixture. Most dimers thus comprise two phenol units which are p,p′ or o,p′-coupled by an ethylene bridge. Remarkably, the bridge is either unsubstituted (as in 3–5, 8, and 9) or contains a –CH2OH substituent (as in 6, 7, and 10–12), whereas a –CH3 substituent was never analyzed. Moreover, unlike the monomers, the dimers always possess at least two hydroxyl groups, making them favourable candidates as building blocks for a broad range of polymers (e.g. polyurethanes, polyesters, polycarbonates).93–100
The DCM lignin oil was further characterized by two dimensional (2D) heteronuclear single quantum coherence (HSQC) NMR analysis (Fig. 2b). This technique is a powerful tool for the identification of lignin structural features like interunit linkages.101–104 Many C–H cross-signals in the HSQC spectrum are well reported in literature like those of the main substructures present in native lignin, viz. p-coumaryl, coniferyl and sinapyl alcohol units, while connected through various interunit linkages such as β-O-4, phenylcoumaran, resinol, β-1, spirodienone, dibenzodioxocin and 4-O-5.101–105 The oxygenated side chain region of the HSQC spectra, represented in the δC/δH region of 50–95/2.5–6 ppm, gives useful information about these interunit linkages. In lignin depolymerization studies, HSQC NMR is often used to examine the cleavage of ether bonds by following the decrease in intensity of C–H correlation signals related to substructures with ether bonds.55,64,66,106 Upon depolymerization however, also new correlation signals appear, attributed to chemical structures in which the original ether bonds between the phenol units are broken, but the C–C bonds remain. Breaking of α-O-4 in a phenylcoumaran unit for example results in a substructure of two phenol units linked by a β-5 bond (Scheme 2). Such structures were also identified by GC/MS in the dimer fraction of the ‘DCM lignin oil’, shown in Fig. 1b (see structures 5, 7, 9, and 11). Unfortunately, only little information is available about C–H correlation signals in a HSQC spectrum of lignin samples solely comprising C–C interunit linkages. Predictions via ChemDraw of δC and δH chemical shifts of a range of lignin substructures (ESI,† Fig. S2 and S4) were therefore performed, and were sufficiently accurate, compared to literature values, to be helpful in the identification of structures and functionalities (ESI,† Table S3). A plot of the predicted δC–δH chemical shift pairs was made to simulate an artificial HSQC spectrum (ESI,† Fig. S3). To facilitate interpretation, a second plot was made indicating the regions in which the Cα–Hα, Cβ–Hβ and Cγ–Hγ correlation signals of the side-chains in the most important structures are present (ESI,† Fig. S4).
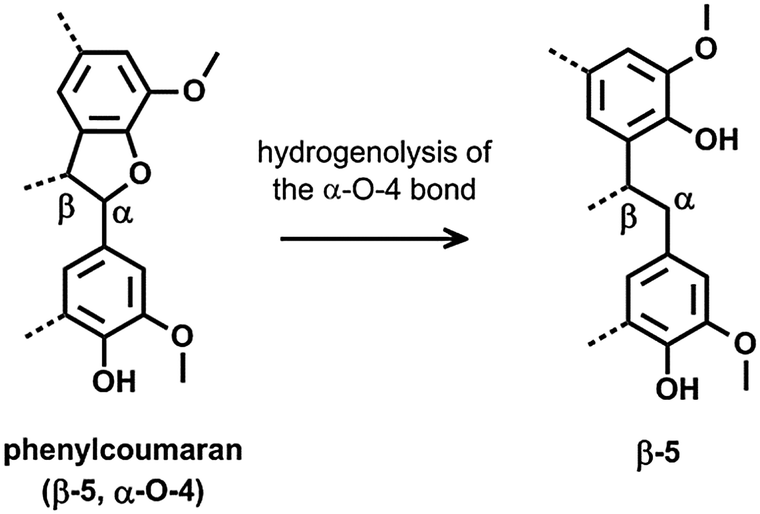 | ||
| Scheme 2 Reductive ring opening of phenylcoumaran (β-5, α-O-4), resulting in a dimeric structure with a β-5 linkage. | ||
In order to characterize the di- and oligomer fractions in the DCM lignin oil, the oil was first extracted with hexane to remove most of the apolar monomers, together with a small part of the short apolar oligomers like the dimers 1, 2, 3, 5, and 9 in Fig. 1b. The separation of the mono- from the oligomeric fraction is clearly demonstrated by GPC analysis (Fig. 2a) of the DCM lignin oil, the hexane extracted phase and the residue after hexane extraction (hexane residue). The corresponding HSQC spectrum of the hexane-extracted phase (ESI,† Fig. S5) shows the expected C–H correlation signals of the earlier identified monomers with mainly propyl and some ethyl side chains as the dominant signals. The HSQC spectra of the DCM lignin oil and the hexane residue, containing most of the oligomers, are displayed in Fig. 2b and c. The correlation signals of ethyl, propyl and propanol side-chains, the methoxy groups and the guaiacyl and syringyl structures are marked in colour. For each fraction, additional 1H-, 13C- and DEPT-NMR spectra are provided in the ESI,† Fig. S6–S8. In the side-chain region of the spectra (Fig. 2b top and c top), the Cα–Hα and Cβ–Hβ correlation signals of substructures with ether bonds are very small or even absent (region marked with α/βether), indicating that most of the ether bonds in β-O-4, phenylcoumaran, resinol and spirodienone structures have indeed been broken, in agreement with the GC/MS structural analysis. Instead, especially for the hexane residue, a number of signals were observed in the δC/δH 22–48/1.4–3.4 ppm region (marked with α/βnon-ether). According to the Chemdraw NMR, these signals can be assigned as Cα–Hα and Cβ–Hβ correlation signals of structures with β-5, β-1 and β–β C–C bonds, but without ether bonds. The δC/δH 58–68/3.2–4.5 ppm region further shows Cγ–Hγ correlation signals of linked (via C–C and ether bonds) or free propanol side chains (marked with γOH, linked and free). As more signals can be observed in this region than in the δC/δH 0.5–1.5/10–20 ppm region, corresponding to Cγ–Hγ correlation signals of linked or free propyl side chains (marked with γlinkedandfree), it is suggested that rather the propanol side-chains instead of the propyl units act as bridging groups between phenol units (like in the dimers 6, 7, and 10–12 in Fig. 1b). The propyl side-chains are mainly present as free side chains in the different compounds. These NMR results corroborate the earlier GC/MS structure analysis, in which propyl-type bridges were also not observed (Fig. 1b). Next to propanol groups, also ethyl side chains represent a significant fraction of the bridges between the phenol units in the oligomer fraction (see dimer structures 3, 4, 5, 8, and 9 in Fig. 1b). These ethyl-bridges are likely formed by Cβ–Cγ bond cleavage in original propanol-type linkages, which may proceed via retro-condensation or direct hydrogenolysis chemistry.59,107,108
The aromatic region of the HSQC spectra (Fig. 2b bottom and c bottom) clearly shows the correlation signals of free guaiacyl- and syringyl-units (marked in color). However, in their vicinity, a set of other signals was observed, especially in the hexane residue, indicative of compounds with varying chemical environments close to the guaiacyl and syringyl C–H entities. This is most likely due to C–C linkages between side-chains, between side-chains and aromatic rings or between aromatic rings. In the spectrum of the hexane residue, three signals were unambiguously assigned to C–H entities of guaiacyl units involved in β-5 and β-1 bonds, in accordance with the literature (Fig. 2c bottom, as the dimer structure 3–12 in Fig. 1b).109
The 2D HSQC NMR analysis showed a high content of hydroxyls in the phenolic oligomers of the hexane residue. Such a high content is of paramount importance to their potential use in the synthesis of e.g., polyurethanes and polyesters.93–96 The quantification of the OH-content, following a reported acetylation method using 1H-NMR analysis (ESI,† Fig. S9),96 demonstrates a remarkably high OH-content of 8.83 mmol g−1, corresponding to 1.47–1.87 OH-groups per phenolic unit, assuming an average phenol monomer MW in the oligomer structure of 166 to 212 g mol−1, respectively. Such a high OH-content thus corroborates the real potential of the produced polar lignin oligomers in several future polymer applications.
Comparison with different lignocellulosic feedstocks
Now that the nature and the benefits of the catalytic system have been demonstrated and the main products have been analyzed, it is equally important to comprehend the impact of the lignocellulose structure variability. Reaction parameters as in entry 5 (Table 1) were used for the comparison of feedstocks. First, the advantage of using a raw lignocellulose material instead of a separated lignin stream is demonstrated. Ethanol organosolv lignin from birch (EOL) was chosen because of its high-purity (viz. sulfur-free, low in residual carbohydrates and ash).37–40 Compared with the former results on birch wood, a low yield of phenolic monomers (3%) and dimers (6%) was obtained (Table 2, entries 1 and 2). Most likely, an altered chemical lignin structure,37–40i.e. a decreased content of ether bonds and an increased amount of C–C bonds compared to protolignin, is responsible for the limited degree of depolymerization with EOL. This result is further supported by GPC analysis of initial and reacted EOL, only showing a small shift towards smaller components (Fig. 3). For the production of high-value chemicals from an isolated lignin like the EOL used in this study, thermochemical depolymerization methods such as pyrolysis110,111 or chemocatalytic methods under more severe conditions49–55 seem more suitable.| Entry | Substrate | Phenolic product yieldse (C%) | Delignificatione (wt%) | Total sugar retentione (C%) | Total sugar polyol yieldf (C%) | ||
|---|---|---|---|---|---|---|---|
| PG + PSe | Total monomers | Dimers | |||||
| a Reaction conditions: 2 g of substrate, 0.3 g of 5% Ru/C, 40 mL of methanol, 3 h, 3 MPa H2 at RT (∼12 MPa at 523 K). b Reaction conditions: carbohydrate pulp fraction + Ru/C catalyst from step 1, 0.5 g of H4[Si(W3O10)4]·xH2O, 50 mL of water, 16 h, 463 K, 5 MPa H2 at RT (∼7 MPa at 463 K). c 1 g of ethanol organosolv lignin from birch (EOL), reaction conditions for the production of EOL are given in the ESI, Table S5.37 d Pine–spruce mixture. e A definition of the presented parameters is provided in the caption of Table 1 and in the ESI. PS/PG ratios are provided in the ESI, Table S4. f Yields are based on the amount of carbon in the obtained carbohydrate fraction (procedure in the ESI). | |||||||
| 1 | Birch | 42 (84) | 50 | 18 | 93 (80) | 81 | 74 |
| 2c | EOL birch | 1.7 (59) | 3 | 6 | — | — | — |
| 3 | Poplar | 33 (75) | 44 | 16 | 86 (65) | 85 | 52 |
| 4 | Softwoodd | 17 (83) | 21 | 15 | 56 (40) | 78 | 63 |
| 5 | Miscanthus | 12 (43) | 27 | 8 | 63 (56) | 85 | 59 |
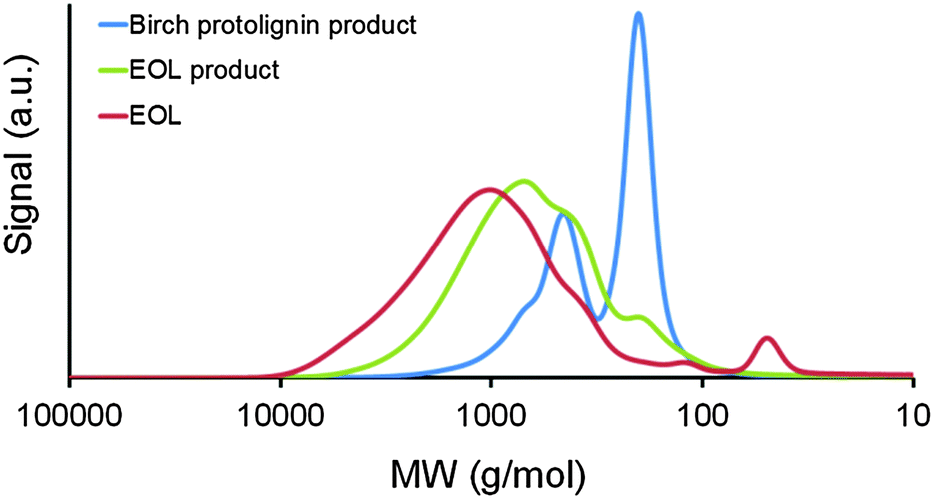 | ||
| Fig. 3 Gel permeation chromatograms of ethanol organosolv lignin (EOL) and the DCM lignin oils, obtained after the hydrogenolysis of EOL and birch sawdust (entries 1 and 2, Table 2). Polystyrene standards were used for calibration. | ||
The proposed biorefinery was further examined on three additional types of lignocelluloses: poplar (Populus × canadensis) as a second hardwood next to birch (Betula pendula), a sawmill rest fraction of pine and spruce representing softwoods, and miscanthus (Miscanthus giganteus) as a perennial grass. All three substrates are fast-growing crops, which are highly relevant in the context of biomass applications.11,26,112 The results are summarized in Table 2. The lignin and sugar compositions of each substrate are provided in the ESI,† Table S4, followed by a more detailed distribution of the monomer (ESI,† Table S5) and dimer products (ESI,† Fig. S10). A difference in product distribution and total monomer yield is immediately apparent.
The hardwoods, birch and poplar, resulted in the highest monomer and dimer yields, corresponding to a very high degree of delignification (Table 2, entries 1 and 3). Since lignin from hardwoods is typically composed of syringyl-(S) and guaiacyl-(G) units, both PS and PG are the dominant phenolic monomers here. In contrast, softwood lignin is mainly composed of G-units, while the lignin of grasses contains a mixture of H-(p-hydroxyphenyl), G- and S-units.32,45,113 Softwood lignin was clearly less susceptible to depolymerization with a moderate degree of delignification of 56%, yielding 21% monomers (Table 2, entry 4). With 15%, the dimer yield was however comparable with that of the hardwoods. As expected, the mono- and dimer products of softwood almost exclusively contained G-units, as opposed to the high S-content in the mono- and dimers from hardwoods (Fig. 4a and ESI,† Tables S4 and S5 and Fig. S10). Softwood conversion also led to higher amounts of 5–5 bonded dimers (ESI,† Fig. S10) in agreement with its nearly exclusive formation from G-moieties.
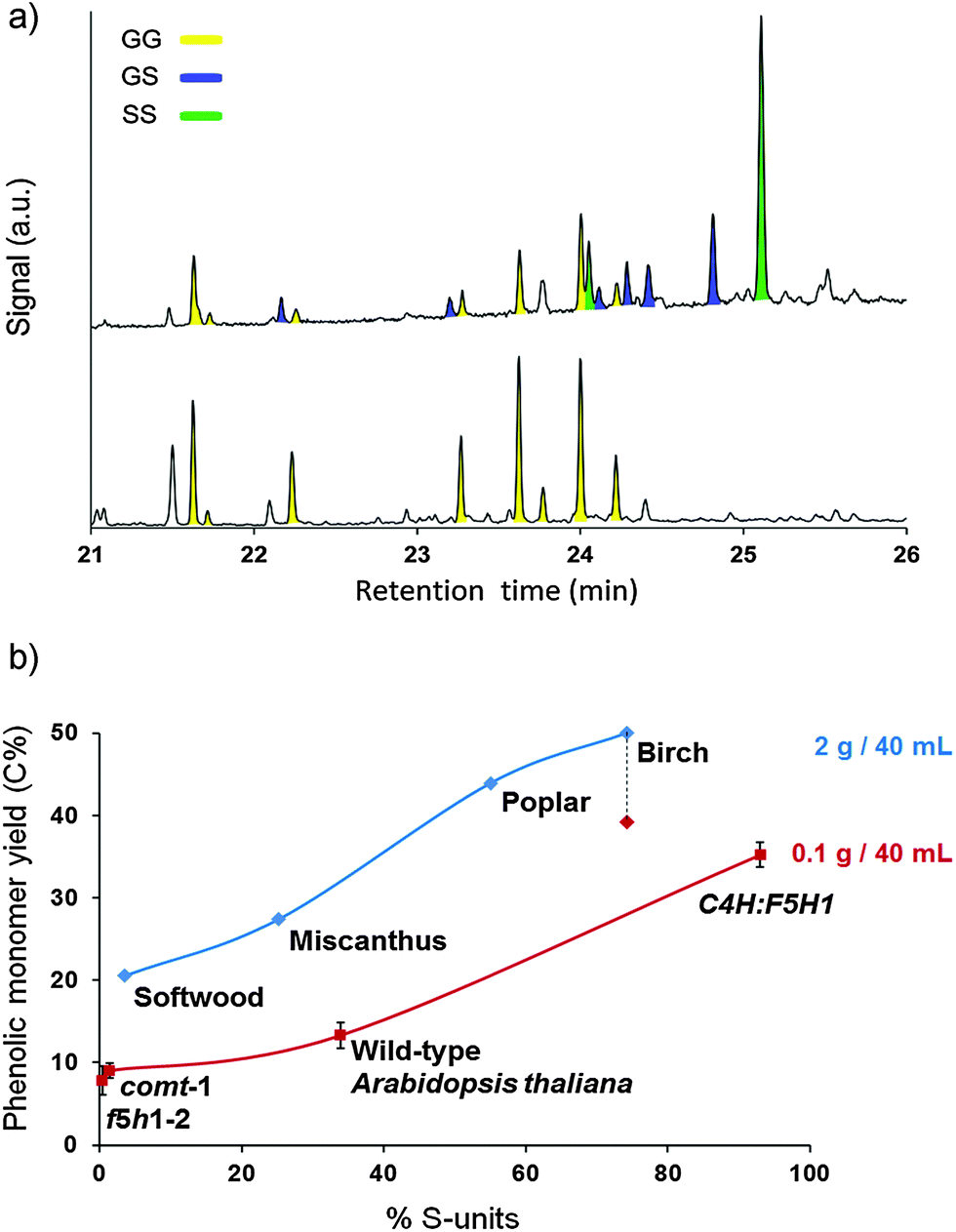 | ||
| Fig. 4 (a) GC of the trimethylsilylated dimer fraction from birch (top) and softwood (bottom). Each color represents signals of dimers with the same guaiacyl–syringyl composition. (b) Phenolic monomer yields as a function of the syringyl content (S%) in lignin for a set of lignocellulose substrates (diamonds) and Arabidopsis thaliana lines (squares). Reactions with Arabidopsis were downscaled and repeated 3 times, and the error bars indicate the standard deviation. For comparison, birch hydrogenolysis was also performed at the small scale (dotted line, red diamond). Reaction conditions: 523 K, 3 MPa H2 at RT (∼12 MPa at 523 K), 3 h, substrate (2 g/0.1 g), Ru/C (0.3 g/0.015 g), 40 mL of methanol. % S-units in lignocellulose substrates via 2D HSQC NMR of DCM lignin oil. % S-units in Arabidopsis samples via thioacidolysis (procedures in the ESI†). | ||
Finally, miscanthus grasses resulted in an intermediate degree of delignification as well as an intermediate monomer yield, with the formation of two specific phenolic monomers, assigned to the methanolysis and side-chain hydrogenation of p-coumaric and ferulic acid (ESI,† Fig. S11). Both acids are typically present in grasses.38,39,113
These results suggest a direct correlation between the lignin building block composition and its tendency to depolymerize into mono-, di- and oligomers. Fig. 4b shows the total phenolic monomer yield after hydrogenolysis of each feedstock as a function of its syringyl (S) content in the DCM lignin oil (ESI,† Table S4). This was determined by integrating the H2,6, G2 and S2,6 correlation signal in the aromatic region of the HSQC spectra.114 It becomes clear that a higher S-content in the DCM lignin oil is directly correlated with a higher phenolic monomer yield as well as a more efficient delignification (Table 2). This is in accordance with earlier results, obtained for Kraft pulping, in which a higher S-content in lignin also resulted in a more efficient wood delignification.115–117
To avoid the influence of species-specific properties rather than the type of lignin building block, similar feedstock tests were conducted on a single species. To that end, Arabidopsis thaliana (arabidopsis) genotypes were used that have lignin with a contrasting S-content. The Arabidopsis genes ferulate 5-hydroxylase1 (F5H1) and caffeic acid O-methyltransferase (COMT) are crucial in the biosynthesis of S-units. Consequently, the corresponding mutants, F5H1-2 and COMT-1, are rich in G-units and have only traces of S-units.70,118,119 On the other hand, over-expression of F5H1 (C4H:F5H1) resulted in plants with high S-content and low G-content.118,120,121 Finally, wild-type Arabidopsis plants have a G/S ratio of about 2/1.70 The lignin content and the monomer composition of each line, determined by thioacidolysis, are shown in the ESI,† Table S6. The monomer product distribution has been added in the ESI,† Table S7. Due to the small sample size of the Arabidopsis material, the hydrogenolysis process was downscaled from gram to sub-gram of feedstock loading and the reactions were performed in triplicate to ensure their reproducibility. Nevertheless, the same trend was obtained with the other natural lignocellulosic feedstocks, thus corroborating the previous assumption that a high S-content in lignin is imperative to obtain high yields to phenolic monomers. The lower absolute yield with the arabidopsis samples is likely due to a feedstock reactor loading effect. Indeed, lowering of the biomass weight (from 2 g to 0.1 g per 40 mL) for the reference reaction with birch wood also resulted in a lower phenolic monomer yield, values shifting from 50% to 39% (Fig. 4). The beneficial effect of S can be attributed to the fact that S-moieties lack free ortho-positions, and therefore they are unable to couple via 5–5 or β-5 C–C bonds. For this reason, a high S% results in a more accessible linear lignin structure with a lower percentage of stable C–C linkages.67,88
Thus, a comparison of the hydrogenolytic results of several lignocellulosic feedstocks emphasizes the importance of a smart feedstock choice. The results suggest that hardwoods and genetically engineered plants with a high S-content are the preferred substrates for the lignin-first biorefinery.
Valorization of carbohydrate pulps to chemicals
In the context of a sustainable and economically viable biorefinery, the valorization of protolignin can only be justified when the remaining carbohydrate pulp is also readily processable towards value-added products. As the recent literature already describes the simultaneous fermentation of hexoses and pentoses towards ethanol,122,123 an enzymatic conversion process can be envisioned in analogy with the next generation bio-ethanol industry. Powder X-ray diffraction patterns of the original birch wood and the isolated carbohydrate pulp after catalytic delignification, compared in the ESI,† Fig. S12, indicate the presence of crystalline cellulose in both samples. Although a larger set of parameters needs to be evaluated, this observation already suggests that paper production might be possible.Next to biofuels and paper, a third valorization option is the chemocatalytic conversion of the pulp towards high-value commodity chemicals.6,7,10,23,24,73,124,125 Here, the presence of the Ru/C catalyst in the carbohydrate pulp was exploited and a conversion towards sugar polyols, based on a bifunctional catalytic system from Geboers et al., was demonstrated.72 Hereto, tungstosilicic acid and water were mixed with an isolated pulp fraction and subsequently heated to 463 K under external H2 pressure. The hydrolytic power of the acid is used to convert cellulose into glucose and hemicellulose into mainly xylose and in smaller amounts into arabinose and mannose. The released sugars are then hydrogenated to their respective sugar alcohols in the presence of the Ru/C catalyst. Fig. 5 shows the obtained yields of sugar polyols as a function of the reaction time. The product distribution and the general chemical structure of the products are displayed as well. After 8 h, a sugar polyol yield of 70% was obtained, starting from the pulp of entry 5, Table 1 (dotted lines), despite the presence of residual lignin. Sorbitol, xylitol and their anhydrous analogues constitute the main product fraction. Mannitol and arabitol as well as the smaller polyols erythritol, threitol, glycerol, propylene glycol and ethylene glycol complete the remaining fraction. A maximal total yield of 74%, accompanied by a shift towards anhydrous products, was achieved at a longer reaction time of 16 h. The valorization potential of the obtained carbohydrate fraction from the other lignocellulose substrate was also demonstrated, resulting in somewhat lower yields between 52 and 63% of sugar polyols (entries 3–5, Table 2).
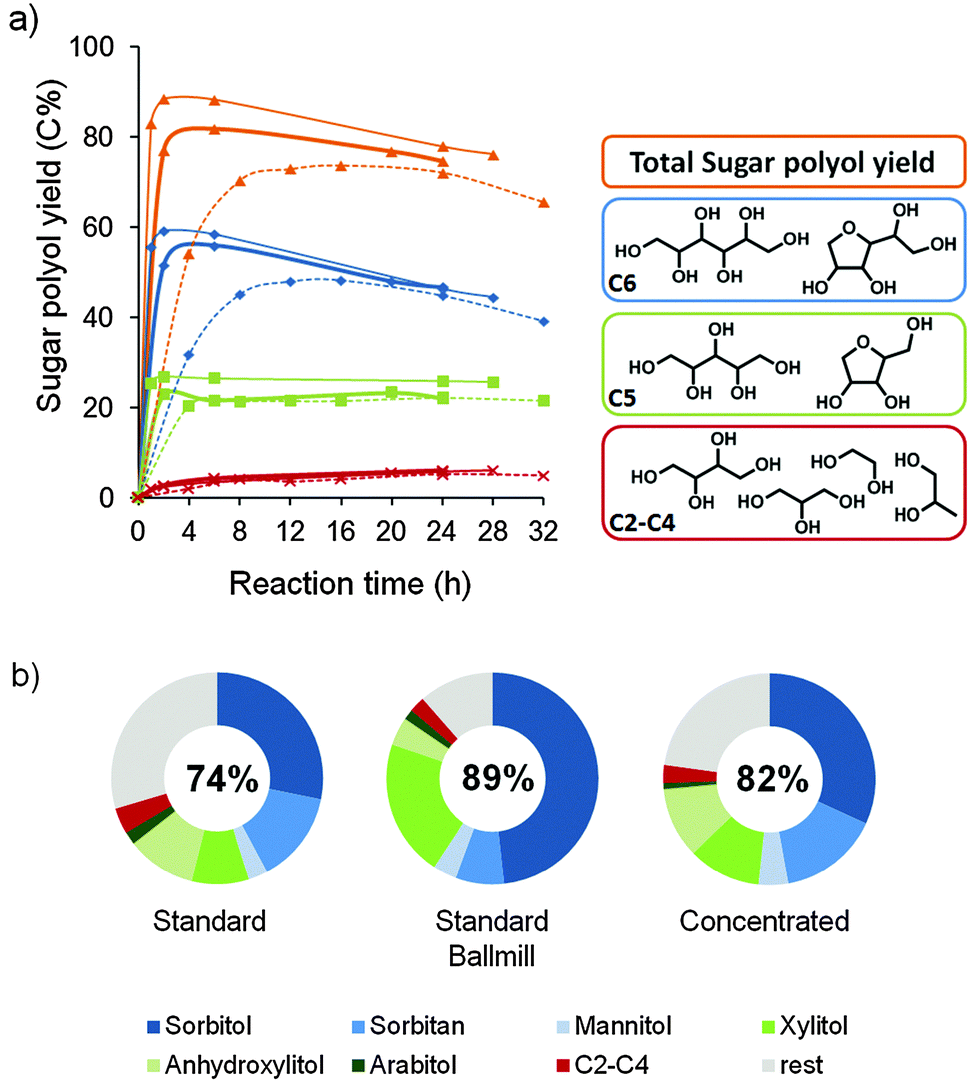 | ||
| Fig. 5 Chemocatalytic conversion of the carbohydrate pulp, obtained after birch hydrogenolysis (reactions Table 1, entries 5/10). Reaction conditions: 0.5/1.66 g of H4[Si(W3O10)4]·xH2O, 50 mL of water, 463 K, 5 MPa H2 at RT (∼7 MPa at 463 K). (a) Time profile of sugar polyol yield on the standard pulp (dotted lines), the ballmilled standard pulp (thin lines) and the pulp obtained after a 10 g concentrated reaction (thick lines), and (b) product distribution in near maximum polyol yield (left to right: after 8 h, 2 h and 2 h reaction), maximum polyol yield is given in the circle (left to right: after 16 h, 2 h, and 6 h). | ||
To further improve the selectivity towards xylitol and sorbitol, the reaction rate was enhanced by subjecting the carbohydrate pulp to a ballmill procedure (ESI†), prior to its catalytic conversion. This procedure is known to improve the reactivity of cellulose towards chemical reactions.126–129 The crystallinity of the carbohydrate pulp was altered, as illustrated by XRD in the ESI,† Fig. S12. The results are represented by the thin lines in Fig. 5. Already after 2 h, a maximal polyol yield of 89% was reached. The product distribution at that time also showed a great improvement in selectivity towards sorbitol and xylitol, which is directly related to the shortened reaction time.
As low biomass concentrations and the use of ballmilling might raise concerns with regard to the feasibility of the process at an industrial scale, an experiment with more concentrated carbohydrate pulp (untreated pulp of reaction 10, Table 1) was carried out, while keeping the Ru/C to acid ratio constant (thick lines, Fig. 5). In line with Geboers et al.,72,128 the use of higher pulp concentration resulted in a substantial increase of the conversion rate, already forming 77% polyols after 2 h, while also a higher maximal sugar polyol yield (82%) and a more selective production of sorbitol and xylitol were obtained. Instead of performing an energy intensive ball-milling procedure prior to catalysis, it is more advisable to work with a higher pulp concentration in water.
To conclude, the catalytic experiments clearly demonstrate the multiple use of Ru/C in the two subsequent reduction steps, as well as a catalytic valorization of the isolated pulp to polyols.
Brief economic assessment
To illustrate the economic valorization potential of the proposed biorefinery scheme, the future revenues from the conversion of birch wood are roughly estimated in Table 3. The calculations are based on the concentrated 600 mL reaction (entry 11, Table 1) for the lignin derived products and the concentrated carbohydrate conversion (thick lines, Fig. 5) for the sugar derived products, combined with current market information. Since the cost of transportation limits the volumes in which woody biomass can be economically collected, processing of such a geographically dispersed feedstock is best accomplished at moderate-sized facilities centered in regions where lignocellulose waste or crops is generated or easily transported to e.g., harbors. We here show that a process volume, similar to that of a medium sized paper mill (∼200 kton lignocellulose per year),130,131 can be sufficient to produce valuable sugar- and lignin-based chemicals in an economically profitable way at a realistic production scale for each product.| Starting material | Products after lignin-first biorefinery (kg) | Target molecules | Theoretical yieldd/selectivity (wt%) | Expected yielde (kg) | Current pricef (euros per ton) | Revenueg (euro) | Expected single plant capacityh (kton per year) | Conventional single plant capacityi (kton per year) |
|---|---|---|---|---|---|---|---|---|
| a 92% cellulose and 55% hemicellulose retention in the obtained carbohydrate fraction. b Liquid product yields from top to bottom: methyl xylose (27 C% of initial hemicellulose) and methyl acetate (quantitative conversion of hemicellulose acetyl groups), both including the weight of incorporated methanol (respectively 14 and 26 kg per ton birch); phenolic monomers (50 wt% of initial lignin), dimers (15–20 wt%) and oligomers (15–20 wt%). c Assuming the valorization of methyl xylose to xylitol. d Theoretical yields (wt product/wt reagent) account for: the production of 2 mol ethanol from 1 mol glucose; the addition or loss of H2, hydrolysis water (and the loss of methanol) in the production of sorbitol, sorbitans and xylitol from cellulose, hemicellulose (and methyl xylose); the removal of methoxy-groups to produce alkylated phenolic monomers. e The expected yield is obtained after multiplication of the product weight after lignin first biorefinery with the theoretical yield and the selectivity (e.g. 370 × 112 × 50 = 207 kg sorbitol from 1 ton birch). f Prices from ICIS (2013–2014) and industry, the price of the sorbitans was set the same as the price of sorbitol, yet likely results in a higher value when used for the production of emulsifying agents or converted to isosorbide, an interesting platform chemical,132 the price of lignin products was estimated based on an average price of phenol formaldehyde resins (1500–2000 euros), but alkylphenols can also be used in higher value applications such as aroma components. g Potential revenues are the product from the expected yield and the current price of each product. In parentheses, revenues were corrected for the price of incorporated H2 (∼10 euros per kg) or methanol (∼350 euros per ton). The hydrogenolytic fractionation was estimated to consume 5 kg of H2 per ton of lignocellulose, adding an additional cost of 50 euros per ton birch. h The expected production capacity of each product was based on an envisioned annual process volume of 200 kton of lignocellulose and was compared for each product with the annual production capacity of a ‘conventional’ production plant.133 i Estimation based on the Sumitomo phenolic resin production plant in Japan.134 | ||||||||
| 1 ton wood (birch, dry) 50–100 euros | ||||||||
| 400 kg of cellulose | 370a | Ethanol (benchmark) | 57/>85 | 180 | 550 | 99 | 37 | >100 |
| Sorbitol | 112/>50 | 207 | 700 | 145 (122) | 54 | 20–100 | ||
| Sorbitans | 100/>20 | 74 | >700 | >52 (43) | 7 | — | ||
| 210 kg of hemicellulose (acetyl free) | 116a | Xylitol | 115/>50 | 67 | 3000 | 200 (191) | 23 | 10–35 |
| 69b | Xylitolc | 93/>90 | 58 | 3000 | 174 (161) | 12 | 10–35 | |
| 30–40 kg of acetyl groups | 64b | Methyl acetate | — | 64 | 1350 | 80 (71) | 13 | 20 |
| 190 kg of lignin | 93b | Alkyl phenols | 70/>80 | 52 | >2000 | >104 (94) | 11 | — |
| 29b | Phenolic dimers | — | 29 | >1600 | >46 | 7 | 4–10i | |
| 41b | Phenolic oligomers | — | 41 | >1600 | >66 | 7 | 4–10i | |
Starting from a substrate cost of 50–100 euros per ton of birch, a significant profit on the total revenue was calculated. For example, with the here obtained yields of cellulose to sorbitol/sorbitans and hemicellulose to xylitol and methyl acetate, a rather conservative price estimation of about 600 euros can be generated from 1 ton of birch wood. This corresponds to an added value of 6 to 12 times the feedstock cost. When the roughly estimated revenues from lignin products like alkylated phenols as well as multifunctional di- and oligomers are taken into account, e.g. as a substitute resource of phenolic resins, the revenue for one ton of wood reach up to 800 euros. This theoretical exercise thus shows that lignin valorization can potentially amount to a 30–40% improvement in the economics of the presented lignocellulose biorefinery.
Though the presented values are based on optimized lab-scale experiments at the sub-liter scale, the results are promising and encouraging for future demonstration at the pilot scale. Such an exercise will allow an estimation of the installation and process costs, which next to the product valorization, will evidently play a key role in the success of a lignin-first biorefinery. In the near future, pilot scale experiments should deliver more accurate data.
Conclusion
A catalytic lignocellulose biorefinery process is presented, valorizing both polysaccharide and lignin components into a handful of chemicals. The selective delignification of lignocellulose in methanol through simultaneous solvolysis and catalytic hydrogenolysis, resulted in a lignin oil, rich in phenolic monomers next to di- and short oligomers. At the same time a processable carbohydrate pulp was obtained, with an almost quantitative retention of the original cellulose and a large fraction of the hemicellulose. Several key parameters, like temperature, reaction time, substrate particle size, reactor loading and the choice of solvent and gas, were examined as a first assessment of the techno-economic feasibility of the biorefinery process. The proposed biorefinery scheme was further investigated using other lignocellulose substrates, including genetically modified lines of Arabidopsis thaliana. The results led to a description of the preferred lignocellulose feedstock, being a feedstock rich in S-type lignin.More specifically, the reductive fractionation of birch sawdust in the presence of Ru/C resulted in a delignification up to 90%, 50% being converted into phenolic monomers and about 20% to a family of phenolic dimers, while retaining 80% of the carbohydrates in a processable pulp. Acetyl groups are completely removed from the hemicellulose backbone as methyl acetate, a relatively safe and environmentally friendly solvent and chemical precursor. The resulting methoxylated alkylphenols can be used in aroma components, anti-oxidants, resin productions, plasticizers, or as platform molecules for aromatics and other value-added chemicals.22,32,42,43,97,135 Their selective defunctionalization may also provide bio-based methanol17,32,68 to compensate solvent losses during biorefining.
Characterization efforts of the dimers in the lignin oil reveal compounds, containing at least two hydroxyls with valorization potential in the resin and polymer industry.97–100 Most dimers consist of phenol units which are p,p′- or o,p-coupled by an ethylene bridge, originating, respectively, from β-1 and phenylcoumaran lignin substructures. These ethylene bridges are either unsubstituted or contain a –CH2OH constituent. The oligomers are short, almost completely free of inter-unit ether bonds and structurally related to the dimers, as evidenced by GPC and 2D HSQC NMR.
Next to the lignin oil, a carbohydrate pulp is obtained, useful for the traditional pulp and paper industry or for biofuel production, but it can also be valorized into bio-based chemicals, like for example sorbitol, xylitol and sorbitans. High yields of these chemicals were achieved by chemocatalytic conversion of the carbohydrate pulp, while reusing the Ru/C catalyst from the hydrogenolysis reaction.
Processing lignocellulosic biomass in the proposed biorefinery thus results in 5 valuable product groups, being C5 and C6 polyols, methyl acetate, alkyl phenolic monomers and some larger phenolic oligomer products, which represent about 80% of the convertible fraction of the lignocellulosic feedstock.
To conclude a brief economic assessment was made as a first evaluation of the economic feasibility of the proposed biorefinery process. High revenues may be obtained and the added value of lignin valorization is shown to be substantial. Though the experiments were run at the lab-scale, they are encouraging to demonstrate the technology on a larger scale. To further improve the process economy, the use of cheaper catalysts, a smart catalyst regeneration as well as a continuous flow design are advised. Inspired by recent articles,63,65,66 additional research is now in progress to develop an inexpensive nickel-based biorefinery process in line with the ‘lignin-first’ concept.
Acknowledgements
This work was performed in the framework of an IAP-PAI network from BELSPO (Federal Agency) and was supported in part by the Multidisciplinary Research Partnership ‘Biotechnology for a Sustainable Economy’ (01MRB510W) of Ghent University and by the IWT-SBO project ARBOREF. S.V.d.B. and B.D. acknowledge the Institute for the promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen) for doctoral fellowships and W.S., R.V., S.-F.K. and T.R. acknowledge doctoral and postdoctoral fellowships from the Research Foundation – Flanders (FWO) and the FWO research project (G.0996.13N). B.L. acknowledges the Industrial Research Fund of KU Leuven. The authors kindly thank Floris van Tilburg from Cradle Crops for the miscanthus sample and Ecobois for the birch, poplar and softwood samples. The authors are also grateful to Karel Duerinckx for the NMR measurements, Joran Verspreet for his help with the sugar analysis and Joost Steverlynck for his assistance with GPC.Notes and references
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- Biorefineries – Industrial Processes and Products, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH, Weinheim, 2006 Search PubMed.
- Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, ed. C. E. Wyman, John Wiley & Sons, Chichester, 2013 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- S. Van De Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS PubMed.
- J. A. Geboers, S. Van de Vyver, R. Ooms, B. Op de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 Search PubMed.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- M. Dusselier, M. Mascal and B. Sels, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, Springer International Publishing, 2014, vol. 353, ch. 544, pp. 1–40 Search PubMed.
- B. Vanholme, T. Desmet, F. Ronsse, K. Rabaey, F. Van Breusegem, M. De Mey, W. Soetaert and W. Boerjan, Front. Plant Sci., 2013, 4, 174 Search PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS PubMed.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 CAS.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Roman-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 CAS.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 CrossRef CAS PubMed.
- V. K. Venkatakrishnan, W. N. Delgass, F. H. Ribeiro and R. Agrawal, Green Chem., 2015, 17, 178–183 RSC.
- R. Agrawal and N. R. Singh, US Pat., US 8217210 B2, 2012 Search PubMed.
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512 CrossRef CAS PubMed.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- J. J. Bozell, J. E. Holladay, D. Johnson and J. F. White, PNNL-16983, Pacific Northwest National Laboratory, Richland, Washington, 2007, p. 79.
- B. Op de Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morre, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240 CAS.
- S. B. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS.
- B. Yang and C. E. Wyman, Biofuels, Bioprod. Biorefin., 2008, 2, 26–40 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709 CrossRef CAS PubMed.
- M. D. Kaufman Rechulski, M. Käldström, U. Richter, F. Schuth and R. Rinaldi, Ind. Eng. Chem. Res., 2015 DOI:10.1021/acs.iecr.5b00224.
- M. Kaldstrom, N. Meine, C. Fares, R. Rinaldi and F. Schuth, Green Chem., 2014, 16, 2454–2462 RSC.
- R. Carrasquillo-Flores, M. Käldström, F. Schüth, J. A. Dumesic and R. Rinaldi, ACS Catal., 2013, 3, 993–997 CrossRef CAS.
- V. Molinari, M. Antonietti and D. Esposito, Catal. Sci. Technol., 2014, 4, 3626–3630 CAS.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- J. Li, G. Henriksson and G. Gellerstedt, Bioresour. Technol., 2007, 98, 3061–3068 CrossRef CAS PubMed.
- T. Q. Yuan, F. Xu and R. C. Sun, J. Chem. Technol. Biotechnol., 2013, 88, 346–352 CrossRef CAS PubMed.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS PubMed.
- P. Sannigrahi, Y. Q. Pu and A. Ragauskas, Curr. Opin. Environ. Sustainability, 2010, 2, 383–393 CrossRef PubMed.
- J. Wildschut, A. T. Smit, J. H. Reith and W. J. J. Huijgen, Bioresour. Technol., 2013, 135, 58–66 CrossRef CAS PubMed.
- S. Bauer, H. Sorek, V. D. Mitchell, A. B. Ibanez and D. E. Wemmer, J. Agric. Food Chem., 2012, 60, 8203–8212 CrossRef CAS PubMed.
- R. El Hage, N. Brosse, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2010, 95, 997–1003 CrossRef CAS PubMed.
- W. J. J. Huijgen, G. Telysheva, A. Arshanitsa, R. J. A. Gosselink and P. J. de Wild, Ind. Crops Prod., 2014, 59, 85–95 CrossRef CAS PubMed.
- E. Dorrestijn, L. J. J. Laarhoven, I. Arends and P. Mulder, J. Anal. Appl. Pyrolysis, 2000, 54, 153–192 CrossRef CAS.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef CAS PubMed.
- J. Lora, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. B. Gandini, Elsevier, Amsterdam, 2008, pp. 225–241 Search PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS PubMed.
- M. Balat and H. Balat, Appl. Energy, 2009, 86, 2273–2282 CrossRef CAS PubMed.
- R. J. A. Gosselink, E. de Jong, B. Guran and A. Abacherli, Ind. Crops Prod., 2004, 20, 121–129 CrossRef CAS PubMed.
- Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Oxford, 2008 Search PubMed.
- W. Xu, S. J. Miller, P. K. Agrawal and C. W. Jones, ChemSusChem, 2012, 5, 667–675 CrossRef CAS PubMed.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191–196 RSC.
- Q. Song, F. Wang and J. Xu, Chem. Commun., 2012, 48, 7019–7021 RSC.
- J. Zhang, H. Asakura, J. van Rijn, J. Yang, P. Duchesne, B. Zhang, X. Chen, P. Zhang, M. Saeys and N. Yan, Green Chem., 2014, 16, 2432–2437 RSC.
- R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 53, 7310–7315 CrossRef CAS PubMed.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS PubMed.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp, G. B. Hunsinger, M. A. Franden, C. W. Johnson, G. Chupka, T. J. Strathmann, P. T. Pienkos and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12013–12018 CrossRef CAS PubMed.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 CAS.
- J. R. Bower, L. M. Cooke and H. Hibbert, J. Am. Chem. Soc., 1943, 65, 1192–1195 CrossRef CAS.
- J. M. Pepper, C. J. Brounstein and D. A. Shearer, J. Am. Chem. Soc., 1951, 73, 3316–3319 CrossRef CAS.
- J. M. Pepper and Y. W. Lee, Can. J. Chem., 1969, 47, 723–727 CrossRef CAS.
- K. Sudo, D. J. Mullord and J. M. Pepper, Can. J. Chem., 1981, 59, 1028–1031 CrossRef CAS.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. T. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- Q. Song, F. Wang, J. Y. Cai, Y. H. Wang, J. J. Zhang, W. Q. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS PubMed.
- C. Li, M. Zheng, A. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 CAS.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- Y. Nakagawa, M. Ishikawa, M. Tamura and K. Tomishige, Green Chem., 2014, 16, 2197–2203 RSC.
- R. Vanholme, K. Morreel, J. Ralph and W. Boerjan, Curr. Opin. Plant Biol., 2008, 11, 278–285 CrossRef CAS PubMed.
- R. Van Acker, R. Vanholme, V. Storme, J. C. Mortimer, P. Dupree and W. Boerjan, Biotechnol. Biofuels, 2013, 6, 46 CrossRef CAS PubMed.
- R. Vanholme, I. Cesarino, K. Rataj, Y. Xiao, L. Sundin, G. Goeminne, H. Kim, J. Cross, K. Morreel, P. Araujo, L. Welsh, J. Haustraete, C. McClellan, B. Vanholme, J. Ralph, G. G. Simpson, C. Halpin and W. Boerjan, Science, 2013, 341, 1103–1106 CrossRef CAS PubMed.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- S. Van de Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549–1558 CrossRef CAS PubMed.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS PubMed.
- R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972–978 RSC.
- J. C. del Rio, J. Rencoret, A. Gutierrez, L. Nieto, J. Jimenez-Barbero and A. T. Martinez, J. Agric. Food Chem., 2011, 59, 11088–11099 CrossRef CAS PubMed.
- C. Lapierre, B. Pollet, B. Monties and C. Rolando, Holzforschung, 1991, 45, 61–68 CrossRef CAS.
- K. Saito and K. Fukushima, J. Wood Sci., 2005, 51, 246–251 CrossRef CAS PubMed.
- C. Gourson, R. Benhaddou, R. Granet, P. Krausz, B. Verneuil, P. Branland, G. Chauvelon, J. F. Thibault and L. Saulnier, J. Appl. Polym. Sci., 1999, 74, 3040–3045 CrossRef CAS.
- C. M. Courtin, H. Van den Broeck and J. A. Delcour, J. Chromatogr. A, 2000, 866, 97–104 CrossRef CAS.
- J. Snelders, E. Dornez, B. Benjelloun-Mlayah, W. J. J. Huijgen, P. J. de Wild, R. J. A. Gosselink, J. Gerritsma and C. M. Courtin, Bioresour. Technol., 2014, 156, 275–282 CrossRef CAS PubMed.
- R. B. Santos, E. A. Capanema, M. Y. Balakshin, H.-m. Chang and H. Jameel, J. Agric. Food Chem., 2012, 60, 4923–4930 CrossRef CAS PubMed.
- J. Rencoret, J. del Río, A. Gutiérrez, Á. Martínez, S. Li, J. Parkås and K. Lundquist, Wood Sci. Technol., 2012, 46, 459–471 CrossRef CAS.
- F. Kerton and R. Marriott, in Alternative Solvents for Green Chemistry (2), ed. F. Kerton and R. Marriott, The Royal Society of Chemistry, Cambridge, 2013, pp. 1–30 Search PubMed.
- K. Weissermel, Industrial Organic Chemistry, ed. K. Weissermel and H.-J. Arpe, Wiley-VCH GmbH & Co., Weinheim, Germany, 2003 Search PubMed.
- G. Roscher, Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH verlag GmbH, Weinheim, 7th edn, 2003, vol. 38, pp. 107–125 Search PubMed.
- H. Cheung, R. S. Tanke and G. P. Torrence, Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH verlag GmbH, Weinheim, 7th edn, 2003, vol. 1, pp. 209–239 Search PubMed.
- X. Zhou, J. Mitra and T. B. Rauchfuss, ChemSusChem, 2014, 7, 1623–1626 CrossRef CAS PubMed.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- H. Chung and N. R. Washburn, Green Materials, 2013, vol. 1, pp. 137–160 Search PubMed.
- X. Pan and J. Saddler, Biotechnol. Biofuels, 2013, 6, 12 CrossRef CAS PubMed.
- Y. Li and A. J. Ragauskas, J. Wood Chem. Technol., 2012, 32, 210–224 CrossRef CAS.
- H. Chung and N. R. Washburn, ACS Appl. Mater. Interfaces, 2012, 4, 2840–2846 CAS.
- A. Gandini and M. N. Belgacem, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. B. Gandini, Elsevier, Amsterdam, 2008, pp. 243–271 Search PubMed.
- B. G. Harvey, A. J. Guenthner, H. A. Meylemans, S. R. L. Haines, K. R. Lamison, T. J. Groshens, L. R. Cambrea, M. C. Davis and W. W. Lai, Green Chem., 2015, 17, 1249–1258 RSC.
- J. N. G. Stanley, M. Selva, A. F. Masters, T. Maschmeyer and A. Perosa, Green Chem., 2013, 15, 3195–3204 RSC.
- J. J. Cash, M. C. Davis, M. D. Ford, T. J. Groshens, A. J. Guenthner, B. G. Harvey, K. R. Lamison, J. M. Mabry, H. A. Meylemans, J. T. Reams and C. M. Sahagun, Polym. Chem., 2013, 4, 3859–3865 RSC.
- J. Rencoret, A. Gutierrez, L. Nieto, J. Jimenez-Barbero, C. B. Faulds, H. Kim, J. Ralph, A. T. Martinez and J. C. del Rio, Plant Physiol., 2011, 155, 667–682 CrossRef CAS PubMed.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, Materials, 2013, 6, 359–391 CrossRef CAS PubMed.
- R. John and L. L. Larry, Lignin and Lignans, CRC Press, 2010, pp. 137–243 Search PubMed.
- S. R. Ralph, J. Ralph and L. L. Landucci, NMR Database of Lignin and Cell Wall Model Compounds, http://ars.usda.gov/Services/docs.htm?docid=10491.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576–591 CAS.
- J. M. W. Chan, S. Bauer, H. Sorek, S. Sreekumar, K. Wang and F. D. Toste, ACS Catal., 2013, 3, 1369–1377 CrossRef CAS.
- J. Gierer, Wood Sci. Technol., 1985, 19, 289–312 CAS.
- Chemical Modification of Lignocellulosic Materials, ed. N. S. D. Hon, CRC Press, 1995, p. 69 Search PubMed.
- K. M. Torr, D. J. van de Pas, E. Cazeils and I. D. Suckling, Bioresour. Technol., 2011, 102, 7608–7611 CrossRef CAS PubMed.
- P. J. De Wild, W. J. J. Huijgen and R. J. A. Gosselink, Biofuels, Bioprod. Biorefin., 2014, 8, 645–657 CrossRef CAS PubMed.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4, 1629–1636 CrossRef CAS PubMed.
- J. S. Yuan, K. H. Tiller, H. Al-Ahmad, N. R. Stewart and C. N. Stewart, Jr., Trends Plant Sci., 2008, 13, 421–429 CrossRef CAS PubMed.
- A. U. Buranov and G. Mazza, Ind. Crops Prod., 2008, 28, 237–259 CrossRef CAS PubMed.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576–591 CAS.
- R. B. Santos, E. A. Capanema, M. Y. Balakshin, H.-M. Chang and H. Jameel, BioResources, 2011, 6, 3623–3637 CAS.
- P. C. Pinto, D. V. Evtuguin and C. P. Neto, Ind. Eng. Chem. Res., 2005, 44, 9777–9784 CrossRef CAS.
- R. B. Santos, H. Jameel, H.-m. Chang and P. W. Hart, BioResources, 2012, 8, 158–171 CrossRef PubMed.
- K. Meyer, A. M. Shirley, J. C. Cusumano, D. A. Bell-Lelong and C. Chapple, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6619–6623 CrossRef CAS.
- R. Vanholme, V. Storme, B. Vanholme, L. Sundin, J. H. Christensen, G. Goeminne, C. Halpin, A. Rohde, K. Morreel and W. Boerjan, Plant Cell, 2012, 24, 3506–3529 CrossRef CAS PubMed.
- R. Vanholme, J. Ralph, T. Akiyama, F. Lu, J. R. Pazo, H. Kim, J. H. Christensen, B. Van Reusel, V. Storme, R. De Rycke, A. Rohde, K. Morreel and W. Boerjan, Plant J., 2010, 64, 885–897 CrossRef CAS PubMed.
- J.-K. Weng, H. Mo and C. Chapple, Plant J., 2010, 64, 898–911 CrossRef CAS PubMed.
- M. Demeke, F. Dumortier, Y. Li, T. Broeckx, M. Foulquié-Moreno and J. Thevelein, Biotechnol. Biofuels, 2013, 6, 1–17 CrossRef PubMed.
- N. V. Sànchez and K. Karhumaa, Biotechnol. Lett., 2014, 1–12 Search PubMed.
- B. Op de Beeck, J. Geboers, S. Van de Vyver, J. Van Lishout, J. Snelders, W. J. J. Huijgen, C. M. Courtin, P. A. Jacobs and B. F. Sels, ChemSusChem, 2013, 6, 199–208 CrossRef CAS PubMed.
- T. Ennaert, J. Geboers, E. Gobechiya, C. M. Courtin, M. Kurttepeli, K. Houthoofd, C. E. A. Kirschhock, P. C. M. M. Magusin, S. Bals, P. A. Jacobs and B. F. Sels, ACS Catal., 2014, 754–768 Search PubMed.
- Q. Zhang and F. Jérôme, ChemSusChem, 2013, 6, 2042–2044 CrossRef CAS PubMed.
- M. Yabushita, H. Kobayashi, K. Hara and A. Fukuoka, Catal. Sci. Technol., 2014, 4, 2312–2317 CAS.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 13, 2167–2174 RSC.
- J. Hilgert, N. Meine, R. Rinaldi and F. Schuth, Energy Environ. Sci., 2013, 6, 92–96 CAS.
- E. Kilby and A. Crèvecoeur, Key Statistics, European Pulp and Paper Industry, CEPI, Confederation of European Paper Industries, Brussels, Belgium, 2013 Search PubMed.
- D. B. McKeever, The United States Woodpulp Industry, Department of Agriculture, Forest Service, Forest Products Laboratory, Madison, US, 1987 Search PubMed.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- S. Ravella, J. Gallagher, S. Fish and R. Prakasham, in D-Xylitol, ed. S. S. da Silva and A. K. Chandel, Springer Berlin Heidelberg, 2012, ch. 13, pp. 291–306 Search PubMed.
- C. I. Corp, in Advanced Materials in Japan, ed. C. I. Corp, Elsevier, Oxford, 1992, pp. 73–86 Search PubMed.
- W. Schutyser, S. Van den Bosch, J. Dijkmans, S. Turner, M. Meledina, G. Van Tendeloo, D. P. Debecker and B. F. Sels, ChemSusChem, 2015 DOI:10.1002/cssc.201403375.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee00204d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |