
Promoted C–C bond cleavage over intermetallic TaPt3 catalyst toward low-temperature energy extraction from ethanol†
Rajesh
Kodiyath
*a,
Gubbala V.
Ramesh
a,
Eva
Koudelkova
ab,
Toyokazu
Tanabe
ac,
Mikio
Ito
d,
Maidhily
Manikandan
ae,
Shigenori
Ueda
f,
Takeshi
Fujita
g,
Naoto
Umezawa
a,
Hidenori
Noguchi
dh,
Katsuhiko
Ariga
h and
Hideki
Abe
*a
aNational Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: KODIYATH.Rajesh@nims.go.jp; ABE.Hideki@nims.go.jp
bDepartment of Physical Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentska 573, CZ-532 10 Pardubice, Czech Republic
cKanagawa University, 3-27 Rokkakubashi, Yokohama, Kanagawa 221-8686, Japan
dGlobal Research Center for Environmental and Energy based on Nanomaterials Science (GREEN), National Institute for Materials Science (NIMS), Namiki 1-1, Tsukuba, 305-0044, Japan
eCrystal Growth Centre, Anna University, Chennai, Tamil Nadu 600-025, India
fSynchrotron X-ray Station at SPring-8, National Institute for Materials Science, 1-1-1 Kouto, Sayo, Hyogo 679-5148, Japan
gWPI Advanced Institute for Materials Research, Tohoku University, Sendai 980-8577, Japan
hWPI International Center for Material Nanoarchitectonics, National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
First published on 23rd January 2015
Abstract
Novel intermetallic TaPt3 nanoparticles (NPs) are materialized, which exhibit much higher catalytic performance than state-of-the-art Pt3Sn NPs for electrooxidation of ethanol. In situ infrared-reflection-absorption spectroscopy (IRRAS) elucidates that the TaPt3 NPs cleave the C–C bond in ethanol at lower potentials than Pt NPs, efficiently promoting complete conversion of ethanol to CO2. Single-cell tests demonstrate the feasibility of the TaPt3 NPs as a practical energy-extraction catalyst for ethanol fuels, with more than two times higher output currents than Pt-based cells at high discharge currents.
Broader contextThe vast populations of countries with burgeoning economies increasingly require environmentally-friendly day-to-day methods of energy generation. Most of the traditional methods rely on petroleum-combustion systems which produce significant quantities of exhaust, causing serious health conditions and air pollution. Ethanol fuels, which can be produced via biochemical routes including fermentation, are of growing importance to establish the desirable petroleum-free economy. However, ethanol fuels are still precluded from broad use except as an additive to petroleum for traditional combustion systems primarily because of the lack of efficient catalysts which promote complete oxidation of ethanol at low temperatures to fully extract chemical energy without heat loss. To address this, we report that intermetallic TaPt3 nanoparticles (NPs), which are materialized for the first time via a wet-chemistry route, exhibit substantially high performance toward the complete electro-oxidation of ethanol at room temperature. The electro-oxidation of ethanol fuels catalyzed by the TaPt3 NPs is accompanied by no toxic exhaust and is more energy-efficient than combustion systems, allowing populations to improve their quality of life and reduce the impact of their emissions on the global environment. |
Small organic molecules (SOMs) including formic acid and alcohols are becoming crucial as environmentally benign fuels for sustainable economy and society.1 In particular, ethanol is of interest because it contains the energetically dense C–C bond and can be produced via biochemical processes, with the possibility of carbon-neutral economy.2,3 However, ethanol fuels are not in widespread use except as an additive to petroleum for traditional combustion systems because of the lack of efficient catalysts to promote complete oxidation of ethanol near room temperature, without accompanying heat loss.3,4
Pt catalysts can efficiently promote complete electrooxidation of ethanol to carbon dioxide (CO2) involving C–C bond cleavage, but have poor long-term activity because of severe catalytic poisoning by one of the reaction intermediates, carbon monoxide (CO poisoning).3b,5 Alloying Pt with late-d-metals or metalloids including Ru, Fe, Co, Ni, Cu or Sn improves both the tolerance to CO poisoning and the catalytic activity toward C1-molecule fuels such as methanol and formic acid, but diminishes the catalytic activity toward complete electrooxidation of ethanol.3c,5b In addition, the traditional binary-alloy catalysts lack long-term stability in repeated use because of surface segregation: the counter elements of Pt readily leach out of the alloy or migrate into the bulk during long-term operation at high overpotentials.6
It is acknowledged that the improved catalytic performance of the late-d-metal–Pt alloys for the C1 molecules is attributed to reaction-active OH admolecules, which are formed over the electropositive late-d-metal atoms in aqueous media and preferentially oxidize reaction intermediates on neighboring Pt atoms (bi-functional mechanism).7 Early-d-metals including Ta may be rational alloy counterparts to Pt because Ta is much more electropositive and oxyphilic than the late-d-metals and can more favorably form OH species to promote the desired, complete electrooxidation of ethanol.8 Furthermore, the Ta–Pt alloys, when atomically ordered in an intermetallic phase of TaPt3, can act as a more stable catalyst than conventional late-d-metal alloys because of their large enthalpy of formation, e.g. ΔHf = −59.5 kJ mol−1 of Ta for TaPt3 compared with ΔHf = −13.6 kJ mol−1 of Fe for Fe–Pt alloys.9 However, synthesis of intermetallic TaPt3 catalysts in the desired forms of nanoparticles or porous materials has proven a challenge because of the extremely oxyphilic nature of Ta metal (Ta(0)).
In this communication, we report the first successful synthesis of intermetallic TaPt3 in the form of nanoparticles (TaPt3 NPs), and demonstrate their much enhanced activity toward electrooxidation of ethanol (EOR) in comparison with pure Pt NPs or state-of-the-art catalytic alloys, Pt3Sn NPs. The TaPt3 NPs also exhibit higher stability to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-times repeated EOR than Pt NPs or the alloy catalysts. Single-cell assembly comprising the TaPt3 NPs as the anode catalyst achieved higher power output than a reference cell comprising Pt catalysts. In situ IRRAS elucidated that the TaPt3 NPs efficiently catalyze C–C cleavage in ethanol at lower onset potentials than Pt NPs to promote complete conversion of ethanol to CO2.
000-times repeated EOR than Pt NPs or the alloy catalysts. Single-cell assembly comprising the TaPt3 NPs as the anode catalyst achieved higher power output than a reference cell comprising Pt catalysts. In situ IRRAS elucidated that the TaPt3 NPs efficiently catalyze C–C cleavage in ethanol at lower onset potentials than Pt NPs to promote complete conversion of ethanol to CO2.
The TaPt3 NPs were synthesized by co-reduction of metal precursors in dry diglyme (see ESI† for the details). Fig. 1a shows powder X-ray diffraction (pXRD) profiles of the as-prepared product and of the products annealed at different temperatures. Broad diffraction peaks are observed for the as-prepared product, indicating low crystallinity and/or small particle size. However, clear diffraction peaks appear when annealing the as-prepared product at high temperatures in a vacuum. The product annealed at 600 °C exhibits five diffraction peaks at 39.7°, 46.2°, 67.3°, 81.2° and 85.6°, corresponding to the 111, 200, 220, 311 and 222 reflections of an fcc-type structure. The product annealed at 600 °C consisted of alloy nanoparticles (Ta–Pt NPs) in which Ta and Pt were statistically distributed over the fcc lattice (Fig. 1b). The calculated lattice parameter, a = 3.932 Å, is larger than that of pure Pt, a = 3.920 Å, showing incorporation of larger Ta atoms in the Pt matrix to form the Ta–Pt phase. As the annealing temperature increases, additional peaks emerge from the intermetallic TaPt3 phase (at 800 °C and 900 °C, Fig. S1†). The pXRD pattern for the product annealed at 1000 °C is fully assigned to intermetallic TaPt3 (NbPt3-type, space group P21/m, a = 4.869 Å, b = 5.537 Å, c = 9.269 Å, β = 100.62°, Fig. 1b).10
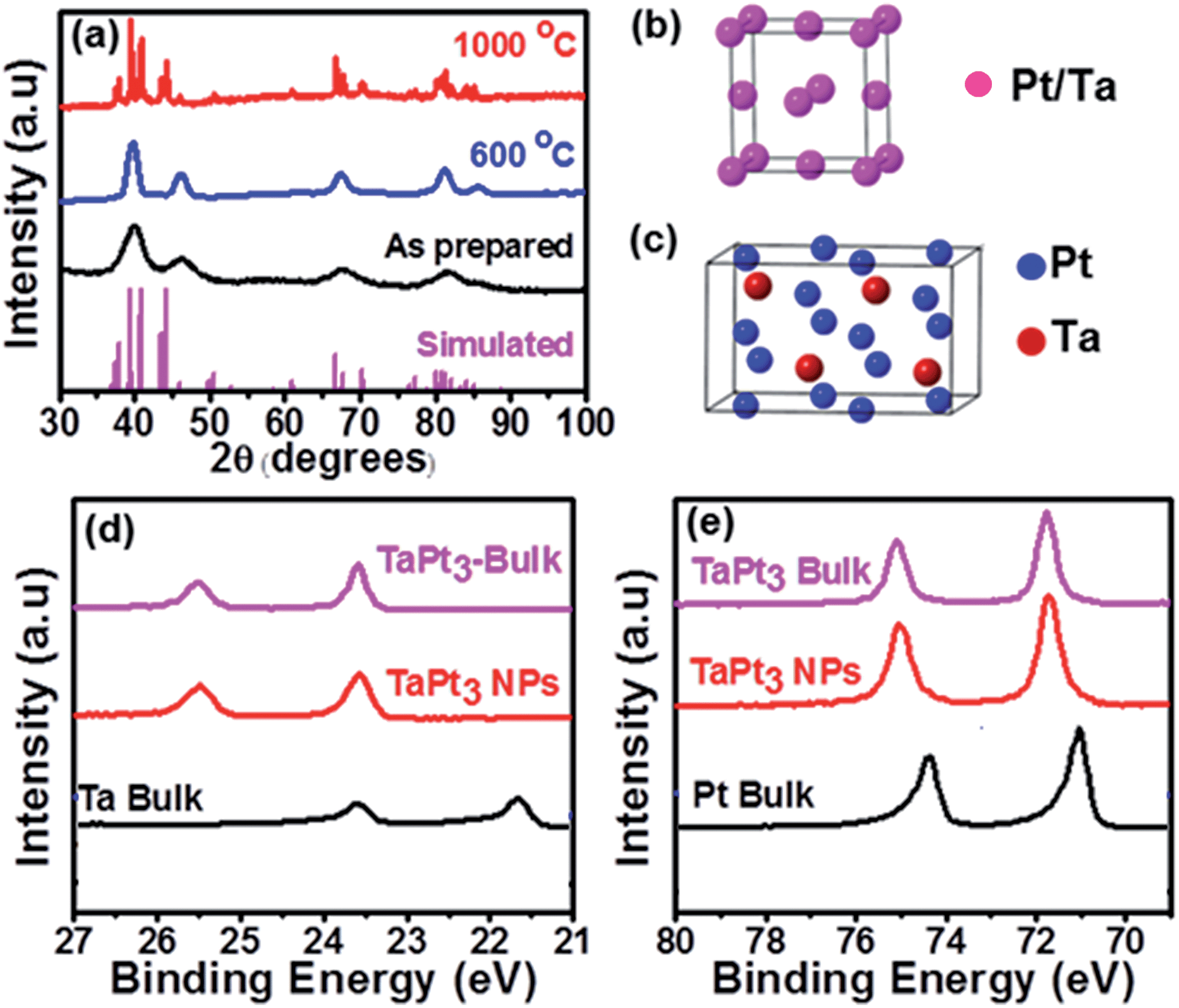 | ||
| Fig. 1 (a) pXRD profiles of Ta–Pt NPs (as-prepared and annealed at different temperatures). For comparison, the simulated pXRD pattern for intermetallic TaPt3 is presented at the bottom. Structural models showing atomic arrangements in the Ta–Pt NPs (b) and TaPt3 NPs (c). HX-PES spectra in the Ta 4f- (d) and Pt 4f- (e) regions for the TaPt3 NPs. HX-PES spectra for bulk Pt, bulk Ta and bulk TaPt3 are also shown as references. | ||
Hard X-ray photoemission spectra (HX-PES) of the TaPt3 NPs show clear emission peaks from the metallic Ta(0) (Fig. 1d). The binding energies of the Ta-core emissions from the TaPt3 NPs are consistent with the values for bulk TaPt3 (Fig. 1d). Note that the Ta-core emissions from the TaPt3 NPs are shifted to higher binding energies with respect to those from pure Ta metal, showing that the valence electrons of Ta atoms are used for formation of Ta–Pt bonds to diminish the screening of the nucleus charge (Table S1†). The formation of the Ta–Pt bonds also causes an increase in the binding energy of the Pt-core emissions from the TaPt3 NPs, with respect to those of pure Pt (Fig. 1e and Table S1†).
Fig. 2a shows that the prepared TaPt3 NPs were agglomerated to form a network structure. The STEM image of a TaPt3 NP and the corresponding FFT pattern indicate that the atoms in the TaPt3 NPs are ordered in the monoclinic NbPt3-type structure, as expected from pXRD (Fig. 2b and c).10 The EDS spectra confirmed that the Ta- to Pt atomic ratio in the TaPt3NPs is 1:3 (Fig. S2†). The compositional mapping over the TaPt3 NPs is presented in Fig. 2d–g. Importantly, the distributions of Pt (red) and Ta (green) are uniform over the TaPt3 NPs, as is evident from the composite image (Fig. 2g). The pXRD-, HX-PES- and TEM/STEM characterization demonstrated that annealing of the as-prepared product at 1000 °C leads to the desired, single-phase TaPt3 NPs.
 | ||
| Fig. 2 (a) TEM- and (b) STEM images of the TaPt3 NPs. The inset shows a simulated atomic arrangement in the TaPt3 NPs, viewed in the 〈201〉 direction. (c) FFT pattern obtained from (b). (d)–(g) EDS mapping of Pt (red), Ta (green) and the composite image for the TaPt3 NPs. | ||
The catalytic activity of the TaPt3 NPs toward the ethanol-electrooxidation reaction (EOR) was tested in comparison with that of the Pt NPs and Pt-alloy NPs (Fig. 3a and b). The electrooxidation currents were normalized with the electrochemical surface area (ECSA, ESI†).11 The electrooxidation currents normalized to the Pt loading weight (mass activity) are shown in ESI as Fig. S3.† The EOR peak-current density of the TaPt3 NPs is three times higher than that of the Pt NPs (Fig. 3a). The EOR onset potential of the TaPt3 NPs, +0.27 V, is 0.17 V lower than that of the Pt NPs, +0.44 V. The TaPt3NPs are superior to the state-of-the-art EOR catalyst, carbon-supported Pt3Sn NPs (Pt3Sn/C), in terms of the higher current density and the lower onset potential (Fig. 3b). One of the reasons for the enhanced activity of the TaPt3 NPs is likely to be the ability of oxyphilic Ta reacting with water to form Ta–OH, which can further oxidize the reaction intermediates of alcohols chemisorbed onto the neighboring Pt atoms.12
 | ||
| Fig. 3 (a) and (b) Cyclic voltammetry profiles for the TaPt3 NPs, Pt NPs and Pt3Sn/C in 1.5 M ethanol solution. (c) Variation of ECSA of the TaPt3 NPs and Pt NPs as a function of potential cycles. (d) I–V profiles and power density profiles of the TaPt3 NPs and Pt NPs, obtained at room temperature in 1 M ethanol solution. | ||
To investigate the long-term stability of the TaPt3 NPs, we repeated potential cycles from −0.17 V to +1.0 V in 0.5 M H2SO4 solution. Fig. 3c depicts the ECSA of the catalysts as functions of the number of cycles. The TaPt3 NPs show good stability to repeated potential cycles: the TaPt3 NPs retain 85% of the initial ECSA even after 10000 cycles. The increase in ECSA of the TaPt3 NPs, which is observed on the first few cycles, may be attributed to removal of contaminants from the catalyst surface, which is evident from the prominent hydrogen adsorption/desorption peaks (Fig. S4†).13 EDS mapping after the stability test further confirmed that the TaPt3 NPs retain their atomic arrangement and chemical composition even after the potential cycles (Fig. S5 and S6†). By clear contrast, the Pt NPs and Pt3Sn/C retain only <50% of the initial ECSA after 10000 cycles, because of agglomeration and/or leaching of NPs (Fig. S7†).14 The results of the stability test demonstrate that alloying of Pt with Ta significantly stabilizes the catalyst against harsh electrochemical conditions, possibly because the strong Ta–Pt bonds inhibit surface segregation and/or leaching during the catalysis.
We examined the feasibility of the TaPt3 NPs as a practical energy-extraction catalyst for ethanol fuels using a direct-ethanol-fuel-cell (DEFC) assembly (Fig. S8 and S9†). Fig. 3d shows the I–V profiles and power–density profiles obtained for the different cell assemblies, each of them loaded the TaPt3 NPs (TaPt3–DEFC) or the Pt NPs (Pt–DEFC).15 The TaPt3–DEFC and the Pt–DEFC exhibited similar performance at low discharge currents. However, both the output potential and power output of TaPt3–DEFC became higher than those of Pt–DEFC when the discharge current exceeded 0.15 mA cm−2 (Fig. S10†). The power density of the TaPt3–DEFC reached 215 μW at 1.6 mA cm−2, almost two times higher than that of the Pt–DEFC. This value was equal to 1/2 of the power density of a DEFC assembly comprising the state-of-the-art DEFC catalyst, Pt3Sn/C, even though the particle size of the TaPt3 NPs (>200 nm) was 100 times larger than that of the supported Pt3Sn NPs (2–3 nm) (Fig. S11 and Table S2†). Note that the TaPt3–DEFC, unlike Pt–DEFC, exhibited no degradation in power density at high current densities >1 mA cm−2, showing inhibited polarization effects at the electrodes and improved tolerance to CO poisoning.16 Indeed, CO-stripping tests (Fig. S12†) and density-functional-theory (DFT) calculations (Fig. S13†) verified that the CO-chemisorption to the TaPt3 surface is significantly weakened, resulting in improved CO-poisoning tolerance which is competitive to that of the Pt3Sn/C (Fig. S12†).
We further performed in situ IRRAS measurements to elucidate the EOR kinetics over the TaPt3 NPs (Fig. 4a–d). Fig. 4a shows a series of IRRAS spectra for the TaPt3 NPs in the CO-stretching region, acquired with increasing potentials (Fig. S14†). When the potential reaches +0.15 V, an anomaly becomes visible on the profile at 2070 cm−1. This anomaly corresponds to the stretching mode of the CO molecules which are generated through cleavage of the C–C bond in ethanol.17 The CO-stretching peak continuously grows with increasing potential up to +0.35 V, resulting in enhanced EOR current in the potential range from +0.15 to +0.35 V (Fig. 4a). The CO-stretching peak gradually diminishes when the potential exceeds +0.40 V and finally becomes invisible at +0.60 V, as a result of the full conversion of CO admolecules to CO2 (Fig. S10†). By contrast, CO evolves over the Pt NPs first at +0.25 V or higher potentials, which is consistent with the literature (Fig. 4b).18 The TaPt3 NPs are more active than Pt catalysts toward the C–C bond cleavage, in terms of the low-onset potential for the CO generation.
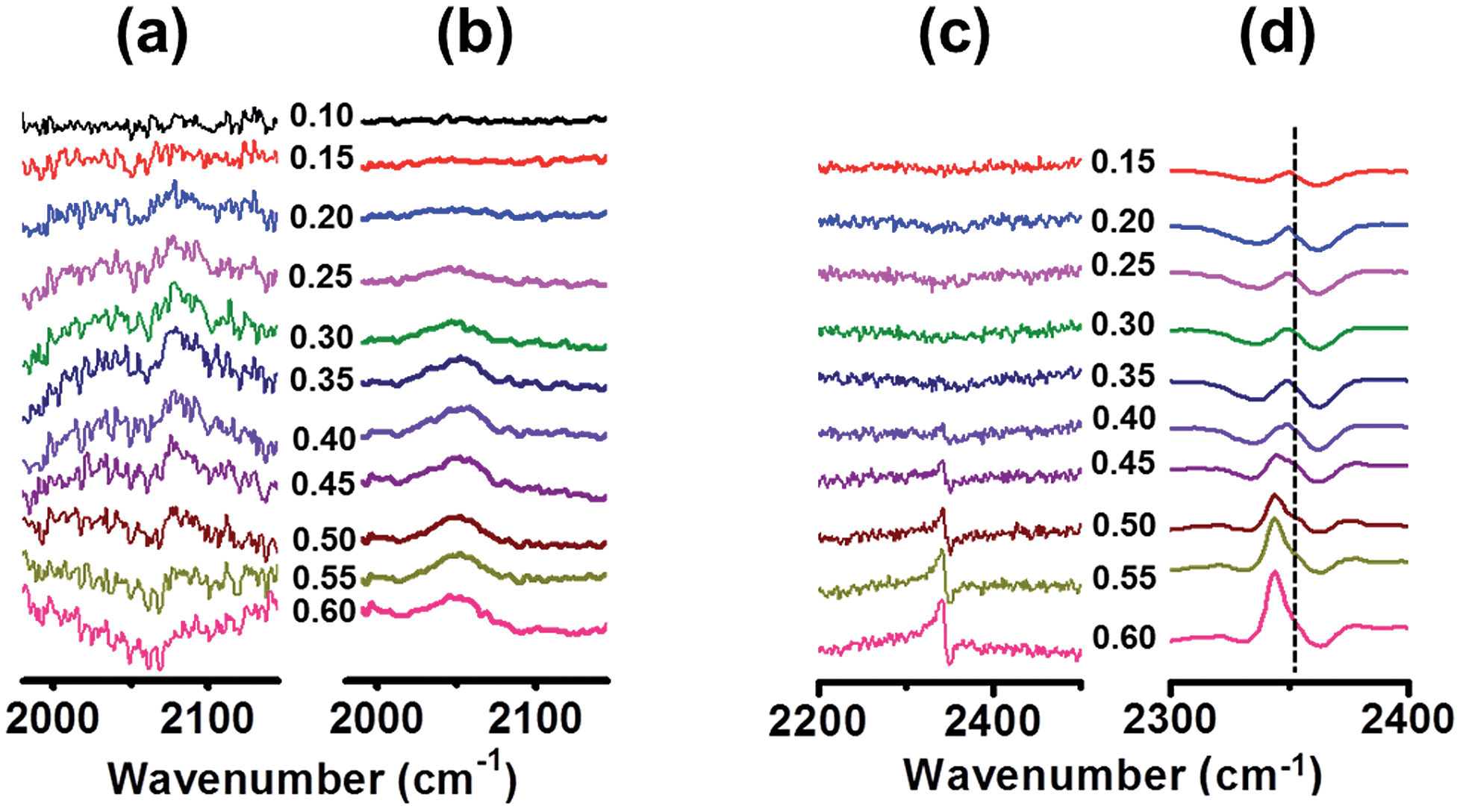 | ||
| Fig. 4 In situ IRRAS spectra (from 1900 to 2500 cm−1) over the TaPt3 NPs (a) and Pt NPs. (b) In situ IRRAS spectra (from 2200 to 2500 cm−1) over the TaPt3 NPs (c) and Pt NPs (d). The broken line in (d) situated at 2350 cm−1 corresponds to the peak position for atmospheric CO2. The vertical axes correspond to Δ absorbance (arbitrary unit). | ||
The TaPt3 NPs efficiently catalyze not only the C–C bond cleavage but also the complete conversion of ethanol to CO2. As shown in Fig. 4c, when the potential exceeds +0.35 V, the asymmetric stretching peak of CO2 appears at 2342 cm−1 over the TaPt3 NPs. The CO2-stretching peak over the Pt NPs first becomes visible at +0.45 V, indicating that the Pt NPs are less active toward the complete ethanol/CO2 conversion than the TaPt3 NPs (Fig. 4d, note that the IRRAS spectra contain background signals from atmospheric CO2 at 2350 cm−1).19 Importantly, CO2 starts to evolve over the TaPt3 NPs at a similar potential to that at which the CO-stretching peak starts to decrease, +0.35 V (compare Fig. 4a and c). In addition, the increase in the CO2-stretching peak at >+0.40 V (Fig. 4c) is proportional to the decrease in the CO-stretching peak (Fig. 4a). The enhanced ethanol/CO2 conversion over the TaPt3 NPs is primarily attributed to electrooxidation of the CO admolecules.
In conclusion, we have successfully developed a high-performance alcohol-electrooxidation catalyst, TaPt3 NPs. The TaPt3 NPs are superior to state-of-the-art binary alloy catalysts in terms of stability and catalytic activity toward electrooxidation of ethanol. In situ IRRAS measurements elucidated that the TaPt3 NPs efficiently promote both C–C bond cleavage in ethanol and complete conversion of ethanol to CO2. Moreover, single-cell tests demonstrated that the TaPt3 NPs act as a highly feasible catalyst for the desirable low-temperature energy extraction from ethanol fuels. The large particle size of the current TaPt3 NPs limits the figure of merit of TaPt3 NPs-based catalysts. However, the particle size of catalysts can be significantly reduced by dispersing the NPs over appropriate electroconductive supports, such as carbon nanoparticles or graphenes (Fig. S15†).20 The molecular kinetics of the promoted C–C bond cleavage and CO2 evolution over the TaPt3 surface are being investigated currently. The developed, high-performance TaPt3 NPs electrocatalyst could help to promote low-temperature energy management based on ethanol fuels, meeting present energy/environmental challenges.
Acknowledgements
This work was preliminarily supported by the JST PRESTO program, the Ministry of Education, Culture, Sports, Science and Technology (MEXT). The HX-PES measurements were performed under the approval of the NIMS Synchrotron X-ray Station (Proposal no. 2012B4609, 2013A4600, 2014A4603). The authors are grateful to HiSOR, Hiroshima University, and JAEA/SPring-8 for the development of HX-PES at BL15XU of SPring-8. We thank Dr Oruganti Anjaneyulu for his help in obtaining Fuel Cell Data.Notes and references
- (a) G. Ertl, H. Knözinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, VCH, Weinheim, 1997 Search PubMed; (b) M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed; (c) H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95 CrossRef CAS PubMed; (d) C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutance and J.-M. Leger, J. Power Sources, 2002, 105, 283 CrossRef CAS; (e) C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183 CrossRef CAS PubMed; (f) N. M. Aslam, M. S. Masdar, S. K. Kamarudin and W. R. W. Daud, APCBEE Proc., 2012, 3, 33 CrossRef CAS PubMed; (g) X. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124 CrossRef CAS PubMed.
- (a) M. Z. F. Kamarudin, S. K. Kamarudin, M. S. Masdar and W. R. W. Daud, Int. J. Hydrogen Energy, 2013, 38, 9438 CrossRef CAS PubMed; (b) A. Brouzgou, A. Podias and P. Tsiakaras, J. Appl. Electrochem., 2013, 43, 119 CrossRef CAS.
- (a) S. Song and P. Tsiakaras, Appl. Catal., B, 2006, 63, 187 CrossRef CAS PubMed; (b) C. Lamy, S. Rousseau, E. M. Belgsir, C. Coutanceau and J. M. Leger, Electrochim. Acta, 2004, 49, 3901 CrossRef CAS PubMed; (c) Z. F. Xu and Y. Wang, J. Phys. Chem. C, 2011, 115, 20565 CrossRef CAS PubMed.
- (a) R. Kavanagh, X. M. Cao, W. F. Lin, C. Hardacre and P. Hu, Angew. Chem., Int. Ed., 2012, 51, 1572 CrossRef CAS PubMed; (b) J. C. M. Silva, L. S. Parreira, R. F. B. De Souza, M. L. Calegaro, E. V. Spinace, A. O. Neto and M. C. Santos, Appl. Catal., B, 2011, 110, 141 CrossRef CAS PubMed.
- (a) F. H. B. Lima and E. R. Gonzalez, Electrochim. Acta, 2008, 53, 2963 CrossRef CAS PubMed; (b) Q. Wang, G. Q. Sun, L. H. Jiang, Q. Xin, S. G. Sun, Y. X. Jiang, S. P. Chen, Z. Jusys and R. J. Behm, Phys. Chem. Chem. Phys., 2007, 9, 2686 RSC.
- (a) S. Alayoglu, P. Zavalij, B. Eichhorn, Q. Wang, A. I. Frenkel and P. Chupas, ACS Nano, 2009, 3, 3127 CrossRef CAS PubMed; (b) D. Xu, Z. Liu, H. Yang, Q. Liu, J. Zhang, J. Fang, S. Zou and K. Sun, Angew. Chem., Int. Ed., 2009, 48, 4217 CrossRef CAS PubMed; (c) X. Ji, K. T. Lee, R. Holden, L. Zhang, J. Zhang, G. A. Botton, M. Couillard and L. F. Nazar, Nat. Chem., 2010, 2, 286 CrossRef CAS PubMed.
- (a) T. Iwasita and E. Pastor, Electrochim. Acta, 1994, 39, 531 CrossRef CAS; (b) F. Vigier, C. Coutanceau, F. Hahn, E. M. Belgsir and C. Lamy, J. Electroanal. Chem., 2004, 563, 81 CrossRef CAS PubMed.
- B. deB. Darwent, NSRDS-NBS, 31, 1970.
- A. R. Miedema, R. Boom and F. R. De Boer, J. Less-Common Met., 1975, 41, 283 CrossRef CAS.
- B. C. Giessen, R. H. Kane and N. J. Grant, Trans. Metall. Soc. AIME, 1965, 233, 855 CAS.
- Y. J. Gu and W. T. Wong, Langmuir, 2006, 22, 11447 CrossRef CAS PubMed.
- M. Krausa and W. Vielstich, J. Electroanal. Chem., 1994, 379, 307 CrossRef.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. Disalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81 CrossRef CAS PubMed.
- (a) Z. Chen, M. Waje, W. Li and Y. Yan, Angew. Chem., Int. Ed., 2007, 46, 4060 CrossRef CAS PubMed; (b) B. Y. Xia, H. B. Wu, Y. Yan and X. W. Lou, J. Am. Chem. Soc., 2013, 135, 9480 CrossRef CAS PubMed.
- J. P. Pereira, D. S. Falcao, V. B. Oliveira and A. M. F. R. Pinto, J. Power Sources, 2014, 256, 14 CrossRef CAS PubMed.
- J. Shim, J. Lee, Y. Ye, J. Hwang, S. K. Kim, T. H. Lim, U. Wiesner and J. Lee, ACS Nano, 2012, 6, 6870 CrossRef CAS PubMed.
- (a) Z. Peng, C. Kisielowski and A. T. Bell, Chem. Commun., 2010, 48, 1854 RSC; (b) C. Zhang, S. Y. Hwang, A. Trout and Z. Peng, J. Am. Chem. Soc., 2014, 136, 7805 CrossRef CAS PubMed.
- J. M. Leger, S. Rousseau, C. Coutanceau, F. Hahn and C. Lamy, Electrochim. Acta, 2005, 50, 5118 CrossRef CAS PubMed.
- (a) T. Herranz, M. Ibanez, J. L. Gomez de la Fuente, F. J. Perez-Alonso, M. A. Pena, A. Cabot and S. Rojas, ChemElectroChem, 2014, 1, 885 CrossRef CAS; (b) N. Erini, R. Loukrakpam, V. Petkov, E. A. Baranova, R. Yang, D. Teschner, Y. Huang, S. R. Brankovic and P. Strasser, ACS Catal., 2014, 4, 1859 CrossRef CAS; (c) A. Kowal, M. Li, M. Shao, K. Sasaki, M. B. Vukmirovic, J. Zhang, N. S. Marinkovic, P. Liu, A. I. Frenkel and R. R. Adzic, Nat. Mater., 2009, 8, 325 CrossRef CAS PubMed; (d) W. P. Zhou, M. Li, C. Koenigsmann, C. Ma, S. S. Wong and R. R. Adzic, Electrochim. Acta, 2011, 56, 9824 CrossRef CAS PubMed.
- (a) S. Guo and S. Sun, J. Am. Chem. Soc., 2012, 134, 2492 CrossRef CAS PubMed; (b) H. Qiu, X. Dong, B. Sana, T. Peng, D. Paramelle, P. Chen and S. Lim, ACS Appl. Mater. Interfaces, 2013, 5, 782 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, pXRD profiles, HX-PES spectra, EDS spectra, details of electrochemical measurements, and DFT simulations are provided. See DOI: 10.1039/c4ee03746d |
| This journal is © The Royal Society of Chemistry 2015 |