
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando electron paramagnetic resonance spectroscopy – formation of mossy lithium on lithium anodes during charge–discharge cycling
Johannes
Wandt
a,
Cyril
Marino
*a,
Hubert A.
Gasteiger
a,
Peter
Jakes
b,
Rüdiger-A.
Eichel
bcd and
Josef
Granwehr
*b
aTechnical Electrochemistry, Technische Universität München, Garching, Germany. E-mail: cyril.marino@psi.ch
bInstitute of Energy and Climate Research (IEK-9), Forschungszentrum Jülich, Jülich, Germany. E-mail: j.granwehr@fz-juelich.de
cRWTH Aachen University, Institute of Physical Chemistry, Aachen, Germany
dJülich Aachen Research Alliance (JARA), Section JARA-Energy, Aachen, Germany
First published on 16th February 2015
Abstract
The formation of mossy lithium and lithium dendrites so far prevents the use of lithium metal anodes in lithium ion batteries. To develop solutions for this problem (e.g., electrolyte additives), operando measurement techniques are required to monitor mossy lithium and dendrite formation during electrochemical cycling. Here we present a novel battery cell design that enables operando electron paramagnetic resonance (EPR) spectroscopy. It is shown that time-resolved operando EPR spectroscopy during electrochemical cycling of a lithium-metal/LiFePO4 (LFP) cell provides unique insights into the lithium plating/dissolution mechanisms, which are consistent with ex situ scanning electron microscopy (SEM) analysis. To demonstrate the viability of the operando EPR method, two cells using different electrolytes were studied. When using an electrolyte containing fluoroethylene carbonate (FEC) additive, a higher reversibility of the lithium anode and reduced formation of micro-structured (mossy/dendritic) lithium were observed.
Broader contextThe expanding market for portable electronic devices and the emerging electric transportation sector create an increasing demand for rechargeable high energy density batteries. One option to significantly increase the energy density would be the use of metallic lithium anodes due to the light weight and very low potential of lithium. Unfortunately, the use of lithium anodes with commonly used liquid aprotic electrolytes has so far been prevented by the formation of micro-structured lithium during battery charge – so called dendritic or mossy lithium – which both consumes active lithium and liquid electrolyte and also poses a serious safety hazard. In this study, we present electrochemical operando EPR (electron paramagnetic resonance) spectroscopy as a novel tool to study the formation of lithium dendrites in real-time and under realistic conditions. As a case study, we investigate the extent of the formation of micro-structured lithium in lithium/lithium iron phosphate cells in the absence or presence of fluoroethylene carbonate additive, which is known to reduce dendrite formation. The results provided by a detailed EPR line shape analysis and supported by ex situ SEM images clearly show that operando EPR spectroscopy is a powerful diagnostic technique, yielding valuable information that is not accessible by commonly used microscopic techniques. |
Introduction
Lithium metal would be an ideal battery anode material due to its high specific capacity (3860 mA h g−1) and its very low potential (−3.04 V vs. standard hydrogen electrode), thus enabling outstanding gravimetric energy densities. A lithium metal anode combined with lithium- and manganese-rich layered metal oxide cathodes would enable an energy density increase of Li-ion battery systems by 50% to ≈300 W h kg−1system compared to currently used graphite anodes.1 The development of a reliable lithium anode is especially critical for new cell chemistries, such as Li–Sulfur and Li–Air, in order to realize their potential gravimetric energy densities.1–4 According to Sion Power, a leading developer of Li–S batteries, the “generation of porous ‘mossy’ Li deposits” is one of “two major mechanisms limiting Li–S cycle life”.5 A variety of strategies to “master the Li–electrolyte interface”,6 ranging from ceramic blocking layers7–9 and advanced charging procedures10 to organic or inorganic electrolyte additives,11,12 have been investigated with only limited success so far.13Despite significant scientific effort and large investments of battery makers over the last 40 years, the lithium metal anode in combination with liquid electrolytes has not been successfully commercialized14 (the only exception are liquid solvent free lithium–polymer batteries). This is mainly due to the formation of micro-structured (mossy/dendritic) lithium during battery charge, which reduces cell life due to irreversible electrolyte consumption15 and also poses a serious safety threat due to the possibility of internal cell shortening.16,17
These persistent problems led to the use of graphite as anode material instead of metallic lithium by Sony in 1991, paving the way for modern Li-ion battery technology.18,6 Graphite can reversibly intercalate and deintercalate lithium ions, thus enabling excellent cycle life over several thousand cycles.19 However, the intercalation potential for lithium ions into graphite is very close to the Li/Li+ potential, which can cause plating of metallic lithium on the graphite particles if the battery is charged at low temperatures or with high C-rates.20–26 Lithium plating on graphite usually takes place in a dendritic morphology and therefore impairs both cell life and safety due to the abovementioned reasons.25,27 Accordingly, researchers have tried to find electrolyte compositions and additives to prevent or at least reduce lithium plating and dendrite formation.28–30 Since lithium plating on graphite is partially reversible at open circuit conditions, detailed studies of dendrite formation require operando rather than in situ/ex situ techniques.
Most studies on lithium plating and dendrite formation are based on microscopy techniques. Optical spectroscopy has been successfully applied in both in situ31,32 and operando33,34 setups but is limited by the low resolution. In contrast, scanning electron microscopy (SEM) shows better resolution but is restricted to polymer electrolyte cells for in situ35 or operando experiments.16 At the current stage, there are only very few analytical techniques available that give quantitative or semi-quantitative information about the occurrence of micro-structured lithium during cell cycling under operando conditions. In 2010, Bhattacharyya and co-workers introduced electrochemical in situ and operando7Li-NMR spectroscopy,36 which has since then also been used by other groups.37–39 NMR spectroscopy can provide valuable insights as exemplified by the operando imaging of lithium dendrites by Chandrashekar et al.37 Still, one disadvantage of operando NMR is the low spectral resolution due to the inability to use magic angle spinning. As another analytical approach that is applicable during electrochemical cell cycling, operando electron magnetic measurements have recently been presented by Gershinsky and co-workers for the investigation of conversion materials such as FeSb2.40
In this work we propose to use operando electron paramagnetic resonance (EPR) spectroscopy as a new analytical technique for the semi-quantitative determination of mossy or dendritic lithium, henceforth referred to as “micro-structured lithium” (as suggested by Bhattacharyya and co-workers36). With EPR spectroscopy, the detection of micro-structured lithium on the anode surface is based on the EPR resonance caused by the conduction electrons in metallic lithium, whereas Li+-ions are EPR inactive. In comparison to NMR spectroscopy, EPR is expected to show a higher sensitivity per unit volume due to the higher gyromagnetic ratio of electron spins and a higher selectivity for dendrite detection due to the roughly ten times smaller skin depth of EPR microwaves in comparison to NMR radiowaves.
A new electrochemical cell setup suitable for operando EPR spectroscopy during electrochemical cycling is introduced. We investigate the morphological changes of a lithium metal anode during cycling of a Li/LiFePO4 (LFP) cell as case study to demonstrate the capabilities of electrochemical operando EPR spectroscopy. First, our new cell design is validated by a comparison of the electrochemical performance with a standard cell design. Next, the evolution of the EPR resonance of metallic lithium is analyzed using a standard electrolyte with or without fluoroethylene carbonate (FEC) additive, which is known to reduce lithium dendrite formation.29,30,41 The results obtained by operando EPR spectroscopy, supported by a detailed EPR lineshape analysis, are confirmed by ex situ scanning electron microscopy (SEM) images.
Materials and methods
Operando cell design
Fig. 1 shows a schematic drawing of the operando EPR cell. A coaxial cell design for electrochemical EPR spectroscopy has previously been suggested by Zhuang et al.42 We use a tubular design with a concentric arrangement of the cell components to meet the geometric constrains of the EPR spectrometer. A central copper wire (1 mm diameter, 99.9%, Alfa Aesar) serves as anode current collector and is wrapped with lithium metal foil (450 μm thickness, 25 mm length, 99.9%, Chemetall, Germany). PTFE heat shrink tubes (Deray PTFE, Autec, Germany) confine the lithium anode on both sides to prevent a short circuit. A glass–fiber separator (250 μm thickness, 40 mm length, glass microfiber filter 691, VWR) is rolled around the lithium anode. The LFP electrode (20 mm length), which is coated directly onto a Celgard separator (C480), is assembled with the separator facing inwards. A helical aluminum wire (0.5 mm diameter, 99.999%, Alfa Aesar) serves as cathode current collector. The cell is enclosed by a quartz glass tube (6 mm outer diameter, 0.5 mm wall thickness, QSIL, Germany). The combination of an electrode coated directly onto the porous separator and the aluminum wire as current collector ensures ionic conduction between anode and cathode without shielding off the microwaves. Electrolyte (500 μL) is added directly onto the glass–fiber separator inside the glass tube using an Eppendorf pipette. The cell is sealed on both ends with capillary wax and dried inside the glove box for several hours. | ||
| Fig. 1 Design of electrochemical operando EPR cell. The surrounding quartz glass tube is omitted for clarity. | ||
Electrode and electrolyte preparation
The LFP electrodes were prepared by gap bar coating using an ink composed of 80 wt% carbon coated LFP (1 μm, 2.5 wt% C, 15 m2 g−1, Clariant), 10 wt% Super C65 (Timcal), 10 wt% PVDF (Kynar HSV900, Arkema) and N-methyl pyrrolidone (99.5%, Sigma-Aldrich) as solvent. The ink was prepared by homogenizing all components in a planetary mixer (ARV-310CE, Thinky) for 20 minutes at 2000 rpm. It was coated either onto a Celgard C480 separator for EPR experiments (250 μm wetfilm thickness, loading: ≈3.9 mgLFP cm−2) or onto an aluminum foil for SEM experiments (350 μm wet film thickness, loading: ca. 5.8 mgLFP cm−2). After coating and solvent evaporation at 60 °C, rectangular electrodes (20 mm × 7 mm) were cut out with a scalpel or round electrodes (10 mm diameter) were punched out with a precision punch (Hohsen, Japan). The electrodes were dried in dynamic vacuum at 95 °C for 12 hours in a glass oven (Büchi, Switzerland). The different LFP loadings for the EPR and SEM experiments are necessary to achieve the same current density (in mA cmLi−2) on the lithium anode due to the concentric cell setup of the operando EPR cell. As electrolyte, 1 M lithium hexafluorophosphate (LiPF6) in a mixture of ethylene carbonate (EC) and ethyl methyl carbonate (EMC) (LP57, BASF) was used either as received or after the addition of 10 wt% of FEC (Solvay Chemistry). Cell assembly and preparation of SEM samples was carried out inside an argon-filled glove box (MBraun, O2 and H2O less than 0.1 ppm).Electrochemical testing
Electrochemical testing was done with a VMP3 potentiostat (Bio-Logic, France). After at least 12 hours at open circuit, the cells were cycled between 2.0 and 4.2 V vs. Li/Li+. The cells were charged in a CCCV mode (CV step until current dropped to 20% of current for C/5 charge) and discharged galvanostatically with the following cycling procedure: (i) one C/5 cycle (≡0.23 mA cmLi−2); (ii) one 1C cycle (≡1.15 mA cmLi−2); (iii) six 3C cycles (≡3.45 mA cmLi−2); (iv) one C/1.5 cycle (≡0.76 mA cmLi−2). The C-rate is calculated with respect to the nominal capacity of the LFP electrode and the current density is normalized to the lithium surface area. Notice that the current density (in mA cm−2) is higher for the lithium electrode than for the LFP electrode due to the concentric cell design.SEM experiment
SEM images were recorded on a JEOL JCM-6000 SEM (secondary electron imaging, 15 kV accelerating voltage). The lithium electrodes (17 mm diameter) for the SEM analysis were cycled against LFP electrodes (10 mm diameter) in a custom-made cell described previously.43 An additional Celgard separator was placed between the lithium anode and the glass–fiber separator to prevent damaging the micro-structured lithium surface by the removal of the glass–fiber separator, since it typically strongly sticks to the lithium surface after very few cycles. In contrast, the Celgard separator does not get penetrated by lithium filaments within the limited number of cycles used in this study, so that it can be removed without damaging the lithium anode surface. This is confirmed by the absence of hydrogen gas evolution upon immersing the removed Celgard separator into water; the same observation was reported by Gallus et al.44 After cycling, the cells were transferred back into the glove box and the lithium electrode was harvested. The lithium electrodes were washed twice with 500 μL dimethyl carbonate (DMC) in order to remove LiPF6 and EC residues and dried in vacuum for 15 minutes. The cells were transferred into the SEM using a conductive carbon tape (Plano, Germany) and a sample holder that limited the contact to ambient atmosphere to a few seconds; a short air contact has been reported to have no impact on the microscopic structure of dendritic lithium surfaces.35 To determine the thickness of the micro-structured lithium layer, a special sample holder was used, in which the lithium electrode was bent down by 90°.EPR spectroscopy
EPR spectra were recorded on a Bruker ElexSys E-540 continuous-wave (cw) X-band EPR spectrometer, equipped with an ER 4108 TMHS resonator operating at 9.897 GHz. Microwave power was set to 1.00 mW. Sweeps were performed with 5 mT width and a center field of 353.2 mT. As is common practice, the cw EPR spectra were recorded as first derivatives of the signal with respect to the external magnetic field B0. The field modulation frequency was set to 100 kHz, and the modulation amplitude was 0.1 mT. EPR spectra were recorded continuously during cell cycling. Each spectrum took 1 min to record, which represented the temporal resolution of the two-dimensional time-resolved EPR data.The EPR signal caused by Fe3+ is generally much wider than the signal from metallic Li.45,46 It could be suppressed by using a relatively low modulation amplitude and by performing a zeroth order baseline correction.
EPR data analysis
A quantitative analysis of EPR spectra from conduction electrons in metals needs to take the diffusion of electrons in and out of the skin of the conductor into account.47 Since the phase of the microwave field used to excite the spins changes as a function of depth into the conductor, the shape of the EPR resonances varies as a function of the geometry and thickness of the conductor. This complicates the quantification of the EPR signal, since the number of contributing spins is not simply proportional to the area under the integrated experimental spectrum, as conventionally assumed for the determination of spin concentrations.48 An exact quantification requires either a calibration with a set of samples that cover the full range of lineshapes observed experimentally or a theoretical model that can be used to fit the experimental resonances. Nonetheless, as long as the signal is dominated by a single resonance and the lineshape does not change drastically, integration of the experimental first derivative spectrum, followed by the calculation of the area under the obtained spectrum, provides at least a relative measure of changes in the number of spins contributing to the signal. If a lineshape change occurs quickly, the continuity of the signal amplitude can be used as a qualitative test whether a complete recalibration is required. To minimize systematic errors caused by deviations of the cell positioning and orientation in the EPR resonator, the amplitude was normalized to the amplitude of the pristine cell at the beginning of electrochemical cycling. Besides its simplicity, this method has the advantage that it is very robust.Theoretical expressions for the lineshape of conduction EPR signals have been derived for flat plates of various thicknesses d47 and for spherical metal particles with different radii a.49 For porous or micro-structured lithium, no theoretical lineshape and amplitude models are currently available. Since the expressions for flat plates and for spherical particles agree with each other in the limit of thicknesses and radii that are either very small or very large compared with the skin depth δ of the metal, we use an empirical approach, validated by comparison with SEM images at selected positions within the cycling protocol, to relate EPR lineshapes with the dimension of the structures of deposited lithium.
For conductor structures that are much smaller or much bigger than δ, the resonance can be represented as a superposition of the absorptive part χ′′ and the dispertive part χ′ of the complex susceptibility,50
χ = χ′′cos(ϕ) + χ′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(ϕ) sin(ϕ) | (1) |
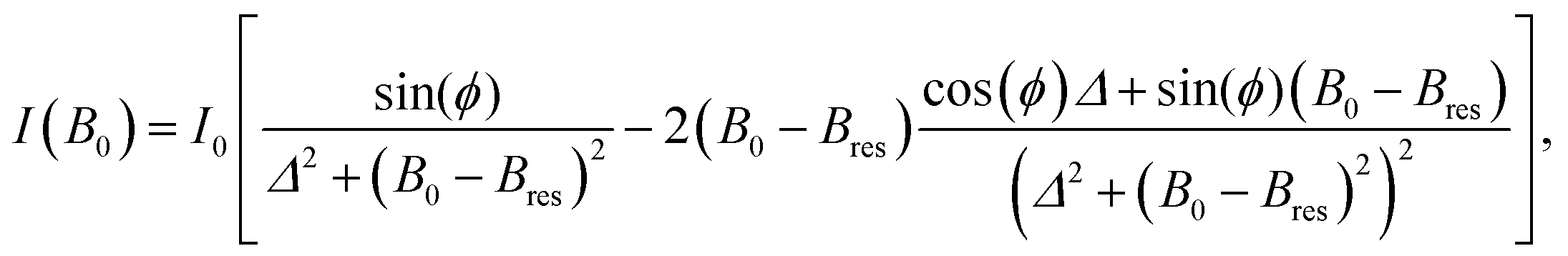 | (2) |
Results
Electrochemistry
The new operando EPR cell design was validated by comparing the voltage profiles and the electrochemical performance with our custom made standard cell design with a spring compressed circular electrode stack, which has been described in a previous publication.43Fig. 2 shows the initial C/5 and the sixth 3C cycle. The C/5 cycles for all cells (Fig. 2a) show a flat voltage plateau around 3.5 V vs. Li/Li+, which is typical for LFP.52 The polarization is low during charge and discharge for both cell designs, with a slightly lower overpotential for the cell with the FEC additive. The 3C cycles (Fig. 2b) show a significantly higher overpotential and lower charging capacity for the operando cell design. In the absence of FEC, both the EPR and the standard cell show a second voltage plateau during 3C discharge, which is caused by changing overpotentials of the lithium counter electrode during the first cycles with higher current density.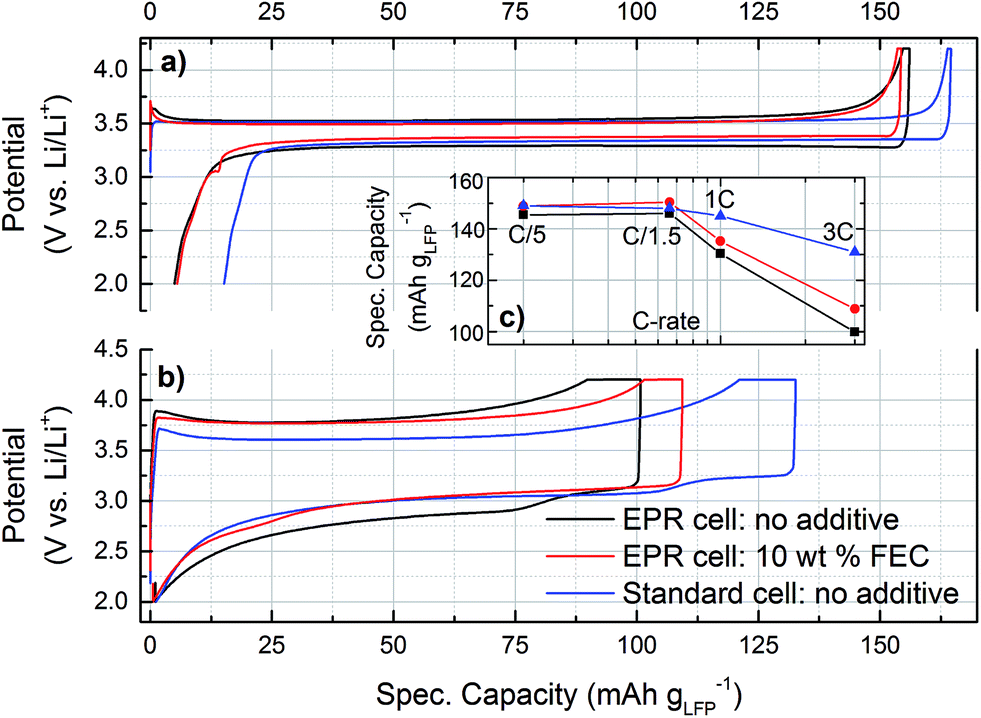 | ||
| Fig. 2 Voltage profiles at (a) C/5 and (b) 3C of the operando EPR cells containing electrolyte without additive (black) or with 10 wt% FEC (red), and of a standard cell design (blue) for comparison. (c) Comparison of the rate capability of these cells. | ||
Fig. 2c compares the rate capability of the operando EPR cells and the standard cell design. They all yield specific capacities of ≈150 mA h g−1LFP at C/5. The moderate deviation from the theoretical specific energy of 170 mA h g−1LFP is within the range that is typically observed in practical LFP cells,52–56 particularly since our LFP electrodes were not optimized for high rates. Above C/1.5, the standard cell performs better, still maintaining ≈130 mA h g−1LFP at 3C compared to only ≈100 mA h g−1LFP for the operando EPR cell. The main reasons for the reduced rate performance of the operando EPR cells are (i) the limited electric contacting of the LFP electrode by the aluminum wire, which increases the ohmic resistance, and (ii) the lack of compression compared to the metal spring compression in the standard cell design.43 While the limited contact area of the LFP electrode with the current collector might negatively affect the homogeneity of the current distribution at high C-rates, this effect would be the same with and without the FEC additive. Despite these minor shortcomings, our operando EPR cell clearly shows essentially identical cycling behavior at low C rates to that of optimized conventional battery cells.
Li-EPR resonance during electrochemical cycling
Fig. 3 shows the evolution of the EPR signal and the voltage curves for the cell with standard electrolyte and with FEC additive. The EPR signal shows a significant difference for both cells already during the first cycle. With standard electrolyte, the Li EPR intensity increases during charge (≡non-uniform lithium plating) and hardly decreases during discharge (≡Li anode dissolution). In contrast, the increase is less pronounced with FEC-containing electrolyte and is completely reversible during the first cycle.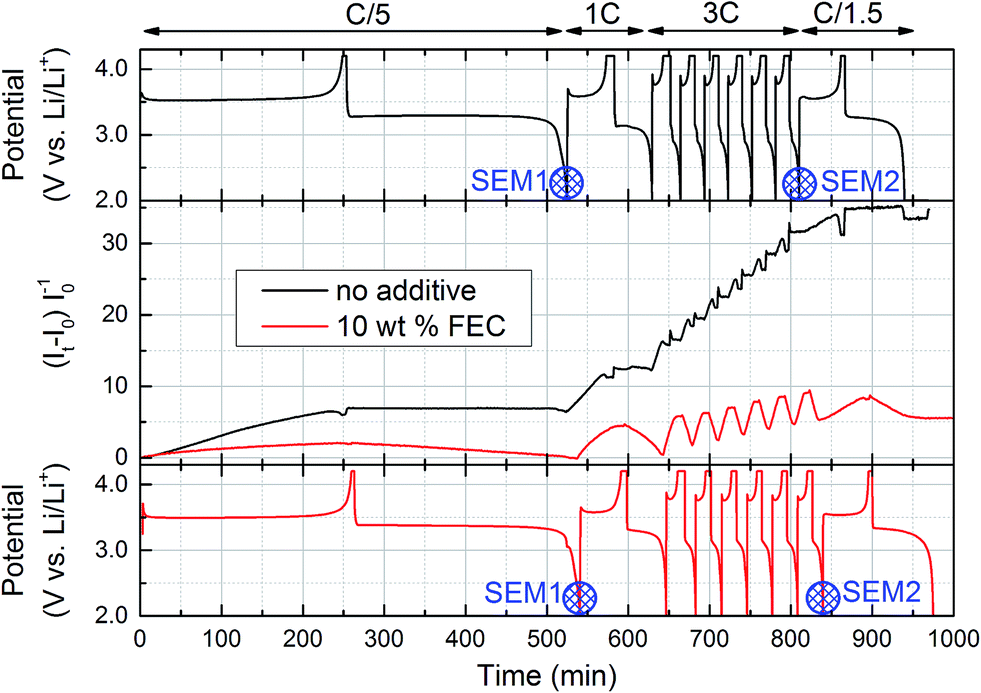 | ||
| Fig. 3 Overview of cycling of operando EPR cells containing electrolyte without additive (black) and with 10 wt% FEC (red). Top and bottom panel: Voltage profiles according to cycling procedure shown above top panel; blue ‘SEM1’ and ‘SEM2’ markers indicate positions where ex situ SEM images of lithium anodes were recorded. Central panel: Normalized intensity of EPR signal, obtained by calculating the area under the numerically integrated experimental first derivate Li spectrum. | ||
For subsequent cycles at higher C-rates, the EPR signal for the cell with standard electrolyte further increases during every charge without significantly decreasing during discharge. With FEC-containing electrolyte, the EPR signal also increases during charge, but decreases again during discharge, implying a better reversibility of the anode processes. Still, during the six 3C-cycles the EPR signal does not completely reverse during discharge, thus also causing a steady increase for the cell containing FEC electrolyte. An interesting detail, highlighting the adequate time resolution of the operando EPR technique, is the kink in the EPR signal with decreasing current density during the constant voltage charging step, observed during all six 3C-charges in FEC electrolyte (see middle panel in Fig. 3). At the end of the cycling procedure, the relative EPR signal with standard electrolyte exceeds the signal from the cell with FEC electrolyte by about a factor seven.
Fig. 4a–c compare the EPR spectra of the cells without and with FEC additive at different times. Both resonances show the asymmetric shape expected for metallic lithium. The shift of the center of the two lines with respect to each other is caused by a different resonance frequency of the resonator when loaded with the two cells. Both pristine cells at the beginning of cycling show a similar linewidth and a lineshape with ϕ ∼ π/2. Without additive, linewidth and asymmetry diminish within about 15 min and then remain fairly constant. With FEC additive, the EPR resonance changes significantly during the first cycle, but it is essentially reversible. Once charging at an increased rate, the shape of the resonance changes to a width and asymmetry similar to the cell without additives within two minutes.
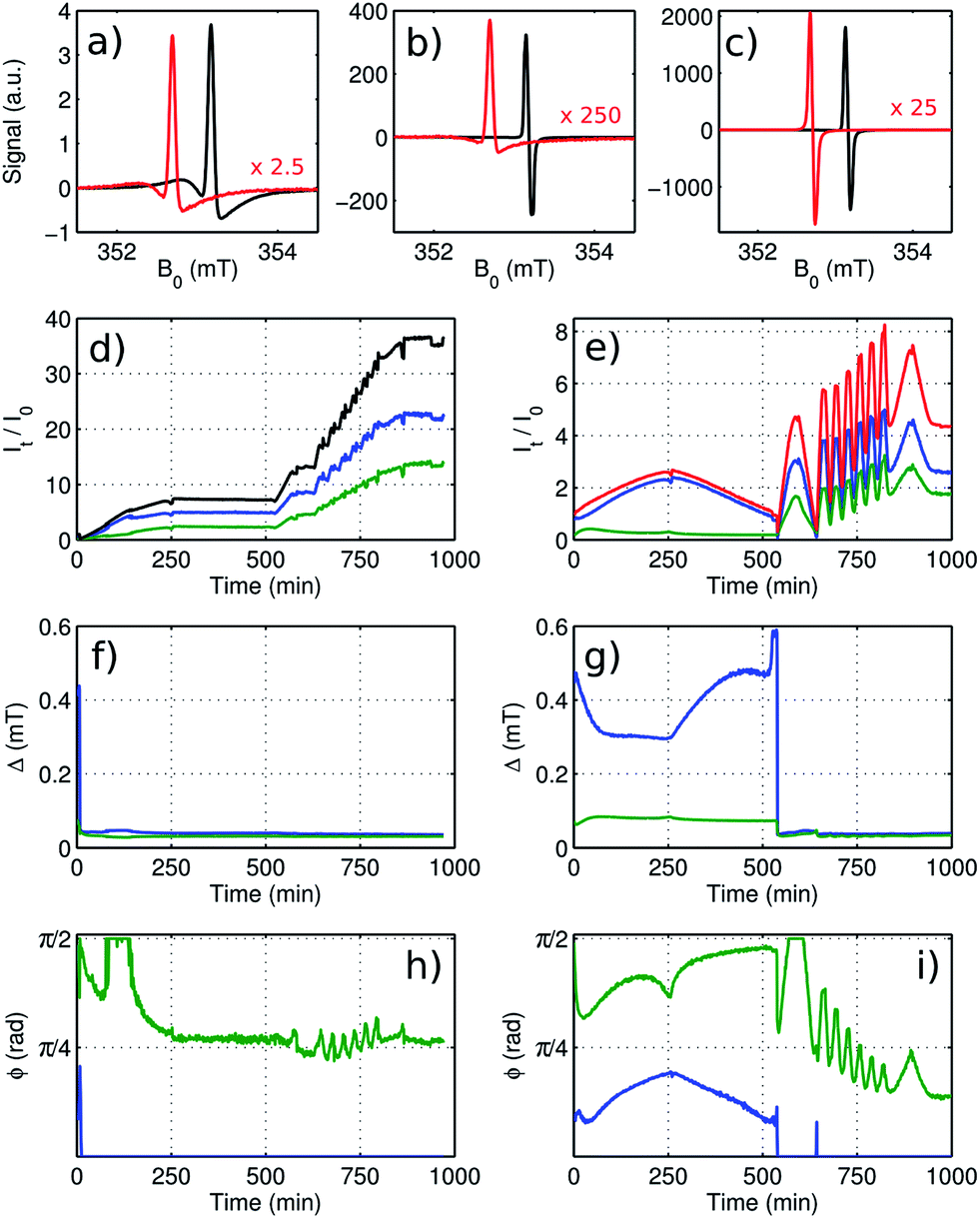 | ||
| Fig. 4 Analysis of time-resolved EPR of metallic Li for cell containing electrolyte without additives (black) and cell containing electrolyte with 10 wt-% FEC (red). The spectra at the beginning of cycling (a), at the end of the first cycle (b), corresponding to position SEM1 in Fig. 3, and at the end of the last 3C cycle (c), corresponding to position SEM2 in Fig. 3, are compared. The red spectra are scaled as indicated in each panel. The resonances were fitted using a model with two phase-shifted Lorentzian lines. The amplitudes for the broader (blue) and for the narrower component (green) are shown for the cell without additives (d) and the FEC-containing cell (e). The sum of both components is shown in black and red for each cell. The width and the asymmetry of each component is shown for the cell without additives (f and h) and for the FEC-containing cell (g and i). | ||
The result of a least-squares fit of the EPR spectra, using a model consisting of two lines according to eqn (2), is shown for the cell with standard electrolyte in Fig. 4d, f and h and for the FEC cell in Fig. 4e, g and i. With a single line, no satisfactory fit was possible. Fig. 4d and e show the amplitude of the two components (green and blue) for the two cells, relative to the sum of both amplitudes at the beginning of cycling. These amplitudes are proportional to the number of contributing spins. The width and the asymmetry for both fitted signal components are shown as a function of time in Fig. 4f and g and Fig. 4h and i respectively. The two components show different characteristics during the first charge–discharge cycle for the FEC cell and for the first few minutes for the cell with standard electrolyte, indicating that these components are caused by two structurally different domains. The narrow component (green color) with Δ ∼ 0.1 mT shows a high asymmetry, ϕ ∼ π/2, which indicates that it is originating from bulk lithium of the anode. The broader component (blue color) with Δ ∼ 0.3–0.5 mT and an intermediate asymmetry, ϕ ∼ π/6, shows a reversible amplitude change by more than a factor two. At the same time the bulk lithium from the anode gets shielded by the additional metal on its surface, causing a signal decrease. Starting with the second cycle for the FEC cell, both linewidths abruptly adjust to a similar value and the amplitudes start to progress synchronously. The same effect is observed more gradually right from the beginning of cycling for the cell with standard electrolyte. This indicates that from that point, a distribution of ϕ rather than multiple distinct components is causing the observed lineshape.
During the first cycle for the cell with FEC additive and during the first few minutes for the cell with standard electrolyte, the asymmetry of the two components varies noticeably, albeit not drastically, and the two amplitudes are not changing synchronously, hence the area of the integrated signal does not lead to quantitative information about the relative number of spins contributing to the signal. Nonetheless, since the signal amplitude is dominated by the broader of the two components (which is not the visually dominant feature in Fig. 4a), the signal amplitudes as obtained by integration and by fitting differ from each other by less than 20%. For subsequent cycles, the lineshapes vary only weakly such that the relative amplitude changes obtained from calculating the area under the integrated experimental spectrum can be taken as semi-quantitative. Therefore this simple and robust method is suitable to quantify the relative growth of the porous layer of metallic lithium on top of the lithium anode during electrochemical cycling.
SEM images
SEM images of cycled lithium electrodes, shown in Fig. 5 and 6, were recorded after the C/5-cycle (position SEM1 in Fig. 3) and after the sixth 3C-cycle (position SEM2 in Fig. 3). | ||
| Fig. 5 Ex situ SEM images of cells containing standard electrolyte (top row) and electrolyte with 10 wt% FEC (bottom row). Images were recorded after the C/5 cycle (a and c) and after the sixth 3C cycle (b and d). Lithium electrodes in (b) and (d) were bent down by 90° using a special sample holder to show the cross section of the micro-structured lithium layer on top of the underlying lithium surface. Notice the different lengthscales of images (a,c and b,d). | ||
 | ||
| Fig. 6 Ex situ SEM images showing specific surface structures of lithium electrodes cycled with FEC additive. (a) Close-up recorded after the initial C/5 cycle. (b) Surface section where a patch of micro-structured lithium had been turned over during sample preparation (after the sixth 3C cycle), revealing the metallic lithium anode below. Craters on the anode surface and the matching connecting lithium particles on the lower side of the micro-structured lithium are indicated by white arrows. (c) Lithium micro-structures forming on the lithium electrode upon cycling, recorded at higher resolution. Pore structures are of micrometer size with sub-micrometer pore wall thickness, which is smaller than the skin depth δ of the microwaves used for the EPR experiments. | ||
After the first cycle (SEM1), differences on the lithium electrode surface can already be detected by eye, as shown in the two insets in Fig. 5a and c. Note that one must only consider the central part of the lithium electrodes, as the LFP counter electrode has a smaller diameter than the lithium electrode (10 mm vs. 17 mm). The electrode containing standard electrolyte is partially covered by micro-structured lithium, which clearly protrudes from the electrode surface (Fig. 5a). The electrode containing FEC electrolyte shows black spots with hardly any three dimensional structure. According to the SEM images, at some of these black spots there is still a very thin layer of residual micro-structured lithium grouped around holes in the dense lithium surface. At other sites, the micro-structured lithium had dissolved completely, leaving behind bare holes. In contrast, the micro-structured lithium layer is thick and intact in the standard electrolyte. At some sites the micro-structured lithium layer was slightly damaged during the SEM sample preparation, revealing holes that would otherwise be covered by the micro-structured lithium film. Several other groups have also reported the occurrence of similar holes on cycled lithium electrodes.38,57–59
After the sixth 3C cycle (SEM2), both electrodes were covered with a thick film of micro-structured lithium. To compare the amount of micro-structured lithium, SEM images were taken from electrodes that had been bent down 90° to expose the cross section of the micro-structured layer, as shown in Fig. 5b and d. Based on these images, the thickness of the micro-structured film was determined to be 66 ± 5 μm with the standard electrolyte and 20 ± 2 μm with the FEC additive.
In the electrode cycled with FEC electrolyte, discrete and uniformly shaped holes with a diameter of about 10 μm could be observed in the cracks (Fig. 5d), whereas the rest of the lithium surface appears to be unchanged. In contrast, the underlying lithium surface in the standard electrolyte was very inhomogeneous and rough, containing large holes and canyon-like structures (not visible in Fig. 5b).
The holes in the lithium anode surface seem to affect the growth of micro-structured lithium, which is supported by two further observations: Fig. 6a shows three holes and a thin layer of residual micro-structured lithium after the first C/5-cycle in FEC electrolyte. It can clearly be seen that the micro-structured lithium grows out of the hole and then spreads over the lithium surface. This mechanism is also consistent with Fig. 6b, where part of the micro-structured lithium film had been separated during SEM sample preparation but was still attached to the electrode, thus revealing its lower side. The lithium particles on the lower side are arranged in the exact same pattern as the holes on the corresponding lithium surface. This further confirms that the micro-structured lithium layer is only connected to the underlying lithium surface by a few contact points through the holes. Limited contact between the micro-structured lithium layer and the underlying lithium surface has already been reported in 1990 by Yamaki et al.60 and has also been observed more recently by Orsini et al.35 and by Steiger et al.34 on cross section SEM images. The porosity of micro-structured lithium is depicted in Fig. 6c with an increased resolution, showing that the pore wall dimensions are in the sub-micrometer range. Very similar structures of the micro-structured lithium layer have been previously reported by other groups.37,57
Discussion
Interpretation of EPR results
The microwave irradiation used to induce electron spin transitions is subject to a finite penetration depth into the ‘bulk’ lithium-metal anode owing to the skin effect. At 9.5 GHz, the skin depth is δ ≈ 1.1 μm,61,62 which is two orders of magnitude smaller than the thickness of the Li anode. Only a small fraction of the Li metal is contributing to the EPR signal, which is therefore proportional to the surface area of the lithium anode rather than to its volume. If, during charging, more lithium is deposited smoothly on the anode, the signal does not change appreciably. The experimentally observed amplitude change can be caused by an increase of the Li metal surface area or by a reduction of the electrical conductivity, which would increase δ. The latter would also cause a significant lineshape change. This is neither observed for the cell without additives after the first few minutes nor during fast cycling for the FEC cell, therefore it is anticipated that the EPR signal largely scales with the surface area. The formation of lithium micro-structures (mossy/dendritic) with a thickness of about 1–2 μm, which is comparable to δ, has been reported35 and was observed in the SEM images (Fig. 6c), with pore wall diameters at least an order of magnitude smaller. Hence the newly formed surface micro-structures are penetrated by the microwave field and, due to the increased surface area and supported by the growth direction perpendicular to the magnetic microwave field lines, contribute to the observed EPR signal increase. The formation of lithium micro-structures is further supported by the change of the line asymmetry from ϕ ∼ π/2 for the pristine cell, which originates from lithium metal that is much thicker than δ, to ϕ ∼ 0 during cell cycling, which indicates a thickness of the micro-structure walls smaller than δ.51The rate of the linewidth change for the FEC cell at the beginning of the 1C cycle shows an approximately five times higher value than the rate for the cell with standard electrolyte at the beginning of the C/5 cycle. This indicates that although the FEC cell is more robust toward the formation of micro-structured lithium, once it starts to build, it becomes the dominant form of lithium deposition. This micro-structured lithium could be considered non-locally as surface lithium with an increased time TD it takes a spin to diffuse through the skin depth. The result would be a decreased linewidth and an increased line symmetry,49 as observed for the FEC cell after the first cycle and for the cell without additives quickly after starting the initial charging. Furthermore, when electrical contact is lost, the porous surface would behave more like isolated lithium metal with small dimensions. Corresponding line widths have been reported to narrow substantially, depending on the size of the structure.63
Comparing EPR and SEM results
For the two electrolytes, both operando EPR spectroscopy and ex situ SEM analysis reveal a different lithium plating/stripping behavior already during the first cycle, which is caused by the improved properties of the solid/electrolyte interface (SEI) in the FEC-containing electrolyte. The FEC additive is known to be reduced and polymerize at lithium potential, thus forming a very flexible SEI layer.29,30,41,64 The latter was supported by a recent study on the chemical composition of the surface film formed on silicon anodes in the presence of FEC by Markevich et al.64 Besides an improved mechanical flexibility, the SEI formed in FEC-containing electrolytes also has an increased ionic conductivity,30 which results in a more homogeneous current distribution.15,58,65 As a consequence, lithium plating in the FEC-containing electrolyte can be expected to occur rather homogeneously in comparison to the standard electrolyte, which is reflected by the less pronounced increase of the EPR signal and the continuously large linewidth during the first charge. Because of the homogeneous and rather compact lithium deposition according to the moderate increase of the EPR signal, most lithium plated during first charge is stripped during first discharge and the EPR signal returns to its initial value after the first cycle for the FEC-containing cell.In contrast, the SEI that forms in the standard carbonate-based electrolyte mainly consists of lithium salts (such as LiF, Li2CO3 and Li2O) formed by irreversible electrolyte reduction.66 Due to its composition the SEI shows poor adhesion to the lithium surface and cannot withstand significant volume changes.15 Accordingly, already during first charge the non-uniform lithium deposition and the corresponding volumetric expansion results in mechanical stress on the SEI at sites of preferred lithium plating; this causes the SEI to break, and fresh lithium is exposed to the electrolyte. Although fresh lithium has been shown to react with the electrolyte within seconds,67 the new SEI on the exposed lithium is thinner than on the rest of the electrode. Further lithium deposition will therefore preferentially take place at these sites, causing the growth of micro-structured (mossy/dendritic) lithium during the first charge. During the subsequent discharge, the EPR signal hardly decreases in the standard electrolyte, which is consistent with SEM images showing a significant amount of residual micro-structured lithium after the first cycle. This is due to the dissolution of ‘bulk’ lithium from the lithium anode, while the main part of the micro-structured lithium is not dissolved.
The dissolution of bulk lithium rather than micro-structured lithium is a direct consequence of the limited contact points between the micro-structured lithium layer and the underlying lithium anode, as shown in Fig. 6b. If the contact points dissolve during discharge before all the micro-structured lithium has been oxidized, the micro-structured lithium layer loses electric contact to the anode and becomes electrochemically inactive, forming so called ‘dead lithium’.57 This mechanism, which is also consistent with the EPR data, is in very good agreement with recent results published by Steiger et al., who follow the development of micro-structured lithium by optical microscopy and also observe limited contact points (“stems”) as previously mentioned.34
The occurrence of non-uniform lithium plating during the first cycle is in accordance with literature. In our experiments, the geometric current density during the initial C/5 cycle was 0.23 mA cm−2Li. Several other groups have also reported non-uniform lithium plating (dendrites, moss or particles) at similar11,59,68 or even lower33 current densities in liquid electrolytes without film-forming additives. Stark el al. observed the formation of discrete lithium particles already during the first couple of seconds of lithium plating in EC/DMC electrolyte,12 which is consistent with the instantaneous increase of the EPR signal on the first charge.
During subsequent cycles the increased current density causes an accumulation of micro-structured lithium in both electrolytes as consistently shown by the increasing EPR signal and by SEM images. It is generally accepted that an increase in charging current density also enhances the formation of micro-structured lithium.35,58,59,65
The lithium plating/stripping process seems to be almost completely irreversible during cell cycling with standard electrolyte due to the lack of EPR signal decrease during discharge phases, whereas it is at least partially reversible with the FEC containing electrolyte. The similarity of the linewidth and asymmetry for the two cells at faster cycling rates indicates that the build-up of the signal-inducing lithium structure is similar for both cells, but the decrease during discharge proceeds differently.
The growth of the micro-structured lithium layer during cycling takes place at the interface with the metallic lithium anode, where new lithium micro-structures growing out of the holes pushes away the old layer of micro-structured lithium.34 This preferential lithium plating on the lithium metal surface is favored over lithium plating on top of the micro-structured layer due to the poor electronic conductivity of the latter.
Conclusions
An operando EPR cell was developed to perform EPR spectroscopy during electrochemical cycling. These measurements provided time-resolved semi-quantitative information about the plating and stripping of metallic lithium on lithium-metal anodes, which is particularly useful for investigating the formation of micro-structured lithium (mossy/dendritic). The method is complementary to optical and electron microscopic techniques that have their strength in monitoring the morphology of the electrode surface in turn.The potential of operando EPR was explored by comparing two cells, the first one containing a standard electrolyte and the other one with an FEC additive to the electrolyte that is known to reduce the formation of micro-structured lithium (mossy/dendritic). Based on the EPR signal intensity and lineshape, it was shown that lithium deposition during electrochemical cell cycling was much more reversible when using FEC additive. The EPR results were confirmed by ex situ SEM images that were recorded at selected positions using identically cycled cells.
Owing to the high sensitivity of the EPR technique, already the early stages of the formation of lithium micro-structures in lithium batteries could be monitored. As compared to operando NMR,36 the EPR experiment is considerably more sensitive, and since microwave frequencies instead of radiofrequencies are used, the skin depth is significantly smaller. Therefore EPR excites a thinner layer on the lithium metal surface, providing information with higher surface localization.
The presented interpretation of operando EPR data was based on the empirical extension of theoretical findings that for flat surfaces and for spherical particles of analogous dimensions the same lineshapes are observed. In addition, very characteristic lineshape changes are observed when going from structures that are large to structures that are small compared with the skin depth of the microwave field used to excite the spins. It was deduced, supported by SEM images, that the same qualitative lineshape changes are observed when changing from a smooth lithium anode with a thickness much larger than the skin depth to porous lithium with wall thickness smaller than the skin depth. In principle, it should be possible to interpret the data quantitatively. A suitable procedure was sketched, yet theoretical expressions of conduction EPR signals from porous metallic lithium must first be derived.
Acknowledgements
We would like to thank Hans Kungl (IEK-9) for very helpful discussions, Magnus Graf (IEK-9) for experimental support with the EPR measurements, Johannes Landesfeind (TUM) for valuable support with the SEM measurements, and Yi-Chun Lu (Chinese University of Hongkong) for contributions to the operando EPR cell design. TUM gratefully acknowledges the financial support by BMW AG and by the Bavarian Ministry of Economic Affairs and Media, Energy and Technology under the auspices of the EEBatt project. IEK-9 gratefully acknowledges funding from the German Federal Ministry of Education and Research (BMBF-project DESIREE, grant number 03SF0477A).References
- K. G. Gallagher, S. Goebel, T. Greszler, M. Mathias, W. Oelerich, D. Eroglu and V. Srinivasan, Energy Environ. Sci., 2014, 7, 1555–1563 CAS.
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2012, 159, R1–R30 CrossRef CAS.
- P. Bruce, S. Freunberger, L. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–30 CrossRef CAS PubMed.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed. Engl., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- Y. Mikhaylik, I. Kovalev, R. Schock, K. Kumaresan, J. Xu and J. Affinito, Meet. Abstr. Electrochem. Soc., 2009, 902, 216 Search PubMed.
- J.-M. Tarascon, Philos. Trans. R. Soc., A, 2010, 368, 3227–3241 CrossRef PubMed.
- J. B. Bates, US Pat., 5314765, 1994 Search PubMed.
- S. J. Visco and Y. S. Nimon, US Pat., 7645543 B2, 2010 Search PubMed.
- S. J. Visco, E. Nimon, B. Katz, US Pat., 7282296, 2007 Search PubMed.
- A. Aryanfar, D. Brooks, B. V Merinov, W. A. Goddard, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. Lett., 2014, 5, 1721–1726 CrossRef CAS PubMed.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J.-G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- J. K. Stark, Y. Ding and P. A. Kohl, J. Electrochem. Soc., 2013, 160, D337–D342 CrossRef CAS.
- J. T. Vaughey, G. Liu and J.-G. Zhang, MRS Bull., 2014, 39, 429–435 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- D. Aurbach, E. Zinigrad, Y. Cohen and H. Teller, Solid State Ionics, 2002, 148, 405–416 CrossRef CAS.
- M. Dollé, L. Sannier, B. Beaudoin, M. Trentin and J.-M. Tarascon, Electrochem. Solid-State Lett., 2002, 5, A286–A289 CrossRef.
- K. J. Harry, D. T. Hallinan, D. Y. Parkinson, A. A. Macdowell and N. P. Balsara, Nat. Mater., 2013, 12, 1–6 Search PubMed.
- K. Ozawa, Solid State Ionics, 1994, 69, 212–221 CrossRef CAS.
- H. Buqa, D. Goers, M. Holzapfel, M. E. Spahr and P. Novák, J. Electrochem. Soc., 2005, 152, A474–A481 CrossRef CAS.
- M. C. Smart, B. V. Ratnakumar, L. Whitcanack, K. Chin, M. Rodriguez and S. Surampudi, IEEE Aerosp. Electron. Syst. Mag., 2002, 17, 16–20 CrossRef.
- P. Arorat, R. E. White and M. Doyle, J. Electrochem. Soc., 1998, 145, 3647–3667 CrossRef.
- S. S. Zhang, K. Xu and T. R. Jow, J. Power Sources, 2006, 160, 1349–1354 CrossRef CAS.
- M. C. Smart and B. V. Ratnakumar, J. Electrochem. Soc., 2011, 158, A379–A389 CrossRef CAS.
- M. Dubarry, C. Truchot, B. Y. Liaw, K. Gering, S. Sazhin, D. Jamison and C. Michelbacher, J. Electrochem. Soc., 2012, 160, A191–A199 CrossRef.
- Z. Li, J. Huang, B. Yann Liaw, V. Metzler and J. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS.
- M. C. Smart, B. L. Lucht, S. Dalavi, F. C. Krause and B. V. Ratnakumar, J. Electrochem. Soc., 2012, 159, A739–A751 CrossRef CAS.
- L. Gireaud, S. Grugeon, S. Laruelle, B. Yrieix and J.-M. Tarascon, Electrochem. Commun., 2006, 8, 1639–1649 CrossRef CAS.
- O. Crowther and A. C. West, J. Electrochem. Soc., 2008, 155, A806–A811 CrossRef CAS.
- R. McMillan, H. Slegr, Z. Shu and W. Wang, J. Power Sources, 1999, 81–82, 20–26 CrossRef CAS.
- R. Mogi, M. Inaba, S.-K. Jeong, Y. Iriyama, T. Abe and Z. Ogumi, J. Electrochem. Soc., 2002, 149, A1578–A1583 CrossRef CAS.
- K. Nishikawa, T. Mori, T. Nishida, Y. Fukunaka, M. Rosso and T. Homma, J. Electrochem. Soc., 2010, 157, A1212–A1217 CrossRef CAS.
- T. Nishida, K. Nishikawa, M. Rosso and Y. Fukunaka, Electrochim. Acta, 2013, 100, 333–341 CrossRef CAS.
- J. Steiger, D. Kramer and R. Mönig, J. Power Sources, 2014, 261, 112–119 CrossRef CAS.
- J. Steiger, D. Kramer and R. Mönig, Electrochim. Acta, 2014, 136, 529–536 CrossRef CAS.
- F. Orsini, A. Du Pasquier, B. Beaudoin and J. M. Tarascon, J. Power Sources, 1998, 76, 19–29 CrossRef CAS.
- R. Bhattacharyya, B. Key, H. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, Nat. Mater., 2010, 9, 504–510 CrossRef CAS PubMed.
- S. Chandrashekar, N. M. Trease, H. J. Chang, L.-S. Du, C. P. Grey and A. Jerschow, Nat. Mater., 2012, 11, 311–315 CrossRef CAS PubMed.
- N. Schweikert, A. Hofmann, M. Schulz, M. Scheuermann, S. T. Boles, T. Hanemann, H. Hahn and S. Indris, J. Power Sources, 2013, 228, 237–243 CrossRef CAS.
- F. Poli, J. S. Kshetrimayum, L. Monconduit and M. Letellier, Electrochem. Commun., 2011, 13, 1293–1295 CrossRef CAS.
- G. Gershinsky, E. Bar, L. Monconduit and D. Zitoun, Energy Environ. Sci., 2014, 7, 2012–2016 CAS.
- A. V. Cresce, S. M. Russell, D. R. Baker, K. J. Gaskell and K. Xu, Nano Lett., 2014, 14, 1405–1412 CrossRef CAS PubMed.
- L. Zhuang and J. Lu, Rev. Sci. Instrum., 2000, 71, 4242–4248 CrossRef CAS.
- S. Meini, M. Piana, N. Tsiouvaras, A. Garsuch and H. A. Gasteiger, Electrochem. Solid-State Lett., 2012, 15, A45–A48 CrossRef CAS.
- D. R. Gallus, R. Schmitz, R. Wagner, B. Hoffmann, S. Nowak, I. Cekic-Laskovic, R. W. Schmitz and M. Winter, Electrochim. Acta, 2014, 134, 393–398 CrossRef CAS.
- P. Jakes, E. Erdem, A. Ozarowski, J. van Tol, R. Buckan, D. Mikhailova, H. Ehrenberg and R.-A. Eichel, Phys. Chem. Chem. Phys., 2011, 13, 9344–9352 RSC.
- E. Erdem, V. Mass, A. Gembus, A. Schulz, V. Liebau-Kunzmann, C. Fasel, R. Riedel and R.-A. Eichel, Phys. Chem. Chem. Phys., 2009, 11, 5628–5633 RSC.
- F. Dyson, Phys. Rev., 1955, 98, 349–359 CrossRef CAS.
- G. R. Eaton, S. S. Eaton, D. P. Barr and R. T. Weber, Quantitative EPR, Springer, Vienna, 2010 Search PubMed.
- R. H. Webb, Phys. Rev., 1967, 158, 225–233 CrossRef CAS.
- V. Sitaram, A. Sharma, S. Bhat, K. Mizoguchi and R. Menon, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 035209 CrossRef.
- G. Feher and A. F. Kip, Phys. Rev., 1955, 98, 337–348 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- K. Zaghib, J. Shim, A. Guerfi, P. Charest and K. A. Striebel, Electrochem. Solid-State Lett., 2005, 8, A207–A210 CrossRef CAS.
- D. Y. W. Yu, K. Donoue, T. Inoue, M. Fujimoto and S. Fujitani, J. Electrochem. Soc., 2006, 153, A835–A839 CrossRef CAS.
- C. Fongy, A.-C. Gaillot, S. Jouanneau, D. Guyomard and B. Lestriez, J. Electrochem. Soc., 2010, 157, A885–A891 CrossRef CAS.
- C. Fongy, S. Jouanneau, D. Guyomard, J. C. Badot and B. Lestriez, J. Electrochem. Soc., 2010, 157, A1347–A1353 CrossRef CAS.
- I. Yoshimatsu, T. Hirai and J. Yamaki, J. Electrochem. Soc., 1988, 135, 2422–2427 CrossRef CAS.
- D. Aurbach, Y. Gofer and J. Langzam, J. Electrochem. Soc., 1989, 136, 3198–3205 CrossRef CAS.
- M. Arakawa, S. Tobishima, Y. Nemoto and M. Ichimura, J. Power Sources, 1993, 43–44, 27–35 CrossRef.
- J. Yamaki, in 40th Int. Soc. Electrochem. Meet., 1990, pp. 150–151 Search PubMed.
- R. B. Lewis and T. R. Carver, Phys. Rev. Lett., 1964, 12, 693–695 CrossRef CAS.
- M. Höhne, Phys. Status Solidi B, 1978, 85, 525–534 CrossRef.
- K. Saiki, T. Fujita, Y. Shimizu, S. Sakoh and N. Wanda, J. Phys. Soc. Jpn., 1972, 32, 447–450 CrossRef CAS.
- E. Markevich, K. Fridman, R. Sharabi, R. Elazari, G. Salitra, H. E. Gottlieb, G. Gershinsky, A. Garsuch, G. Semrau, M. A. Schmidt and D. Aurbach, J. Electrochem. Soc., 2013, 160, A1824–A1833 CrossRef CAS.
- J. Yamaki, S. Tobishima, K. Hayashi, K. Saito, Y. Nemoto and M. Arakawa, J. Power Sources, 1998, 74, 219–227 CrossRef CAS.
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS.
- M. Odziemkowski and D. E. Irish, J. Electrochem. Soc., 1992, 139, 3063–3074 CrossRef CAS.
- N. Schweikert, R. Heinzmann, A. Eichhöfer, H. Hahn and S. Indris, Solid State Ionics, 2012, 226, 15–23 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |