
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly tunable fluorinated trispyrazolylborates [HB(3-CF3-5-{4-RPh}pz)3]− (R = NO2, CF3, Cl, F, H, OMe and NMe2) and their copper(I) complexes†
Thomas F.
van Dijkman
a,
Maxime A.
Siegler
b and
Elisabeth
Bouwman
 *a
*a
aLeiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA, Leiden, the Netherlands. E-mail: bouwman@chem.leidenuniv.nl
bDepartment of Chemistry, Johns Hopkins University, MD 21218, Baltimore, USA
First published on 16th November 2015
Abstract
The ethene and carbon monoxide adducts of copper(I) with seven trispyrazolylborate ligands ([HB(3-CF3-5-{4-RPh}pz)3]−; R = NO2 (4a), CF3 (4b), Cl (4c), F (4d), H (4e), OMe (4f) and NMe2 (4g)) were synthesized and characterized. The ligands were synthesized from their corresponding pyrazoles and sodium tetrahydridoborate and were obtained as solvent adducts of their sodium salts after workup. When the pyrazole with the most electron-withdrawing substituent (R = NO2) is used the asymmetric ligand [HB(3-CF3-5-{4-NO2Ph}pz)2(3-{4-NO2Ph}-5-CF3pz)]− (4a′) is formed as the major product. Copper(I) complexes with ethene or CO as a co-ligand were prepared in good yields and were structurally characterized using 1H NMR, 13C NMR and infrared spectroscopy. Single crystal X-ray crystallography analyses revealed the structures of Na4a′, Na4b, four copper ethene complexes and four copper carbonyl complexes. The structures of the copper(I) complexes show CuI ions in pseudo-tetrahedral coordination environments consisting of three nitrogen atoms of the trispyrazolylborate ligand and the carbonyl or η2-coordinated ethene ligands, with nearly identical coordination environments around the CuI ion. The compound [Na(4a′)(H2O)]n crystallizes as one-dimensional chains with intermolecular Na⋯O2N interactions. The sodium ions were found in severely distorted octahedral geometries with three nitrogen atoms from the trispyrazolylborate ligand, one aqua ligand and two oxygen atoms from the nitro group of an adjacent molecule. The compound [Na2(4b)2(μ-H2O)2] crystallizes as a centrosymmetric water-bridged dimer: two five-coordinate square-pyramidal sodium ions each are coordinated facially by three nitrogen atoms from a trispyrazolylborate ligand and two bridging water ligands. Below the base of the pyramidal structure one intermolecular and two intramolecular Na⋯F short contacts are present. The 1H and 13C NMR spectra of the copper–ethene complexes show signals for the ethene ligands in the range of 4.84–4.96 ppm and 84.9–86.8 ppm respectively. The infrared spectra of the carbonyl complexes show CO stretching frequencies in the range of 2096–2120 cm−1. Both the NMR signals for the ethene ligands and infrared signals for the carbonyl ligands were found to show good correlations with the Hammett σp parameters of the substituents on the phenyl rings of the ligands.
Introduction
Trispyrazolylborates (commonly referred to as scorpionate ligands1) form a highly versatile class of ligands that were pioneered by Trofimenko in the 1960s. They have since been developed from the simple unsubstituted Tp− into ligands of increasing complexity and scope.2–7 The great diversity in steric and electronic properties available in trispyrazolylborate ligands allows for the optimization of complexes for specific purposes such as catalysis and biomimetic structural and functional models. The properties of trispyrazolylborate ligands can be changed by systematic modification of the substituents on the pyrazolyl groups or, though much less common, on the central boron atom. At present a wide variety of the facially coordinating tridentate ligands is known with substituents ranging from alkyl groups to aryl groups (both aromatic like phenyl groups and heteroaromatic such as the thienyl group) as well as phosphines, esters and amides.8–13 Due to the structurally and electronically similar properties of the pyrazole and imidazole rings trispyrazolylborate ligands offer an interesting avenue towards the modelling of biological systems that incorporate multiple histidine residues coordinating facially to the metal center in the active site. A metal that is encountered often in such sites is copper, in enzymes and proteins such as hemocyanin, cytochrome-c oxidase, ceruloplasmin, superoxide dismutase, laccase and many others.14 Copper offers advantages besides being biologically relevant; the ethene and carbonyl complexes of copper(I) offer relatively simple systems to characterize the steric and electronic properties of novel trispyrazolylborate ligands. Such complexes have a (pseudo) tetrahedral coordination environment in which a single copper(I) ion is facially coordinated by a single trispyrazolylborate ligand and one ethene or carbonyl ligand which allows for unambiguous characterization of the steric and electronic properties of the trispyrazolylborate ligand. Unfortunately comparison of the properties of trispyrazolylborate ligands typically results in their differences being ascribed to a combination of steric and electronic factors without detailed knowledge of the contributions of each factor. To facilitate the comparison of the steric and electronic factors a series of isosteric, purely electronically varied trispyrazolylborates would be useful. By keeping the steric properties of the binding pocket around the metal center constant and varying only the electronic properties of the trispyrazolylborate ligands a spectrochemical series of isosteric trispyrazolylborate ligands could be created. Such a spectrochemical series of trispyrazolylborate ligands could be used to separately estimate the optimal electronic properties of a trispyrazolylborate ligand in a catalytic or biomimetic system prior to optimization of the steric factors. To create a spectrochemical trispyrazolylborate ligand series strict regiochemical control during ligand synthesis is required. The coordination environment around the metal must vary as little as possible while the electronic properties of the ligand are varied. In this regard trispyrazolylborate ligand synthesis offers a convenient handle in that the 3-positions of the pyrazolyl rings are typically occupied by the bulkiest substituent. Exceptions to this situation occur if the pyrazole also carries a particularly electron-withdrawing substituent like a trifluoridomethyl group, in which case the electron-withdrawing group assumes the 3-position instead. The resulting electron-withdrawing trispyrazolylborate ligands have a special place in copper(I) chemistry because they allow for the synthesis and isolation of otherwise thermally and oxidatively unstable copper(I) complexes with ligands like ethene and carbon monoxide. Examples of such ligands include [Tp(CF3)2]−, [Tp(CF3)3]−, [TpCF3,Ph]−, [Ttz(CF3)2]− and [TpCF3]−.15–18 The [TpCF3,RCu(C2H4)] complexes of these ligands are air stable as are the corresponding carbonyl complexes which have infrared CO stretching frequencies close to that of free CO indicating particularly electron-poor copper(I) centers. Both effects are unusual for copper(I) trispyrazolylborate complexes and can be ascribed to the electron-withdrawing effects of the fluorinated substituents on the pyrazole moieties. The trifluoridomethyl groups thus tune the electronic properties of the ligand to a great extent while adding only a limited amount of steric bulk and a chemically inert binding pocket around the copper(I) center. With the aim of synthesizing isosteric, electronically different trispyrazolylborate ligands the ligand [TpCF3,Ph]− in particular is interesting because of its potential for chemical modification of the phenyl rings while retaining the structure of the binding pocket surrounding the copper(I) center. Delocalization in the aromatic rings allows for efficient charge transfer from substituents on the distal phenyl rings to the copper(I) ion while conformational differences between the resulting complexes are limited by the steric bulk of the crowded phenyl groups. Particularly substitution on the para-position of the phenyl rings would result charge transfer without significant structural modification to the ligands. Even though the substituents would be placed relatively far away from the copper(I) ions the charges of the substituents could be transferred quite effectively by resonance effects in the conjoined aromatic rings. In this work the effect of para-substitution on the phenyl rings of [TpCF3,Ph]− was examined; by judicious selection of electron donating and withdrawing substituents a spectroscopic series was created (Table 1). Substituents were chosen that offer a broad range of electronic properties (based on their Hammett σp parameters) and are tolerant to the strongly reducing conditions required for the synthesis of these trispyrazolylborate ligands.19 To study the upper limit of electron withdrawing effects the particularly electron withdrawing nitro substituent was also included despite its sensitivity to reducing conditions.| R |
σ
p![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Ligand | [Tp CF 3,4-RPh CuL] | ||
|---|---|---|---|---|---|
| L = C2H4 | L = CO | ||||
a The third pyrazole ring is schematically shown as [N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N].
b Taken from ref. 18.
c The major product is an asymmetric isomer, indicated with a′, see text. N].
b Taken from ref. 18.
c The major product is an asymmetric isomer, indicated with a′, see text.
|
|||||

|
NO2c |
0.78 | 4a/4a′ | 5a′ | 6a′ |
| CF3 | 0.54 | 4b | 5b | 6b | |
| Cl | 0.23 | 4c | 5c | 6c | |
| F | 0.06 | 4d | 5d | 6d | |
| H | 0 | 4e | 5e | 6e | |
| OMe | −0.27 | 4f | 5f | 6f | |
| NMe2 | −0.83 | 4g | 5g | 6g | |
Results
Synthesis
The ligands used in this work were synthesized from 4′-substituted acetophenones (1a–g) in a multistep procedure (see Scheme 1). The 4-substituted acetophenones were converted into 4′-substituted benzoyltrifluoridoacetates (2a–g) in a Claisen condensation with ethyl trifluoridoacetate in THF or diethyl ether. Typically the use of diethyl ether gave higher yields and less side products but the more polar THF was sometimes required to keep reaction mixtures from becoming too viscous. The 4′-substituted benzoyltrifluoridoacetates were then converted into the corresponding pyrazoles (3a–g) by a two-step condensation reaction with hydrazine hydrate which resulted in product mixtures comprising the desired pyrazole and (partially) hydrated intermediates which were dehydrated by refluxing in dilute hydrochloric acid; without the additional dehydration step the reaction is typically incomplete. Alternative methods were less successful; reflux in toluene under Dean–Stark conditions required much longer reaction times and vacuum thermolysis frequently led to co-sublimation of incompletely dehydrated products. The pyrazoles can be purified readily by means of vacuum sublimation to yield pure and dry products that can be used immediately for ligand synthesis. | ||
| Scheme 1 (a) CF3COOEt, KOtBu/NaOEt, Et2O, rt; (b) N2H4·H2O, EtOH, reflux; (c) 3 N HCl, reflux; (d) NaBH4, Ar either at 180 °C (melt reaction) or in refluxing 4-methylanisole. | ||
The pyrazoles were converted into the trispyrazolylborate ligands by heating in the presence of NaBH4 (Scheme 1). A reaction temperature of 180 °C was used in a solvent-free reaction as reported for the synthesis of Na4e except in the cases of Na4a and Na4f for which 4-methylanisole was used as a solvent.16 In the case of Na4f the solvent was used because the melting point of 3g is too high to let the reaction proceed smoothly at 180 °C and higher temperatures led to excessive scorching. For Na4a the solvent-free synthesis resulted in the formation of large amounts of byproducts, whereas stepwise heating in 4-methylanisole facilitated the formation of the monopyrazolylborate and bispyrazolylborate intermediates at lower reaction temperatures before raising the reaction temperature to 180 °C. This was not possible without the use of a solvent as the melting point of 3a is too high (171 °C). As evident from the 1H and 13C NMR spectra of the sodium salt and the complexes of [4a]− the nitro-substituted ligand formed almost exclusively as the asymmetric species in which two of the pyrazole rings have the trifluoridomethyl group in the 3-position and one pyrazole ring is attached “in reverse” with the trifluoridomethyl group in the 5-position (see Scheme 2). This asymmetric isomer will be referred to as 4a′.
 | ||
| Scheme 2 The asymmetric structure of the major fraction [4a′]−. | ||
All trispyrazolylborate ligands were isolated as solvent adducts of their sodium salts, typically incorporating solvents such as acetone, THF or diethyl ether in stoichiometric ratios. Instances of mixed solvent adducts were also observed such as in Na4d·(Et2O)0.5(acetone)0.5. Examples of similar solvent adducts in other fluorinated trispyrazolylborate ligands have been described.17,20,21 Na4b formed as the water adduct even when recrystallized in the presence of suitable solvents or mixtures thereof. All ligand salts exchanged some solvent with adventitious water upon standing in air.
The ethene and carbonyl copper(I) complexes (respectively 5a′, 5b–g and 6a′, 6b–g, see Table 1) were synthesized by stirring the sodium salts of the trispyrazolylborate ligands Na4a′, Na4b–g and CuI in DCM in an atmosphere of ethene or carbon monoxide. It was found that if CuI was added to an ethene or carbon monoxide saturated solution of the ligand in DCM the synthesis could be performed with minimal further use of Schlenk techniques. Workup consisted of filtration to remove NaI and evaporation of the solvent in vacuo to yield the complexes as white powders which were recrystallized from DCM/pentane to yield the pure products. Most of the isolated ethene or carbonyl copper(I) complexes are air stable for at least six months in the solid state. In solution in dichloromethane 5a′, 6a′, 5g and 6g slowly turn green over the course of hours with associated loss of the ethene or carbonyl ligands (as evident from their NMR spectra) if exposed to air. Complexes 5b–g and 6b–g show good stability in the presence of air, light and moisture and can be manipulated without special precautions. The complexes 5a′ and 6a′ were found to be less stable over time and had to be kept under argon to avoid decomposition. Aside from DCM the complexes were found to be slightly soluble in n-pentane, n-hexane and cyclohexane and to have good solubility in benzene, toluene, THF, 1,4-dioxane, chloroform and acetonitrile. A general trend appears to be that the more polar complexes (those incorporating methoxy and dimethylamino substituents) are less soluble in the more apolar solvents although even 5g and 6g are somewhat soluble in n-hexane. Coordinating solvents (acetonitrile, acetone and THF) cause decomposition of the complexes over the course of hours to days; decomposition is slowest for the most Lewis acidic complexes.
Single crystal X-ray crystallography
Colorless single crystals of Na4a′, Na4b, 5b, 5c, 5d, 5f, 6b, 6c, 6d and 6f were obtained by slow evaporation of DCM solutions at −20 °C and were characterized by single crystal X-ray crystallography. Crystallographic data are given in Tables S1–S3.† Selected distances and bond angles are given in Table 2 for the sodium salts of 4a′ and 4b, in Table 3 for the ethene complexes, and in Table 4 for the carbonyl complexes. Projections of the structures of Na4a′ and Na4b are shown in Fig. 1 and 2. As representative examples of the copper(I) complexes projections of the structures of 5b and 6b are shown in Fig. 3; projections of the other structures are provided in the ESI.†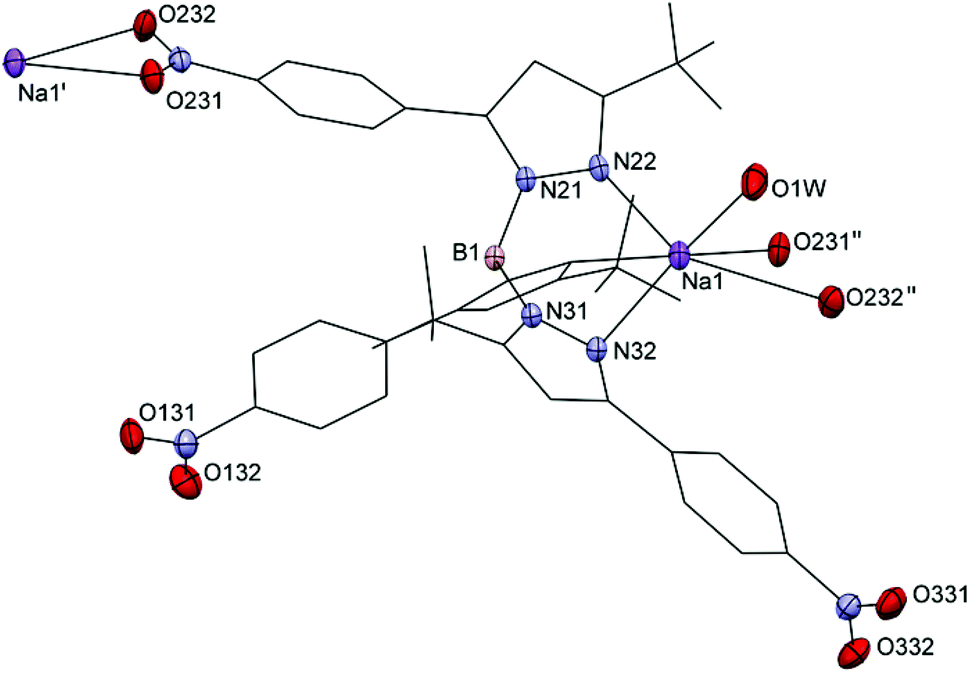 | ||
| Fig. 1 Projection of part of the structure of [Na(4a′)(H2O)]n with displacement ellipsoids plotted at the 50% probability level at 110(2) K. For clarity major parts of the ligand are shown in wireframe and hydrogen atoms have been omitted. Symmetry operations ′ = [⅔ − x, y − ½, ⅔ − z], ′′ = [⅔ − x, y + ½, ⅔ − z]. | ||
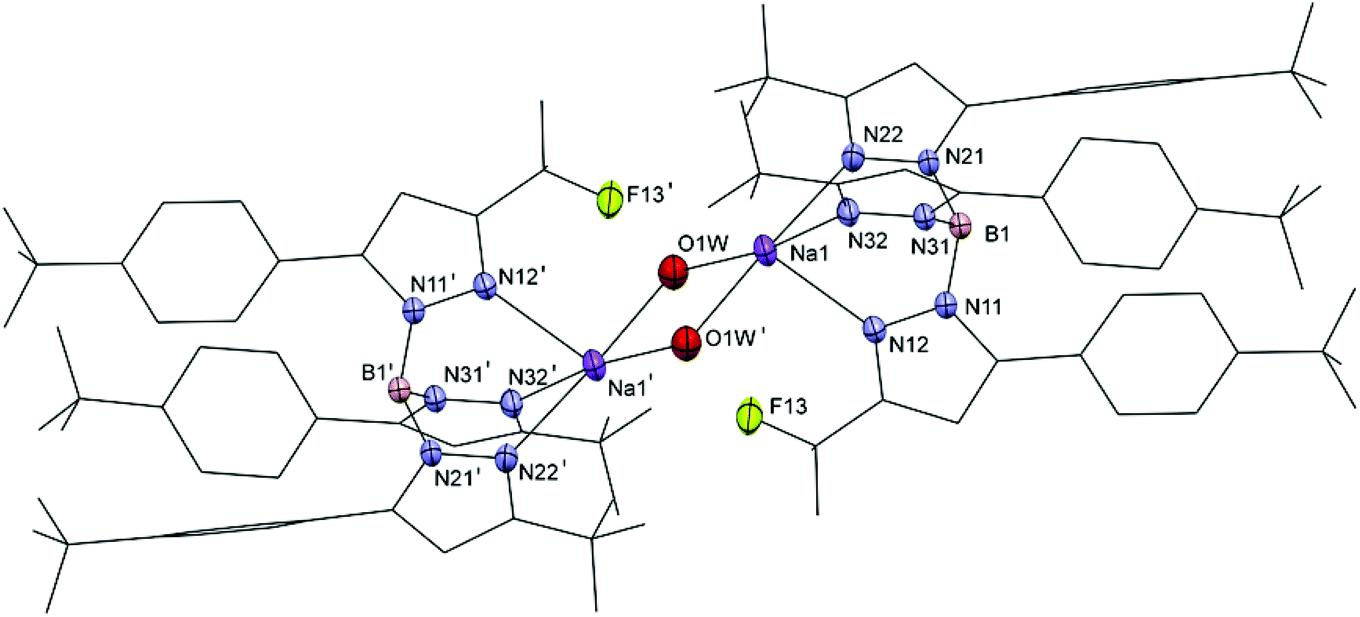 | ||
| Fig. 2 Projection of the structure of [Na2(4b)2(μ-H2O)2] with displacement ellipsoids plotted at the 50% probability level at 150(2) K. For clarity major parts of the ligand are shown in wireframe and hydrogen atoms have been omitted. Symmetry operation ′ = [−x, 1 − y, −z]. | ||
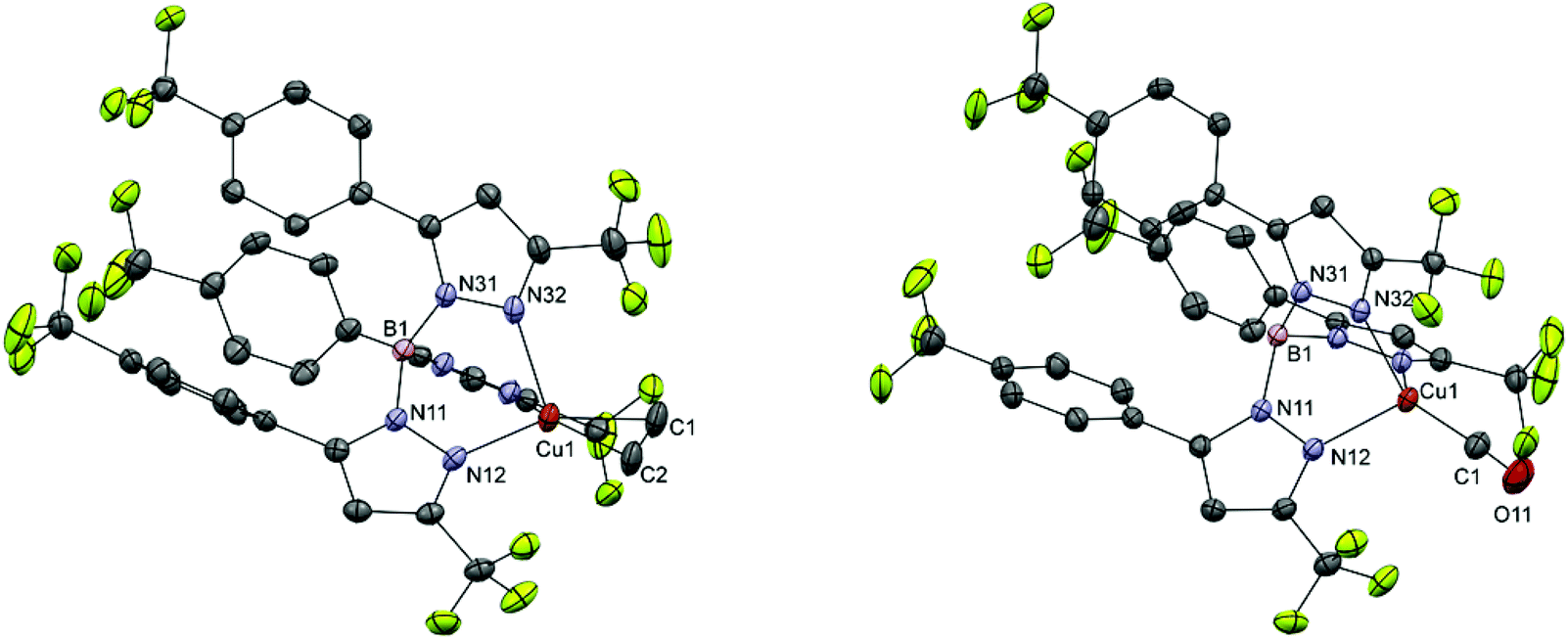 | ||
| Fig. 3 Projections of the structures of 5b (left) and 6b (right) with displacement ellipsoids plotted at the 50% probability level at 110(2) K. For clarity hydrogen atoms have been omitted. Projections of the structures of the other ethene and carbonyl complexes are available in the ESI.† | ||
| Bond distances (Å) | Na4a′ | Na4b | Bond angles (°) | Na4a′ | Na4b |
|---|---|---|---|---|---|
| Na1–N12 | 2.4731(15) | 2.4595(12) | O1W–Na1–O1W′ | 82.03(4) | |
| Na1–N22 | 2.4351(15) | 2.4440(12) | O1W–Na1–N12 | 99.18(6) | 104.30(4) |
| Na1–N32 | 2.5737(15) | 2.4355(12) | O1W–Na1–O231′ | 86.28(6) | |
| Na1–O1W | 2.2581(16) | 2.4512(12) | O1W–Na1–O232′ | 67.35(5) | |
| Na1–O1W′ | 2.4274(12) | O1W′–Na1–N12 | 100.89(4) | ||
| N11–N12 | 1.3566(19) | 1.3605(14) | O1W′–Na1–N32 | 99.69(4) | |
| N21–N22 | 1.3603(18) | 1.3589(15) | O1W–Na1–N22 | 103.78(6) | 104.57(4) |
| N31–N32 | 1.3551(19) | 1.3590(15) | N22–Na–N32 | 81.30(5) | 73.79(4) |
| Na1⋯F12 | 3.625(2) | N12–Na–F13′ | 162.1(3) | ||
| Na1⋯F13′ | 2.930(12) | O231′–Na1–O231′ | 46.64(4) | ||
| Na1⋯F21′ | 4.144(1) | ||||
| Na1⋯F23 | 3.197(4) | ||||
| Na1⋯F33 | 3.116(2) |
| Bond distances (Å) | ||||
|---|---|---|---|---|
| 5b | 5c | 5d | 5f | |
| CCethene |
1.357(4) | 1.311(14) | 1.350(3) | 1.351(8) |
| Cu–Cethene | 2.034(2) | 2.019(9) | 2.0428(16) | 2.054(5) |
| 2.051(5) | 2.035(9) | 2.0455(16) | 2.046(5) | |
| Cu–N12 | 2.0450(15) | 2.094(3) | 2.0255(14) | 2.026(4) |
| Cu–N22 | 2.0369(15) | 2.070(3) | 2.0164(13) | 2.046(4) |
| Cu–N32 | 2.2465(16) | 2.137(4) | 2.2964(13) | 2.224(4) |
| N11–N12 | 1.3626(19) | 1.356(5) | 1.3685(17) | 1.360(5) |
| N21–N22 | 1.3619(19) | 1.355(5) | 1.3648(17) | 1.363(6) |
| N31–N32 | 1.3631(19) | 1.363(5) | 1.3682(17) | 1.358(5) |
| N11–B1 | 1.553(2) | 1.558(5) | 1.553(2) | 1.564(5) |
| N21–B1 | 1.554(2) | 1.561(5) | 1.552(2) | 1.539(6) |
| N31–B1 | 1.545(2) | 1.537(6) | 1.545(2) | 1.551(6) |
| Bond angles (°) | ||||
|---|---|---|---|---|
| 5b | 5c | 5d | 5f | |
| a Bond distances and bond angles are only provided for one of the two crystallographically independent molecules. | ||||
| C–Cu–C | 38.79(11) | 37.7(4) | 38.55(7) | 38.5(2) |
| Cu–CC |
71.3(2) | 70.5(5) | 70.83(10) | 70.5(3) |
| 69.9(2) | 71.8(5) | 70.62(10) | 71.1(3) | |
| N12–Cu1–N22 | 92.55(6) | 89.58(14) | 90.77(5) | 94.64(15) |
| N22–Cu1–N32 | 87.03(6) | 90.60(13) | 90.77(5) | 88.25(14) |
| N12–Cu1–N32 | 89.81(5) | 90.17(13) | 85.68(5) | 87.01(14) |
| C1–Cu1–N12 | 149.26(9) | 126.2(4) | 115.04(7) | 148.49(18) |
| C1–Cu1–N22 | 112.92(9) | 111.5(3) | 147.96(7) | 110.75(18) |
| C1–Cu1–N32 | 107.70(9) | 135.8(4) | 108.95(7) | 111.30(18) |
| C2–Cu1–N12 | 111.18(9) | 111.8(3) | 151.13(6) | 110.68(18) |
| C2–Cu1–N22 | 147.41(11) | 148.9(3) | 111.15(6) | 144.25(19) |
| C2–Cu1–N32 | 114.02(12) | 110.8(3) | 111.57(7) | 116.93(18) |
a
| Bond distances (Å) | ||||
|---|---|---|---|---|
| 6b | 6c | 6d | 6f | |
| C1–O11 | 1.124(2) | 1.126(4) | 1.123(8) | 1.130(4) |
| Cu1–C1 | 1.7998(19) | 1.800(3) | 1.791(6) | 1.793(3) |
| Cu–N12 | 2.0642(15) | 2.069(2) | 2.0552(19) | 2.038(2) |
| Cu–N22 | 2.0496(15) | 2.056(2) | 2.062(2) | |
| Cu–N32 | 2.0505(15) | 2.057(3) | 2.075(2) | |
| N11–N12 | 1.3571(19) | 1.354(3) | 1.364(2) | 1.353(3) |
| N21–N22 | 1.3623(19) | 1.358(3) | 1.367(3) | |
| N31–N32 | 1.3625(19) | 1.360(3) | 1.358(3) | |
| N11–B1 | 1.548(2) | 1.553(3) | 1.553(2) | 1.560(3) |
| N21–B1 | 1.557(2) | 1.549(3) | 1.554(3) | |
| N31–B1 | 1.553(2) | 1.546(4) | 1.554(3) | |
| Bond angles (°) | ||||
|---|---|---|---|---|
| 6b | 6c | 6d | 6f | |
| a Bond distances and bond angles are only provided for one of the two crystallographically independent molecules. | ||||
| Cu1–C1–O11 | 176.0(2) | 177.4(3) | 180.0 | 178.3(3) |
| C1–Cu1–N12 | 118.66(8) | 122.17(11) | 125.13(5) | 126.66(11) |
| C1–Cu1–N22 | 129.95(8) | 127.99(11) | 121.77(12) | |
| C1–Cu1–N32 | 126.11(7) | 124.75(13) | 125.74(11) | |
| N12–Cu1–N22 | 90.60(6) | 90.14(9) | 90.19(7) | 91.65(8) |
| N22–Cu1–N32 | 88.85(6) | 90.58(8) | 90.23(7) | |
| N32–Cu1–N12 | 91.85(6) | 90.37(8) | 90.31(8) | |
Na4a′ crystallized as a coordination polymer with bridging NO2⋯Na+ interactions (with bidentate κ2-O,O′ coordination). The sodium ions are in a severely distorted octahedral coordination sphere comprising three nitrogen atoms from the ligand 4a′, two oxygen atoms from a nitro group of an adjacent complex and a water molecule, which is stabilized by hydrogen bridges to the nearby nitro group oxygen atoms. The Na–N distances range from 2.4351(15) to 2.5737(15) Å, the Na–Oaqua distance is 2.25581(16) Å and the Na–O2N distances are 2.5291(14) and 2.8633(14) Å. The severely distorted octahedral coordination sphere on the sodium ion in Na4a′ appears to be complemented by an intramolecular Na⋯FC contact between the sodium ion and a nearby trifluoridomethyl group. The upper limit to what may be considered as short contacts (and thus potentially dative bonds) between a hard donor/acceptor pair such as sodium ions and fluorine atoms in fluoridocarbons is somewhat vague as it depends on the ionic and covalent radii that are used. Using the ionic radius of six-coordinate Na+ ions reported by Shannon et al. (1.02 Å) Plenio et al. suggested an upper limit to the interatomic distance between Na+ ions and fluorine atoms in fluoridocarbons at 3.07 Å.22,23 It was further pointed out that Na⋯FC contacts typically have shallow potential wells, which means small outside forces such as those present in crystals because of packing can cause significant changes to the interatomic distance between the sodium ion and the fluorine atom. The distances observed for the Na⋯FC contacts in Na4a′ are 3.625(2) Å and 4.144(1) Å, considerably longer than the upper limit of 3.07 Å even when crystal packing effects are taken into account. The proximity of the trifluoridomethyl groups is therefore not considered to constitute actual bonding between the sodium ion and the fluorine atoms but merely dipolar interactions.
Na4b crystallized as a dimer comprising two sodium ions in distorted square-pyramidal coordination geometries. The sodium ions are each coordinated facially by three nitrogen atoms of the trispyrazolylborate ligand and by two bridging water molecules (Oaqua). The Na–N distances range from 2.4355(12) Å to 2.4595(12) Å and the Na–Oaqua distances are 2.4274(12) Å and 2.4512(12) Å. trans to the apical position are one intermolecular and two intramolecular Na⋯FC short contacts, at distances of respectively 2.930(12), 3.197(4) and 3.116(2) Å. With the foregoing considerations regarding Na⋯FC short contacts in mind it appears reasonable to consider both the intermolecular and intramolecular Na⋯FC distances in the structure of Na4b as short contacts.
The crystal structures of Na4a′ and [Na(4e)(H2O)]n contain non-bridging water ligands while the crystal structures of Na4b and [Na2(TpCF3,CH3)2(μ-H2O)2] are dinuclear and contain bridging water molecules. The Na–Oaqua distance in the crystal structure of Na4a′ of 2.2558(16) Å is comparable to the distance observed in [Na(4e)(H2O)]n (2.245(2) Å) but shorter than the distance in [Na2(TpCF3,CH3)2(μ-H2O)2] (2.417(2) Å), which is closer to the Na–Oaqua distance found in Na4b (2.4274(12) Å and 2.4512(12) Å).16
Complexes 5b, 5c, 5d, 5f, 6b, 6c, 6d and 6f all have highly similar coordination environments around the copper(I) center. In every case the copper(I) center is coordinated by three nitrogen atoms from the trispyrazolylborate ligand and either the carbon atom of the carbonyl ligand or the ethene ligand in the typical η2 coordination. Bond lengthening or contraction in the carbonyl and ethene ligands as a result of π-backbonding interactions are not evident from the bond lengths observed in the crystal structures and are assumed to be obscured by crystal packing effects. The Cu–N distances in the ethene complexes (5b, 5c, 5d and 5f) range from 2.0164(13) to 2.2964(13) Å with in all cases two shorter bonds (2.0164(13)–2.094(3) Å) and one longer bond (2.173(4)–2.2964(13) Å); such asymmetry in the Cu–N bond lengths was also observed in other [CuI(TpR,R′)(C2H4)] complexes.24,25 The Cu–N bond lengths in the carbonyl complexes (6b, 6c, 6d and 6f) fall in the range 2.038(2) Å–2.075(2) Å and do not feature the two ranges of Cu–N bond lengths observed in the ethene complexes. The unequal Cu–N bond lengths in the ethene complexes are attributed to the symmetry of ethene. Ethene ligands have a two-fold symmetry while the carbonyl ligands have full rotational symmetry. This means that while the carbonyl copper complexes can retain the approximate threefold symmetry of the trispyrazolylborate ligand, whereas the ethene complexes cannot. The resulting pseudo-twofold symmetry observed in the solid state of the ethene complexes means that π-backbonding into the π* orbitals of the ethene ligands can not occur with equal contributions from all of the pyrazole rings but mostly from two of the three rings. The resulting discrepancy in charge transfer from the rings to the π*-orbitals of the ethene ligand explains the presence of two short Cu–N bonds and one long Cu–N bond in complexes 5b–g. The compounds 6b, 6c and 6f crystallized with pseudo threefold rotational symmetry with Cu–C–O angles ranging from 176.0(2) to 178.3(3)°. Complex 6d crystallized with proper threefold rotational symmetry. The elongated thermal ellipsoid for the oxygen atom of the carbonyl ligand suggests that this ligand is most likely disordered as the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond may not be perfectly aligned along the threefold axis. The deviation from rotational symmetry in 6b, 6c and 6f is small and might be the result of crystal packing effects.
O bond may not be perfectly aligned along the threefold axis. The deviation from rotational symmetry in 6b, 6c and 6f is small and might be the result of crystal packing effects.
NMR spectroscopy
The 1H and 13C-NMR spectra of compounds 5a′, 5b–g and 6a′, 6b–g were recorded in deuterated DCM as it was found to be the only solvent in which the ethene complexes are all soluble and air-stable over longer periods of time (more than 30 minutes). CDCl3, acetone-d6, THF-d8, benzene-d6 and DMSO-d6 caused solutions of the copper ethene compounds to turn green after some time. The coordinating solvents DMSO-d6, acetone-d6, THF-d8 and benzene-d6 (a π-donor solvent) all appeared to be competing with the ethene ligands as peaks for free ethene grow over time on 1H NMR. The complexes with the less Lewis basic trispyrazolylborate ligands are more stable in solution. Complexes 5b–g gave very similar spectra with no evidence of anisotropy in the signal for the ethene ligand at room temperature. Considering the asymmetry observed in the crystal structures of complexes 5b–g splitting of the ethene peaks or at least some peak broadening could be expected. The absence of such effects appears to indicate fluxional behavior of the ethene ligand within the NMR timescale. In the NMR spectra of 5b–g the ethene ligands were detected as sharp singlets with chemical shifts between 4.81 and 4.96 ppm which are upfield from the chemical shift of free ethene (5.40 ppm in DCM-d2) but not as far upfield as the signals observed in complexes of non-fluorinated trispyrazolylborate ligands like [TpMe2Cu(C2H4)] (4.41 ppm in DCM-d2).26 The ethene protons of 5a′ showed as a singlet at 4.44 ppm which was assigned to the asymmetric isomer; a small singlet at 4.99 ppm in the same spectrum was tentatively assigned to the ethene protons of the complex 5a incorporating the symmetric isomer [4a]−. The ratio of the integrals of the ethene protons of the symmetric 5avs. the asymmetric species 5a′ was approximately 1:20. In all ethene complexes peak broadening in the presence of free ethene was not observed, indicating that the exchange of coordinated ethene, if at all, does not occur within the NMR time scale. Solutions of all ethene complexes in DCM-d2 show no peak for free ethene even upon prolonged standing. The 1H-NMR signals for the borohydrides were observed in the sodium compounds Na4a–g as well as the carbonyl and ethene complexes as broadened singlets with chemical shifts in the range 4.3–4.9 ppm. In principal the resonances of the borohydrides should be present as overlapping quartets and smaller septets as a result of splitting by 10B (I = 3) and 11B (I = 3/2). Typically, however, these resonances are observed as (broadened) quartets only in relatively symmetric environments. In less symmetric environments the quadrupoles of the boron nuclei are more pronounced and the borohydride resonances broaden to broad singlets without distinguishable splitting as is the case in this work.27
In the 13C-NMR spectra of complexes 6a′ and 6b–g no signals were observed for the CO ligands even after increasing the relaxation delay; we ascribe the lack of signals for CO to the low natural abundance of 13CO and peak broadening.28,29 The shifts of the resonances trispyrazolylborate ligands show only small differences between the ethene and the carbonyl complexes. For complexes 5b–g the change in the 1H-NMR and 13C-NMR shifts of the ethene protons and carbon atoms respectively is linearly correlated with the σp values of the para-substituents on the phenyl groups (R2 = 96% for 1H and R2 = 97% for 13C; see Fig. 4).
 | ||
| Fig. 4 1H chemical shifts (squares, red trend line. 5a′ was not included in the trend line) and 13C chemical shifts (triangles, blue trend line) of the ethene ligands vs. the Hammett σp parameters of the substituents on the trispyrazolylborate ligands in complexes 5a–g and 5a′. | ||
Infrared spectroscopy
The IR spectra of complexes 5a′, 5b–g and 6a′, 6b–g were recorded in the solid state at 1 cm−1 resolution. Complexes 6a′, 6b–g have CO stretching frequencies in the range 2096–2120 cm−1, close to that of free CO (2143 cm−1). This indicates that the compounds have relatively Lewis-acidic copper(I) ions compared to other, similar, copper(I) carbonyl complexes like [TpMe2CuCO] (2066 cm−1) and [TpPh2CuCO] (2080 cm−1, see Table 5 for more examples). The CO stretching frequencies correlate linearly (R2 = 95%) with the σp values of the substituents on the phenyl rings for compounds, with the exception of 6a′ and 6g (see Fig. 5). For complex 6a′ this is likely because the structure of [4a′]− is asymmetric and thus poorly comparable to the other ligands in the series, in analogy with the ethene resonances in the NMR spectra of compound 5a′. The CO stretching frequency observed in complex 6g is significantly higher than the value predicted based on the trend line in Fig. 5 (approximately 2076 cm−1) for which no obvious explanation is available. Possibly unusual crystal packing effects cause the CO stretching frequency to be different than predicted, but as all attempts to grow crystals failed this hypothesis remains untested. Unfortunately infrared measurements on a sample of 6g in DCM were inconclusive as no clear signal was observed until evaporation caused solid 6g to precipitate. CO stretching typically shows itself in infrared spectroscopy as sharp absorptions due to the symmetry of the vibration. One possible explanation for the absence of an obvious CO stretching peak in solution is a lowering of the symmetry of the bonding environment surrounding the carbonyl ligand. Such lowering of local symmetry could be the result of out-of-axis vibrations of the carbonyl ligand, somewhat similar to motion of the ethene ligands in the NMR experiments and presumably have similarly low barriers. We surmise that liberation of the carbonyl ligand from the copper ion is considerably more pronounced in solution compared to the solid state and that as a result peak broadening causes the weak signal to be drowned out by noise. | ||
| Fig. 5 Infrared CO stretching frequencies (red squares) and BH stretching frequencies (blue triangles) of complexes 6a′, 6b–gvs. the Hammett σp parameters of the substituents on the trispyrazolylborate ligands. The red linear trend line was calculated including all points except 6a′ and 6g (R2 = 0.95). The blue linear trend line was calculated including all complexes 6a′, 6b–g (R2 = 0.74). | ||
| Compound | C2H4a (ppm) |
ν CO (cm−1) |
ν
BHf (cm−1) |
CC (Å) |
CO (Å) |
Ref. | |
|---|---|---|---|---|---|---|---|
| 13C | 1H | ||||||
| a DCM-d2. b CDCl3. c C6D6. d Asymmetric isomer. e Symmetric isomer. f As observed in the Cu(I) CO complexes. | |||||||
| Free C2H4/CO | 123.2 | 5.40 | 2143 | 1.3384(10) | 1.13078(9) | 30, 31 | |
| [Tp(CF3)2CuL] | 89.1b | 4.96b | 2137 | 2634 | 1.325(9) | 1.110(5) | 18, 20, 32 |
| [Cu(4a′)L]d | 86.4 | 4.44 (4.99e) | 2105 | 2611 | This work | ||
| [Cu(4b)L] | 86.8 | 4.96 | 2120 | 2620 | 1.342(3) | 1.124(2) | This work |
| [Cu(4c)L] | 86.4 | 4.91 | 2113 | 2616 | 1.340(9) | 1.126(4) | This work |
| [TpCF3,MeCuL] | 2107 | 2575 | 1.122(3) | 33 | |||
| [Cu(4d)L] | 86.1 | 4.91 | 2103 | 2616 | 1.350(3) | 1.123(8) | This work |
| [TpCF3,PhCuL] | 85.8 | 4.91 | 2101 | 2639 | 1.30(1) | 1.113(5) | This work, 16 |
| [TpCF3CuL] | 85.8c | 4.89c | 2100 | 2507 | 1.34(1) | 1.126(5) | 18,20 |
| [Cu(4f)L] | 85.6 | 4.81 | 2096 | 2636 | 1.351(8) | 1.130(4) | This work |
| [Cu(4g)L] | 84.9 | 4.84 | 2096 | 2647 | This work | ||
| [TpMe2CuL] | 4.41 | 2066 | 2500 | 1.329(9) | 26,34 | ||
| [TpPh2CuL] | 81.6b | 3.53 | 2080 | 2635 | 1.381(19) | 1.08(1) | 8,35 |
The B–H moieties of the trispyrazolylborate ligands feature IR stretching frequencies that are typically found as small, broad peaks around 2600 cm−1 (see Table 5 and Fig. 5) in Lewis-basic trispyrazolylborate complexes. In order to compare the B–H stretching frequencies of ligands 4a–g the carbonyl complexes 6a′ and 6b–g and other, structurally similar, carbonyl complexes were compared. The pseudo-threefold axial symmetries observed in such carbonyl complexes mean that the comparison is not likely to be distorted by local asymmetry. The B–H stretching frequencies of complexes 6a′ and 6b–g were observed in the region 2611–2647 cm−1. The B–H stretching frequencies show considerably less linearity vs. the σp parameters of the substituents of the trispyrazolylborate ligands 4a′ and 4b–g (R2 = 0.74) than the CO stretching frequencies. Intriguingly, compared to the unexpectedly high CO stretching frequency the B–H stretching frequencies of 6a′ and 6g appear almost exactly on the trend line in Fig. 5. The most Lewis-basic trispyrazolylborate ligands have the highest B–H stretching frequencies as donation of electron density towards the borohydride increase the strength of the hydridic bond.
Discussion
In this work seven new fluorinated trispyrazolylborate ligands ([TpCF3,4-RPh]−, R = NO2, CF3, Cl, F, H, MeO and NMe2) and their copper(I) complexes with carbon monoxide and ethene were prepared. The ligands form an isosteric spectrochemical series based on the electronic properties of the substituents placed on the 4-position of the phenyl rings. The pyrazoles 3a–g were readily prepared from ethyl trifluoridoacetate and 4′-substituted acetophenones using a Claisen condensation, followed by cyclization with hydrazine. The synthesis of the trispyrazolylborate ligands requires fine-tuning of the conditions depending on the specific pyrazole using solventless reactions whenever possible; 4-methylanisole was used as a solvent if temperature control was required or the melting point of the pyrazole was too high. To the best of our knowledge [4a′]− is only the second example in the literature of a trispyrazolylborate ligand to include the synthetically challenging nitro group, the first being [TpNO2]−.36The trispyrazolylborate ligands studied in this work all formed as their symmetric isomers with the trifluoridomethyl groups in the 3-positions of the pyrazole rings, except [4a′]− which formed as a mixture of symmetric and asymmetric species. The major product was the asymmetric species in which one of the pyrazole rings is connected to the boron center with the nitrogen atom adjacent to the phenyl ring. The regioselectivity usually observed in the synthesis of trispyrazolylborate ligands is a result of the relative steric and electronic properties of the substituents on the 3 and 5 positions of the pyrazole, which influence the nucleophilicity of the nitrogen atoms. Typically the bulkiest substituent assumes the 3 positions of the pyrazole rings in the trispyrazolylborate anion. However, if one of the substituents is considerably more electron withdrawing than the other it will assume the 3 position even if the other substituent is bulkier. This regioselectivity is pronounced in ligands with clearly sterically or electronically differentiated substituents like [TpPh,Me]−, [TpCF3,Me]− and [TpCF3]− but breaks down when the steric and electronic differences are small such as in [TpiPr,Me]− which formed a 4:1 mixture of symmetric and the asymmetric isomers.37 Evidently the strongly electron withdrawing nitro group in [4a′]− causes the electronic disparity between the trifluoridomethyl groups and phenyl rings to diminish sufficiently to shift the thermodynamic equilibrium of the system so that the asymmetric isomer is favored over the symmetric isomer. The absence of the formation of asymmetric isomers in the other ligands in the series puts an upper limit on the σp parameter of the substituents that can be used to synthesize symmetric [TpCF3,4R-Ph]− ligands between +0.54 (CF3) and +0.78 (NO2).
The good correlation between the chemical shifts of the ethene protons in complexes 5b–gvs. σp of the substituents on the phenyl rings of the ligands was used to predict the chemical shift of the ethene protons of the symmetric complex 5a. Based on the trend line in Fig. 4 and the σp parameter of the nitro group the signal for the symmetric complex 5a was predicted to be around 4.98 ppm and indeed a weak singlet was found at 4.99 ppm. Comparison of the integrals of the ethene protons in the symmetric and asymmetric complexes gave an approximate 1:20 ratio. Unfortunately attempts to locate the infrared CO stretching frequency of the symmetric isomer of 6a using the σp parameter of the nitro group and the trend line in Fig. 5 were unsuccessful as the predicted CO stretching frequency for the symmetric isomer 6a at 2128 cm−1 would be obscured by the much stronger absorption at 2105 cm−1 of the asymmetric isomer 6a′.
More difficult to explain is the unexpectedly high CO stretching frequency observed for 6g, which was predicted to be at 2076 cm−1 but instead was found at 2096 cm−1, the same value as found for 6f. It appears implausible that the CO stretching frequencies of 6f and 6g are equal as an effect of equal amounts of π-backbonding interactions as the 1H and 13C chemical shifts of the ethene ligands in 5f and 5g almost exactly conform to the predicted values. Unfortunately the higher than expected CO stretching frequency of 6g cannot be conclusively explained by crystal packing effects as infrared spectroscopy on 6g in solution was inconclusive and all attempts at crystallization failed.
With the exceptions of 6a and 6g the predictability of the chemical shifts of the ethene protons in the ethene complexes and CO stretching frequencies in the carbonyl complexes with respect to the σp parameters of the substituents of the phenyl rings in the trispyrazolylborate ligands is excellent. The ability of the ligands to propagate the electron donating or withdrawing effects of the substituents on the phenyl rings over as many as eight bonds and almost one nanometer, is remarkable. The explanation for these long range effects is found in the ability of the ligands to propagate charges by means of resonance structures. Further underscoring the importance of resonance effects is the observation that the B–H stretching frequencies observed in the carbonyl complexes 6a′ and 6b–g are much less predictable than the CO stretching frequencies because the nitrogen atom adjacent to the borohydride is not part of these resonance structures. We conclude that indeed the π-backbonding ability of the copper(I) centers can be modified extensively through substitution of the trispyrazolylborate ligands without significantly affecting the steric properties of the binding pocket surrounding the metal center.
Conclusions
In this work the synthesis and characterization of copper(I) complexes comprising a spectrochemical series of trispyrazolylborate ligands and ethene or carbon monoxide have been described. The ethene and carbonyl complexes described in this work are essentially isostructural around their copper(I) centers while their electronic properties vary significantly and predictably based on the Hammett σp parameters of the substituents placed on the trispyrazolylborate ligands. The spectroscopic data reveal good correlations between the chemical shifts of the ethene protons and CO stretching frequencies vs. the Hammett σp parameters. Notable exceptions to the, otherwise predictable, compounds in this work were the ligand [4a′]− and the carbonyl complex 6g. The ligand [4a′]− formed almost exclusively as an asymmetric isomer with one of the pyrazole rings counter-rotated so as to have its 4′-nitrophenyl substituent in the 3-position instead of the trifluoridomethyl group. We attribute the asymmetry of [4a′]− to the strongly electron withdrawing properties of the nitro group. Copper complex 6g shows a surprisingly high CO stretching frequency. That this aberrant result is not due to trend-breaking properties of the ligand is evident from the good correlation between the predicted and found values in the corresponding ethene complex 5g. The results presented in this work underscore the usefulness of combining the use of more than a single ancillary ligand probe with the systematic study of this promising and important class of ligands.Experimental
General information
All manipulations of air-sensitive compounds were performed in an atmosphere of purified argon gas using standard Schlenk techniques. All solvents were purchased from commercial sources and reagent grade. Solvents used for air-sensitive manipulations were dried and deaerated using a PureSolv MD 5 Solvent Purification System and stored on 3 Å molecular sieves under argon. When appropriate, glassware was flame dried in vacuo immediately prior to use. 1H and 13C NMR spectra were recorded on a Bruker DPX300 spectrometer (300 MHz for 1H and 75.44 MHz for 13C); Bruker DMX400 spectrometer (400 MHz for 1H and 100.6 MHz for 13C); Bruker Avance AV500 spectrometer (500 MHz for 1H, 160 MHz for 11B and 126 MHz for 13C) or Bruker Avance 600 (600 MHz for 1H, 193 MHz for 11B and 151 MHz for 13C). Chemical shifts are given in ppm and referenced using the deuterated solvents as internal references for 1H and 13C.3813C spectra were recorded using 1H-decoupling. Elemental analyses were performed using a Perkin Elmer 2400 series II CHNS/O analyzer or by the Microanalytical laboratory Kolbe in Germany. IR spectra were recorded on a Perkin Elmer UATR Two FT-IR spectrometer set to a resolution of 1 cm−1. HRMS spectra were recorded on a Thermo Scientific LTQ Orbitrap XL high resolution FT-MS system in MeCN. Intermediates 2a, 2b, 2d and 2f were synthesized according to a literature procedure using KOt-Bu in THF instead of sodium in methanol.392d was purified by conversion to the copper(II) diketonate instead of column chromatography as described below.Single crystal X-ray crystallography
All reflection intensities were measured at 110(2) K or 150(2) K (only for Na4b) using a SuperNova diffractometer (equipped with Atlas detector) with Cu Kα radiation (λ = 1.54178 Å) under the program CrysAlisPro (Versions 1.171.36.32/1.171.37.31 Agilent Technologies, 2013/2014). The same program CrysAlisPro was used to refine the cell dimensions and for data reduction. All structures were solved with the program SHELXS-2013/2014 (Sheldrick, 2008) and were refined on F2 with SHELXL-2013/2014.40 Analytical numeric absorption corrections based on a multifaceted crystal model were applied using CrysAlisPro. The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). The H atoms were placed at calculated positions (unless otherwise specified) using the instructions AFIX 43 with isotropic displacement parameters having values 1.2 times Ueq of the attached C atoms. The H atoms attached to C1/C2, C3/C4 (ethene), and to B1 and B2 were found from difference Fourier maps, and their atomic coordinates were refined freely. The H atoms attached to the water molecules (only for Na4a′ and Na4b) and to the B atoms were found from difference Fourier maps, and their coordinates were refined freely using the DFIX instructions. For 5b, 5c, 5d and 5f, the C–H and H⋯H distances of the ethene molecules were restrained using the DFIX instructions. Their isotropic temperature factors were fixed for the ethene molecules (1.2 times Ueq of the attached C atoms).Ligand and complex synthesis
2c (23.0 g, 93.4 mmol) was dissolved in 100 mL n-PrOH and hydrazine hydrate (5.0 mL, 103 mmol) was added dropwise. The reaction was heated to reflux for 2 hours. The reaction was then cooled to 0 °C in an ice bath. 10.5 mL 37% HCl was added after which the ice bath was removed and the reaction was heated to 100 °C for 30 minutes. The reaction was cooled down to room temperature and the mixture was diluted with 600 mL cold water. The product was collected by filtration and washed with water (approx. 1 L). The product was dried in vacuo and purified by vacuum sublimation (160 °C, 10−4 atm) to yield the product as a white solid. Yield 20.4 g (88%). M.p. 151–153 °C. 1H NMR (300 MHz, CDCl3) δ 13.09 (s, 1H), 7.46 (d, J = 8.6 Hz, 2H), 7.38 (d, J = 8.5 Hz, 2H), 6.64 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 144.55, 143.45 (q, J = 37.2 Hz), 135.62, 129.59, 126.87, 126.26, 120.97 (q, J = 268.9 Hz), 101.23 (q, J = 1.6 Hz).
2g (12.7 g, 50 mmol) was dissolved in 125 mL EtOH and cooled to 0 °C in an ice bath. Hydrazine hydrate (2.7 mL, 56 mmol) was added dropwise, when the addition was complete the ice bath was removed and the reaction was stirred at room temperature for 30 minutes before the reaction was heated to reflux for approx. 17 hours. The solvent was evaporated in vacuo and 100 mL 3 M HCl was added. The solution was heated to reflux for 5 minutes, cooled to room temperature and diluted with 500 mL aqueous saturated NaHCO3 solution and 100 mL DCM. The layers were separated and the aqueous fraction was extracted with DCM (2 × 50 mL). The combined organic fractions were washed with water (2 × 100 mL) and brine (1 × 100 mL) and dried over Na2SO4. The solvent was removed in vacuo and the product was recrystallized from hot MeOH/water to yield a white solid. Yield (10.2 g, 80%). M.p. 179–181 °C. 1H NMR (300 MHz, DMSO) δ 12.03 (s, 1H), 7.45 (d, J = 8.9 Hz, 2H), 6.73 (d, J = 8.9 Hz, 2H), 6.62 (s, 1H), 3.00 (s, 6H). 1H NMR (300 MHz, Acetone-d6) δ 12.86 (s, 1H), 7.66 (d, J = 9.1 Hz, 2H), 6.85–6.78 (m, 3H), 3.00 (d, J = 1.6 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 151.07, 145.70, 143.79 (q, J = 35.7 Hz), 126.79, 121.54 (q, J = 268.9 Hz), 115.91, 112.44, 99.50, 40.35.
C30H16BF9N9O6−) 780.11674 found 780.11778.
C33H16BF18N6−) 849.12366 found 849.12078.
:1 mixture of the Et2O and THF adducts. Yield 3.2 g (62%). M.p. 192 °C (decomposition). 1H NMR (400 MHz, CDCl3) δ 7.00 (d, J = 8.4 Hz, 6H), 6.77 (d, J = 8.4 Hz, 6H), 6.44 (s, 3H), 4.57 (bs, 1H), 3.92 (ddd, J = 6.5, 4.2, 2.7 Hz, 2H, coord. THF), 3.55 (q, J = 7.0 Hz, 2H, coord. Et2O), 2.02–1.96 (m, 2H, coord. THF), 1.24 (t, J = 7.0 Hz, 3H, coord. Et2O). 13C NMR (100 MHz, Acetone-d6) δ 150.38, 143.25, 134.56, 131.69, 131.07, 128.52, 121.44, 104.67. HRMS (ESI neg.) m/z calcd For [M−] (C30H16BCl3F9N6−) 747.04459 found 747.04770.
C30H16BF12N6−) 699.13324 found 699.13033.
C33H25BF9N6O3−) 735.19320 found 735.19424.
C36H34BF9N9−) 774.28810 found 774.28907.
General method for synthesis of the copper(I) ethene complexes (5a′, 5b–g)
Na4a′ or Na4b–g (50 mg) was dissolved in 5 mL dry, degassed DCM. The solution was bubbled with ethene for 2 minutes. CuI (1.05 eq.) was then added and the solution was saturated/bubbled with ethene for another 2 minutes. The flask was then stoppered and left to stir at medium speed overnight to form a white suspension. The suspension was filtered using a syringe filter (0.45 μm PTFE) and the solvent was removed in vacuo to yield the products as white powders. The complexes could be further purified by recrystallization from DCM/pentane at −20 °C under an ethene atmosphere to yield colorless blocks except for 5a′ which was brown.General method for the synthesis of the copper(I)–CO complexes (6a′, 6b–g)
Na4a′ or Na4b–g (50 mg) was dissolved in dry, degassed DCM. The solution was bubbled with argon for 2 minutes and placed in a glass-lined autoclave (volume 12 mL). CuI (1.05 eq.) was added and the autoclave was closed and purged with dry nitrogen gas. The autoclave was then pressurized with carbon monoxide to at least 5 atm and left to stir at room temperature for 20 hours. The autoclave was then bubbled with dry nitrogen gas to remove unreacted carbon monoxide. The resulting white suspension was filtered using a syringe filter (0.45 μm, PTFE) and the solvent was removed in vacuo to leave behind the product as a white powder except in the case of 6a′ which was brown.Acknowledgements
This work is supported by NanoNextNL, a micro and nanotechnology consortium of the Government of the Netherlands and 130 partners. We thank Jos van Brussel for CHN analysis; Gerwin Spijksma for HRMS and Fons Lefeber and Karthick Sai Sankar Gupta are acknowledged for assistance with the NMR experiments.References
- For an explanation of the nomenclature of poly(pyrazolyl)borates see p. 5 of ref. 4.
- S. Trofimenko, J. Am. Chem. Soc., 1966, 88, 1842–1844 CrossRef CAS.
- S. Trofimenko, J. Am. Chem. Soc., 1967, 89, 3170–3177 CrossRef CAS.
- S. Trofimenko, Scorpionates: The Coordination Chemistry of Poly(pyrazolyl)borate Ligands, Imperial College Press, London, 1999 Search PubMed.
- C. Pettinari, Scorpionates II: Chelating Borate Ligands, Imperial College Press, London, 2008 Search PubMed.
- S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS.
- H. V. R. Dias and C. J. Lovely, Chem. Rev., 2008, 108, 3223–3238 CrossRef CAS PubMed.
- N. Kitajima, K. Fujisawa, C. Fujimoto, Y. Morooka, S. Hashimoto, T. Kitagawa, K. Toriumi, K. Tatsumi and A. Nakamura, J. Am. Chem. Soc., 1992, 114, 1277–1291 CrossRef CAS.
- M. A. Casado, V. Hack, J. A. Camerano, M. A. Ciriano, C. Tejel and L. A. Oro, Inorg. Chem., 2005, 44, 9122–9124 CrossRef CAS PubMed.
- B. S. Hammes, M. W. Carrano and C. J. Carrano, J. Chem. Soc., Dalton Trans., 2001, 1448–1451 RSC.
- J. C. Calabrese, P. J. Domaille, S. Trofimenko and G. J. Long, Inorg. Chem., 1991, 30, 2795–2801 CrossRef CAS.
- C. Pérez Olmo, K. Böhmerle, G. Steinfeld and H. Vahrenkamp, Eur. J. Inorg. Chem., 2006, 2006, 3869–3877 CrossRef.
- S. Kealey, N. J. Long, P. W. Miller, A. J. P. White and A. D. Gee, Dalton Trans., 2008, 2677–2679 RSC.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- X. Kou, J. Wu, T. R. Cundari and H. V. R. Dias, Dalton Trans., 2009, 915–917 RSC.
- H. V. R. Dias and T. Goh, Polyhedron, 2004, 23, 273–282 CrossRef CAS.
- N. B. Jayaratna, I. I. Gerus, R. V. Mironets, P. K. Mykhailiuk, M. Yousufuddin and H. V. R. Dias, Inorg. Chem., 2013, 52, 1691–1693 CrossRef CAS PubMed.
- H. V. R. Dias, H.-J. Kim, H.-L. Lu, K. Rajeshwar, N. R. de Tacconi, A. Derecskei-Kovacs and D. S. Marynick, Organometallics, 1996, 15, 2994–3003 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- H. V. R. Dias, H. L. Lu, H. J. Kim, S. A. Polach, T. Goh, R. G. Browning and C. J. Lovely, Organometallics, 2002, 21, 1466–1473 CrossRef CAS.
- W. A. King, G. P. A. Yap, C. D. Incarvito, A. L. Rheingold and K. H. Theopold, Inorg. Chim. Acta, 2009, 362, 4493–4499 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- H. Plenio, Chem. Rev., 1997, 97, 3363–3384 CrossRef CAS PubMed.
- C. Martín, J. M. a. Muñoz-Molina, A. Locati, E. Alvarez, F. Maseras, T. s. R. Belderrain and P. J. Pérez, Organometallics, 2010, 29, 3481–3489 CrossRef.
- H. V. R. Dias, X. Y. Wang and H. V. K. Diyabalanage, Inorg. Chem., 2005, 44, 7322–7324 CrossRef CAS PubMed.
- J. S. Thompson, R. L. Harlow and J. F. Whitney, J. Am. Chem. Soc., 1983, 105, 3522–3527 CrossRef CAS.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- K. Fujisawa, T. Ono, Y. Ishikawa, N. Amir, Y. Miyashita, K.-i. Okamoto and N. Lehnert, Inorg. Chem., 2006, 45, 1698–1713 CrossRef CAS PubMed.
- M. Kujime, T. Kurahashi, M. Tomura and H. Fujii, Inorg. Chem., 2007, 46, 541–551 CrossRef CAS PubMed.
- J. L. Duncan, Mol. Phys., 1974, 28, 1177–1191 CrossRef CAS.
- W. Gordy, Microwave Spectroscopy, Wiley, New York, 1953 Search PubMed.
- H. V. R. Dias and H.-L. Lu, Inorg. Chem., 1995, 34, 5380–5382 CrossRef CAS.
- K. Fujisawa, M. Yoshida, Y. Miyashita and K.-i. Okamoto, Polyhedron, 2009, 28, 1447–1454 CrossRef CAS.
- M. I. Bruce and A. P. P. Ostazewski, J. Chem. Soc., Dalton Trans., 1973, 2433–2436 RSC.
- Unpublished results.
- M. Pellei, G. Papini, G. G. Lobbia, S. Ricci, M. Yousufuddin, H. V. Rasika Dias and C. Santini, Dalton Trans., 2010, 39, 8937–8944 RSC.
- M. Cano, J. V. Heras, S. Trofimenko, A. Monge, E. Gutierrez, C. J. Jones and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 1990, 3577–3582 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- S. Büttner, A. Riahi, I. Hussain, M. A. Yawer, M. Lubbe, A. Villinger, H. Reinke, C. Fischer and P. Langer, Tetrahedron, 2009, 65, 2124–2135 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1430362–1430371. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04006j |
| This journal is © The Royal Society of Chemistry 2015 |