
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extending motifs in lithiocuprate chemistry: unexpected structural diversity in thiocyanate complexes†
Andrew J.
Peel
a,
Madani
Hedidi
bc,
Ghenia
Bentabed-Ababsa
c,
Thierry
Roisnel
d,
Florence
Mongin
b and
Andrew E. H.
Wheatley
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: aehw2@cam.ac.uk; Fax: (+)44 (0)1223 336362
bChimie et Photonique Moléculaires, Institut des Sciences Chimiques de Rennes, UMR 6226, Université de Rennes 1-CNRS, Bâtiment 10A, Campus de Beaulieu, 35042 Rennes, France
cLaboratoire de Synthèse Organique Appliquée, Faculté des Sciences, Université d'Oran 1 Ahmed Ben Bella, BP 1524 El M'Naouer, 31000 Oran, Algeria
dCentre de Diffractométrie X, Institut des Sciences Chimiques de Rennes, UMR 6226, Université de Rennes 1-CNRS, Bâtiment 10B, Campus de Beaulieu, 35042 Rennes, France
First published on 4th November 2015
Abstract
The new area of lithio(thiocyanato)cuprates has been developed. Using inexpensive, stable and safe CuSCN for their preparation, these complexes revealed Lipshutz-type dimeric motifs with solvent-dependent point group identities; planar, boat-shaped and chair shaped conformers are seen in the solid state. In solution, both Lipshutz-type and Gilman structures are clearly seen. Since the advent in 2007 of directed ortho cupration, effort has gone into understanding the structure-reactivity effects of amide ligand variation in and alkali metal salt abstraction from Lipshutz-type cuprates such as (TMP)2Cu(CN)Li2(THF) 1 (TMP = 2,2,6,6-tetramethylpiperidide). The replacement of CN− with SCN− is investigated presently as a means of improving the safety of lithium cuprates. The synthesis and solid state structural characterization of reference cuprate (TMP)2Cu(CN)Li2(THP) 8 (THP = tetrahydropyran) precedes that of the thiocyanate series (TMP)2Cu(SCN)Li2(L) (L = OEt29, THF 10, THP 11). For each of 9–11, preformed TMPLi was combined with CuSCN (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the presence of sub-stoichiometric Lewis base (0.5 eq. wrt Li). The avoidance of Lewis basic solvents incurs formation of the unsolvated Gilman cuprate (TMP)2CuLi 12, whilst multidimensional NMR spectroscopy has evidenced the abstraction of LiSCN from 9–11 in hydrocarbon solution and the in situ formation of Gilman reagents. The synthetic utility of 10 is established in the selective deprotometalation of chloropyridine substrates, including effecting transition metal-free homocoupling in 51–69% yield.
:1) in the presence of sub-stoichiometric Lewis base (0.5 eq. wrt Li). The avoidance of Lewis basic solvents incurs formation of the unsolvated Gilman cuprate (TMP)2CuLi 12, whilst multidimensional NMR spectroscopy has evidenced the abstraction of LiSCN from 9–11 in hydrocarbon solution and the in situ formation of Gilman reagents. The synthetic utility of 10 is established in the selective deprotometalation of chloropyridine substrates, including effecting transition metal-free homocoupling in 51–69% yield.
Introduction
Enhanced methods for the regioselective functionalization of aromatics that avoid the complications associated with the use of traditional main group organometallic bases1 are of ongoing interest. This search led, in 1999,2 to the development of the first of what have become known as ‘synergic bases’3 of the type RmM(NR′2)nAM (R = alkyl; m = 0, 2, 3; M = less polarizing metal; NR′2 = amide; n = 1, 2, 3; AM = (more polarizing) alkali metal). These have, for example, previously incorporated M = Al,4 Cd,5 Mg,6 Mn7 and Zn,8 and have shown hitherto unachievable potential in anionic activation9 and templated polymetalation.10 In a similar vein, new lithium cyanocuprates11 have been central to the development of directed ortho cupration (DoC)12 (Fig. 1).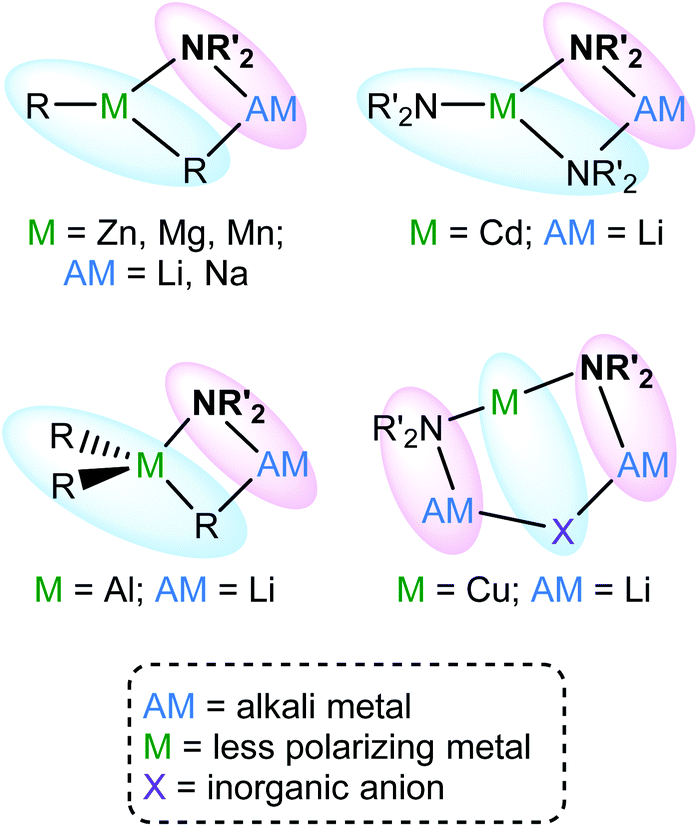 | ||
| Fig. 1 Known structural motifs in synergic base chemistry. | ||
Following the inception of lithium cuprate chemistry13 through the development of R2CuLi,14 attempts have focused on enhancing reactivity. Modifications have taken two major forms. Firstly, lithium amidocuprates have been developed, offering often unique reactivities as well as the potential of the amido group as a non-transferable ligand and as a chiral auxiliary.15 Secondly, there has been a focus on the incorporation of LiCN within lithium cuprates,16 the presumption being that the cyano group would be transferred to Cu to give a higher order (tricoordinate) copper centre.17 The issue of the Cu-sequestering of cyanide has been discussed at length in the literature, though calculations,18 spectroscopy19 and X-ray diffraction20 have increasingly pointed to the retention of lower order (dicoordinate) copper. This was noted too in the recently developed field of DoC transformations, with the 2:1 reaction of TMPLi (TMP = 2,2,6,6-tetramethylpiperidide) and CuCN giving complexes that could be characterized by X-ray diffraction. Results revealed that in the solid state such Lipshutz formulation cuprates were dimers based on (TMP)2Cu(CN)Li2(L) (L = OEt21, THF 2)12,21 monomers that clearly lacked Cu–CN interactions (Scheme 1).
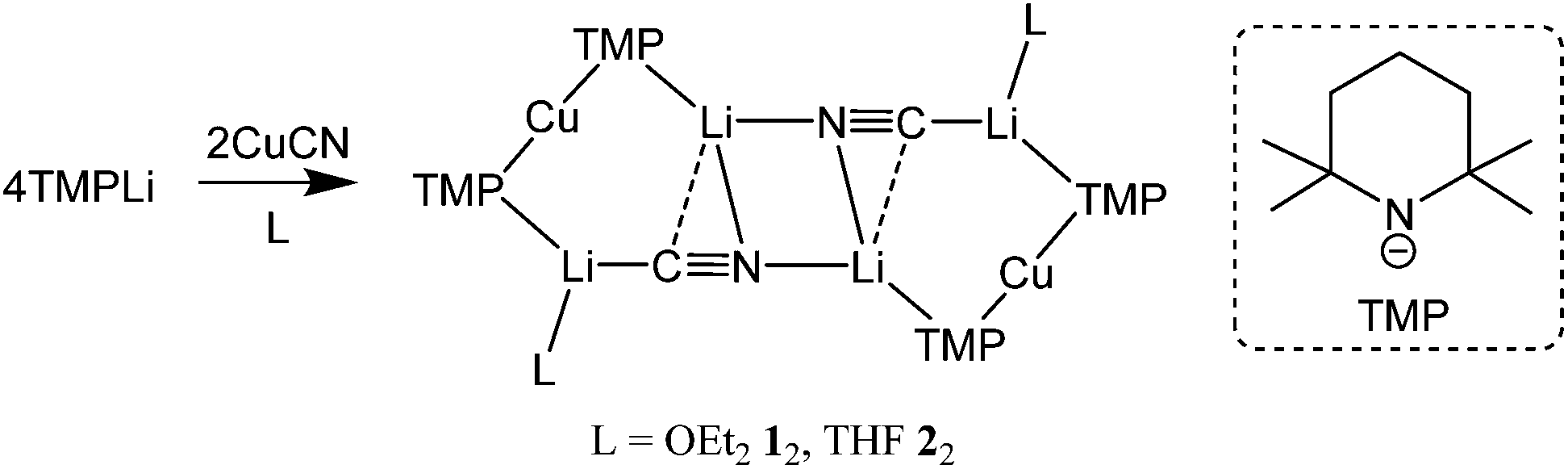 | ||
| Scheme 1 Formation of the dimers of cuprates 1 and 2. | ||
Finally, the reaction of CuCN, RLi and TMPLi established that the inclusion of cyanide in the cuprate structure was by no means guaranteed, by furnishing the externally solvated amido(organyl) monomers RCu(TMP)Li(L)n (R = Ph, L = THF, n = 3 3; R = Me, L = TMEDA = N,N,N′,N′-tetramethylethylenediamine, n = 1 4).22 This observation took on added importance when recent DFT analysis suggested the in situ conversion of cyanide-incorporating cuprates to reactive Gilman reagents as the precursor to DoC reaction.23
Recent studies have sought to extend the principles established by the isolation of 1 and 2 to the use of general inorganic anions. This has led to the isolation and full characterization of (TMP)2Cu(X)Li2(L) (X = Cl 5, Br 6, I 7, L = OEt2, THF).23,24 These species have been viewed as being Lipshutz-type by virtue of their demonstrating essentially the same structural principles as 1 and 2. They have been successfully tested in the deprotonative metalation of halopyridines,25 notably in the course of azafluorenone synthesis.24 Herein we extend this principle further, introducing the use of the thiocyanate anion as a non-toxic but potentially synthetically useful analogue of the cyanide components in 1 and 2. Preliminary results reveal novel variations in thiocyanatocuprate structure as a function of solvent both in the solid state and in solution.
Results and discussion
Solid state analysis
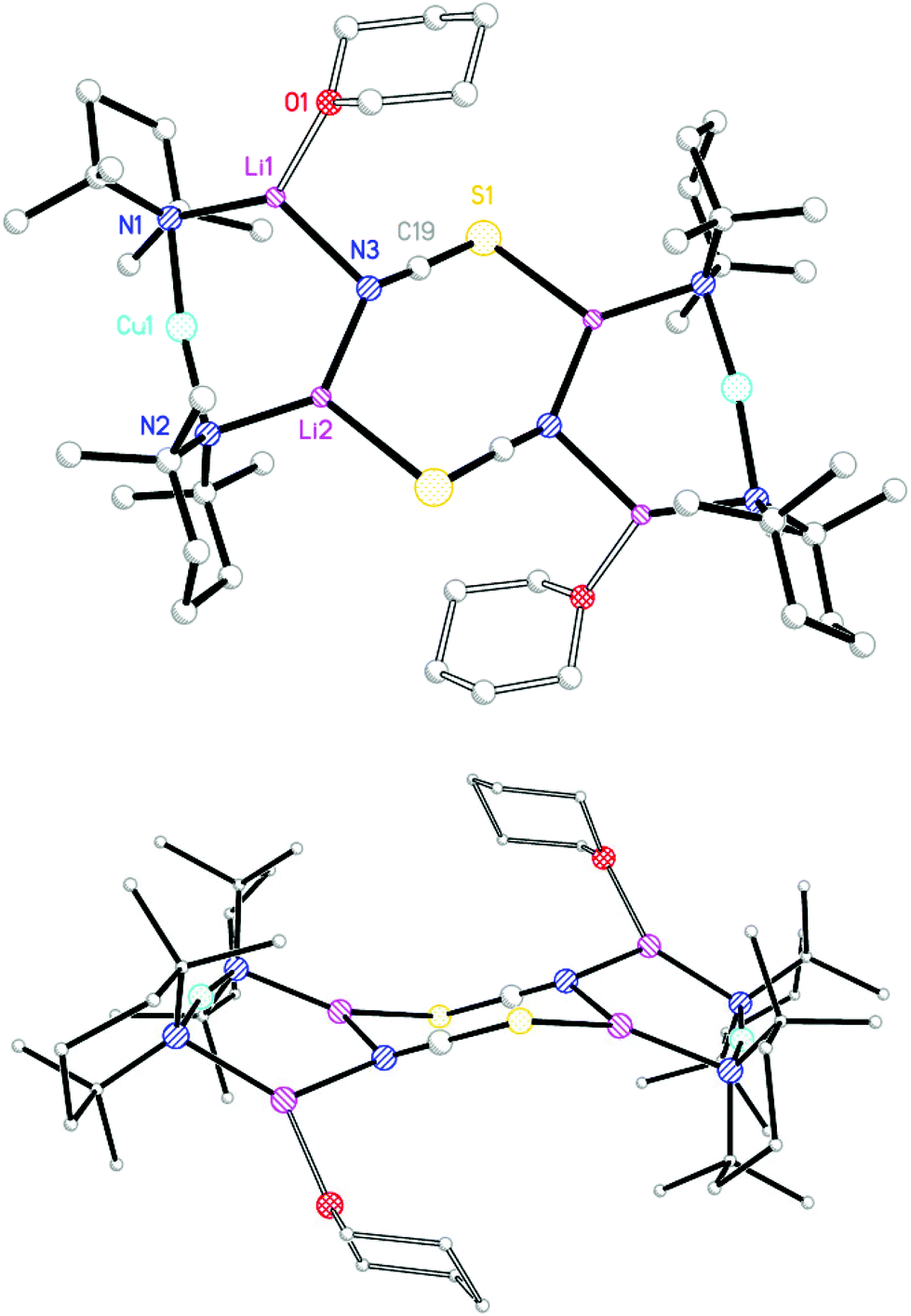
With this study aiming to probe new thiocyanatocuprate bases solvated by THF, OEt2 and THP (= tetrahydropyran) it was first necessary to complete the series of cyanocuprates 1,12221 and (TMP)2Cu(CN)Li2(THP) 8. To do this a hexane solution of TMPLi containing also THP (0.5 eq. wrt Li) was added to a suspension of CuCN (0.5 eq. wrt Li) in toluene. Following the addition of hexane the mixture was heated to reflux and then filtered to give a pale-straw coloured solution from which block-like crystals could be obtained (Scheme 2).
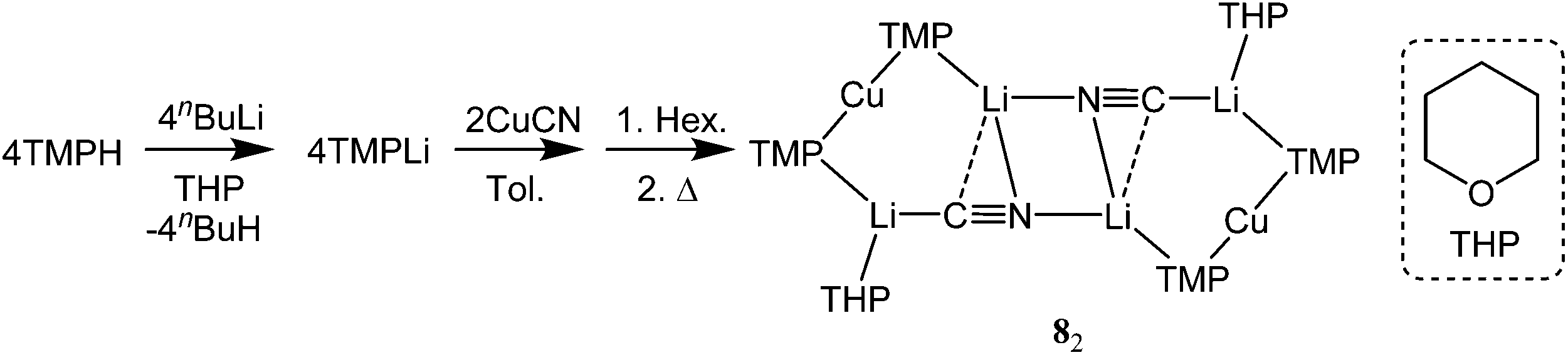 | ||
| Scheme 2 Synthesis of 8. | ||
The product was shown by 1H NMR spectroscopy to incorporate TMP and THP in a 2:1 ratio, suggesting a formulation analogous to that previously reported for 1 and 2. Corroboration of this view came from X-ray diffraction, which established the product to be 8 and to be the analogue of 1 and 2 (Fig. 2). All three structures proved to be relatively flat. IR spectroscopy on 8 revealed a dominant C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching mode at
N stretching mode at ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 2104.3 cm−1, with a signal developing at 2138.3 cm−1 upon air exposure (see ESI, Fig. S1†), which compared closely with prior work.22
= 2104.3 cm−1, with a signal developing at 2138.3 cm−1 upon air exposure (see ESI, Fig. S1†), which compared closely with prior work.22
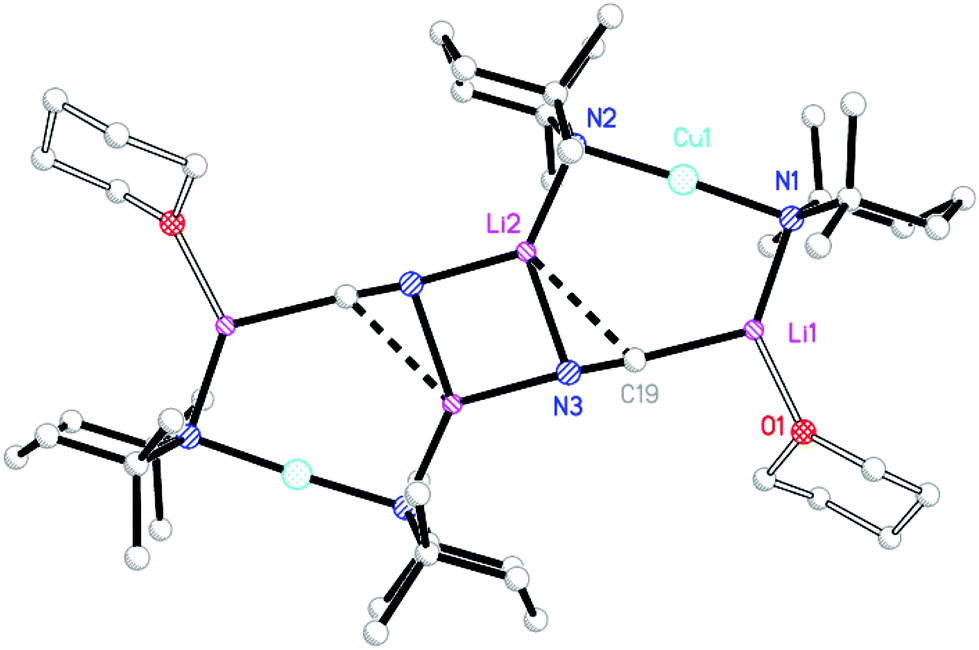 | ||
| Fig. 2 Molecular structure of 82. H-atoms and ligand disorder omitted. Selected bond lengths (Å) and angles (°): N1–Cu1 1.950(3), N2–Cu1 1.926(3), N1–Li1 1.981(7), N2–Li2 1.951(7), C19–Li1 2.111(13), N3–Li2 2.171(13), N3–Li2A 2.047(14), Cu1–N1–Li1 90.9(2), Cu1–N2–Li2 97.2(2), N1–Li1–C19 122.3(5), N2–Li2–N3 135.2(5), N2–Li2–N3A 130.5(5). | ||
Moving to the employment of CuSCN in an attempt to render a safer analogue of OEt2-solvate 1, a low temperature solution of TMPH in Et2O/toluene was treated with nBuLi. The resulting solution was transferred to a suspension of CuSCN in toluene. The mixture was heated to reflux, turning from pale cream to grey-black, whereupon filtration gave a yellow solution. Storage at room temperature gave a low yield of needle-like crystals after 1 day, which dissolved with further standing, and after several days were replaced with crystals of pseudo-rhombic habit in low yield. 1H NMR spectroscopy revealed these to comprise TMP and OEt2 in a 2:1 ratio. Though 13C NMR spectroscopy failed to suggest the presence of SCN, this ligand was obviously incorporated, with IR spectroscopy revealing peaks at 2064.6 (s) and 1996.6 (m) cm−1 (Fig. S2†). Air exposure of the sample for 30 s. incurred the growth of a signal at 2107.1 cm−1, which compared with 2066 and 2070 cm−1 for calculated free [SCN]− and LiSCN, respectively.26,27 Overall, these data suggest the presence of [S–CN]− moieties. This spectroscopic suggestion of a (TMP)2Cu(SCN)Li2(OEt2) formulation was verified by X-ray diffraction, which established an essentially flat dimer 92 (Scheme 3 and Fig. 3). The thiocyanatocuprate was found to be based on an essentially planar 8-membered (LiNCS)2 metallocyclic core (the maximal deviation of any atom from a mean plane defined by the four Li+ ions and the two thiocyanate ligands being just 0.12 Å). The suggestion from vibrational spectroscopy that formally Li–S-bonded [S–CN]− moieties are present27 is perhaps most clearly reinforced crystallographically by the shortness of the N3–C19 distance (1.149(3) Å), though at 1.631(3) Å S1–C19 hints at some level of delocalization in the anionic ligand. Formal, S-anion behaviour is also suggested by the significantly inequivalent N3–Li1 and N3–Li2 distances (2.250(5) and 1.998(5) Å, respectively); these relative lengths are consistent with a single N-based lone pair bisecting the Li1–N3–Li2 angle but favouring interaction with Li2 on grounds of electrostatic directionality (Fig. 3a).28
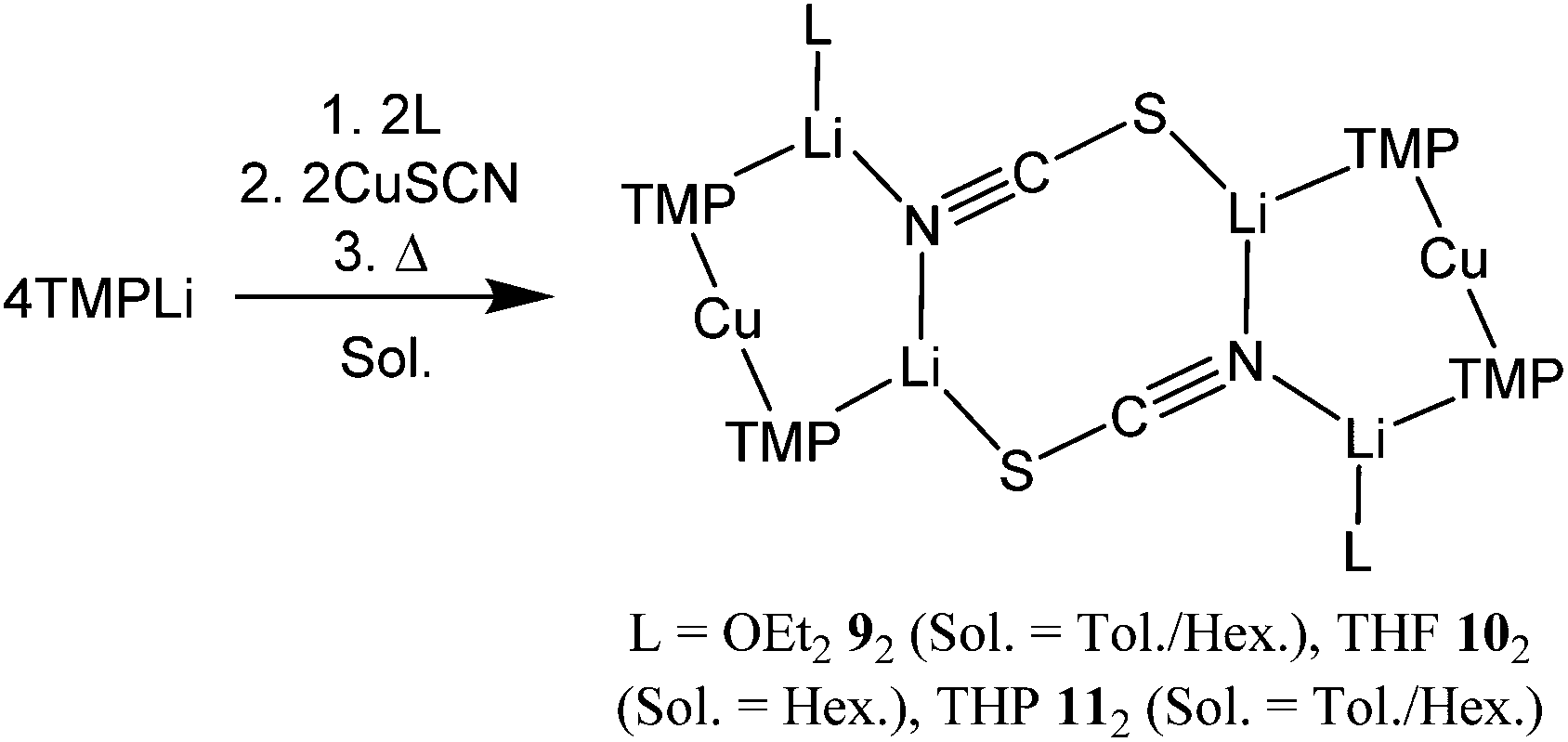 | ||
| Scheme 3 Synthesis of 9–11. | ||
 | ||
| Fig. 3 (a) Molecular structure of 92. H-atoms omitted. Selected bond lengths (Å) and angles (°): N1–Cu1 1.9175(18), N2–Cu1 1.9074(18), N1–Li1 2.015(4), N2–Li2 1.970(4), S1A–Li2 2.518(4), N3–Li1 2.250(5), N3–Li2 1.998(5), N3–C19 1.149(3), S1–C19 1.631(3), Cu1–N1–Li1 83.82(15), Cu1–N2–Li2 89.46(15), N1–Li1–N3 123.7(2), N2–Li2–N3 128.7(2), Li1–N3–C19 110.7(2), Li2–N3–C19 140.0(2), S1A–Li2–N3 113.17(19), Li2–S1A–C19A 103.92(13); (b) side-on view emphasising the essentially flat dimer core. | ||
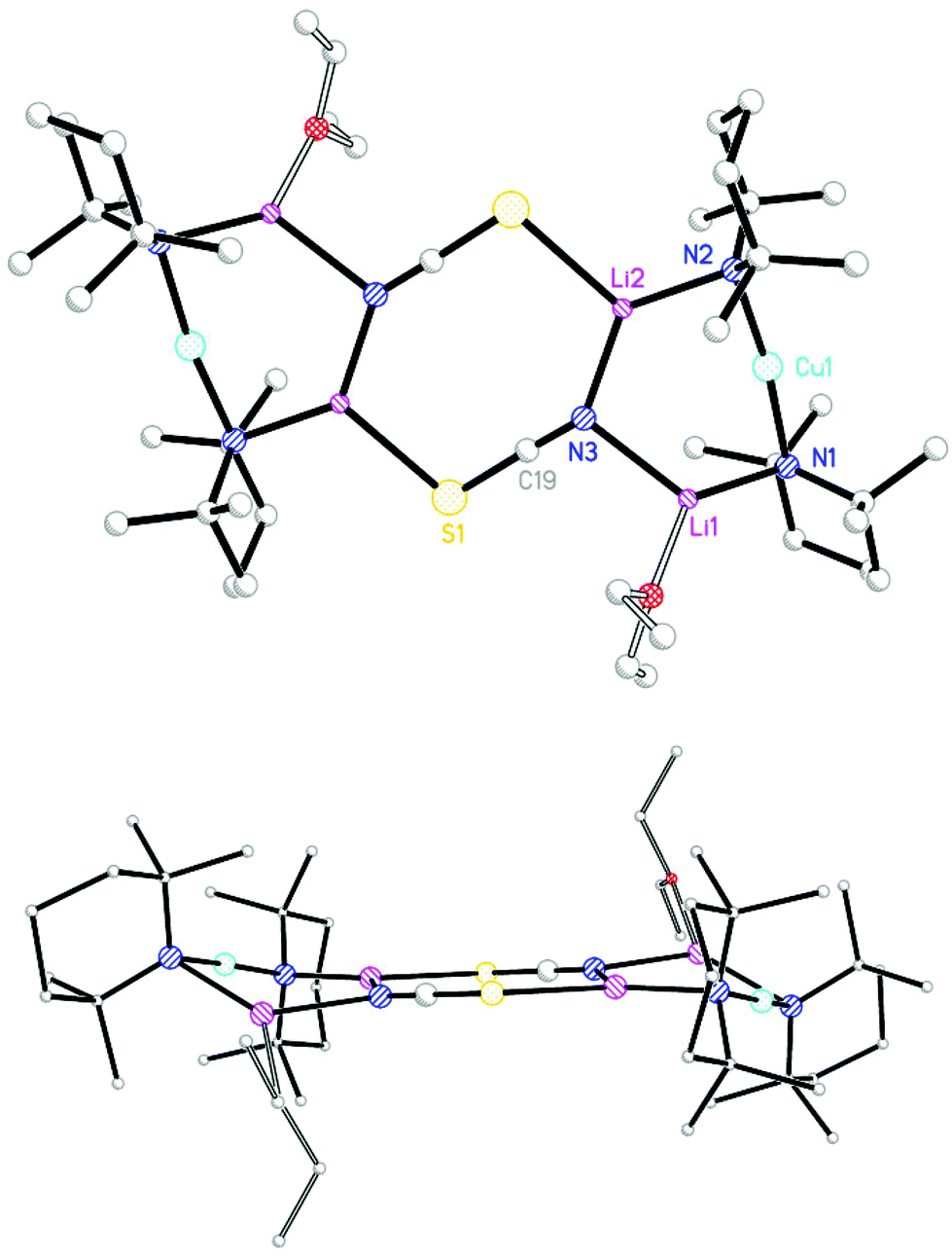
Having obtained the OEt2-solvate of (TMP)2Cu(SCN)Li2 and established its essential planarity, THF was introduced to probe whether the structure remained fundamentally unchanged (cf. 1vs.2). TMPLi in hexane and THF (0.5 eq. wrt Li) was added to CuSCN, allowing the isolation of colourless prismatic crystals (Scheme 3). NMR spectroscopy revealed that, as for 9, these crystals incorporated TMP and Lewis base in a 2:1 ratio and a SCN group (δ 141.5 ppm by 13C NMR spectroscopy). Corroboration of the last point came from IR spectroscopy, with two peaks seen at 2050.4 (s) and 1998.0 (m) cm−1 (Fig. S3,†cf. 2064.6 and 1996.6 cm−1 in 92), the signals being replaced by a peak at 2105.7 cm−1 after air exposure for 30 s. X-ray diffraction was undertaken, with data revealing a dimer based on (TMP)2Cu(SCN)Li2(THF) 10. However, in contrast to the structure of 92, 102 exhibited a novel boat conformation (Fig. 4) based on the aggregation of two crystallographically independent monomers (of which one will be representatively discussed) and in which the geometry at sulfur is fundamentally altered relative to that seen in 92. The result is that the two sulfur centres in 102 constitute the hinge about which deviation from planarity of the dimer operates. In spite of this, diffraction fails to reveal any significant difference in the bond lengths associated with sulfur (cf. S1–C19 1.631(3), S1–Li2A 2.518(4) Å in 92 and S2–C24 1.625(3), S2–Li1 2.500(6) Å in 102). This suggested equivalent thiocyanate anion structure to that seen in 92 even if the angle at sulfur now dramatically constricted on account of the dimer folding along the S⋯S vector (Li2–S1A–C19A 103.92(13)° in 92 compares with Li1–S2–C24 91.72(17)° in 102). The two 6-membered Cu-containing CuLi2N3 rings in 102 are essentially planar (angles at Li1 and N3 sum to 359.9° and 359.6°, respectively).
 | ||
| Fig. 4 (a) Molecular structure of 102. H-atoms omitted. Selected bond lengths (Å) and angles (°): N1–Cu1 1.908(2), N2–Cu1 1.906(2), N1–Li1 1.977(5), N2–Li2 1.995(6), S2–Li1 2.500(6), N3–Li1 2.071(6), N3–Li2 2.105(6), N3–C1 1.159(4), S1–C1 1.627(3), Cu1–N1–Li1 87.45(18), Cu1–N2–Li2 87.66(18), N1–Li1–N3 130.7(3), N2–Li2–N3 128.9(3), Li1–N3–C1 125.0(3), Li2–N3–C1 124.8(3), S2–Li1–N3 108.8(2), Li1–S2–C24 91.72(17); (b) view emphasising the boat-shaped dimer core. | ||
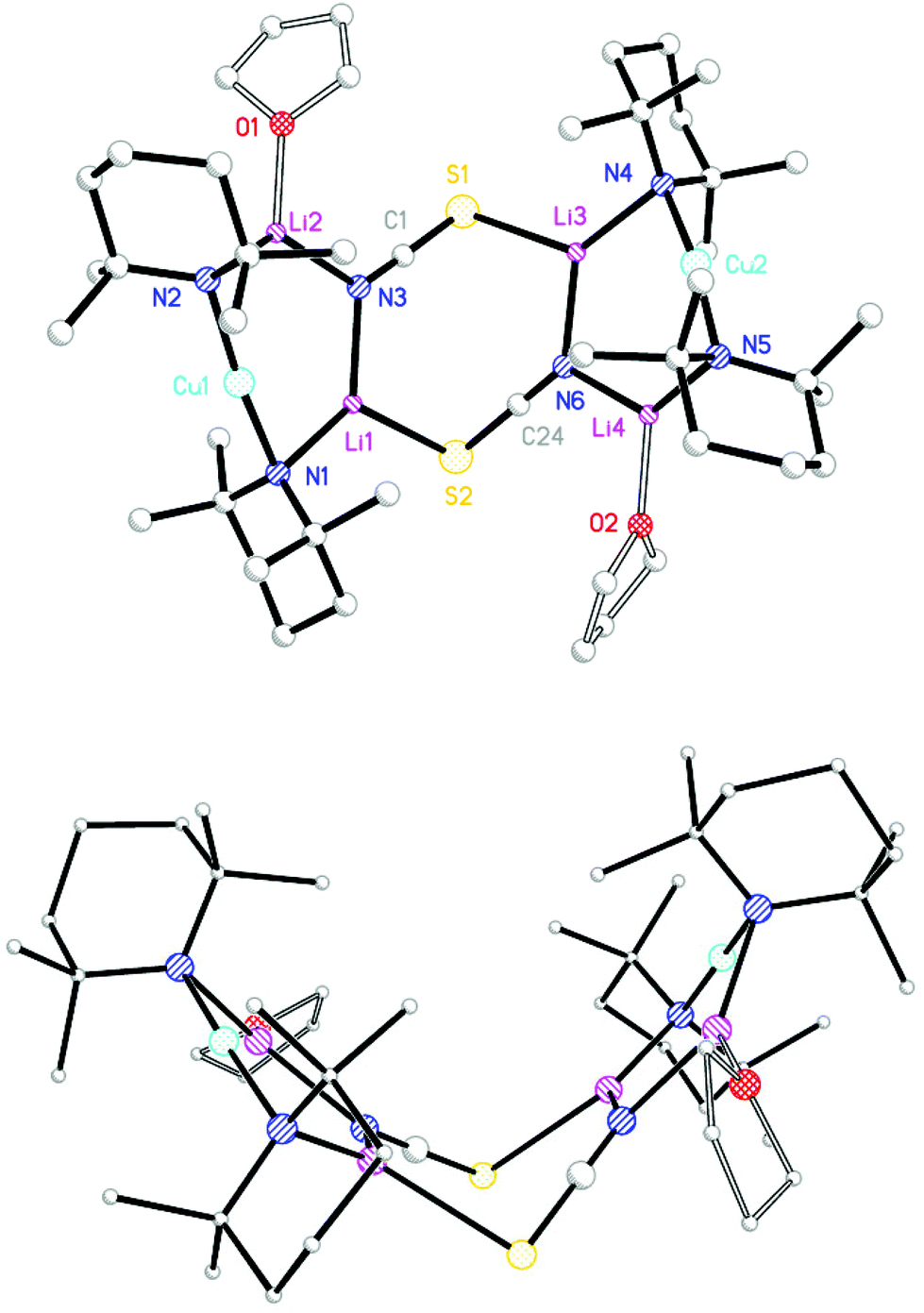
Having established significantly different geometries for 92 and 102 attention switched to use of the Lewis base THP. TMPLi in hexane/toluene and THP (0.5 eq. wrt Li) was reacted with CuSCN, leading to the isolation of a modest yield of colourless blocks (Scheme 3). NMR spectroscopy revealed the presence therein of TMP and THP in a 2:1 ratio. Meanwhile, IR spectroscopy demonstrated signals attributable to SCN at 2063.2 (s) and 2006.5 (w) cm−1, these being replaced by peaks at 2173.7 and 2108.5 cm−1 upon air exposure for 30 s. (Fig. S4†). X-ray diffraction confirmed the expected formulation of the product as the dimer of (TMP)2Cu(SCN)Li2(THP) 11 but revealed a structure (Fig. 5) that, in contrast to the structures of 92 and 102, exhibited a clear chair conformation ostensibly by virtue of the significant displacement (by ±0.43 Å) of Li2 and its symmetry equivalent from the mean plane described by the Li1–N3–C19–S1 fragment and its symmetry analogue. In order to allow this to occur not only S1 but also N3 necessarily deviate from planarity (in contrast to the geometry of N3 in 102), with angles at nitrogen now summing to 351.6°. This small deviation from planarity (presumably limited to maximize the electrostatic directionality of nitrogen) is reflected also in the geometry at S1 for which, at 96.09(12)°, the Li–S–C angle is intermediate between those seen in the two previous structures (103.92(13)° in 92, 91.72(17)° in 102). Diffraction reveals bond lengths of S1–C19 1.632(3), S1–Li2A 2.464(4) Å associated with sulfur, which are essentially the same as those seen in 92 and 102 and point to a common thiocyanate ligand electronic structure.
 | ||
| Fig. 5 (a) Molecular structure of 112. H-atoms omitted. Selected bond lengths (Å) and angles (°): N1–Cu1 1.9204(17), N2–Cu1 1.9148(17), N1–Li1 1.990(4), N2–Li2 1.968(4), S1A–Li2 2.464(4), N3–Li1 2.164(5), N3–Li2 2.005(4), N3–C19 1.162(3), S1–C19 1.632(3), Cu1–N1–Li1 110.75(19), Cu1–N2–Li2 88.84(13), N1–Li1–N3 123.4(2), N2–Li2–N3 128.5(2), Li1–N3–C19 106.7(2), Li2–N3–C19 134.5(2), S1A–Li2–N3 107.90(18), Li2A–S1–C19 96.09(12); (b) the chair-shaped dimer core. | ||
The solvent sensitivity of thiocyanatocuprate formation was also investigated by eliminating external Lewis bases. As described above, reaction of TMPLi (4 mmol) with CuSCN (2 mmol) in bulk hydrocarbon doped with L (= OEt2, THF, TMP; 0.5 eq. wrt Li) gave (TMP)2Cu(SCN)Li2(L) 9–11. However, crystallographic analysis of the product revealed that the avoidance of donor solvent afforded a convenient and clean route to material which demonstrated a single signal at δ 0.90 ppm by 7Li NMR spectroscopy. Crystallography subsequently revealed previously reported (TMP)2CuLi 12,23 which was a dimer in the solid state, with IR spectroscopy corroborating the absence of SCN ligands (see ESI Fig. S5†). However, the suggestion from 7Li NMR spectroscopy that omitting Lewis base avoided contamination of the product with minor impurities23 led us to undertake further re-characterization, obtaining a simple 13C NMR spectrum of the Gilman cuprate (see below) which served to aid our interpretation of the more complex behaviour of 9–11 in solution.
NMR spectroscopy
The improved synthesis of 12 made available clean NMR spectra of the Gilman cuprate (TMP)2CuLi, with 13C and 7Li NMR spectra obtained in d6-benzene that could then be deployed in order to deconvolute the solution behaviour of thiocyanates 9–11. The 13C NMR spectrum revealed a simple set of signals attributable to [TMP]− with singlets due to the 2,6-, 3,5- and 4-positions of the rings seen at δ 54.2, 42.1 and 19.2 ppm, respectively, and two Me signals located at δ 40.1 and 34.5 ppm (Fig. 6d). Comparison with the analogous spectrum obtained for diethyl ether complex 9 (at a concentration of ∼20 mg/0.7 mL d6-benzene, Fig. 6a) revealed it to be dominated by essentially identical signals, with a small amount of decomposition to give TMPH peaks, with minor traces of Lipshutz-type cuprate (see below) also manifest. These data strongly suggest ostensible conversion from the Lipshutz-type structure seen crystallographically for this system (Fig. 3a) to a Gilman formulation in solution.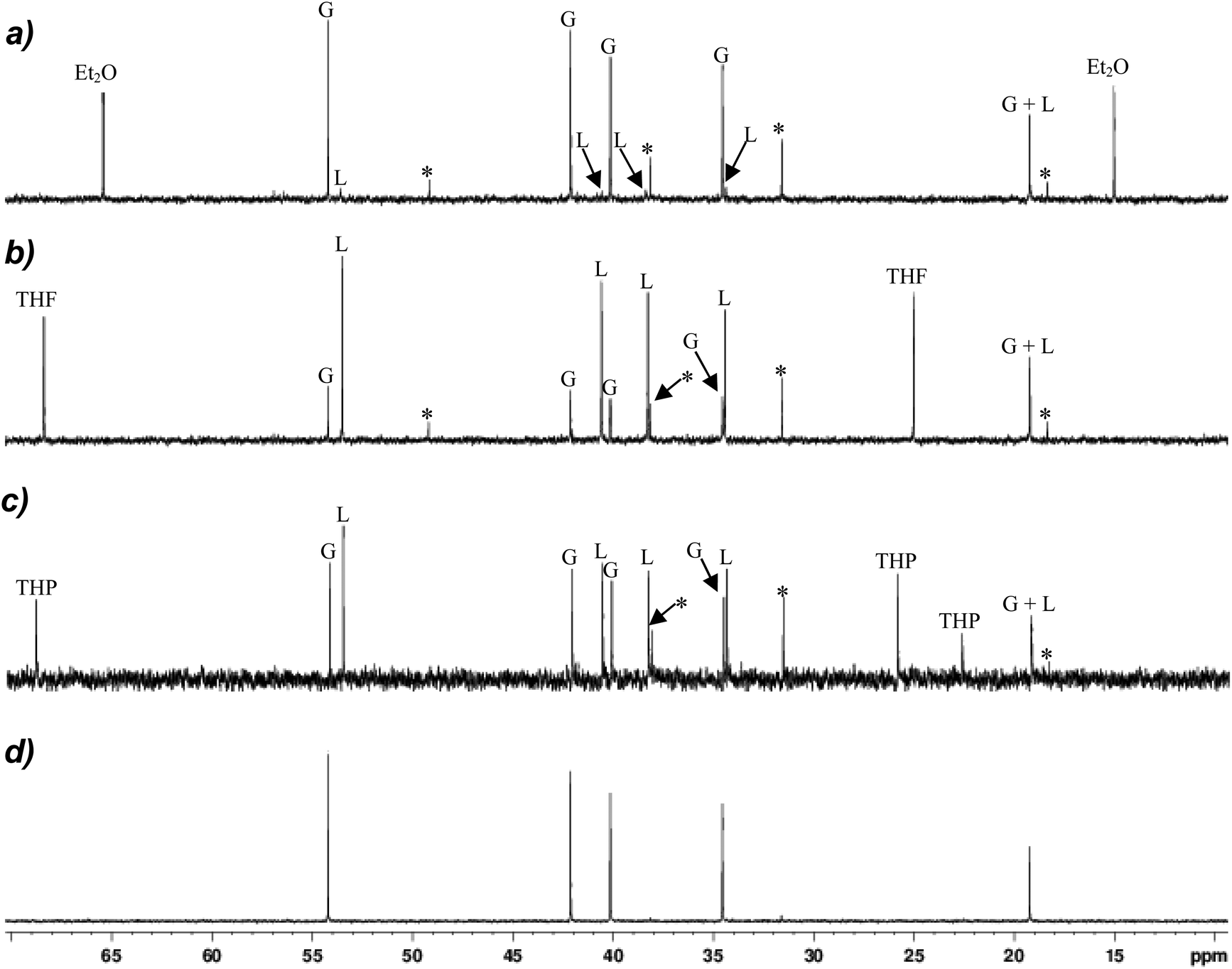 | ||
| Fig. 6 13C NMR spectroscopic data obtained in d6-benzene for (a) 9, (b) 10, (c) 11, (d) 12 (*TMPH, G = Gilman, L = Lipshutz-type). Sample concentrations for 9–11 were 20 mg/0.7 mL. | ||
Moving to the 13C NMR spectroscopic data for THF-solvate 10 at the same concentration, a more complicated system is revealed. (The SCN component itself can be located at δ 141.5 ppm for this system in a 50 mg/0.7 mL sample, Fig. S11.†) It is immediately obvious that whilst signals attributable to the amide ligands in the Gilman cuprate remain, they no longer represent the dominant species in solution (Fig. 6b). Instead, major signals are now observed at the high-field side of each Gilman resonance (at δ 53.5, 40.6, 38.3, 34.4 ppm) and, in one case (δ 19.2 ppm), coincident with it. These are attributed to the corresponding carbon atoms in the Lipshutz-type structure, which appears to be substantially retained in solution, with an approximate Gilman:Lipshutz-type ratio of 1:3 suggested. Equivalently concentrated THP complex 11 (20 mg/0.7 mL in d6-benzene) revealed a similar picture to that demonstrated by 10, albeit the distribution of Gilman and Lipshutz-type species is approaching equivalence (Fig. 6c). Moreover, similar to 10, the analysis of a more concentrated sample (50 mg/0.7 mL in d6-benzene) located the thiocyanate resonance at δ 141.7 ppm (Fig. S11†).
Coincident with the near-quantitative in situ conversion of Lipshutz-type crystalline 9 to a Gilman cuprate in d6-benzene (Fig. 6a) the deposition of a white powder was observed in the NMR tube to which 9 (20 mg/0.7 mL) had been added. Based on a control experiment in which pre-isolated Lipshutz-type cuprate was dissolved in benzene and the resulting white deposit analyzed by IR spectroscopy, we attribute this observation to LiSCN precipitation (Fig. S6†). 7Li NMR spectroscopy on this sample therefore accorded no signal attributable to LiSCN. Rather, it revealed a dominant peak precisely matching the δ 0.90 ppm Gilman cuprate peak in 12 (Fig. 7d) accompanied by the development of a minor high-field signal at δ 0.65 ppm (Fig. 7a). Consistent with the 13C NMR spectroscopic data, the high-field signal can be attributed to the retention of a small amount of Lipshutz-type cuprate in hydrocarbon solution, with the relative integrations (of 1 and 0.4) suggesting an approximate Gilman:Lipshutz-type ratio of 5:1.
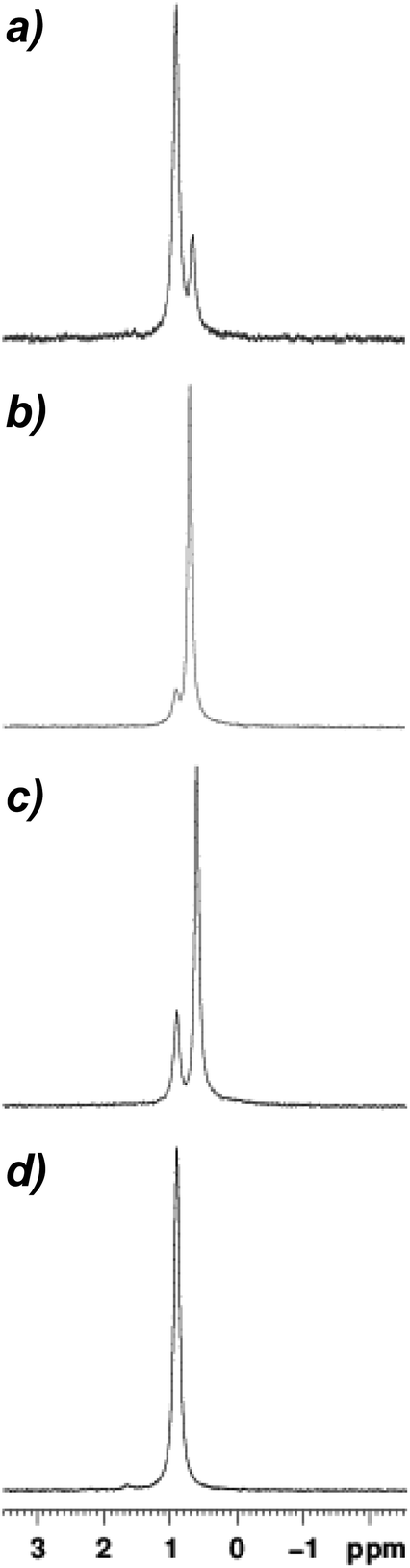 | ||
| Fig. 7 7Li NMR spectroscopic data obtained in d6-benzene for (a) 9, (b) 10, (c) 11, (d) 12. Sample concentrations for 9–11 were 20 mg/0.7 mL. | ||
Moving to 10 in solution it is immediately apparent that the 7Li NMR spectrum revealed a significant change in behaviour, with the high-field signal at δ 0.71 ppm now dominant (Fig. 7b). Taken together with 13C NMR spectroscopic data, this confirms the attribution of the high-field signal as retained Lipshutz-type cuprate and emphasizes the solvent dependence of LiSCN abstraction, with a Gilman:Lipshutz-type ratio of 0.2:1 calculated based on the 7Li NMR data – which substantiates the 13C NMR spectrum in Fig. 6b. Fig. 7c reinforces this view, with the Gilman:Lipshutz-type ratio of 0.6:1 calculated from 7Li NMR data for 11 correlating with the appearance of Fig. 6c.
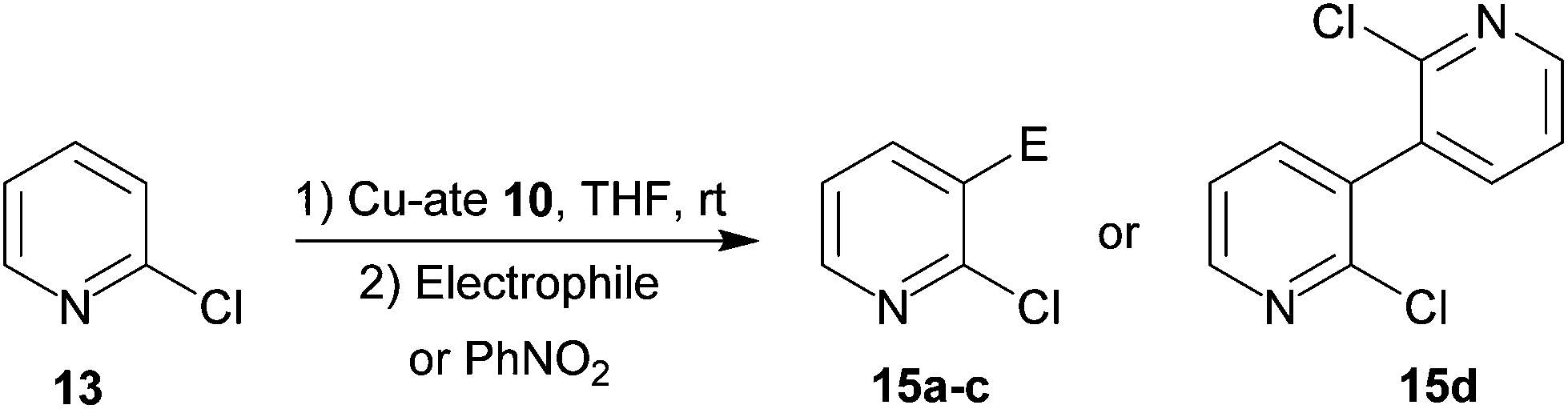
Chloropyridine derivatization
Based upon recent work on the use of halide-containing Lipshutz-type cuprates to facilitate the synthesis of azafluorenone frameworks24 it was decided to test the reactivity of new thiocyanatocuprates in the selective elaboration of halopyridines. The in situ preparation of 10 was therefore undertaken using THF solvent and the resulting mixture was tested in the selective deprotometalation of both 2-chloropyridine 13 and 2,3-dichloropyridine 14. Under the conditions employed it was expected that reaction of 13 would occur at the aromatic 3-position, while the employment of two adjacent acidifying halogens would promote attack of the 4-position in the dichlorinated substrate.Results of the use of in situ-generated 10 in the selective metalation of 13 are reported in Table 1. Under the conditions employed, reaction afforded the 3-substituted derivatives 15a–c in 46–71% yields after subsequent trapping with 4-methoxybenzoyl chloride, methyl iodide or phenyl disulfide, respectively (Table 1, entries 1–3 and Fig. S13†). In the light of cyanocuprate 2 having been shown to promote the quantitative homocoupling of N,N-diisopropylbenzamide in the presence of the oxidant PhNO2,12 so obviating the need for the inclusion of an additional transition metal-based catalyst, it was decided to test the efficacy of 10 in this respect as a safer alternative to the cyanocuprate. Accordingly, the formation of 15d was observed in 69% yield (entry 4).
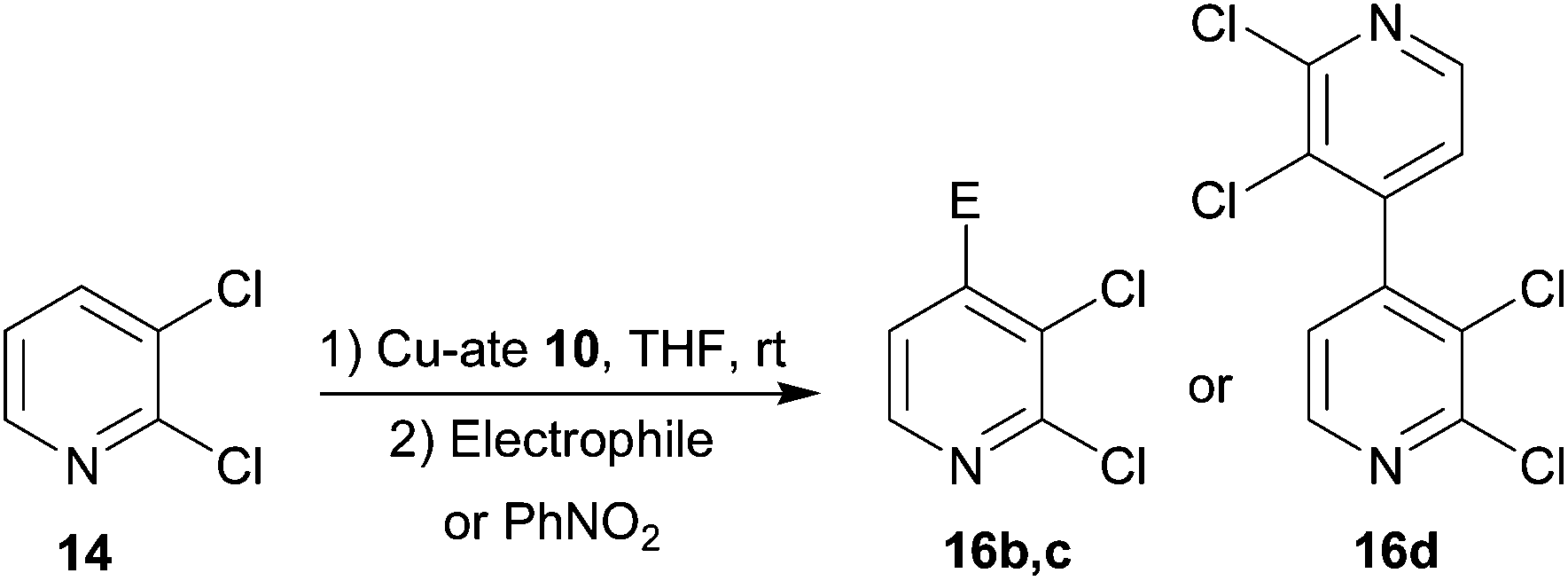
The response of 2,3-dichloropyridine 14 was tested next (Table 2). This is known to undergo lithiation at the 4-position in THF using either diisopropylamidolithium or butyllithium at −75 °C.29 More recently, the putative bases (TMP)3ZnLi and (TMP)2CuLi were used to generate 4-iodo and 4-keto products from 2,3-dichloropyridine.25 Results suggested essentially similar performance of 10 in the selective iodination and acylation of C4 in 2,3-dichloropyridine to that seen for C3 in 2-chloropyridine (Table 2, entries 1, 2 and Fig. S13†). Meanwhile, homocoupling now proceeded to give 16c in 51% yield (entry 3).
Conclusions
The preparation of (TMP)2Cu(CN)Li2(THP) 8 served to extend the family of TMP-incorporating lithiocuprates based on the formulation (TMP)2Cu(CN)Li2(L) (L = OEt21, THF 2) and to establish the prevalence of approximately flat dimers for these complexes in the solid state.12,21 The use of thiocyanate instead of cyanide was then probed to investigate the former as a convenient, cheap and safe alternative that avoids redirecting reactivity in ways recently described when cyanide has been replaced by halide.13 The resulting complexes showed interesting structural variability; the use of Et2O incurring an essentially planar dimer akin to those of 1, 2 and 8 but based on a (LiNCS)2 core, while THF and TMP gave boat and chair conformers, respectively. These aggregates were best viewed as incorporating Li[S–CN] moieties. Consistent with recently developed theoretical views on cuprate reactivity,23 solid state structures were not necessarily retained in solution, with at least some Gilman cuprate formation noted for each thiocyanate system in benzene. Lastly, preliminary synthetic investigations successfully applied 10 to the selective deprotometalation and homocoupling of halopyridines, with ongoing work seeking to establish the extent of the synthetic portfolio of these new reagents.
Experimental section
General synthetic and analytical details
Reactions and manipulations were carried out under an inert atmosphere of dry nitrogen, using standard double manifold and glove-box techniques. Solvents were distilled off sodium (toluene) or sodium–potassium amalgam (Et2O, THF, hexane) immediately prior to use. Copper(I) thiocyanate and tetrahydropyran (THP, Sureseal) were purchased from Aldrich and 2,2,6,6-tetramethylpiperidine (TMPH) was purchased from Alfa Aesar. The amine was stored over molecular sieve (4 Å) while the other chemicals were used as received. nBuLi (2.5 mL, 1.6 M in hexanes) was purchased from Acros and used as received. IR spectra were collected on a Bruker Alpha FT IR spectrometer. NMR data were collected on a Bruker Avance III HD 500 MHz Smart Probe FT NMR spectrometer (500.200 MHz for 1H, 125.775 MHz for 13C, 194.397 for 7Li). Spectra were obtained at 25 °C and chemical shifts are internally referenced to d6-benzene and calculated relative to TMS except for 7Li, for which an external reference was used (1 M LiCl in D2O). Chemical shifts are expressed in δ ppm. The following abbreviations are used: br = broad, m = multiplet, s = singlet, G = Gilman cuprate, L = Lipshutz-type cuprate.Crystallographic details
| 8 2 | 9 2 | 10 2 | 11 2 | |
|---|---|---|---|---|
| Formula | C48H92Cu2Li4N6O2 | C46H92Cu2Li4N6O2S2 | C46H88Cu2Li4N6O2S2 | C48H92Cu2Li4N6O2S2 |
| M | 940.11 | 980.21 | 976.18 | 1004.23 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/c | P21/c |
| a | 9.0869(4) | 16.1549(5) | 15.1170(7) | 15.8974(7) |
| b | 11.3341(5) | 11.4981(3) | 14.7117(7) | 8.1755(3) |
| c | 13.6040(6) | 15.2650(5) | 25.3458(11) | 22.0201(9) |
| α | 86.210(2) | 90 | 90 | 90 |
| β | 76.776(2) | 98.687(2) | 104.763(2) | 100.199(2) |
| γ | 82.219(2) | 90 | 90 | 90 |
| V | 1350.49(10) | 2802.96(15) | 5450.7(4) | 2816.7(2) |
| Z | 1 | 2 | 4 | 2 |
| ρ calcd | 1.156 | 1.161 | 1.190 | 1.184 |
| μ | 1.258 | 1.908 | 1.962 | 1.912 |
| Data | 13834 |
29571 |
73948 |
41304 |
| Unique data | 4736 | 4914 | 9650 | 4945 |
| R int | 0.0324 | 0.0513 | 0.0610 | 0.0495 |
| θ (°) | 3.339–66.540 | 2.767–66.719 | 3.023–66.831 | 2.824–66.556 |
| wR2 | 0.1896 | 0.1043 | 0.1294 | 0.0929 |
| R | 0.0624 | 0.0409 | 0.0474 | 0.0360 |
| GoF | 1.072 | 1.016 | 1.030 | 1.037 |
| Parameters | 393 | 326 | 575 | 297 |
| Peak/hole (e Å–3) | 0.925/−0.446 | 0.538/−0.251 | 0.717/−0.556 | 0.545/−0.394 |
2104.3 cm−1 (m).
2064.6 cm−1 (s), 1996.6 cm−1 (m).
2050.4 cm−1 (s), 1998.0 cm−1 (m).
2063.2 cm−1 (s), 2006.5 cm−1 (w).
Synthesis and characterization of chloropyridine derivatives
A stirred solution of LiTMP was prepared at 0 °C by sequentially treating THF (5 mL) with 2,2,6,6-tetramethylpiperidine (0.68 mL, 4 mmol) and nBuLi (2.5 mL, 1.6 M hexanes solution, 4 mmol). This reagent was then treated with copper(I) thiocyanate (0.24 g, 2 mmol). The mixture was stirred for 15 min at 0 °C before the introduction of 2-chloropyridine (13, 0.19 mL, 2 mmol) or 2,3-dichloropyridine (14, 0.30 g, 2 mmol). After 2 h. at RT, the electrophile (4 mmol) was added either as a neat liquid or as a solution in THF (5 mL). The mixture was stirred overnight at RT before addition of a 1 M aqueous solution of NaOH (20 mL) and extraction with Et2O (2 × 20 mL). After washing the organic phase with an aqueous saturated solution of NH4Cl (10 mL) and drying over anhydrous Na2SO4, the solvent was evaporated under reduced pressure and the product isolated after purification by flash chromatography on silica gel (the eluent is given in the product description).2-Chloro-3-pyridyl 4-methoxyphenyl ketone 15a was prepared from 13 by using 4-methoxybenzoyl chloride as the electrophile and was isolated (eluent: 8:2 heptane/AcOEt) in 46% yield as a yellow powder: mp 79 °C. The product was identified from a previous report.24
2-Chloro-3-methylpyridine 15b was prepared from 13 by using methyl iodide as the electrophile and was isolated (eluent: 9:1 heptane/AcOEt) as a yellow oil (estimated yield: 65%). The product was identified from a previous report.36 2-Chloro-3-(phenylsulfanyl)pyridine 15c (see Fig. S14†) was prepared from 13 by using phenyl disulfide as the electrophile and was isolated (eluent: 9:1 heptane/AcOEt) in 71% yield as a greenish powder: mp 70–72 °C; IR(ATR): 689, 725, 745, 795, 909, 1021, 1029, 1060, 1146, 1382, 1434, 1474, 1547, 1736, 2927, 3062 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.02 (dd, J = 7.8 and 4.5 Hz, 1H), 7.08 (dd, J = 7.8 and 1.8 Hz, 1H), 7.41–7.56 (m, 3H), 7.48–7.54 (m, 2H), 8.13 (dd, J = 4.5 and 1.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 122.8 (CH), 129.6 (CH), 130.1 (2CH), 130.4 (C), 134.8 (2CH), 136.1 (C), 136.3 (CH), 145.9 (CH), 147.9 (C); X-ray diffraction data (CCDC-1427011): C11H8ClNS, M = 221.69, monoclinic P21/a (I.T.#14), a = 9.0933(3), b = 11.0308(4), c = 10.0368(4) Å, β = 94.6760(10)°, V = 1003.40(6) Å3, Z = 4, ρcalcd = 1.468 g cm−3, μ = 0.543 mm−1 (a final refinement on F2 with 2298 unique intensities and 127 parameters converged at wR(F2) = 0.0716 (R(F) = 0.0287) for 2067 observed reflections with I > 2σ(I). 2,2′-Dichloro-3,3′-bipyridine 15d was prepared from 13 by using nitrobenzene (oxidative agent) and was isolated (eluent: 7:3 heptane/AcOEt) in 69% yield as a yellow powder: mp 118–120 °C. The product was identified by comparison with a commercial product.
2,3-Dichloro-4-methylpyridine 16b was prepared from 14 by using methyl iodide as the electrophile and was isolated (eluent: 9:1 heptane/AcOEt) as a yellow oil (estimated yield: 58%). The product was identified from a previous report.37 2,3-Dichloro-4-(phenylsulfanyl)pyridine 16c (see Fig. S15†) was prepared from 14 by using phenyl disulfide as the electrophile and was isolated (eluent: 9:1 heptane/AcOEt) in 62% yield as a yellow powder: mp 98 °C; IR(ATR): 690, 742, 750, 793, 1038, 1204, 1355, 1422, 1441, 1551, 1736, 3059 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.34 (d, J = 5.4 Hz, 1H), 7.40–7.51 (m, 5H), 7.82 (d, J = 5.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 119.2 (CH), 125.1 (C), 128.1 (C), 130.3 (2CH), 130.6 (CH), 135.8 (2CH), 145.7 (CH), 149.1 (C), 153.3 (C); X-ray diffraction data (CCDC-1427012): C11H7Cl2NS, M = 256.14, monoclinic P21 (I.T.# 4), a = 7.4916(7), b = 8.4263(9), c = 8.7496(9) Å, β = 93.708(4)°, V = 551.18(10) Å3, Z = 2, ρcalcd = 1.543 g cm−3, μ = 0.740 mm−1 (a final refinement on F2 with 2275 unique intensities and 136 parameters converged at wR(F2) = 0.0788 (R(F) = 0.0299) for 2187 observed reflections with I > 2σ(I). Flack parameter = −0.02(7). 2,3,2′,3′-Tetrachloro-4,4′-bipyridine 16d was prepared from 14 by using nitrobenzene (oxidative agent) and was isolated (eluent: 8:2 heptane/AcOEt) in 51% yield as a white powder: mp 200–202 °C. The product was identified from a previous report.38
Acknowledgements
This work was supported by the U.K. EPSRC through grant EP/J500380/1 (A. P.) and the Ministère de l'Enseignement Supérieur et de la Recherche scientifique Algérien (M. H.). F. M. thanks the Institut Universitaire de France and also Thermo Fisher for the generous gift of 2,2,6,6-tetramethylpiperidine. Thanks go also to Dr Andrew Bond (University of Cambridge) for help with crystallographic analysis. Detailed supporting data for this paper are available at the University of Cambridge data repository (see https://www.repository.cam.ac.uk/handle/1810/252480). These include some data in the file formats fid (NMR spectroscopic data) and cif (crystallographic data), which can be opened using the programs TopSpin 3.2 (or similar) and Mercury CSD 3.5 (or similar), respectively.References
- (a) H. W. Gschwend and H. R. Rodriguez, Org. React., 1979, 26, 1 CAS; (b) P. Beak and V. Snieckus, Acc. Chem. Res., 1982, 15, 306 CrossRef CAS; (c) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS; (d) T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297 CrossRef CAS; (e) M. Schlosser, in Organometallics in Synthesis: A Manual, ed. M. Schlosser, Wiley, New York, 2nd edn, 2002, ch. 1 Search PubMed; (f) F. García, M. McPartlin, J. V. Morey, D. Nobuto, Y. Kondo, H. Naka, M. Uchiyama and A. E. H. Wheatley, Eur. J. Org. Chem., 2008, 644–647 CrossRef.
- Y. Kondo, M. Shilai, M. Uchiyama and T. Sakamoto, J. Am. Chem. Soc., 1999, 121, 3539 CrossRef CAS.
- (a) R. E. Mulvey, Organometallics, 2006, 25, 1060 CrossRef CAS; (b) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802 CrossRef CAS PubMed; (c) R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743 CrossRef CAS PubMed; (d) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794 CrossRef CAS PubMed; (e) F. Mongin and M. Uchiyama, Curr. Org. Chem., 2011, 15, 2340 CrossRef CAS; (f) R. E. Mulvey, Dalton Trans., 2013, 42, 6676 RSC; (g) A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470 CrossRef CAS PubMed; (h) F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563 CrossRef CAS PubMed.
- (a) M. Uchiyama, H. Naka, Y. Matsumoto and T. Ohwada, J. Am. Chem. Soc., 2004, 126, 10526 CrossRef CAS PubMed; (b) H. Naka, M. Uchiyama, Y. Matsumoto, A. E. H. Wheatley, M. McPartlin, J. V. Morey and Y. Kondo, J. Am. Chem. Soc., 2007, 129, 1921 CrossRef CAS PubMed; (c) J. García-Álvarez, E. Hevia, A. R. Kennedy, J. Klett and R. E. Mulvey, Chem. Commun., 2007, 2402 RSC; (d) H. Naka, J. V. Morey, J. Haywood, D. J. Eisler, M. McPartlin, F. García, H. Kudo, Y. Kondo, M. Uchiyama and A. E. H. Wheatley, J. Am. Chem. Soc., 2008, 130, 16193 CrossRef CAS PubMed.
- (a) K. Snégaroff, J. M. L'Helgoual'ch, C. Bentabed-Ababsa, T. T. Nguyen, F. Chevallier, M. Yonehara, M. Uchiyama, A. Derdour and F. Mongin, Chem. – Eur. J., 2009, 15, 10280 CrossRef PubMed; (b) K. Snégaroff, S. Komagawa, M. Yonehara, F. Chevallier, P. C. Gros, M. Uchiyama and F. Mongin, J. Org. Chem., 2010, 75, 3117 CrossRef PubMed; (c) F. Mongin and M. Uchiyama, Curr. Org. Chem., 2011, 15, 2340 CrossRef CAS; (d) G. Dayaker, A. Sreeshailam, D. V. Ramana, F. Chevallier, T. Roisnel, S. Komagawa, R. Takita, M. Uchiyama, P. R. Krishna and F. Mongin, Tetrahedron, 2014, 70, 2102 CrossRef CAS.
- (a) R. E. Mulvey, V. L. Blair, W. Clegg, A. R. Kennedy, J. Klett and L. Russo, Nat. Chem., 2010, 2, 588 CrossRef CAS PubMed; (b) S. E. Baillie, V. L. Blair, E. Hevia and A. R. Kennedy, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, m249 CAS.
- J. Garcia-Álvarez, A. R. Kennedy, J. Klett and R. E. Mulvey, Angew. Chem., Int. Ed., 2007, 46, 1105 CrossRef PubMed.
- (a) M. Uchiyama, T. Miyoshi, Y. Kajihara, T. Sakamoto, Y. Otani, T. Ohwada and Y. Kondo, J. Am. Chem. Soc., 2002, 124, 8514 CrossRef CAS PubMed; (b) W. Clegg, S. H. Dale, E. Hevia, G. W. Honeyman and R. E. Mulvey, Angew. Chem., Int. Ed., 2006, 45, 2370 CrossRef CAS PubMed; (c) M. Uchiyama, Y. Matsumoto, D. Nobuto, T. Furuyama, K. Yamaguchi and K. Morokuma, J. Am. Chem. Soc., 2006, 128, 8748 CrossRef CAS PubMed; (d) Y. Kondo, J. V. Morey, J. M. Morgan, P. R. Raithby, D. Nobuto, M. Uchiyama and A. E. H. Wheatley, J. Am. Chem. Soc., 2007, 129, 12734 CrossRef CAS PubMed; (e) W. Clegg, J. Garcia-Álvarez, P. Garcia-Álvarez, D. V. Graham, R. W. Harrington, E. Hevia, A. R. Kennedy, R. E. Mulvey and L. Russo, Organometallics, 2008, 27, 2654 CrossRef CAS; (f) A. R. Kennedy, J. Klett, R. E. Mulvey and D. S. Wright, Science, 2009, 326, 706 CrossRef CAS PubMed.
- (a) G. Wittig, Angew. Chem., 1958, 70, 65 CrossRef; (b) W. Tochtermann, Angew. Chem., Int. Ed. Engl., 1966, 5, 351 CrossRef CAS.
- A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834 CrossRef PubMed.
- R. P. Davies, Coord. Chem. Rev., 2011, 255, 1226 CrossRef CAS.
- S. Usui, Y. Hashimoto, J. V. Morey, A. E. H. Wheatley and M. Uchiyama, J. Am. Chem. Soc., 2007, 129, 15102 CrossRef CAS PubMed.
- P. J. Harford, A. J. Peel, F. Chevallier, R. Takita, F. Mongin, M. Uchiyama and A. E. H. Wheatley, Dalton Trans., 2014, 43, 14181 RSC.
- (a) H. Gilman, R. Jones and L. Woods, J. Org. Chem., 1952, 17, 1630 CrossRef CAS; (b) H. House, W. Respess and G. Whitesides, J. Org. Chem., 1966, 31, 3128 CrossRef CAS.
- (a) P. Reiss and D. Fenske, Z. Anorg. Allg. Chem., 2000, 626, 1317 CrossRef CAS; (b) R. P. Davies, S. Hornauer and P. B. Hitchcock, Angew. Chem., Int. Ed., 2007, 46, 5191 CrossRef CAS PubMed; (c) R. M. Gschwind, Chem. Rev., 2008, 108, 3029 CrossRef CAS PubMed.
- G. Whitesides, W. Fisher, J. San Filipo, R. Bashe and H. House, J. Am. Chem. Soc., 1969, 91, 4871 CrossRef CAS.
- (a) B. H. Lipshutz, R. S. Wilhelm and D. M. Floyd, J. Am. Chem. Soc., 1981, 103, 7672 CrossRef CAS; (b) B. H. Lipshutz, S. Sharma and E. L. Ellsworth, J. Am. Chem. Soc., 1990, 112, 4032 CrossRef CAS; (c) B. Lipshutz and B. James, J. Org. Chem., 1994, 59, 7585 CrossRef CAS.
- (a) S. Bertz, J. Am. Chem. Soc., 1990, 112, 4031 CrossRef CAS; (b) J. Snyder, D. Spangler, J. Behling and B. Rossiter, J. Org. Chem., 1994, 59, 2665 CrossRef CAS; (c) J. Snyder and S. Bertz, J. Org. Chem., 1995, 60, 4312 CrossRef CAS; (d) T. L. Stemmler, T. M. Barnhart, J. E. Penner-Hahn, C. E. Tucker, P. Knochel, M. Bohme and G. Frenking, J. Am. Chem. Soc., 1995, 117, 12489 CrossRef CAS.
- G. van Koten and J. G. Noltes, J. Am. Chem. Soc., 1979, 101, 6593 CrossRef CAS.
- (a) G. van Koten and J. G. Noltes, J. Chem. Soc., Chem. Commun., 1972, 940 RSC; (b) G. van Koten, J. T. B. H. Jastrzebski, F. Muller and C. H. Stam, J. Am. Chem. Soc., 1985, 107, 697 CrossRef CAS; (c) C. Kronenberg, J. Jastrzebski, A. Spek and G. van Koten, J. Am. Chem. Soc., 1998, 120, 9688 CrossRef; (d) G. Boche, F. Bosold, M. Marsch and K. Harms, Angew. Chem., Int. Ed., 1998, 37, 1684 CrossRef CAS.
- P. J. Harford, A. J. Peel, J. P. Taylor, S. Komagawa, P. R. Raithby, T. P. Robinson, M. Uchiyama and A. E. H. Wheatley, Chem. – Eur. J., 2014, 20, 3908 CrossRef CAS PubMed.
- J. Haywood, J. V. Morey, A. E. H. Wheatley, C.-Y. Liu, S. Yasuike, J. Kurita, M. Uchiyama and P. R. Raithby, Organometallics, 2009, 28, 38 CrossRef CAS.
- S. Komagawa, S. Usui, J. Haywood, P. J. Harford, A. E. H. Wheatley, Y. Matsumoto, K. Hirano, R. Takita and M. Uchiyama, Angew. Chem., Int. Ed., 2012, 51, 12081 CrossRef CAS PubMed.
- (a) N. Marquise, P. J. Harford, F. Chevallier, T. Roisnel, A. E. H. Wheatley, P. C. Gros and F. Mongin, Tetrahedron Lett., 2013, 54, 3154 CrossRef CAS; (b) N. Marquise, P. J. Harford, F. Chevallier, T. Roisnel, V. Dorcet, A.-L. Gagez, S. Sablé, L. Picot, V. Thiéry, A. E. H. Wheatley, P. C. Gros and F. Mongin, Tetrahedron, 2013, 69, 10123 CrossRef CAS.
- K. Snégaroff, T. T. Nguyen, N. Marquise, Y. S. Halauko, P. J. Harford, T. Roisnel, V. E. Matulis, O. A. Ivashkevich, F. Chevallier, A. E. H. Wheatley, P. C. Gros and F. Mongin, Chem. – Eur. J., 2011, 17, 13284 CrossRef PubMed.
- (a) L. H. Jones, J. Chem. Phys., 1956, 25, 1068 Search PubMed; (b) L. H. Jones, J. Chem. Phys., 1958, 28, 1234 CrossRef CAS.
- O. Reckeweg, A. Schulz, B. Blachkowski, T. Schleid and F. J. DiSalvo, Z. Naturforsch., B: Chem. Sci., 2014, 69, 17 CAS.
- K. Gregory, P. v. R. Schleyer and R. Snaith, Adv. Inorg. Chem., 1991, 37, 47 CrossRef CAS.
- E. Marzi, A. Bigi and M. Schlosser, Eur. J. Org. Chem., 2001, 1371 CrossRef CAS.
- T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3 CrossRef PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- M. Mallet, J. Organomet. Chem., 1991, 406, 49 CrossRef CAS.
- J. K. Laha, S. M. Barolo, R. A. Rossi and G. D. Cuny, J. Org. Chem., 2011, 76, 6421 CrossRef CAS PubMed.
- M. Abboud, V. Mamane, E. Aubert, C. Lecomte and Y. Fort, J. Org. Chem., 2010, 75, 3224 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1429483–1429486, 1427011 and 1427012. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03882k |
| This journal is © The Royal Society of Chemistry 2016 |