
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly nucleophilic dipropanolamine chelated boron reagents for aryl-transmetallation to iron complexes†
Jay J.
Dunsford
,
Ewan R.
Clark
and
Michael J.
Ingleson
*
School of Chemistry, University of Manchester, M13 9PL, UK. E-mail: Michael.ingleson@manchester.ac.uk
First published on 29th October 2015
Abstract
New aryl- and heteroarylboronate esters chelated by dipropanolamine are synthesised directly from boronic acids. The corresponding anionic borates are readily accessible by deprotonation and demonstrate an increase in hydrocarbyl nucleophilicity in comparison to other common borates. The new borates proved competent for magnesium or zinc additive-free, direct boron-to-iron hydrocarbyl transmetallations with well-defined iron(II) (pre)catalysts. The application of the new borate reagents in representative Csp2–Csp3 cross-coupling led to almost exclusive homocoupling unless coupling is performed in the presence of a zinc additive.
Introduction
Nucleophilic organoboron compounds are crucial in modern synthesis, particularly in transition metal catalysed carbon–carbon bond formation such as the synthetically ubiquitous Suzuki–Miyaura (SM) reaction.1–3 In the SM reaction the transfer of a hydrocarbyl group from boron to a transition metal is an essential step and one that has been extensively studied for palladium systems.4 Extending the SM reaction from palladium catalysis to base metal catalysts, with a particular emphasis on iron is important to reduce our current reliance upon expensive and toxic palladium catalysts.5,6 The unique reaction pathways accessible to iron catalysis6–10 (both one and two electron manifolds) also represent a considerable driving force for the advancement of this area. Recent advances in iron catalysed SM type cross-coupling have been significant, particularly in the employment of anionic borate nucleophiles, [B(Ar)4]−,11,12 [RB(pin)(tBu)]− (pin = pinacolato, R = aryl/alkenyl)13,14 and [R′BR3]− (R′ = Alkyl or Aryl).15 However, in contrast to Pd catalysed SM, where ArB(OR)2 and a range of bases in aqueous/ether media readily effect boron to Pd transmetallation, transmetallation to iron is more challenging and analogous conditions do not effect hydrocarbyl transfer. Instead boron to iron transmetallation requires organometallic activating agents (e.g.tBuLi or ArMgX) in examples employing pinacolboronate esters or triorganoboranes.13–15 Furthermore, additives such as MgBr2, ZnCl2 or Zn(Ar)2 are required for good heterocoupling yields in the Fe catalysed SM reaction, with additive free systems providing inferior heterocoupling outcomes.14 These additives are proposed to facilitate hydrocarbyl transmetallation, although their specific role(s) in this process have yet to be defined. Hydrocarbyl transmetallation from boron-to-iron16 employing modular boronate reagents that are readily accessible, particularly from the widely available boronic acids, and are activated for transmetallation using simple bases (e.g., Group I M[OR] salts) would be attractive for iron catalysed SM protocols.17 Herein, we describe our efforts toward this goal, principally through the design of highly nucleophilic borate reagents and their application in boron-to-iron hydrocarbyl transmetallation.Results and discussion
Our recent investigations on boron-to-iron hydrocarbyl transmetallation, employing [Fe(NHC)2X2] (NHC = N-heterocyclic carbene, X = Cl or OMe) systems with arylborate nucleophiles afforded key observations of significance to our current investigation.18 Specifically, that alkoxide transfer from arylborates of the general formulae, [ArB(eg)(OMe)]− (eg = ethylene glycolato) occurs in preference to aryl transfer (Fig. 1). Considering this, we set out to design a new class of highly nucleophilic borates that incorporate tethered, trianionic ligands and are accessible from boronic acids and Group I M[OR] salts, with the intention of facilitating hydrocarbyl transfer in preference to alkoxide transfer (Fig. 1).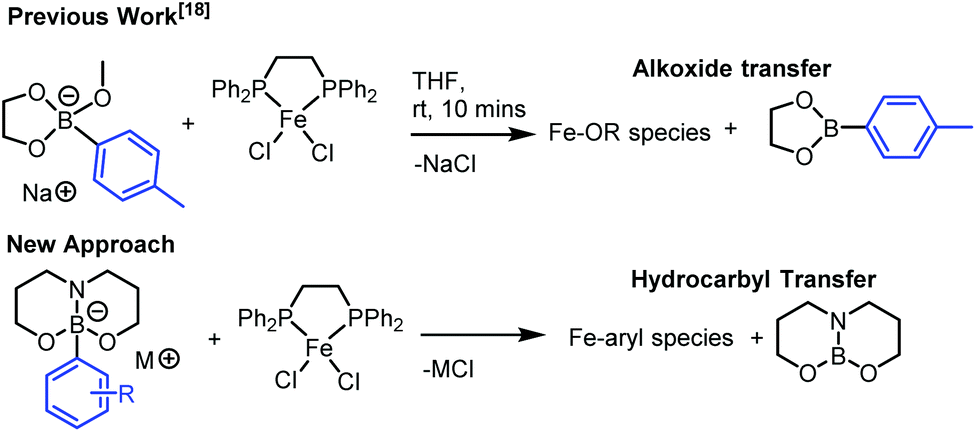 | ||
| Fig. 1 Proposal to favour hydrocarbyl transmetallation over alkoxide transfer to iron from boronate reagents. | ||
Borate reagents incorporating tethered, trianionic ligands are known,19 with the triolborates particularly noteworthy,20 however these are non-productive in Fe catalysed SM couplings.12 Borates incorporating a trianionic 6,6-bicyclic chelate are rare,21 but are attractive as they will afford a reduction in ring strain in the conjugate 3-coordinate boronate ester formed post hydrocarbyl transfer relative to 5,5-bicyclic and 6,6,6-tricyclic (e.g. triolboronate) systems. This would facilitate the planarisation of the neutral 3-coordinate boronate ester by-product and thus its stabilisation through increased π-orbital overlap, an outcome enhanced further by the greater multiple bond character present in B-NR2 relative to B-OR; both factors were envisaged to enhance the nucleophilicity of the anionic hydrocarbyl-borate.
Combining 1.1 equiv. of dipropanolamine (which can be easily prepared on multi-gram scales from cheap, commercially available reagents)22 with phenylboronic acid (1 equiv.) in THF afforded the desired neutral boronate ester 1a within 10 minutes at ambient temperature. The poor solubility of 1a in THF allows for its isolation by filtration and subsequent recrystallisation from hot acetone affords analytically pure 1a in 89% yield (9.74 g) (Fig. 2). The rigid nature of the 6,6-bicyclic chelate is evident in the 1H NMR spectra of 1a through the diastereotopic nature of the CH2 protons within the chelate backbone, a feature also observed in related 5,5-bicyclic systems.23 It is noteworthy that 5,5-bicyclic analogues demonstrate fluxional behaviour at temperatures above 40 °C, whereas 1a is considerably more conformationally rigid, demonstrating no fluxional behaviour up to 110 °C (in d6-DMSO), analogous to the extremely robust MIDA boronate esters.24 This simple esterification protocol can be extended to a number of aryl- or heteroarylboronic acids, affording neutral boronate esters 1a–1q in good to excellent isolated yields (97–72%) (Fig. 2).
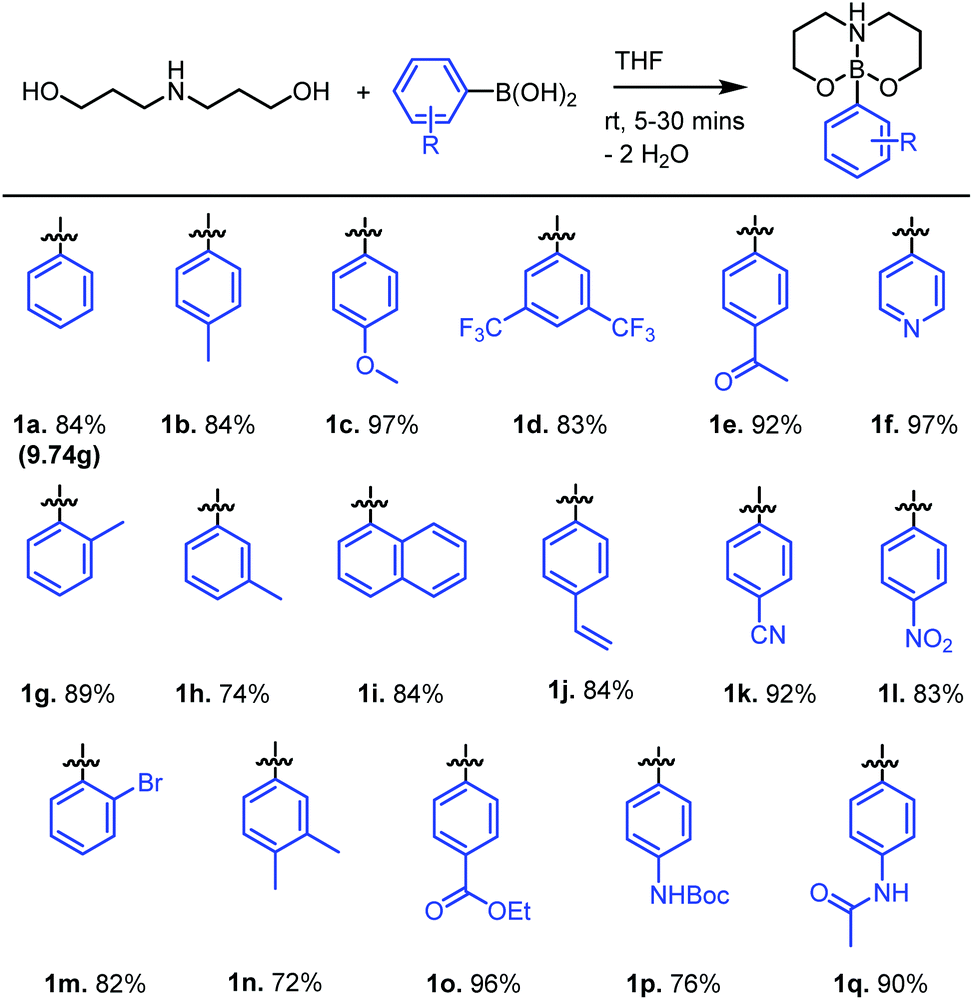 | ||
| Fig. 2 Synthesis of dipropanolamine chelated neutral boronate esters 1a–1q. | ||
Generation of the anionic borates is simple as exemplified by the formation of K[2a] which can be achieved in quantitative yield via deprotonation of 1a with a variety of bases (KH, tBuOK, KOMe) in anhydrous THF.25 The employment of KOH leads only to the recovery of neutral boronate ester, 1a. This observation correlates with the fact that K[2a] slowly hydrolyses to 1a (and presumably KOH) with prolonged storage in the solid or solution state under air. With methods for the generation of the anionic borates in hand, we next focused upon calculating relative phenyl ion affinities (PhIAs) to assess the relative nucleophilicity of K[2a] and other common borate anions. PhIAs were calculated from the isodesmic reactions between tetraphenylborate and the appropriate neutral boron Lewis acid (Table 1). This methodology is analogous to previous approaches for calculating hydride and chloride ion affinities (HIAs and CIAs) of boron Lewis acids, where calculations of HIA correlated well with hydride transfer reactivity.26,27 Analysis of the calculated PhIA values revealed neutral borane 3 to have a substantially lower PhIA (hence K[2a] will provide a better source of Ph−) than other common borate reagents tested (Table 1). The alleviation of ring strain in 3 relative to the 5,5-bicyclic analogue derived from diethanolamine (5,5-ONO, entry 4) and to a greater extent the 6,6,6-tricyclic containing triolborane (entry 1) does indeed have a considerable effect on PhIA and thus the energetics of aryl transfer.
|
|
|||
|---|---|---|---|
| Entry | Lewis acid | Abbreviation | PhIAa (kcal mol−1) |
| a PhIAs calculated at the M06-2X/6-311G+(d,p) level of theory with incorporation of a dichloromethane PCM solvent model. b In parenthesis the PhIA is calculated using atomic coordinates from the solid state structure of K[2a]. | |||
| 1 |
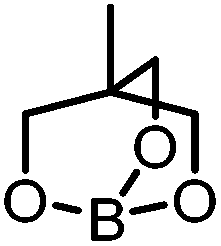
|
Triolborane | −22.8 |
| 2 | BF3 | — | −16.3 |
| 3 | BPh3 | — | 0.0 |
| 4 |
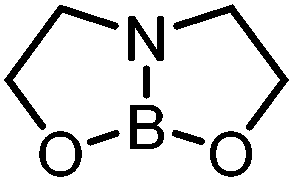
|
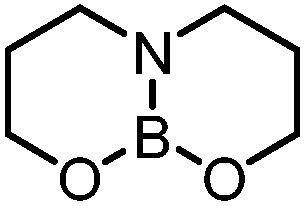
5,5-ONO | +17.0 |
| 5 | B(OMe)3 | — | +22.7 |
| 6 |
|
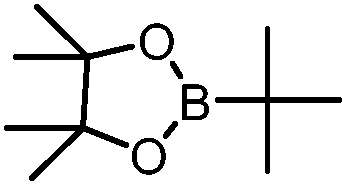
B(Pin)(tBu) | +23.2 |
| 7 |
|
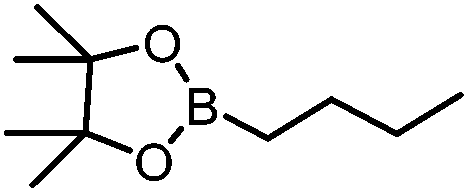
B(Pin)(nBu) | +23.9 |
| 8 |
|
3 (This work) | +36.3 (+31.3)b |
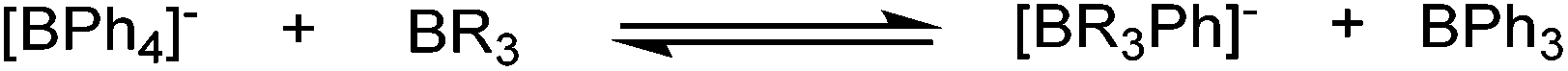
The trends observed through calculated PhIAs were probed experimentally by phenyl ion transfer reactions. Initial studies commenced with K[2a]/3 and boron reagents possessing considerably different calculated PhIAs. The addition of K[2a] to triolborane in THF resulted in slow dissolution of triolborane (which is poorly soluble in THF), with initial 11B NMR spectra (after ca. 10 minutes at 20 °C) showing a mixture of K[2a] (δ11B = 2.5) and a new species (δ11B = 4.4) which is tentatively attributed to reversible Lewis adduct formation between K[2a] and triolborane, presumably by coordination of the N or O nucleophilic sites in K[2a] to the Lewis acidic boron centre of the triolborane. Nevertheless, heating this mixture at 60 °C for 18 h resulted in complete disappearance of both of these signals, full dissolution of the triolborane and formation of 3 (δ11B = 20.0) and [Ph-triolborate]− (δ11B = 3.6) as the major species. The new boron species at δ11B = 20 was confirmed as the expected neutral 3-coordinate boron by-product of hydrocarbyl transmetallation via the independent synthesis and characterization of 3 from dipropanolamine and Me2S·BH3.25 The reaction of K[2a] with Et2O-BF3 proceeded more rapidly with no starting materials observed after <10 min at 20 °C (by 11B NMR spectroscopy), instead 3 and [PhBF3]− are the major species observed, along with an unidentified intermediate (δ11B = 0.1) observed at short times which is fully consumed on heating to 60 °C.
It is more significant in the context of this investigation to experimentally benchmark the PhIA of 3 against boron Lewis acids whose Ph-borate anions transmetallate to Fe (with or without additives). However, attempts to experimentally confirm the relative PhIA of BPh3 and 3 were frustrated by solubility issues (in THF and DCM) along with the formation of an intermediate (δ11B = 0.8) that persisted even on heating to 60 °C for 18 h, this species frustrated crystallisation attempts and is tentatively assigned to a Lewis adduct between K[2a] and BPh3. In contrast, combination of K[2a] and B(OMe)3 resulted in rapid phenyl transfer at 20 °C (with a short lived intermediate again observed, δ11B = 3.1), to ultimately form 3 and [PhB(OMe)3]−, confirming the relative PhIA of 3 and B(OMe)3 (Scheme 1). The combination of K[2a] and B(Pin)(nBu) resulted in only a minor amount of 3 formed after 16 h at 60 °C. We attribute the limited phenyl transfer to a kinetically slow process as opposed to an equilibrium favouring B(Pin)(nBu) and K[2a] as the reverse reaction of 3 with [PhB(Pin)(nBu)]− resulted in minimal formation of [2a]− after 16 h at 60 °C.
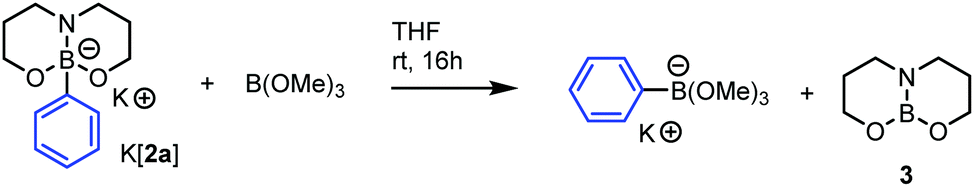 | ||
| Scheme 1 Phenyl ion transfer reaction from K[2a] to trimethoxyborane. | ||
Encouraged by the calculated PhIA value of 3 and the stoichiometric phenyl ion transfer experiments, we moved on to probe whether high hydrocarbyl nucleophilicity in [2a]− would translate to facile boron-to-iron hydrocarbyl transmetallation with common iron(II) (pre)catalysts. We began this investigation with the (pre)catalyst, [Fe(dppe)Cl2]n4 (dppe = 1,2-bis(diphenylphosphino)ethane) (4 was found by X-ray diffraction studies to exist as a 1D coordination polymer in the solid state on recrystallisation from THF).28In situ generation of K[2a] with KH (1 equiv.) in anhydrous THF followed by the addition of 4 (0.5 equiv.) and dppe (0.5 equiv.) then agitation at ambient temperature led to no observable reaction (by 11B{1H} NMR spectroscopy, with K[2a] the only signal observed) after 1 h. However, heating the reaction mixture for 10 minutes at 60 °C in THF (or in toluene) led to the pale green reaction mixture becoming deep red in colour. Analysis of the reaction mixture by 1H NMR spectroscopy was uninformative, however the 11B{1H} NMR spectra revealed the complete disappearance of the 4-coordinate signal of K[2a] and the presence of a single new boron containing species in the three-coordinate region at δ11B 23.9 ppm attributed to 3.29 Isolated samples of K[2a] generated from tBuOK or KOMe produced analogous outcomes, provided all protic by-products from boronate generation (tBuOH/MeOH) were removed prior to use due to their detrimental effect on these specific iron complexes. The requirement for raised temperatures for conversion of K[2a] to 3 on addition of 4/dppe is in contrast to the reaction of Li[(Ph)B(pin(tBu)] (5) with 4 (0.5 equiv. + 0.5 equiv of dppe) which in our hands was found to react within minutes at ambient temperature in the absence of MgBr2 to produce tBuBPin (by 11B NMR spectroscopy). Thus whilst [2a]− is calculated to be thermodynamically a more nucleophilic source of Ph− than 5 it is kinetically slower to react by phenyl transfer with the Fe species present in solution under these conditions.
With the in situ11B{1H} NMR data being consistent with hydrocarbyl loss from [2a]− we sought evidence for the formation of arylated iron species. Most notably the generation of significant quantities of biphenyl (ca. 0.5 equiv.) post aryl transfer was confirmed by analysis of the reaction mixture by GC-MS against a calibrated internal standard,25 indicative of Fe-Ph species.14b Consumption of all K[2a] on addition of 0.5 equiv. of 4 (by 11B{1H} NMR spectroscopy) suggests the transfer of two aryl equivalents to iron, although the transfer of just one aryl equivalent to Fe and formation of an adduct between an Fe species and a second equivalent of [2a] (which may not be observable in the 11B NMR spectra) is also feasible. We disfavour a reaction stoichiometry other than 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as increasing the ratio of K[2a]:Fe from 2:1 to 3:1 and to 4:1 resulted in an increasing amount of K[2a] persisting in solution (by 11B NMR spectroscopy). Analogous outcomes were observed on combining K[2c] (or K[2d]) and 4 in a 2:1 ratio, with significant biaryl formed in each case (by GC-MS versus an internal standard). Repeated efforts to isolate any Fe species from transmetallation to iron complexes with K[2a] were unsuccessful in our hands.
:1 as increasing the ratio of K[2a]:Fe from 2:1 to 3:1 and to 4:1 resulted in an increasing amount of K[2a] persisting in solution (by 11B NMR spectroscopy). Analogous outcomes were observed on combining K[2c] (or K[2d]) and 4 in a 2:1 ratio, with significant biaryl formed in each case (by GC-MS versus an internal standard). Repeated efforts to isolate any Fe species from transmetallation to iron complexes with K[2a] were unsuccessful in our hands.
[Fe(dpbz)2Cl2], 6 (0.5 equiv.) (dpbz = 1,2-bis(diphenylphosphino)benzene) reacted analogously with K[2a] or K[2b], with two equivalents of borate consumed per Fe centre (by 11B NMR spectroscopy) and significant biphenyl or bi-tolyl (ca. 0.5 equiv. by GC-MS) formed again indicating significant transmetallation from boron to iron. The previously reported iron(I) complex, [Fe(dpbz)2(p-Tol)] 730 (Fig. 3) was observed from this reaction mixture by X-band continuous-wave EPR measurements at 120 K further confirming successful boron to Fe transmetallation. The spectrum of 7 is in excellent agreement with previous X-band CW-EPR measurements obtained for 7 by Bedford and co-workers30 and with an independently prepared sample of 7 obtained from the reaction of 6 with p-TolMgBr (2 equiv.) (Fig. 3). Whilst the presence of EPR and NMR spectroscopy silent Fe complexes cannot be precluded from these reactions the formation of significant quantities of the homocoupled biaryl compounds and the observation of 7 by EPR spectroscopy combined do confirm significant boron to iron hydrocarbyl transmetallation using [2x].
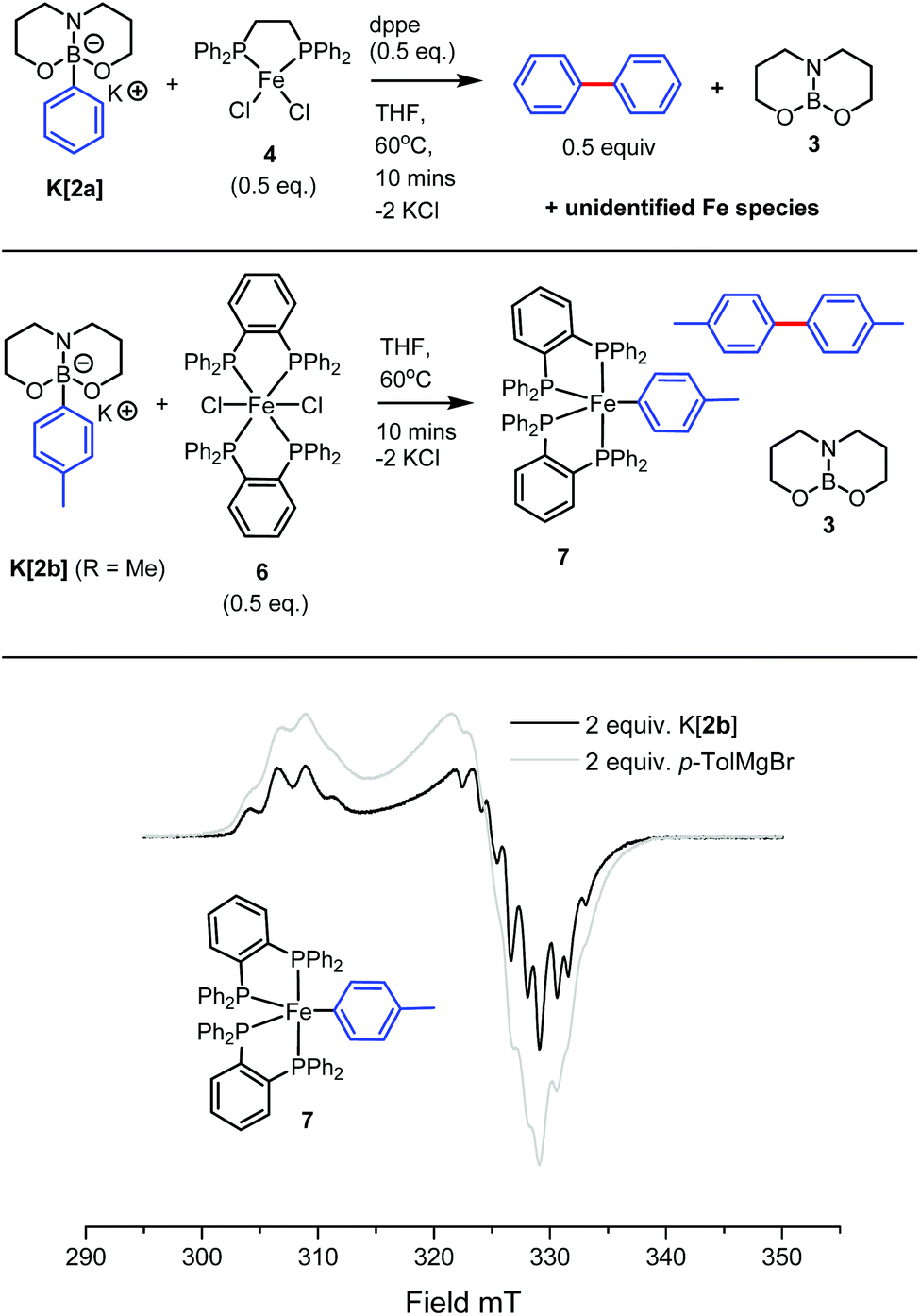 | ||
| Fig. 3 Top, reaction of K[2a] and 4 (0.5 equiv.). Middle, the formation of the Fe(I) complex 7 from K[2b] and 6 (0.5 equiv.). Bottom, comparison of the CW-EPR spectra recorded at 120 K of in situ generated 7 from either K[2a] (black) or p-TolMgBr (grey). | ||
With boron-to-iron hydrocarbyl transmetallation confirmed we moved on to evaluating the efficiency of K[2a] in iron catalysed Csp2–Csp3 SM cross-coupling. Employing K[2a] and 3-methoxybenzyl bromide in the presence of 4 + dppe (both at 10 mol% loading) led to the complete consumption of 3-methoxybenzyl bromide and almost exclusive formation of 1,2-bis(3-methoxyphenyl)ethane with only a very minor amount of heterocoupled product (by GC-MS). This observation is in contrast to the outcomes reported employing the tBuLi activated arylpinacol boronate esters, such as 5, or Grignard activated triorganoboranes, which under similar conditions, in the presence of magnesium or zinc additives led predominantly to heterocoupling products.14,15 Analogous homocoupling outcomes to that observed through the use of K[2a] were found in the employment of the lithium salt Li[2a], hence the difference in reactivity between M[2a] and 5 is not cation derived. Furthermore, repeating the cross coupling reactions using Li[2a] with MgBr2 additive, led to homocoupling again being the dominant pathway.
Diarylzinc co-catalysts are used widely in Fe SM catalysis, but it is important to note that species formulated as aryl2Zn are actually often ionic zincates, and this can significantly impact reactivity in cross-coupling.31 For example, the previously reported attempted synthesis of (p-tol)2Zn led instead to the zincate [Mg(THF)4Br2(Zn(p-tol)2)2]n, 8 (on recrystallisation, Scheme 2, inset).32 Using 10 mol% of a stock solution of 8 (equating to a total of 0.08 mmol of tolyl nucleophile) and 5 mol% 4/dppe resulted in the heterocoupling product from K[2a] and cyclo-heptylbromide being the major product (Scheme 2). However, the product distribution from this reaction is significantly less selective towards heterocoupling than cross coupling cycloheptylbromide using [tBuB(Pin)Ar]− (or [Ar(iPr)BBN]−) with magnesium or zinc additives.14,15 Repeating the coupling of cyclo-heptylbromide with K[2a] in the absence of 8, with or without MgBr2 as an additive, led to no heterocoupling product formation under a range of conditions. The omission of 4 also resulted in no heterocoupling confirming the importance of Fe under these conditions.
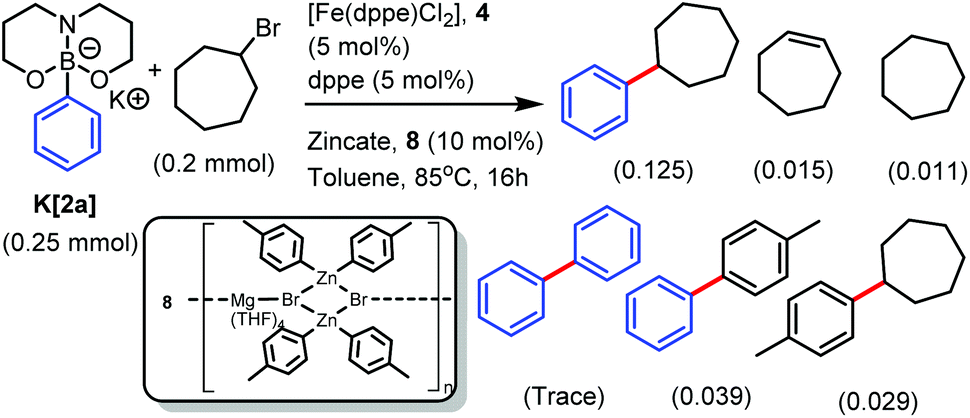 | ||
| Scheme 2 Zincate mediated Fe catalysed SM cross coupling using K[2a]. | ||
The disparity between magnesium and zinc additives led us to investigate the direct reaction between MX2 (M = Zn or Mg, X = Br or Cl) and K[2a]. Combination of MgBr2 and K[2a] in THF resulted in no observable reaction at 20 °C and 60 °C (after 24 h by 11B NMR spectroscopy). In contrast, K[2a] reacts rapidly with ZnCl2 with complete consumption of K[2a] and formation of 3 as the only new observable boron containing product after 5 h in THF at 20 °C or after 30 min in toluene at 60 °C (by 11B NMR spectroscopy). This is suggestive of transmetallation from boron in [2a]− to ZnCl2 but not to MgBr2, consistent with the disparate additive performance in Fe catalysed cross coupling reactions using K[2a]. Related transmetallation from boron to ZnCl2 has been previously observed, e.g. using [sBu3B(p-tolyl)][Mg(solv)Cl],15b which is a boron based aryl nucleophile also effective in Fe catalysed SM reactions. It is notable that less nucleophilic borates such as [PhBF3]− and [PhBtriolborane]− give no heterocoupling under Fe catalysed SM conditions even in the presence of zinc additives.12 In control reactions 2 equivalents of [PhBF3]− (or [PhBtriolborane]−) were heated in THF at 60 °C for 16 h with ZnCl2. Subsequent analysis of these reaction mixtures in d6-DMSO (in which all boron species, e.g., [PhBY3]− and the conjugate neutral borane, are soluble) revealed no evidence for transmetallation to zinc with [PhBF3]− (or [PhBtriolborane]−) being the only major boron product observed (by 11B NMR spectroscopy).
Conclusions
In summary, neutral aryl- and heteroarylboronate esters incorporating a 6,6-bicyclic chelate and their corresponding anionic borates can be readily synthesised from the parent boronic acids. The novel borate species demonstrate enhanced hydrocarbyl nucleophilicities in comparison to other common arylborates, as assessed by PhIA calculations and stoichiometric phenyl ion transfer experiments. The new borates undergo additive-free, direct boron-to-iron hydrocarbyl transmetallations with simple bis-phosphine ligated iron(II) (pre)catalysts. However, the application of the new boronates to iron catalysed Csp2–Csp3 cross-coupling led predominantly to homocoupling in the absence of Zn additives, highlighting the inherent intricacies associated with iron-catalysed Suzuki–Miyaura type Csp2–Csp3 cross-couplings. Presumably, the kinetics of aryl transfer to active Fe species (either direct or via aryl-Zn species) has to be optimal to match other steps involved in the heterocoupling cycle and without zinc additives the transfer of aryl from boron in [2a] to key Fe species is non-optimal.Experimental
General experimental details
Unless otherwise stated, all manipulations were carried out using standard Schlenk techniques under argon, or in an MBraun UniLab glovebox, under an atmosphere of argon (<0.1 ppm O2/H2O). Unless otherwise indicated, solvents were distilled from appropriate drying agents: tetrahydrofuran (potassium); toluene (potassium); n-hexane (NaK); and dichloromethane (CaH2). Tetrahydrofuran and dichloromethane were stored over activated 3 Å molecular sieves while toluene and n-hexane were stored over potassium mirrors. 3,3′-azanediylbis(propan-1-ol),22 [Fe(dppe)Cl2],33 [Fe(dpbz)2Cl2],11 and [Fe(dpbz)2(p-Tol)],30 were all prepared according to previously reported literature procedures. All other compounds were purchased from commercial sources and used as received. NMR spectra were recorded on Bruker AvanceIII-400, Bruker AvanceII-500 or Bruker Ascend-400 spectrometers. Chemical shifts are reported as dimensionless δ values and are frequency referenced relative to residual protio-impurities in the NMR solvents for 1H and 13C{1H} respectively, while 11B{1H}, 19F{1H} and 7Li shifts are referenced relative to external BF3-etherate, hexafluorobenzene and LiCl, respectively. Coupling constants J are given in Hertz (Hz) as positive values regardless of their real individual signs. The multiplicity of the signals are indicated as “s”, “d”, “t” “pent”, “sept” or “m” for singlet, doublet, triplet, pentet, septet or multiplet, respectively. High resolution mass spectra (HRMS) were recorded on a Waters QTOF mass spectrometer. GC-MS analysis was performed on an Agilent Technologies 7890A GC system equipped with an Agilent Technologies 5975C inert XL EI/CI MSD with triple axis detector. The column employed was an Agilent J&W HP-5 ms ((5%-Phenyl)-methylpolysiloxane) of dimensions: length, 30 m; internal diameter, 0.250 mm; film, 0.25 μm. Microanalysis was performed by Mr Stephen Boyer at the London Metropolitan University microanalytical service. X-band CW-EPR spectra were recorded at 120 K on a Bruker EMXmicro spectrometer operating at 9.35 GHz field modulation, 2 mW microwave power and equipped with a high sensitivity Bruker cavity (ER 4119HS).General procedure for the synthesis of 1a–1q
Under ambient conditions, with no additional precautions taken to exclude air or moisture, a round bottom flask was charged with 3,3′-azanediylbis(propan-1-ol) (1.1 equiv.) and tetrahydrofuran. To this stirred solution was added the appropriate boronic acid (1 equiv.) and the reaction mixture stirred at ambient temperature. Typically, the desired products 1a–1q began to deposit from solution after stirring for between 5 and 30 minutes, however in examples where this was not the case, stirring was continued at ambient temperature for 16 h. Isolation of the insoluble material by filtration followed by washing with cold tetrahydrofuran typically afforded 1a–1q as colourless free-flowing solids of sufficient purity to be used without further purification. Alternatively, 1a–1q can be recrystallised from hot acetone.10-phenyloctahydro-[1,3,2]oxazaborinino[2,3-b][1,3,2]oxazaborinin-5-ium-10-uide21 (1a): Prepared according to the general procedure. Phenylboronic acid (6.10 g, 50.0 mmol) and 3,3′-azanediylbis(propan-1-ol) (7.33 g, 55.0 mmol) in tetrahydrofuran (200 mL) afforded 1a as a free-flowing white solid (9.74 g, 89%). 1H NMR (400 MHz, DMSO-d6, 298 K): 7.39 (2H, d, J = 6.7 Hz, o-CH); 7.19 (2H, t, J = 7.5 Hz, m-CH); 7.09 (1H, t, J = 7.5 Hz, p-CH); 5.90 (1H, bs, NH); 3.76–3.70 (2H, m, CH2); 3.50–3.44 (2H, m, CH2); 3.09–3.02 (2H, m, CH2); 2.86–2.80 (2H, m, CH2); 1.81–1.72 (2H, m, CH2); 1.51–1.42 ppm (2H, m, CH2). 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): 131.8; 126.9; 125.5; 59.7; 46.0; 24.3 ppm. 11B{1H} NMR (128.4 MHz, DMSO-d6, 298 K): 3.2 ppm. HRMS (ESI) m/z: calculated for [M + H]+, C12H19BNO2+, 220.1509, found: 220.1516. Crystals suitable for X-ray diffraction were grown by the slow evapouration of a concentrated acetone solution of 1a at ambient temperature.
See ESI† for further details for 1b–1q synthesised via the general procedure.
General procedure for the synthesis of M[2a] (M = K or Li)
A flame dried Schlenk tube was charged with 1a (1 equiv.) and dried under vacuum for ca. 1 h prior to the addition of anhydrous THF or toluene (X mL) under an argon atmosphere. To this stirred suspension was added the appropriate base (1 equiv.) and the reaction mixture stirred at ambient temperature until the solution became homogeneous. Removal of the solvent generally afforded a pale residue which could be triturated with anhydrous n-hexane to afford M[2a] (M = K or Li) as a free-flowing white solid after isolation by filtration and drying under vacuum (see Table S1† for isolated yields).K[2a]: Prepared according to the general procedure. 1a (750.0 mg, 3.42 mmol) and KH (136.8 mg, 3.42 mmol) in tetrahydrofuran (10 mL) afforded K[2a] as a free-flowing white solid (862 mg, 98%).1H NMR (400 MHz, THF-d8, 298 K): 7.54 (2H, d, J = 6.9 Hz, o-CH); 7.09 (2H, t, J = 7.2 Hz, m-CH); 6.96 (1H, t, J = 7.5 Hz, p-CH); 3.61–3.56 (2H, m, CH2); 3.52–3.43 (2H, m, CH2); 3.34–3.25 (2H, m, CH2); 2.74–2.66 (2H, m, CH2); 1.88–1.75 (2H, m, CH2); 1.36–1.26 (2H, m, CH2) ppm. 13C{1H} NMR (100 MHz, THF-d8, 298 K): 135.1; 127.1; 125.0; 62.6; 51.8; 31.1 ppm. 11B{1H} NMR (128.4 MHz, THF-d8, 298 K): 2.5 ppm. Anal Calcd for C12H18BKNO2: C, 56.04; H, 6.66; N, 5.45. Found: C, 55.84; H, 6.49; N, 5.39. Crystals suitable for X-ray diffraction were grown by the slow evaporation of a concentrated THF solution of K[2a] at ambient temperature.
Computational details
Calculations were performed using the Gaussian09 suite of programmes.34 Structures were pre-optimised at the HF/3-21G level followed by optimisation at the M06-2X/6-311G+(d,p) level35 with inclusion of a PCM model for solvent correction (DCM).36 Structures were confirmed as minima by frequency analysis and the absence of imaginary frequencies.Acknowledgements
We gratefully acknowledge the Royal Society (for the award of a University Research Fellowship for M. J. I.), the European Research Council under Framework 7 (J. J. D. grant agreement no. 305868), the Leverhulme Trust (E. R. C.) and the EPSRC (grant number EP/K039547/1) for funding. We also acknowledge the EPSRC UK National Service for Computational Chemistry Software (NSCCS), the EPSRC UK National Electron Paramagnetic Resonance facility at the University of Manchester, Mr Daniel Sells for assistance with EPR measurements and Professor David Collison for helpful discussions.References
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- For a review discussion the transmetallation in palladium catalysed Suzuki–Miyaura see: A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS PubMed.
- E. Negishi, Hand-book of Organopalladium Chemistry for Organic Synthesis, Wiley, New York, 2002 Search PubMed.
- For recent reviews on Fe catalysed cross coupling see: (a) R. B. Bedford and P. B. Brenner, Top. Organomet. Chem., 2015, 50, 19–46 CrossRef; (b) O. L. Kuzmina, A. K. Steib, A. Moyeux, G. Cahiez and P. Knochel, Synthesis, 2015, 1696–1705 CAS; (c) E. Nakamura, T. Hatakeyama, S. Ito, K. Ishizuka, L. Ilies and M. Nakamura, Iron-Catalyzed Cross-Coupling Reactions, Org. React., 2013, 83(1), 1–210 Search PubMed.
- C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- B. Plietker, Iron Catalysis in Organic Chemistry, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- (a) M. J. Ingleson and R. A. Layfield, Chem. Commun., 2012, 48, 3579–3589 RSC; (b) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed.
- For select recent articles in this area see: (a) S. L. Daifuku, M. H. Al-Afyouni, B. E. R. Snyder, J. L. Kneebone and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, 9132–9143 CrossRef CAS PubMed; (b) M. H. Al-Afyouni, K. L. Fillman, W. W. Brennessel and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, 15457–15460 CrossRef CAS PubMed; (c) Y. Liu, J. Xiao, L. Wang and L. Deng, Organometallics, 2015, 34, 599–605 CrossRef CAS; (d) A. Hedstrom, Z. Izakian, I. Vreto, C.-J. Wallentin and P. O. Norrby, Chem. – Eur. J., 2015, 21, 5946–5953 CrossRef PubMed; (e) G. Bauer, M. D. Wodrich, R. Scopelliti and X. L. Hu, Organometallics, 2015, 34, 289–298 CrossRef CAS; (f) J. A. Przyojski, K. P. Veggeberg, H. D. Arman and Z. J. Tonzetich, ACS Catal., 2015, 5, 5938–5946 CrossRef CAS; (g) D. Gaertner, A. L. Stein, S. Grupe, J. Arp and A. J. von Wangelin, Angew. Chem., Int. Ed., 2015, 54, 10545–10459 CrossRef CAS PubMed; (h) O. M. Kuzmina, A. K. Steib, S. Fernandez, W. Boudout, J. T. Markiewicz and P. Knochel, Chem. – Eur. J., 2015, 8242–8249 CrossRef CAS PubMed.
- R. B. Bedford, E. Carter, P. M. Cosgwell, J. N. Harvey, N. J. Gower, M. F. Haddow, D. M. Murphy, E. C. Neeve and J. Nunn, Angew. Chem., Int. Ed., 2013, 52, 1285–1288 CrossRef CAS PubMed.
- R. B. Bedford, M. A. Hall, G. R. Hodges, M. Huwe and M. C. Wilkinson, Chem. Commun., 2009, 6430–6432 RSC.
- For [(alkenyl)B(pin)(tBu)]− see: T. Hashimoto, T. Hatakeyama and M. Nakamura, J. Org. Chem., 2012, 77, 1168–1173 CrossRef CAS PubMed.
- For [(aryl)B(pin)(tBu)]− see: (a) T. Hatakeyama, T. Hashimoto, Y. Kondo, Y. Fujiwara, H. Seike, H. Takaya, Y. Tamada, T. Ono and M. Nakamura, J. Am. Chem. Soc., 2010, 132, 10674–10676 CrossRef CAS PubMed; (b) S. L. Daifuku, J. L. Kneebone, B. E. R. Snyder and M. L. Neidig, J. Am. Chem. Soc., 2015, 137, 11432–11444 CrossRef CAS PubMed; (c) R. B. Bedford, P. B. Brenner, E. Carter, T. W. Carvell, P. M. Cogswell, T. Gallagher, J. N. Harvey, D. M. Murphy, E. C. Neeve, J. Nunn and D. R. Pye, Chem. – Eur. J., 2014, 20, 7935–7938 CrossRef CAS PubMed; (d) R. B. Bedford, T. Gallagher, D. R. Pye and W. Savage, Synthesis, 2015, 1761–1765 CrossRef CAS.
- For [R3BR′]− see: (a) T. Hatakeyama, T. Hashimoto, K. K. A. D. S. Kathriarachchi, T. Zenymo, H. Seike and M. Nakamura, Angew. Chem., Int. Ed., 2012, 51, 8834–8837 CrossRef CAS PubMed; (b) R. B. Bedford, P. B. Brenner, E. Carter, J. Clifton, P. M. Cogswell, N. J. Gower, M. F. Haddow, J. N. Harvey, J. A. Kehl, D. M. Murphy, E. C. Neeve, M. L. Neidig, J. Nunn, B. E. R. Snyder and J. Taylor, Organometallics, 2014, 33, 5767–5780 CrossRef CAS.
- For a rare example of direct boron-to-iron transmetallation employing a low-coordinate iron hydride species see: Y. Yu, W. W. Brennessel and P. L. Holland, Organometallics, 2007, 26, 3217–3226 CrossRef CAS PubMed.
- For comments on the importance of boron-to-iron transmetallation see: J. A. Gladysz, R. B. Bedford, M. Fujita, F. P. Gabbaï, K. I. Goldberg, P. L. Holland, J. L. Kiplinger, M. J. Krische, J. Louie, C. C. Lu, J. R. Norton, M. A. Petrukhina, T. Ren, S. S. Stahl, T. D. Tilley, C. E. Webster, M. C. White and G. T. Whiteker, Organometallics, 2014, 33, 1505–1527 CrossRef CAS.
- J. J. Dunsford, I. A. Cade, K. L. Fillman, M. L. Neidig and M. J. Ingleson, Organometallics, 2014, 33, 370–377 CrossRef CAS.
- For an overview of boron transmetallating agents see: A. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- For specific examples using triolborates in palladium catalysis see: (a) Y. Yamamoto, M. Takizawa, X.-Q. Yu and N. Miyaura, Angew. Chem., Int. Ed., 2008, 47, 928–931 CrossRef CAS PubMed; (b) S. Sakashita, M. Takizawa, J. Sugai, H. Ito and Y. Yamamoto, Org. Lett., 2013, 15, 4308–4311 CrossRef CAS PubMed.
- For one example of a neutral borane containing a 6,6-bicyclic chelate see: R. Csuk, H. Hönig and C. Romanin, Monatsh. Chem., 1982, 113, 1025–1035 CrossRef CAS.
- C. Granier and R. Guilard, Tetrahedron, 1995, 51, 1197–1208 CrossRef CAS.
- M. K. Reilly and S. D. Rychnovsky, Synlett, 2011, 2392–2396 CAS.
- E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 14084–14085 CrossRef CAS PubMed.
- See ESI.†.
- (a) E. R. Clark, A. Del Grosso and M. J. Ingleson, Chem. – Eur. J., 2013, 19, 2462–2466 CrossRef CAS PubMed; (b) E. R. Clark and M. J. Ingleson, Organometallics, 2013, 32, 6712–6717 CrossRef CAS.
- For neutral boranes see: M. T. Mock, R. G. Potter, D. M. Camaioni, J. Li, W. G. Dougherty, W. S. Kassel, B. Twamley and D. L. DuBois, J. Am. Chem. Soc., 2009, 131, 14454–14465 CrossRef CAS PubMed.
- Crystals suitable for X-ray diffraction were obtained by heating a concentrated THF solution of 4 at 70 °C followed by slow cooling. See ESI† for the polymeric structure of 4. During the course of this work an analogous structure was published see: D. Patel, A. Wooles, A. D. Cornish, L. Steven, E. S. Davies, D. J. Evans, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2015, 44, 14159 RSC.
- 11B{1H} NMR shifts of 3 vary over the range 20–24 ppm due to presence of paramagnetic Fe complexes. In the absence of paramagnetic metal complexes it δ11B in THF is: 20.0 ppm.
- C. J. Adams, R. B. Bedford, E. Carter, N. J. Gower, M. F. Haddow, J. N. Harvey, M. Huwe, M. Angeles-Carter, S. M. Mansell, C. Mendoza, D. M. Murphy, E. C. Neeve and J. Nunn, J. Am. Chem. Soc., 2012, 134, 10333–10336 CrossRef CAS PubMed.
- For the formation and role of zincates in cross-coupling see: (a) E. Hevia, J. Z. Chua, P. García-Álvarez, A. R. Kennedy and M. D. McCall, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5294–5299 CrossRef CAS PubMed; (b) L. Jin, C. Liu, J. Liu, F. Hu, Y. Lan, A. S. Batsanov, J. A. K. Howard, T. B. Marder and A. Lei, J. Am. Chem. Soc., 2009, 131, 16656–16657 CrossRef CAS PubMed; (c) L. C. McCann and M. G. Organ, Angew. Chem., Int. Ed., 2014, 53, 4386–4389 CrossRef CAS PubMed; (d) L. C. McCann, H. N. Hunter, J. A. C. Clyburne and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 7024–7027 CrossRef CAS PubMed; (e) T. D. Bluemke, W. Clegg, P. Garcia-Alvarez, A. R. Kennedy, K. Koszinowski, M. D. McCall, L. Russo and E. Hevia, Chem. Sci., 2014, 5, 3552–3562 RSC; (f) R. B. Bedford, M. Huwe and M. C. Wilkinson, Chem. Commun., 2009, 600–602 RSC.
- J. J. Dunsford, E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2015, 54, 5688–5692 CrossRef CAS PubMed.
- D. J. Evans, P. B. Hitchcock, G. J. Leigh, B. K. Nicholson, A. C. Niedwieski, F. S. Nunes and J. F. Soares, Inorg. Chim. Acta, 2001, 319, 147–158 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision C1), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- http://comp.chem.umn.edu/info/DFT.htm .
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental, computational and X-ray crystallographic details and all relevant NMR spectra. CCDC 1010524, 1010526 and 1058782. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03835a |
| This journal is © The Royal Society of Chemistry 2015 |