
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two RhIII-substituted polyoxoniobates and their base-induced transformation: [H2RhNb9O28]6− and [Rh2(OH)4Nb10O30]8−†
Jung-Ho
Son
*a and
Willam H.
Casey
ab
aDepartment of Chemistry, University of California, Davis, One Shields Ave. Davis, CA 95616, USA. E-mail: junghoson@gmail.com
bDepartment of Chemistry, Department of Earth and Planetary Sciences, University of California, Davis, One Shields Ave. Davis, CA 95616, USA
First published on 9th November 2015
Abstract
Two new rhodium-substituted polyoxoniobates, [H2RhNb9O28]6− (RhNb9) and [Rh2(OH)4Nb10O30]8− (Rh2Nb10) are reported. The two distinct RhIII-substituted niobate clusters behave differently when the pH is raised with TMAOH: the Rh2Nb10 is stable until pH ∼ 12.7, but RhNb9 dissociates to form RhNb5 and RhNb10, similar to some of our other metal-substituted niobates, such as the MNb9 ions (M = Cr or Mn), which transform to MNb10 when the solution pH is raised.
Transition-metal (TM) – substituted polyoxometalates are an important class of materials. The TM-substitution can add catalytic function to the cluster and the polyoxometalate framework adds great redox properties, especially for polyoxo-molybdates and -tungstates.1 In the polyoxoniobate system, a series of TM-substituted decaniobate-type [MNb9O28]x− (MNb9, M = Cr–Ni) have been synthesized recently2 and add to the Ti-, V- and Cu-substituted polyoxoniobates that were previously known.3 In these studies, the substitution is limited to the early transition metals. For the heavier transition metals, there have been structures reported for Re(CO)3-, CpRh- or Pt-coordinated (capped) hexaniobates (Nb6).4 However, substitution of 2nd- or 3rd-row transition metals in the polyoxoniobates as atoms internal to the structure, rather than as capping atoms, has not yet been reported to our knowledge, although among the group V polyoxometalates, [H2PtIVV9O28]5− is known in the polyoxovanadate system.5
Here we report the synthesis, structure, characterization and photocatalytic H2 evolution study of two RhIII-substituted niobates. The two compounds reported here have the stoichiometry: [H2RhNb9O28]6− (RhNb9) and [Rh2(OH)4Nb10O30]8− (Rh2Nb10) as tetramethylammonium (TMA) salts (Fig. 1). The structures of these clusters resemble those of two CrIII-substituted niobates, [H2CrNb9O28]6− (CrNb9) and [Cr2(OH)4Nb12O30]8− (Cr2Nb10) that we previously described.2,6
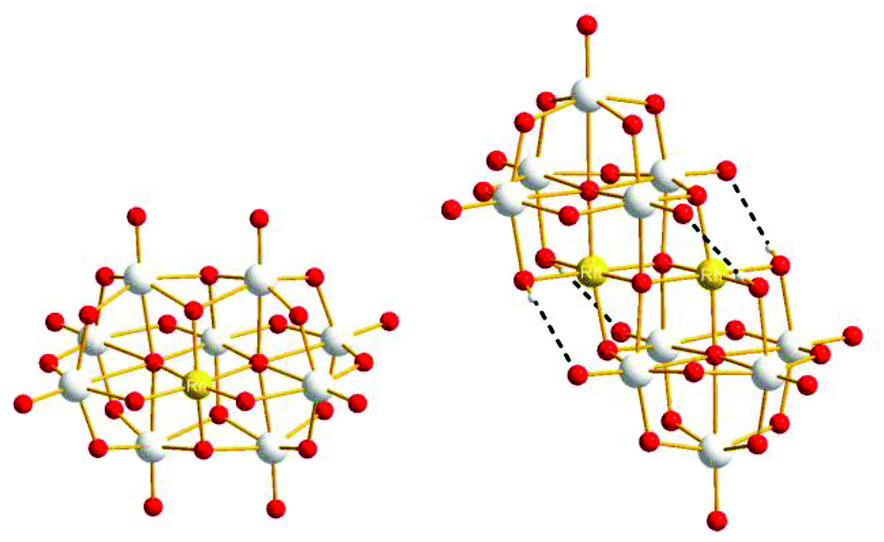 | ||
| Fig. 1 Ball-and-stick model of RhNb9 (left) and Rh2Nb10 (right) (Nb: gray, Rh: gold, O: red). Intramolecular hydrogen bonds are shown with dashed line in Rh2Nb10. | ||
Substitution of RhIII in the polyoxoniobate structure was challenging. Our previous methods employed for MNb9 (M = Cr–Ni) generally showed low yield for rhodium substitution (less than 1%).2 The low yield might be a result from the notoriously slow reaction rate of ligand substitutions at the RhIII center.7 When we attempted to circumvent the slow kinetics with temperature, we found that some RhIII was reduced to Rh0 as a gray or black powder mixed with the crude product at the hydrothermal reaction conditions when the temperature was higher than 120 °C. In order to solve this problem, we added hydrogen peroxide in the reaction mixture to prevent reduction of RhIII. Correspondingly the yields were improved when H2O2 was added (40% and 7% for Rh2Nb10 and RhNb9, respectively). Hydrogen peroxide also might have helped to dissociate the rather stable Nb6 or Nb10 ions and facilitate the formation of RhIII-substituted structures.8
The solution after hydrothermal reaction was typically a mixture of Nb10, Nb6, RhNb9 and Rh2Nb10 ions, as found by electrospray-ionization mass spectra (ESI-MS). We took advantage of the slightly different solubility of each compound to facilitate separation and purification of the Rh-substituted molecules. After washing with isopropanol, the product was extracted with ethanol. The ethanol extract was a mixture of Rh2Nb10 and Nb6 ions, and the precipitate that remained after ethanol extraction was a mixture of Nb10 and RhNb9. The RhNb9 was separated from Nb10 by extraction with ethanol/methanol mixture. Mild heating of the ethanol extract for a few hours caused condensation of more soluble Nb6 into less soluble Nb10 precipitate. The ethanolic orange solution that remained after this heating step consisted of mostly Rh2Nb10. The crystalline products of Rh2Nb10 and RhNb9 were obtained after solvent evaporation.
In the crystal structure of RhNb9, RhIII is substituted at the central metal site so that it does not possess a terminal oxo group, as we also observed in the MNb9O28 (M = Cr–Ni) series.2 The RhIII metal is disordered among the two central sites due to the centrosymmetry, and the sum of RhIII occupancy in those two sites is 1.12, which agrees with stoichiometry of RhNb9. Bond valence sum (BVS) calculation of the Rh site is (3.03), indicating the oxidation state of RhIII. The BVS values of two Rh–μ2-O–Nb (1.37 and 1.38) are much lower than other bridging oxygen atoms, which suggest that those are protonated, similarly to the substituted MNb9 (M = Cr–Ni).2 The structure of Rh2Nb10 is similar to Cr2Nb10, and it can be described as two RhNb5 Lindqvist-type clusters fused by two μ4-O atoms linking two RhIII and two μ3-O atoms linking RhIII and NbV. The oxidation state of rhodium in Rh2Nb10 is also RhIII, as determined by BVS calculation (2.95). The Rh–O bond lengths in Rh2Nb10 are longer and more regular (2.0245(16)–2.0605(16) Å) than Cr–O bonds in Cr2Nb10 (1.9428(13)–2.0131(12) Å). In the structure of Rh2Nb10, four protons are found on the four Rh–μ2-O–Nb, like in the structure of Cr2Nb10.6 Those protons form intramolecular hydrogen bonds to the neighbouring Nb![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (H⋯O distances of 2.309 and 2.386 Å). The ESI-MS spectra of the RhNb9 and Rh2Nb10 agree with their assigned stoichiometries (Fig. S1†).
O (H⋯O distances of 2.309 and 2.386 Å). The ESI-MS spectra of the RhNb9 and Rh2Nb10 agree with their assigned stoichiometries (Fig. S1†).
The pH-dependent stability of the RhIII-substituted niobate clusters were studied by using ESI-MS. When titrated with TMAOH, the golden yellow color of Rh2Nb10 solution did not change until highly basic conditions (pH ∼ 12.9, Fig. S2†), and most of the Rh2Nb10 clusters remained intact for months at this strongly basic condition, as checked by ESI-MS (Fig. S3†). When the solution of RhNb9 was titrated with TMAOH to this condition, the solution color slowly changed from orange to faint yellow overnight (Fig. S2†). The ESI-MS spectra of the solution after one day (Fig. 2) indicated dissociation of RhNb9 to RhNb5 and Nb6. Also, formation of a new RhNb10 was detectable via ESI-MS, which could have formed by self-assembly of dissociated fragments (Fig. 2). It is most likely that this RhNb10 would have a similar structure of previously reported [H2MnIVNb10O32]8− (MnNb10), in view of their similar ESI-MS pattern.9 This observation spurred us to further investigate other TM-substituted polyoxoniobate clusters. We added 50 mg of TMAOH·5H2O to each aqueous solution containing 30 mg of MNb9 (M = Ti, Cr–Ni) and Cr2Nb10 clusters to make pH ∼ 12.6 and monitored the solution by using ESI-MS (Fig. S4–S10†). The Cr2Nb10 clusters were stable at this pH for a long period of time, like Rh2Nb10 (Fig. S4†). The TiNb9, CrNb9 and MnNb9 clusters changed in a few days at this high-pH condition. Some Ti2Nb8 clusters,3a,c,d formed after a week when the pH of TiNb9 was increased (Fig. S5†). Considerable amount of CrNb10 formed after a few days from the CrNb9 solution at high pH (Fig. S6†). This result shows that CrIII- and RhIII-substituted polyoxoniobates are not only structurally similar, but also transform via similar pathways at high pH (Scheme 1). The color of MnNb9 solution changed from purple to brown with time, suggesting oxidation of MnIII, and ESI-MS spectra after 19 days showed formation of small amount of MnIVNb10 (Fig. S7†). However, species such as MnNb5 or CrNb5 was not detectable, which suggests that they are unstable. Other MNb9 clusters (MFe–Ni) were relatively stable at the high pH, but small amount of Nb6 as a decomposition product was detected (Fig. S8–S10†). Thus M2Nb10 (M = RhIII or CrIII) seems to be more stable than MNb9 at high pH. This higher stability of M2Nb10 is partly attributable to the existence of intramolecular hydrogen bonds, which hold the structure together, perhaps making it less susceptible to base hydrolysis.
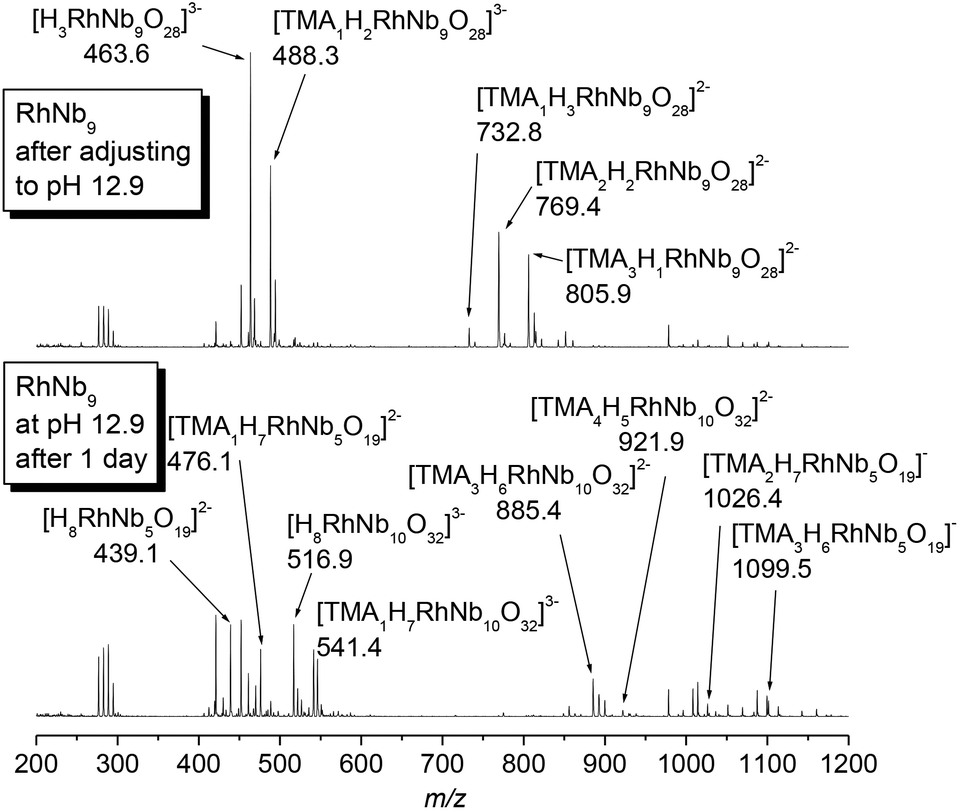 | ||
| Fig. 2 Change of ESI-MS spectra of RhNb9 when the solution pH was adjusted to 12.9. | ||
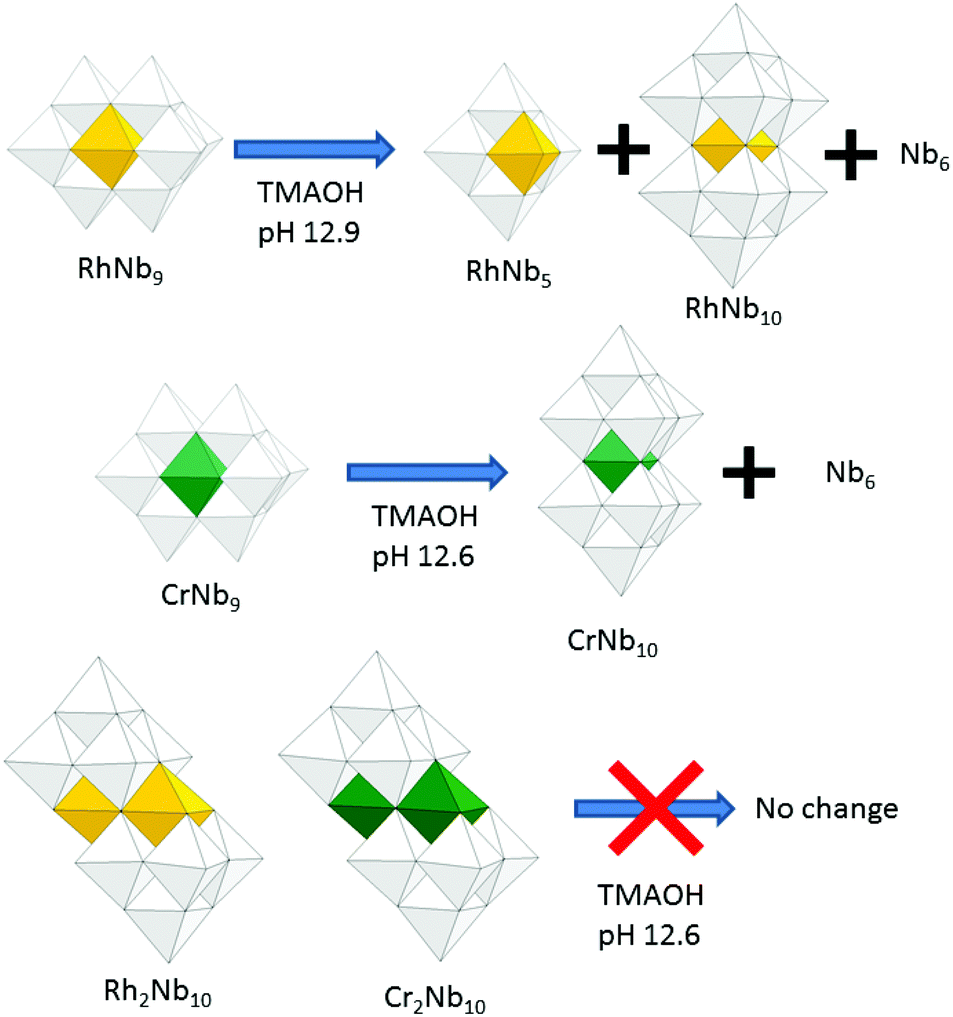 | ||
| Scheme 1 Base-induced transformation of MNb9 and M2Nb10 (M = Rh, Cr). | ||
When titrated with acid, Rh2Nb10 was evident in the ESI-MS spectra until pH 4.0, and RhNb9 was stable until pH 4.5 (Fig. S11 and S12†), although we recognize that the kinetics of dissociation may be suppressed by inclusion of the RhIII. Both solutions formed hydrous niobium-oxide precipitate below those pH values, which could form without dissociating the structures. On the other hand, we note that the stability window of Rh2Nb10 (4 < pH < 13) is similar to Cr2Nb10.6 The RhNb9 exhibited a wider stability range (4.5 < pH < 12) than other MNb9 clusters (M = Cr–Ni) in general.
The UV-Vis spectra of RhNb9 and Rh2Nb10 are shown in Fig. 3. The Rh2Nb10 shows about twice the absorption of visible light relative to the RhNb9 ion, as expected from the stoichiometry of the clusters. The absorption band of Rh2Nb10 (440 nm) is more blue shifted compared to that of RhNb9 (475 nm), which is responsible for the slightly different colors of the solutions of Rh2Nb10 (golden yellow) and RhNb9 (orange-red). These absorption bands correspond to 1A1g to 1T1g or 1T2g transition of RhIII.10 The RhNb9 and Rh2Nb10 clusters were also characterized by using FT-IR (Fig. S13†). The FT-IR spectrum of Rh2Nb10 show similar feature to that of Cr2Nb10 and that of RhNb9 is similar to those of MNb9, which reflect their structural similarity.
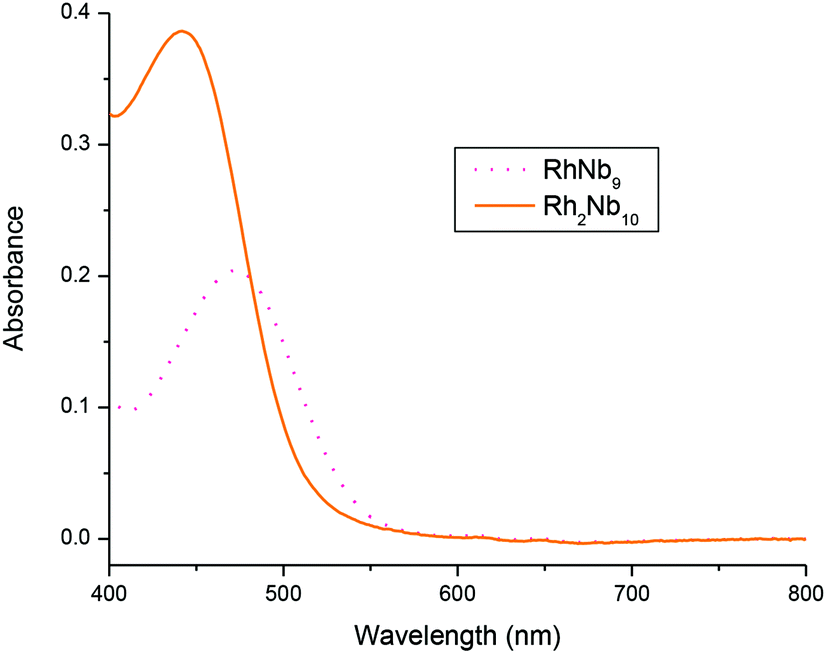 | ||
| Fig. 3 UV-Vis spectra of 2 mM solution of Rh2Nb10 and RhNb9 without background electrolyte. | ||
TM-substituted polyoxometalates, including polyoxoniobates, have recently been actively studied for use in the water-splitting reaction to generate H2 and/or O2 for energy applications.11 We studied H2 evolution from RhNb9 and Rh2Nb10 ions as a continuation of our previous H2-evolution study of the MNb9 ions (M = Cr–Ni). When irradiated with visible light, similarly to the MNb9 ions, neither RhNb9 nor Rh2Nb10 solutions (20% v/v methanol/water) evolved H2. Methanol was used as a sacrificial oxidant. The solution did not evolve H2 without methanol. When a full spectrum from Xe lamp (without UV filter) was employed, however, H2 evolution was observed (50 μmol g−1 h−1 and 43 μmol g−1 h−1, for RhNb9 and Rh2Nb10, Fig. 4). After irradiation, the originally orange-red or yellow solution of RhNb9 and Rh2Nb10 turned greenish brown with small amount of colloids, which probably indicate partial reduction of RhIII to Rh0. We have found such colloids in our previous work and are not surprised by them.2 The RhII is known to exhibit a green color.10 We do not know the amount of RhIII that has been reduced, but comparison of the peak intensities in ESI-MS spectra of the solution before and after irradiation indicated that most of the clusters remained intact (Fig. S14 and S15†). The UV-Vis spectra of Rh2Nb10 before and after the irradiation also did not change considerably, but absorption of RhNb9 solution increased after irradiation, undoubtedly due to the presence of the colloids mentioned above (Fig. S16 and S17†). In the previous H2 evolution study of the MNb9 ions (M = Cr–Ni),2 we found that a significant amount of MNb9 decomposed into Nb6 and Nb10, with corresponding changes in the UV-Vis spectrum due to the colloid formation. Among them, formation of colloids from NiNb9 and CoNb9 positively affected H2 evolution, while colloid formation from other MNb9 (M = Cr–Fe) did not increase the amount of H2 evolution. In our previous work we have found that the colloids were mixed TM–niobium oxide and they were amorphous, as determined by XRD, TEM and EDX. The relative lack of Nb6 and Nb10 decomposition products s after irradiation in the present RhNb9 and Rh2Nb10 suggests that these rhodium RhNb9 and Rh2Nb10 clusters are more stable, perhaps only kinetically so, under the irradiation of intense light when compared to MNb9 (M = Cr–Ni). We also compared H2-evolution activity of the clusters when H2PtCl6 was added as a cocatalyst. In the existence of Pt, Nb10 solution showed ∼20 fold increase in the H2 evolution (1385 μmol g−1 h−1) and amount of precipitate was negligible, but H2 evolution from RhNb9 and Rh2Nb10 after adding H2PtCl6 showed only about 3 fold increases (167 and 152 μmol g−1 h−1, respectively) and conspicuous gray-black precipitate formed in the solution (Fig. 4). This presumed Pt–Rh–NbOx precipitate must be responsible for the slight increase of the H2 evolution, but we did not attempt to characterize it further. The different precipitation behavior of Nb10 and RhNb9/Rh2Nb10 after photocatalytic reaction might be due to their different stabilities upon addition of acidic H2PtCl6. And the lower H2 evolution activity of RhNb9/Rh2Nb10 compared to Nb10 is likely due to the reduced amount of dissolved clusters in solution caused by precipitation, as seen in ESI-MS (Fig. S14 and S15†).
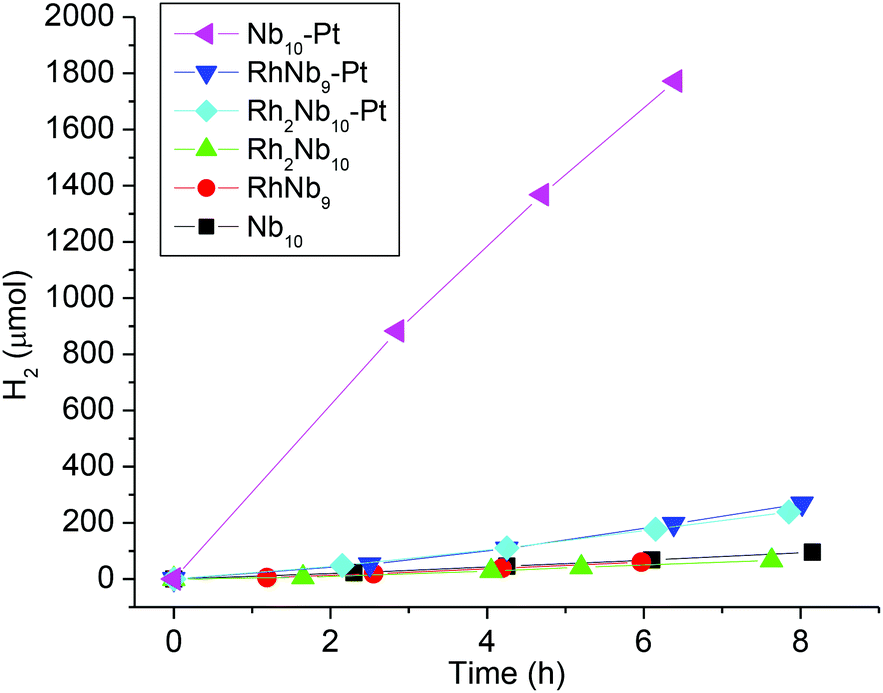 | ||
| Fig. 4 Comparison of H2-evolution activity from the methanol/water solutions (20% v/v) of RhNb9, Rh2Nb10 and Nb10, with and without H2PtCl6. | ||
Conclusions
Two types of new rhodium-substituted polyoxoniobates were synthesized and isolated. The evidences of base-promoted transformation of RhNb9 to RhNb5 and RhNb10 suggest a new synthetic strategy for new polyoxoniobates. Such a reaction can be a useful post-synthetic pathway for new polyoxoniobates, instead of commonly employed hydrothermal reaction in the polyoxoniobate chemistry. The transformation of MnNb9 and CrNb9 at high pH shows that the stabilities of each TM-substituted decaniobate are different, even if they form similar dissociation products at high pH. The RhIII-substituted polyoxoniobates discussed here are important because these can serve as feedstock to make even more TM-substituted polyoxoniobates, such as RhNb5 which might have terminal Rh–OH groups.We thank Jiarui Wang and Prof. Frank E. Osterloh for H2 evolution measurement. This work was supported by an NSF CCI grant through the Center for Sustainable Materials Chemistry, number CHE-1102637.
Notes and references
- (a) L. C. W. Baker, V. S. Baker, K. Eriks, M. T. Pope, M. Shibata, O. W. Rollins, J. H. Fang and L. L. Koh, J. Am. Chem. Soc., 1966, 88, 2329 CrossRef CAS; (b) L. C. W. Baker and J. S. Figgis, J. Am. Chem. Soc., 1970, 92, 3794–3797 CrossRef CAS; (c) T. J. R. Weakley, J. Chem. Soc., Dalton Trans., 1973, 341–346 RSC; (d) C. L. Hill and R. B. Brown Jr., J. Am. Chem. Soc., 1986, 108, 536–538 CrossRef CAS PubMed; (e) M. Faraj and C. L. Hill, J. Chem. Soc., Chem. Commun., 1987, 1487–1489 RSC; (f) J. Hu and R. C. Burns, J. Mol. Catal. A: Chem., 2002, 184, 451–464 CrossRef CAS; (g) J.-H. Choi, J. K. Kim, D. R. Park, T. H. Kang, J. H. Song and I. K. Song, J. Mol. Catal. A: Chem., 2013, 371, 111–117 CrossRef CAS.
- (a) J.-H. Son, J. Wang and W. H. Casey, Dalton Trans., 2014, 43, 17928–17933 RSC; (b) J.-H. Son, C. A. Ohlin and W. H. Casey, Dalton Trans., 2013, 42, 7529–7533 RSC.
- (a) M. Nyman, L. J. Criscenti, F. Bonhomme, M. A. Rodriguez and R. T. Cygan, J. Solid State Chem., 2003, 176, 111–119 CrossRef CAS; (b) C. A. Ohlin, E. M. Villa, J. C. Fettinger and W. H. Casey, Dalton Trans., 2009, 2677–2678 RSC; (c) E. M. Villa, C. A. Ohlin, J. R. Rustad and W. H. Casey, J. Am. Chem. Soc., 2009, 131(45), 16488–16492 CrossRef CAS PubMed; (d) E. M. Villa, C. A. Ohlin and W. H. Casey, J. Am. Chem. Soc., 2010, 132(4), 5264–5272 CrossRef CAS PubMed; (e) G. Guo, Y. Xu, J. Cao and C. Hu, Chem. Commun., 2011, 47, 9411–9413 RSC; (f) P. Huang, C. Qin, X.-L. Wang, C.-Y. Sun, G.-S. Yang, K.-Z. Shao, Y.-Q. Jiao, K. Zhou and Z.-M. Su, Chem. Commun., 2012, 48, 103–105 RSC; (g) J.-H. Son, C. A. Ohlin, E. C. Larson, P. Yu and W. H. Casey, Eur. J. Inorg. Chem., 2013, 1748–1753 CrossRef CAS; (h) Y.-T. Zhang, P. Huang, C. Qin, L.-K. Yan, B.-Q. Song, Z.-X. Yang, K.-Z. Shao and Z.-M. Su, Dalton Trans., 2014, 43, 9847–9850 RSC; (i) P. Huang, E.-L. Zhou, X.-L. Wang, C.-Y. Sun, H.-N. Wang, Y. Xing, K.-Z. Shao and Z.-M. Su, CrystEngComm, 2014, 16, 9582–9585 RSC; (j) J.-Y. Niu, G. Chen, J.-W. Zhao, P.-T. Ma, S.-Z. Li, J.-P. Wang, M.-X. Li, Y. Bai and B.-S. Ji, Chem. – Eur. J., 2010, 16, 7082–7086 CrossRef CAS PubMed.
- (a) A. V. Besserguenev, M. H. Dickman and M. T. Pope, Inorg. Chem., 2001, 40(11), 2582–2586 CrossRef CAS PubMed; (b) P. A. Abramov, M. N. Sokolov, A. V. Virovets, S. Floquet, M. Haouas, F. Taulelle, E. Cadot, C. Vicent and V. P. Fedin, Dalton Trans., 2015, 44(5), 2234–2239 RSC; (c) P. A. Abramov, C. Vicent, N. B. Kompankov, A. L. Gushchin and M. N. Sokolov, Chem. Commun., 2015, 51, 4021–4023 RSC.
- U. Lee, H.-C. Joo, K.-M. Park, S. S. Mal, U. Kortz, B. Keita and L. Nadjo, Angew. Chem., Int. Ed., 2008, 47, 793–796 CrossRef CAS PubMed.
- J.-H. Son, C. A. Ohlin and W. H. Casey, Dalton Trans., 2012, 41, 12674–12677 RSC.
- (a) J. S. Loring, J. Rosenqvist and W. H. Casey, J. Colloid Interface Sci., 2004, 274(1), 142–149 CrossRef CAS PubMed; (b) J. R. Houston, P. Yu and W. H. Casey, Inorg. Chem., 2005, 44(14), 5176–5182 CrossRef CAS PubMed.
- C. A. Ohlin, E. M. Villa, J. Fettinger and W. H. Casey, Angew. Chem., Int. Ed., 2008, 47, 8251–8254 CrossRef CAS PubMed.
- J.-H. Son and W. H. Casey, Dalton Trans., 2013, 42, 13339–13342 RSC.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley-Interscience, New York, 6th edn, 1999 Search PubMed.
- (a) Z. Zhang, Q. Lin, D. Kurunthu, T. Wu, F. Zuo, S.-T. Zheng, C. J. Bardeen, X. Bu and P. Feng, J. Am. Chem. Soc., 2011, 133, 6934–6937 CrossRef CAS PubMed; (b) P. Huang, C. Qin, Z.-M. Su, Y. Xing, X.-L. Wang, K.-Z. Shao, Y.-Q. Lan and E.-B. Wang, J. Am. Chem. Soc., 2012, 134, 14004–14010 CrossRef CAS PubMed; (c) Z.-L. Wang, H.-Q. Tan, W.-L. Chen, Y.-G. Li and E.-B. Wang, Dalton Trans., 2012, 41, 9882–9884 RSC; (d) J.-H. Son, J. Wang, F. E. Osterloh, P. Yu and W. H. Casey, Chem. Commun., 2014, 50, 836–838 RSC; (e) H. Lv, J. Song, H. Zhu, Y. V. Geletii, J. Bacsa, C. Zhao, T. Lian, D. G. Musaev and C. L. Hill, J. Catal., 2013, 307, 48–54 CrossRef CAS; (f) H. Lv, W. Guo, K. Wu, Y. V. Geletii, Z. Chen, S. M. Lauinger, J. Bacsa, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 14015–14018 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, ESI-MS, FT-IR, UV-Vis data. CCDC 1417431 and 1417432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03797b |
| This journal is © The Royal Society of Chemistry 2015 |