
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclometalated Ir(III) complexes of deprotonated N-methylbipyridinium ligands: effects of quaternised N centre position on luminescence†
Benjamin J.
Coe
*a,
Madeleine
Helliwell
a,
James
Raftery
a,
Sergio
Sánchez
a,
Martyn K.
Peers
b and
Nigel S.
Scrutton
b
aSchool of Chemistry, The University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: b.coe@manchester.ac.uk
bManchester Institute of Biotechnology, Faculty of Life Sciences, The University of Manchester, 131 Princess Street, Manchester M1 7DN, UK
First published on 20th October 2015
Abstract
Six new tricationic IrIII complexes of cyclometalating ligands derived from 1-methyl-2-(2′-pyridyl)pyridinium or 1-methyl-4-(2′-pyridyl)pyridinium are described. These complexes of the form [IrIII(C^N)2(N^N)]3+ (C^N = cyclometalating ligand; N^N = α-diimine) have been isolated and characterised as their PF6− and Cl− salts. Four of the PF6− salts have been studied by X-ray crystallography, and structures have been obtained also for two complex salts containing MeCN and Cl− or two Cl− ligands instead of N^N. The influence of the position of the quaternised N atom in C^N and the substituents on N^N on the electronic/optical properties are compared with those of the analogous complexes where C^N derives from 1-methyl-3-(2′-pyridyl)pyridinium (B. J. Coe, et al., Dalton Trans., 2015, 44, 15420). Voltammetric studies reveal one irreversible oxidation and multiple reduction processes which are mostly reversible. The new complexes show intramolecular charge-transfer absorptions between 350 and 450 nm, and exhibit bright green luminescence, with λmax values in the range 508–530 nm in both aqueous and acetonitrile solutions. In order to gain insights into the factors that govern the emission properties, density functional theory (DFT) and time-dependent DFT calculations have been carried out. The results confirm that the emission arises largely from triplet excited states of the C^N ligand (3LC), with some triplet metal-to-ligand charge-transfer (3MLCT) contributions.
Introduction
Cyclometalated IrIII complexes have attracted attention for many years, with a primary focus being their fascinating optical emission properties.1–7 Such species show high stability, large quantum yields and long-lived excited states, due to strong spin–orbit coupling. Therefore, they are of interest for uses in a wide range of fields including organic light-emitting diodes (OLEDs),8–10 light-emitting electrochemical cells,11–13 chemosensors and biological imaging.14–20 Most of the reported complexes fall into two classes; neutral homoleptic IrIII(C^N)3 and heteroleptic IrIII(C^N)2(L–X) (C^N = cyclometalating ligand; L–X = monoanionic ancillary ligand),8,9,21–32 and monocationic heteroleptic species [IrIII(C^N)2(L–L)]+ (L–L = neutral bidentate ligand).7,11–13,33–40 The emission properties of the latter are tuned readily, and various strategies have been adopted. These include changing the degree of π-conjugation in the ligands,34 incorporating electron-rich S-heterocycles,41–44 or most commonly, functionalising C^N or L–L to adjust the energies of the metal and ligand orbitals.1,36,37,44 The electronic properties of substituents and their positioning with respect to the coordinating carbon of C^N influence greatly the energy of the emission.In the context of luminescence, IrIII complexes with C^N ligands derived from pyridinium species were apparently unknown until we reported those of deprotonated 1-methyl-3-(2′-pyridyl)pyridinium (3,2′-C^N).45 The only previous account of structurally related species concerns catalytic studies with hydride complexes that are unsuitable for luminescence.46 A number of reports of cyclometalated complexes of N-methylbipyridinium species with Pd/Pt47 or Ru48 have appeared. The bright blue or blue-green emission and aqueous solubility of our IrIII 3,2′-C^N species45 suggests potential uses in highly efficient OLEDs and/or bio-sensing/imaging. Since the use of IrIII complexes in the latter context16–20,49 is often restricted by poor water solubility,19 increasing their charge from the usual +1 to +3 is beneficial. This structural novelty opens doors for designing further complexes of C^N ligands based on quaternised bipyridinium units with attractive electronic and optical properties. In the previously published complexes [IrIII(C^N)2(N^N)]3+ (N^N = α-diimine), both the nature and energy of the emission are highly influenced by substituents on the ancillary N^N ligand.45 Density functional theory (DFT) calculations show that the blue emission observed when N^N = 2,2′-bipyridyl (bpy) or 4,4′-(tBu)2bpy is mainly triplet ligand-centred (3LC) with some triplet metal-to-ligand charge-transfer (3MLCT) character from C^N. On the other hand, the blue-green emission observed when N^N = 4,4′-(CF3)2bpy has 3L′C with some 3ML′CT nature, due to efficient inter-ligand energy transfer to N^N (L′).
In addition to changing the substituents on N^N, it is of interest to study the effects on the emission properties of varying the location of the quaternised N centre in the C^N ligand. Here, we present a series of new IrIII complexes related to those described recently, but with the C^N ligands derived instead from 1-methyl-2-(2′-pyridyl)pyridinium of 1-methyl-4-(2′-pyridyl)pyridinium. Using as the ancillary ligand bpy, 4,4′-(tBu)2bpy or 4,4′-(CF3)2bpy allows detailed comparisons and reveals the importance of the position of the quaternised centre in achieving blue emission.
Experimental section
Materials and procedures
The compound 1-methyl-3-(2′-pyridyl)pyridinium hexafluorophosphate and the complex salts 4P–6P and 4Cl–6Cl were synthesised as described previously.45 All other reagents were obtained commercially and used as supplied. Products were dried overnight in a vacuum desiccator (silica gel) prior to characterisation. In each case, the bold number refers to the complex cation, while the counter-anions are denoted by P for PF6− or C for Cl−.General physical measurements
1H NMR spectra were recorded on a Bruker UltraShield AV-400 spectrometer, with all shifts referenced to residual solvent signals and quoted with respect to TMS. Elemental analyses were performed by the Microanalytical Laboratory, University of Manchester, and UV–vis spectra were obtained by using a Shimadzu UV-2401 PC spectrophotometer. Mass spectra were recorded by using +electrospray on a Micromass Platform II spectrometer. Cyclic voltammetric measurements were performed by using an Ivium CompactStat. A single-compartment cell was used with a silver/silver chloride reference electrode (3 M NaCl, saturated AgCl) separated by a salt bridge from a 2 mm disc platinum working electrode and platinum wire auxiliary electrode. Acetonitrile was used as supplied from Sigma-Aldrich (HPLC grade), and [NnBu4]PF6 (Fluka, electrochemical grade) was used as the supporting electrolyte. Solutions containing ca. 1.5 × 10−4 M analyte (0.1 M [NnBu4]PF6) were deaerated by purging with N2. All E1/2 values were calculated from (Epa + Epc)/2 at a scan rate of 100 mV s−1. Steady-state emission and excitation spectra were recorded on an Edinburgh Instruments FP920 Phosphorescence Lifetime Spectrometer equipped with a 5 W microsecond pulsed xenon flashlamp. Lifetime data were recorded following excitation with an EPL 375 picosecond pulsed diode laser (Edinburgh Instruments), using time-correlated single photon counting (PCS900 plug-in PC card for fast photon counting). Lifetimes were obtained by tail fitting on the data obtained, or by a reconvolution fit using a solution of Ludox® in the scatterer, and the quality of fit judged by minimisation of reduced chi-squared and residuals squared. Quantum yields were measured upon excitation at 420 nm by using a SM4 Integrating Sphere mounted on an Edinburgh Instruments FP920 Phosphorescence Lifetime Spectrometer.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 4 × 5 mL), and dried to give a bright red powder: 331 mg (68%). δH (400 MHz, CD3CN) 10.20 (2 H, ddd, J = 5.7, 1.7, 0. 7 Hz), 8.51 (2 H, d, J = 8.4 Hz), 8.16 (2 H, ddd, J = 8.5, 7.6, 1.7 Hz), 8.10 (2 H, dd, J = 5.9, 0.8 Hz), 7.74 (2 H, ddd, J = 7.6, 5.7, 1.3 Hz), 7.26 (2 H, dd, J = 7.9, 1.2 Hz), 7.11 (2 H, dd, J = 7.9, 5.9 Hz), 4.58 (6 H, s).
:2, 15 mL) was added to [{IrIII(1-Me-2,2′-bpy)2Cl}2][PF6]4 (100 mg, 0.058 mmol), 2,2′-bipyridyl (30 mg, 0.192 mmol) and AgPF6 (37 mg, 0.146 mmol) under argon, and the mixture heated at 100 °C for 36 h. After cooling to room temperature, the solution was filtered through Celite to remove AgCl and the solvents evaporated. The residue was disolved in a minimum volume of acetone and an excess of [NnBu4]Cl added. The precipitate was filtered off, washed with acetone and purified by column chromatography on Sephadex SP C-25, eluting with 0.025–0.075 M NaCl in acetone/water (1:1). The main yellow band was evaporated to dryness, cold methanol was added and NaCl removed by filtration. The filtrate was evaporated to dryness, and the residue dissolved in cold water. NH4PF6 was added and the precipitate filtered off, washed with ice-cold water and dried to give a bright yellow powder: 77 mg (58%). δH (400 MHz, CD3CN) 8.60 (4 H, d, J = 8.3 Hz), 8.42 (2 H, dd, J = 6.0, 0.9 Hz), 8.26 (2 H, td, J = 8.0, 1.5 Hz), 8.19 (2 H, ddd, J = 8.6, 7.7, 1.6 Hz), 7.94 (2 H, ddd, J = 5.5, 1.5, 0.7 Hz), 7.90 (2 H, ddd, J = 5.7, 1.6, 0.6 Hz), 7.57 (2 H, ddd, J = 7.7, 5.5, 1.3 Hz) 7.46 (2 H, ddd, J = 7.7, 5.7, 1.2 Hz), 7.38 (2 H, dd, J = 7.9, 6.0 Hz), 7.21 (2 H, dd, J = 7.9, 1.3 Hz), 4.65 (6 H, s). ES-MS: m/z = 417 [M − 2PF6]2+, 344 [M − 3PF6]2+, 230 [M − 3PF6]3+. Anal. Calcd (%) for C32H28F18IrN6P3·H2O: C, 33.7; H, 2.6; N, 7.4. Found: C, 33.8; H, 2.3; N, 7.3.
:3, 4 × 5 mL), and dried to give a bright red powder: 331 mg (68%). δH (400 MHz, CD3CN) 10.20 (2 H, ddd, J = 5.7, 1.7, 0. 7 Hz), 8.51 (2 H, d, J = 8.4 Hz), 8.16 (2 H, ddd, J = 8.5, 7.6, 1.7 Hz), 8.10 (2 H, dd, J = 5.9, 0.8 Hz), 7.74 (2 H, ddd, J = 7.6, 5.7, 1.3 Hz), 7.26 (2 H, dd, J = 7.9, 1.2 Hz), 7.11 (2 H, dd, J = 7.9, 5.9 Hz), 4.58 (6 H, s).
:2, 15 mL) was added to [{IrIII(1-Me-2,2′-bpy)2Cl}2][PF6]4 (100 mg, 0.058 mmol), 2,2′-bipyridyl (30 mg, 0.192 mmol) and AgPF6 (37 mg, 0.146 mmol) under argon, and the mixture heated at 100 °C for 36 h. After cooling to room temperature, the solution was filtered through Celite to remove AgCl and the solvents evaporated. The residue was disolved in a minimum volume of acetone and an excess of [NnBu4]Cl added. The precipitate was filtered off, washed with acetone and purified by column chromatography on Sephadex SP C-25, eluting with 0.025–0.075 M NaCl in acetone/water (1:1). The main yellow band was evaporated to dryness, cold methanol was added and NaCl removed by filtration. The filtrate was evaporated to dryness, and the residue dissolved in cold water. NH4PF6 was added and the precipitate filtered off, washed with ice-cold water and dried to give a bright yellow powder: 77 mg (58%). δH (400 MHz, CD3CN) 8.60 (4 H, d, J = 8.3 Hz), 8.42 (2 H, dd, J = 6.0, 0.9 Hz), 8.26 (2 H, td, J = 8.0, 1.5 Hz), 8.19 (2 H, ddd, J = 8.6, 7.7, 1.6 Hz), 7.94 (2 H, ddd, J = 5.5, 1.5, 0.7 Hz), 7.90 (2 H, ddd, J = 5.7, 1.6, 0.6 Hz), 7.57 (2 H, ddd, J = 7.7, 5.5, 1.3 Hz) 7.46 (2 H, ddd, J = 7.7, 5.7, 1.2 Hz), 7.38 (2 H, dd, J = 7.9, 6.0 Hz), 7.21 (2 H, dd, J = 7.9, 1.3 Hz), 4.65 (6 H, s). ES-MS: m/z = 417 [M − 2PF6]2+, 344 [M − 3PF6]2+, 230 [M − 3PF6]3+. Anal. Calcd (%) for C32H28F18IrN6P3·H2O: C, 33.7; H, 2.6; N, 7.4. Found: C, 33.8; H, 2.3; N, 7.3.
Compounds 7P–9P were prepared and purified in a manner identical to 1P–3P by reacting the appropriate ligands with [{IrIII(4,2′-C^N)2Cl}2][PF6]4 instead of [{IrIII(2,2′-C^N)2Cl}2][PF6]4.
X-Ray crystallography
Single crystals of 1P·1.5Me2CO·0.25Et2O, 3P·4MeCN and 7P·MeCN·Et2O were grown by slow diffusion of diethyl ether vapour into solutions in acetone or acetonitrile. For 2P·2C4H8O2, slow evaporation of an acetonitrile/1,4-dioxane solution was used. An attempt to grow crystals of the dimeric precursor [{IrIII(2,2′-C^N)2Cl}2][PF6]4 by slow evaporation of a nitromethane/MeCN solution gave instead the monometallic compound [IrIII(2,2′-C^N)2Cl(MeCN)][PF6]2·MeNO2 (10P·MeNO2), while [IrIII(4,2′-C^N)2Cl2]PF6·2MeNO2·C4H8O2 (11P·2MeNO2·C4H8O2) was obtained when attempting to crystallise [{IrIII(4,2′-C^N)2Cl}2][PF6]4 by slow evaporation of a nitromethane/1,4-dioxane solution. Data were collected on Oxford Diffraction XCalibur 2 or Bruker APEX CCD X-ray diffractometers by using MoKα radiation (λ = 0.71073 Å), and processed by using the Oxford Diffraction CrysAlis Pro or Bruker SMART software packages. The structures were solved by direct methods by using SIR-200450 or SHELXS-97,51 and refined by full-matrix least-squares on all data by using SHELXL-97. All other calculations were carried out by using the SHELXTL package.52
All non-hydrogen atoms were refined anisotropically (except for those of a disordered solvent molecule in 1P·1.5Me2CO·0.25Et2O) and hydrogen atoms were included in idealised positions by using the riding model, with thermal parameters 1.2 times those of aromatic parent carbon atoms, and 1.5 times those of methyl parent carbons. The asymmetric unit of 1P·1.5Me2CO·0.25Et2O contains the complex cation, three disordered PF6− anions, an ordered acetone molecule, a disordered acetone at 0.5 occupancy and a disordered diethyl ether molecule at 0.25 occupancy. All non-H atoms were refined anisotropically, except for the disordered diethyl ether; some restraints were applied for the disordered atoms. H atoms were included in calculated positions, except those of the disordered diethyl ether, which were omitted. The asymmetric unit of 2P·2C4H8O2 contains the complex cation, three PF6− anions and two 1,4-dioxane molecules. The C–O distances in one 1,4-dioxane molecule had to be restrained to 1.4 Å and the displacement parameters for the six ring atoms were refined by using the RIGU and DELU commands. The asymmetric unit of 3P·4MeCN contains the complex cation, three PF6− anions, one of which is disordered, and four acetonitrile molecules, three of which are disordered. Restraints were applied to the F atoms of the disordered PF6−. Crystallographic data and refinement details are presented in Table 1.
| 1P·1.5Me2CO·0.25Et2O | 2P·2C4H8O2 | 3P·4MeCN | 7P·MeCN·Et2O | 10P·MeNO2 | 11P·2MeNO2·C4H8O2 | |
|---|---|---|---|---|---|---|
| Empirical formula | C37.50H39.50F18IrN6O1.75P3 | C42H42F24IrN6O4P3 | C48H56F18IrN10P3 | C38H41F18IrN7OP3 | C25H26ClF12IrN6O2P2 | C28H34Cl2F6IrN6O6P |
| FW | 1229.38 | 1435.92 | 1400.14 | 1238.89 | 960.11 | 958.68 |
| Crystal appearance | Yellow plate | Yellow block | Yellow plate | Green block | Orange rod | Violet block |
| Crystal system | Triclinic | Monoclinic | Triclinic | Monoclinic | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c |
P |
P21/n |
P |
P21/n |
| a/Å | 13.3456(19) | 14.2871(4) | 12.2738(19) | 12.2122(7) | 8.9237(12) | 13.8628(7) |
| b/Å | 13.3584(19) | 19.2131(5) | 12.369(2) | 21.6961(12) | 12.9355(18) | 11.1945(6) |
| c/Å | 13.788(2) | 37.1997(9) | 19.218(3) | 17.2606(9) | 15.352(2) | 22.7606(11) |
| α/° | 70.073(2) | 90 | 98.396(3) | 90 | 72.425(2) | 90 |
| β/° | 87.824(3) | 93.462(1) | 101.854(3) | 101.776(1) | 82.518(3) | 94.945(2) |
| γ/° | 80.281(2) | 90 | 97.282(2) | 90 | 72.619(2) | 90 |
| U/Å3 | 2277.2(6) | 10192.7(5) |
2787.3(8) | 4477.1(4) | 1610.7(4) | 3519.0(3) |
| Z | 2 | 8 | 2 | 4 | 1 | 4 |
| T/K | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) | 180(2) |
| μ/mm−1 | 3.152 | 7.200 | 2.586 | 3.207 | 4.433 | 9.906 |
| Cryst size/mm | 0.55 × 0.40 × 0.20 | 0.19 × 0.15 × 0.08 | 0.45 × 0.40 × 0.10 | 0.40 × 0.30 × 0.20 | 0.45 × 0.20 × 0.20 | 0.17 × 0.15 × 0.12 |
| Reflns collected | 13319 |
34713 |
16345 |
25575 |
9396 | 15947 |
| Independent reflns (Rint) | 9112 (0.0162) | 8853 (0.0470) | 11170 (0.0196) |
9101 (0.0330) | 6387 (0.0158) | 6493 (0.0623) |
| θ max/° (completeness) | 25.00 (98.3) | 66.59 (98.4) | 26.44 (97.2) | 25.00 (99.6) | 26.41 (96.6) | 67.00 (96.2) |
| Reflns with I > 2σ(I) | 8651 | 8415 | 10511 |
7424 | 6080 | 5993 |
| GOF on F2 | 1.042 | 1.072 | 1.025 | 1.013 | 1.067 | 1.019 |
| Final R1, wR2 [I > 2σ(I)] | 0.0314, 0.0818 | 0.0380, 0.0944 | 0.0311, 0.0765 | 0.0377, 0.0845 | 0.0331, 0.0844 | 0.0595, 0.1665 |
| (All data) | 0.0332, 0.0831 | 0.0401, 0.0958 | 0.0337, 0.0780 | 0.0500, 0.0902 | 0.0352, 0.0857 | 0.0621, 0.1703 |
| Peak and hole/eÅ−3 | 1.754, −0.798 | 1.51, −1.10 | 1.726, −1.258 | 1.356, −0.731 | 1.705, −0.934 | 2.369, −1.658 |
Theoretical studies
DFT and time-dependent DFT (TD-DFT) calculations were undertaken on the complex cations 1–3 and 7 by using Gaussian 09.53 Geometry optimisations of the singlet ground (S0) and first triplet excited (T1) states and subsequent TD-DFT calculations were carried out by using the M06 functional54 with the Def2-QZVP55,56 basis set and pseudopotential on Ir and Def2-SVP57 on all other atoms. MeCN was used as CPCM solvent model.58,59 Using these parameters, the first 100 excited singlet states were calculated and simulated UV–vis absorption spectra were convoluted with Gaussian curves of fwhm 3000 cm−1 by using GaussSum.60Results and discussion
Synthesis and characterisation
The new complexes 1–3 and 7–9 (Fig. 1) were synthesised by following the procedure used previously for 4–6.45 The cyclometalated chloride-bridged dimeric intermediates were isolated in crude form only, but identified by 1H NMR spectroscopy. Reacting these dimers with the appropriate N^N ligand in the presence of AgPF6 affords the hexafluorophosphate salts 1P–3P and 7P–9P which were isolated after purification by column chromatography on Sephadex SP C-25. Yields are in the range ca. 30–60%. The chloride salts 1C–3C and 7C–9C were prepared in near quantitative yields by anion metathesis with [NnBu4]Cl in acetone. The identities and purities of the new complex salts are confirmed by 1H NMR spectroscopy, elemental analyses and +electrospray mass spectrometry, and in several cases also by X-ray crystallography (see below). Portions of representative 1H NMR spectra for the bpy-containing complex salts 1P, 4P and 7P are depicted in Fig. 2, showing large changes as the position of the quaternised N atom varies.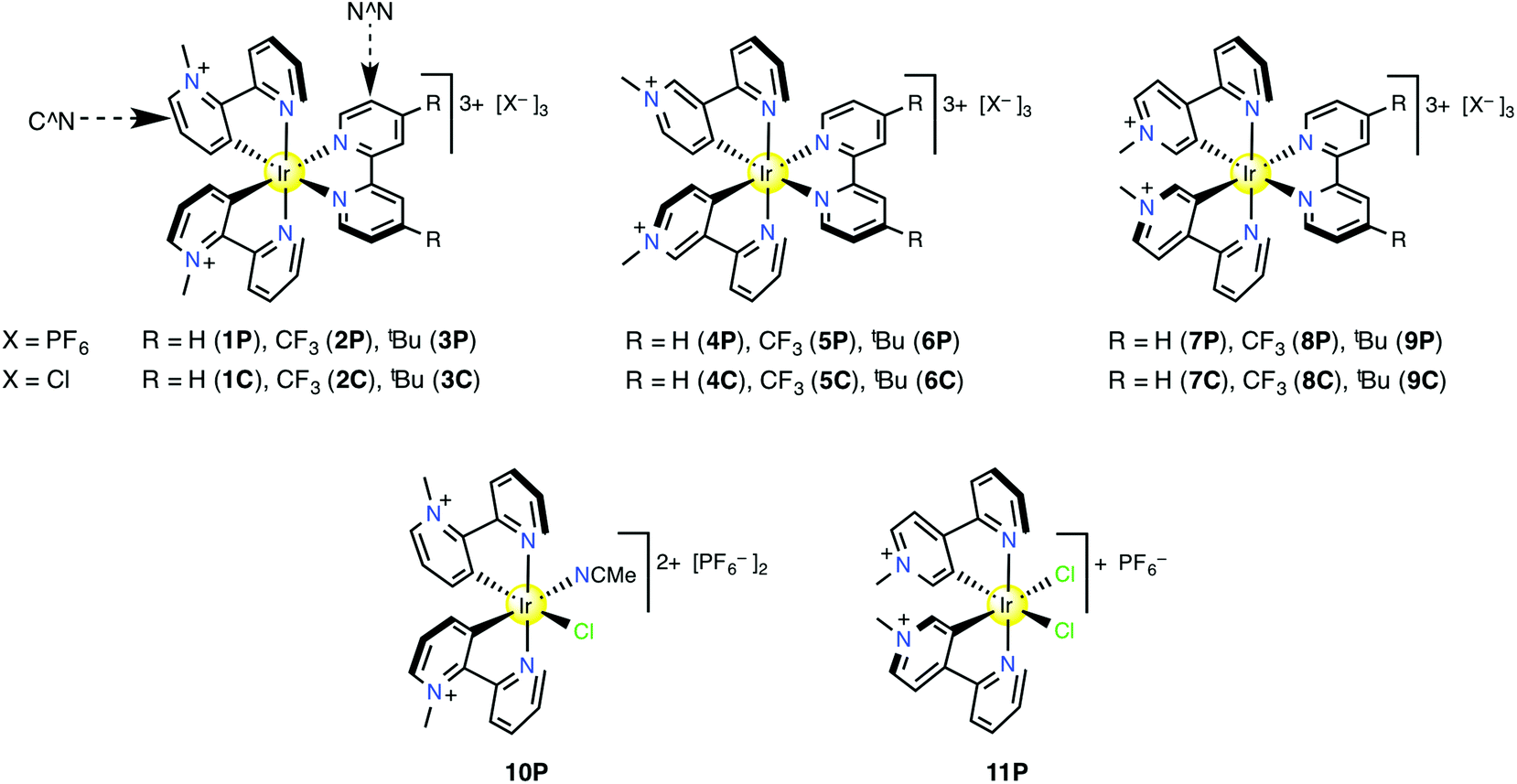 | ||
| Fig. 1 Structures of the studied tris-chelate complex salts and the bis-chelates characterised by X-ray crystallography only. | ||
 | ||
| Fig. 2 Aromatic regions of the 1H NMR spectra of the PF6− salts of complexes 1 (blue), 4 (red), and 7 (green) recorded at 400 MHz in CD3CN. The asterisks denote the signals attributed via COSY studies to the protons of the N^N ligand. | ||
In order to further characterise the system, 13C NMR experiments were carried out for selected compounds 1P, 4P and 7P in CD3CN (see the ESI, Table S1†). The signals were assigned via HMQC and HMBC spectroscopy. As expected, the 13C NMR signals arising from the cyclometalating ring show the most variability as the quaternised N atom moves around. Notably, the cyclometalated carbon atom in 4P (175.08 ppm) is deshielded when compared with 1P (157.05 ppm) and 7P (161.66 ppm). This difference is attributed to the electron-withdrawing effect of the quaternised N atom located in the para-position in 4P. The observed low field chemical shift is similar to those found for the carbenic carbon atom in related IrIII complexes of N-heterocyclic carbene ligands,49b,61–64 and gives an indication of the carbene-like character of the cyclometalated C atom in complexes 4–6.
Structural studies
Single crystals were obtained for solvated forms of 1P–3P and 7P, and also unexpectedly for the chloride complexes in 10P and 11P. Crystallographic data and refinement details are summarised in Table 1. Representions of the molecular structures of the complex cations are shown in Fig. 3, and selected distances and angles are presented in Table 2. The structures of solvates of 4P and 6P have been reported previously,45 and data are included here for comparison purposes. | ||
| Fig. 3 Representations of the molecular structures of the solvates of 1P–3P, 7P, 10P and 11P with the anions, solvent molecules and H atoms removed for clarity (50% probability ellipsoids). Element colours: C = grey; N = blue; F = green; Cl = cyan; Ir = yellow. | ||
| Salt | 1P | 2P | 3P | 4P , | 6P | 7P | 10P | 11P | |
|---|---|---|---|---|---|---|---|---|---|
| a Data taken from ref. 45. b For the two independent complex cations in the asymmetric unit. c trans with respect to the Cl− ligand. d trans with respect to the MeCN ligand. | |||||||||
| Distances (Å) | |||||||||
| Ir–C | 2.002(4) | 2.016(4) | 1.993(3) | 1.99(2) | 1.88(2) | 1.93(2) | 2.011(5) | 1.994(4)c | 1.998(6) |
| Ir–C | 2.003(4) | 2.007(4) | 2.002(3) | 2.01(2) | 1.96(2) | 2.006(9) | 2.014(5) | 1.995(5)d | 1.984(6) |
| Ir–NC^N | 2.043(3) | 2.046(3) | 2.041(3) | 2.04(2) | 2.05(2) | 2.06(1) | 2.049(4) | 2.026(4) | 2.055(5) |
| Ir–NC^N | 2.044(3) | 2.048(3) | 2.042(3) | 2.07(2) | 2.05(2) | 2.044(9) | 2.052(4) | 2.045(4) | 2.060(5) |
| Ir–NN^N | 2.128(3) | 2.135(3) | 2.113(3) | 2.14(2) | 2.14(2) | 2.127(7) | 2.124(4) | ||
| Ir–NN^N | 2.133(3) | 2.136(4) | 2.129(3) | 2.00(2) | 2.10(2) | 2.128(8) | 2.140(4) | ||
| Ir–NMeCN | 2.112(4) | ||||||||
| Ir–Cl | 2.458(1) | 2.457(1) | |||||||
| Ir–Cl | 2.470(1) | ||||||||
| Angles (°) | |||||||||
| C–Ir–C | 87.3(2) | 86.6(2) | 88.5(1) | 85.8(9) | 82.2(8) | 84.4(7) | 86.7(2) | 90.0(2) | 91.5(2) |
| C–Ir–NN^N | 172.1(1) | 173.5(1) | 175.7(1) | 174.4(8) | 177.4(8) | 175.4(7) | 176.3(2) | ||
| C–Ir–NN^N | 97.3(2) | 97.2(1) | 100.0(1) | 100.1(9) | 101.8(8) | 99.6(7) | 96.4(2) | ||
| C–Ir–NN^N | 175.3(1) | 176.3(2) | 170.6(1) | 172.8(8) | 175.4(8) | 175.8(4) | 176.3(2) | ||
| C–Ir–NN^N | 99.0(1) | 99.7(2) | 95.3(1) | 97.9(8) | 99.8(8) | 99.5(4) | 100.0(2) | ||
| C–Ir–NMeCN | 178.1(2) | ||||||||
| C–Ir–Cl | 90.8(1) | 87.3(2) | |||||||
| C–Ir–Cl | 174.0(2) | ||||||||
| C–Ir–NMeCN | 89.6(2) | ||||||||
| C–Ir–Cl | 178.2(1) | 174.9(2) | |||||||
| C–Ir–Cl | 91.6(2) | ||||||||
| C–Ir–NC^N | 79.9(2) | 79.7(2) | 80.0(1) | 80.5(8) | 81.3(6) | 80.6(4) | 79.5(2) | 79.8(2) | 80.1(2) |
| C–Ir–NC^N | 97.1(2) | 93.1(2) | 96.5(1) | 95.5(8) | 95.1(7) | 94.5(4) | 95.7(2) | 97.4(2) | 95.5(2) |
| C–Ir–NC^N | 97.6(1) | 95.1(2) | 96.2(1) | 97.3(8) | 92.1(7) | 91.8(6) | 96.1(2) | 95.7(2) | 95.8(2) |
| C–Ir–NC^N | 79.3(1) | 79.7(2) | 79.6(1) | 79.8(8) | 80.2(6) | 84.9(7) | 80.1(2) | 80.1(2) | 80.3(2) |
| NC^N–Ir–NC^N | 175.8(1) | 171.4(1) | 174.7(1) | 175.3(7) | 171.9(6) | 174.3(3) | 174.1(1) | 175.0(1) | 174.1(2) |
| NC^N–Ir–NN^N | 94.5(1) | 98.2(1) | 97.5(1) | 94.8(6) | 97.1(6) | 97.7(3) | 96.4(1) | ||
| NC^N–Ir–NN^N | 84.4(1) | 87.5(1) | 89.4(1) | 87.8(8) | 86.8(7) | 86.4(3) | 86.8(1) | ||
| NC^N–Ir–NN^N | 88.8(1) | 87.6(1) | 86.2(1) | 89.4(7) | 90.7(6) | 87.3(3) | 87.9(1) | ||
| NC^N–Ir–NN^N | 99.0(1) | 100.0(1) | 95.2(1) | 95.4(7) | 97.0(7) | 97.3(4) | 98.1(1) | ||
| NN^N–Ir–NN^N | 76.5(1) | 76.6(1) | 76.5(1) | 76.6(6) | 76.2(6) | 76.5(3) | 77.0(1) | ||
| NC^N–Ir–NMeCN | 98.1(2) | ||||||||
| NC^N–Ir–Cl | 84.3(1) | 88.9(1) | |||||||
| NC^N–Ir–Cl | 94.5(2) | ||||||||
| NC^N–Ir–NMeCN | 86.0(2) | ||||||||
| NC^N–Ir–Cl | 98.6(1) | 94.9(2) | |||||||
| NC^N–Ir–Cl | 90.1(2) | ||||||||
| NMeCN–Ir–Cl | 89.7(1) | ||||||||
| Cl–Ir–Cl | 90.04(5) | ||||||||
All of the tris-chelate complexes in 1P–4P, 6P and 7P exhibit a pseudooctahedral geometry around the Ir centre with two cyclometalating bipyridinium ligands and one bidentate bpy, 4,4′-(tBu)2bpy or 4,4′-(CF3)2bpy ligand. The strong trans effects of the C-donor fragments affect the structures in two important ways. First, these units adopt a cis geometry, so the pyridyl rings of the C^N ligands are trans disposed. Second, the Ir–N distances to the N^N ligand are extended by ca. 0.07–0.09 Å when compared with those to the C^N ligand in all cases, except for one of the independent complexes in 4P. The Ir–N distances to the N^N ligand are not affected significantly by varying the R substituents. The bite angle of N^N is essentially constant at ca. 77°, while that of the C^N ligand ranges from ca. 82–89°. Inspection of the bond distances within the cyclometalating rings does not reveal any clear evidence for quinoidal character in 4P or 6P that might be expected due to their carbene-like nature indicated by other physical measurements. The structures broadly resemble those reported for related monocationic IrIII complexes, although the C^N bite angles are a little larger than those observed typically (ca. 80–81°).34,36,39,65
The fortuitously obtained structure of 11P is relatively unusual. Various chloride-bridged dimeric IrIII complexes with C^N derived from 2-phenylpyridine (ppy) or its derivatives have been characterised crystallographically.66 However, apparently the only reported structure of a related monometallic dichloride complex is that of cyclometalated 2-(2,4-difluorophenyl)pyridine, crystallised as a monoanion with the cation [IrIII(C^N)2(bpym)]+ (bpym = 2,2′-bipyrimidine) containing the same ligand.67 The average Ir–Cl distance in this known compound of 2.493(4) Å is slightly longer than the corresponding distance in 11P (2.464(1) Å, Table 2). Several neutral complexes IrIII(C^N)2Cl(MeCN) have been reported previously,66a,68 showing Ir–NMeCN distances similar to or slightly longer than that in 10P. The Ir–C distances in 10P are indistinguishable, revealing no variation in structural trans effect between MeCN and Cl−, despite the quite different bonding properties of these two ligands. Comparing the geometric parameters for the 2,2′-C^N complex in 10P with those of the related complexes in 1P–3P shows that replacing the N^N ligand with MeCN and Cl− causes only minor changes which are mostly insignificant. The same applies for replacing N^N with Cl− in the 4,2′-C^N complexes in 7P and 11P.
Electrochemistry
The results of cyclic and differential pulse voltammetric measurements on the PF6− salts 1P–9P recorded in acetonitrile are shown in Table 3. Cyclic voltammograms of 1P–3P are shown in Fig. 4. All potentials are quoted in V with respect to the Ag–AgCl reference electrode. | ||
| Fig. 4 Cyclic voltammograms depicting the reduction processes of complex salts 1P (blue), 2P (green) and 3P (red) recorded at 100 mV s−1 in acetonitrile 0.1 M in [NnBu4]PF6 at 295 K. | ||
| E or E1/2, V vs. Ag–AgCl (ΔEp, mV) | |||||
|---|---|---|---|---|---|
| Salt (isomer)b | IrIV/III | 3+/2+ | 2+/1+ | 1+/0 | 0/1− |
| a Solutions ca. 1.5 × 10−4 M in analyte and 0.1 M in [NnBu4]PF6 at a 2 mm disc platinum working electrode with a scan rate of 100 mV s−1. Ferrocene internal standard E1/2 = 0.44 V, ΔEp = 70–90 mV. b Denotes the position with respect to the quaternised N atom of the N-coordinated 2′-pyridyl ring. c E pa for an irreversible oxidation process determined via differential pulse voltammetry: potential increment = 2 mV; amplitude = 50 mV; pulse width = 0.01 s. d Data taken from ref. 45. e E pc for an irreversible reduction process. | |||||
| 1P (2) | 2.18c | −0.78 (60) | −0.94 (70) | −1.38 (70) | −1.65 (80) |
| 2P (2) | 2.29c | −0.72 (60) | −0.86 (70) | −1.03 (60) | −1.56 (90) |
| 3P (2) | 2.16c | −0.78 (60) | −0.94 (70) | −1.45 (70) | −1.65 (70) |
| 4P (3)d | 2.36c | −1.13e | −1.26 (60) | −1.52e | — |
| 5P (3)d | 2.53c | −0.72 (70) | −1.26e | −1.44e | −1.65 (br) |
| 6P (3)d | 2.34c | −1.18e | −1.36e | −1.56e | — |
| 7P (4) | 2.19c | −0.83 (70) | −0.99 (60) | −1.41 (70) | — |
| 8P (4) | 2.30c | −0.73 (70) | −0.90 (60) | −1.07 (70) | — |
| 9P (4) | 2.16c | −0.82 (80) | −0.98 (80) | −1.48 (80) | −1.76 (80) |
Each compound shows an irreversible oxidation process in the region ca. 2.2–2.5 V, which might be formally assigned to an IrIV/III couple. The relatively high potentials when compared with related complexes of ppy6,11–13,33–39,42 are attributable to the presence of the electron-deficient pyridinium units. DFT calculations (see below) show that the C^N ligands contribute to the HOMO significantly. With a given bipyridinium isomer, the Epa value increases in the order N^N = 4,4′-(tBu)2bpy < bpy < 4,4′-(CF3)2bpy, showing a modest influence (130–190 mV) of the R substituents. As N^N is kept constant, the Epa value increases in the order C^N = 2,2′- ≈ 4,2′- < 3,2′-, revealing that the complexes in which the quaternised N atom is located para to the Ir centre are the most difficult to oxidise (by 180–240 mV).
The reductive regions include multiple processes (Fig. 4). For the 2,2′-C^N complexes in 1P–3P, four reversible waves are observed, corresponding with sequential one-electron reductions to give a monoanionic final product. The E1/2 values are similar for the complexes of bpy (1) and 4,4′-(tBu)2bpy (3), while those for the complex of 4,4′-(CF3)2bpy (2) are lower by 60–420 mV, because the electron-withdrawing influence of the –CF3 groups facilitates reduction. Similar behaviour is shown by the 4,2′-C^N complexes in 7P–9P, except that 7 and 8 display only three waves instead of four. For both 2,2′- and 4,2′-C^N series, the 1+/0 E1/2 value shows the largest dependence on R, suggesting that this third reduction is localised on the N^N ligand. Therefore, the first and second reductions may be assigned to the C^N ligands. In contrast, the reductive behaviour of the 3,2′-C^N complexes in 4P–6P is much less well-defined, with most of the processes being irreversible. Also, sharp anodic return peaks are observed for 1 and 3, indicative of adsorption onto the electrode surface. The nature of these data preclude the discernment of any further trends.
Electronic absorption spectroscopy
The absorption spectral data for the PF6− salts of the new complexes 1–3 and 7–9 recorded in acetonitrile are shown in Table 4, together with those for 4–6.45 Corresponding data for the Cl− salts in water are collected in Table S2,† and spectra of 1P–4P and 7P are shown in Fig. 5. | ||
| Fig. 5 UV–vis absorption spectra of complex salts 1P (blue), 4P (red) and 7P (green) (a); 1P (blue), 2P (green) and 3P (red) (b); recorded in acetonitrile at 295 K. | ||
| Salt (isomer)b | λ max, nm (ε, 103 M−1 cm−1) | E max (eV) | Assignment |
|---|---|---|---|
| a Solutions ca. 1 × 10−5–2 × 10−4 M; ε values are the averages from measurements made at three or more different concentrations (with ε showing no significant variation). b Denotes the position with respect to the quaternised N atom of the N-coordinated 2′-pyridyl ring. c Data taken from ref. 45. | |||
| 1P (2) | 212sh (53.1) | 5.85 | π → π* |
| 257 (34.4) | 4.82 | π → π* | |
| 297 (33.3) | 4.18 | π → π* | |
| 354 (9.4) | 3.50 | d → π* | |
| 437sh (2.8) | 2.84 | d → π* | |
| 2P (2) | 212 (48.2) | 5.85 | π → π* |
| 263 (32.3) | 4.71 | π → π* | |
| 298 (29.7) | 4.16 | π → π* | |
| 350sh (10.2) | 3.54 | d → π* | |
| 426sh (3.4) | 2.91 | d → π* | |
| 3P (2) | 213 (60.4) | 5.82 | π → π* |
| 258 (35.2) | 4.81 | π → π* | |
| 299 (34.0) | 4.15 | π → π* | |
| 312sh (28.9) | 3.97 | d → π* | |
| 355 (9.1) | 3.49 | d → π* | |
| 445sh (2.3) | 2.79 | d → π* | |
| 4P (3)c | 237 (54.3) | 5.23 | π → π* |
| 255sh (48.5) | 4.86 | π → π* | |
| 302 (25.9) | 4.11 | π → π* | |
| 313 (24.5) | 3.96 | d → π* | |
| 352sh (5.3) | 3.52 | d → π* | |
| 5P (3)c | 237 (52.7) | 5.23 | π → π* |
| 259 (52.1) | 4.79 | π → π* | |
| 308 (25.2) | 4.03 | π → π* | |
| 317 (24.3) | 3.91 | d → π* | |
| 350sh (5.8) | 3.54 | d → π* | |
| 6P (3)c | 237 (58.0) | 5.23 | π → π* |
| 260 (49.9) | 4.77 | π → π* | |
| 300 (25.4) | 4.13 | π → π* | |
| 312 (25.9) | 3.97 | d → π* | |
| 355sh (5.5) | 3.49 | d → π* | |
| 7P (4) | 254 (44.8) | 4.88 | π → π* |
| 280sh (32.2) | 4.43 | π → π* | |
| 307 (28.1) | 4.04 | π → π* | |
| 357 (6.8) | 3.47 | d → π* | |
| 392sh (3.8) | 3.16 | d → π* | |
| 435sh (1.9) | 2.85 | d → π* | |
| 8P (4) | 252 (46.2) | 4.92 | π → π* |
| 295 (32.7) | 4.20 | π → π* | |
| 307 (31.7) | 4.04 | π → π* | |
| 350sh (7.8) | 3.54 | d → π* | |
| 389 (5.0) | 3.19 | d → π* | |
| 425sh (2.6) | 2.92 | d → π* | |
| 9P (4) | 234 (44.6) | 5.30 | π → π* |
| 256 (50.1) | 4.84 | π → π* | |
| 299 (31.7) | 4.15 | π → π* | |
| 309 (30.0) | 4.01 | π → π* | |
| 358 (7.2) | 3.46 | d → π* | |
| 394sh (3.8) | 3.15 | d → π* | |
| 440sh (1.9) | 2.82 | d → π* | |
All of complexes 1–9 show intense bands below 320 nm which are assigned to π → π* and high energy MLCT transitions involving both the C^N and N^N ligands. Weaker bands are observed also, with tails extending up to ca. 480 nm in some cases. The lowest energy (LE) band is clearly blue-shifted in the 3,2′-C^N complexes 4–6 (λmax for shoulders ≈ 350–360 nm) when compared with the new complexes 1–3 and 7–9 (λmax for shoulders ≈ 425–445 nm) (Fig. 5a). DFT shows that this band in 4–6 has 1MLCT character with some 1ML′CT and also 1LL′CT contributions for 4 and 5 (L = C^N; L′ = N^N).45 The slight shifts (ca. 0.1 eV) when R changes (Fig. 5b) are consistent with the largely 1MLCT assignment. The observed blue-shifts when varying the structure of the C^N ligands are attributable to destabilisation of their π* orbitals because the para-position of the quaternised N increases its neutral carbene character. Therefore, the energy gap of the MLCT transition increases in 4–6 with respect to their isomeric counterparts 1–3 and 7–9. The absorption spectra are almost unaffected by changing the counter-anions and solvent (Tables 4 and S2†).
Luminescence properties
The emission spectral data for the PF6− and Cl− salts of the new complexes 1–3 and 7–9 recorded in deoxygenated and oxygenated acetonitrile or water are shown in Table 5, together with those for 4–6.45 Spectra of 1P, 4P and 7P are shown in Fig. 6, while those of the other compounds are in the ESI (Fig. S1 and S2†). Changing the counter-anion and solvent has only slight effects on the excitation profiles that remain constant in all cases while monitoring at all the emission maxima. | ||
| Fig. 6 Emission (full lines; excitation at 350 nm) and excitation (dashed lines) spectra of the bpy-containing complex salts 1P (blue), 4P (green) and 7P (red) recorded in acetonitrile at 295 K. | ||
| Salt (isomer)b |
λ
emc (nm) |
τ (μs) | ϕ (%) | ||
|---|---|---|---|---|---|
| deox | ox | deox | ox | ||
| a Solutions 5 × 10−5 M in acetonitrile for PF6− salts, water for Cl− salts. b Denotes the position with respect to the quaternised N atom of the N-coordinated 2′-pyridyl ring. c With excitation over the range 350–440 nm for 1–3 and 7–9, and 315–400 nm for 4–6. deox = deoxygenated; ox = oxygenated. d Estimated experimental errors ±10%. e Data taken from ref. 45. | |||||
| 1P (2) | 526, 550 | 1.3 | 0.9 | 33 | 16 |
| 1C (2) | 527, 555 | 3.9 | 2.3 | 59 | 29 |
| 2P (2) | 519, 540 | 1.4 | 0.8 | 34 | 15 |
| 2C (2) | 518, 545 | 4.1 | 1.8 | 44 | 20 |
| 3P (2) | 530, 556 | 2.0 | 0.9 | 38 | 23 |
| 3C (2) | 530, 558 | 3.9 | 2.1 | 43 | 25 |
| 4P (3)e | 444, 474max, 504, 548 | 3.5 | 1.2 | 24 | 4.7 |
| 4C (3)e | 442, 470max, 504, 547 | 12.1 | 3.9 | 27 | 9.6 |
| 5P (3)e | 466, 494max, 525, 574 | 3.8 | 1.5 | 43 | 16 |
| 5C (3)e | 462, 494max, 529, 575 | 4.3 | 2.5 | 42 | 24 |
| 6P (3)e | 440, 470max, 502, 546 | 3.8 | 1.2 | 43 | 10 |
| 6C (3)e | 440, 468max, 500, 547 | 9.5 | 2.9 | 45 | 14 |
| 7P (4) | 520, 548 | 2.0 | 1.2 | 30 | 15 |
| 7C (4) | 518, 549, 609 | 3.9 | 2.2 | 60 | 32 |
| 8P (4) | 510, 540 | 2.9 | 1.4 | 44 | 22 |
| 8C (4) | 508, 540, 592 | 4.7 | 2.7 | 56 | 33 |
| 9P (4) | 522, 555 | 2.3 | 1.0 | 44 | 18 |
| 9C (4) | 522, 553, 613 | 3.4 | 1.9 | 52 | 27 |
All of the spectra are structured, especially for complexes 4–6,45 indicating significant ligand contributions to the luminescence. The profiles and emission energies of 1–3 and 7–9 are similar and red-shifted when compared to 4–6 (Fig. 6, S1 and S2†). The importance of the position of the quaternised N is evident; when it is located meta to the cyclometalating C atom, green emission is observed (λmax = 508–530 nm), but when it is positioned para to the C, blue or blue-green emission arises (λmax = 468–494 nm). This difference is attributable to the almost carbene-like character and consequent relative orbital energies in 4–6. Various other types of Ir complexes emit in the green region, e.g. [IrIII(ppy-PBu3)3][PF6]2 [ppy-PBu3 = cyclometalated 2-(5-tri-n-butylphosphoniumphenyl)pyridine]69 and monocationic complexes of 4,4′-(tBu)2bpy with –SF5 substituents on the C^N ligands.70
The influence of the R substituents in the 2,2′-C^N (1–3) and 4,2′-C^N (7–9) complexes is small. The emission energies follow the expected trend R = tBu < H < CF3, but with a difference of only ca. 0.05 eV between the extremes (Table 5). This effect can be attributed to the stabilisation of the metal orbitals caused by placing electron-withdrawing groups on the ancillary ligand. These results suggest that in all of these new complexes the emission has a mainly 3LC character involving the C^N ligand with some 3MLCT contribution. By contrast, for the 3,2′-C^N complexes, such a situation pertains for 4 and 6, but 5 behaves differently and gives N^N-based emission.45 Therefore, in that series the emission energy is decreased (and the bands red-shifted) for the –CF3 derivative with respect to the other complexes.
When keeping N^N constant, the quantum yields (Φ) of 1–3 and 7–9 are generally similar to, or a little larger than those of 4–6 (Table 5), showing that moving the quaternised N to a meta-position with respect to the cyclometalating C does not facilitate non-radiative decay. In contrast to the almost invariant emission energies, τ always increases and Φ increases in most instances (sometimes markedly) on moving from a PF6− salt in acetonitrile to its Cl− analogue in water. In all cases, both τ and Φ increase substantially on deoxygenation, consistent with emission originating from triplet excited states.
Theoretical calculations
DFT and TD-DFT calculations have been carried out on the complexes 1–3 and 7 by using the M06 functional with the Def2-QZVP basis set on Ir and Def2-SVP on all other atoms, as used for 4–6 previously.45The optimised structures in the ground state agree well with the data obtained from X-ray crystallography (see the ESI, Table S3†). Selected MOs are depicted in Fig. S3–S6,† and the orbital compositions are shown in Table S4.† The frontier orbitals HOMO−2 to LUMO+2 are essentially invariant within the cations 1–3 and 7. The HOMO−1 and HOMO−2 are mainly centered on the Ir atom (67–72%) with some contribution from the C^N ligand (16–22%), while the HOMO has almost the same contribution from both fragments (Ir 47–55%; C^N 43–51%). The LUMO and LUMO+1 are based on C^N almost completely (96–98%), while the LUMO+2 is located on N^N (95–97%).
The S0 → S1 transition energies calculated in acetonitrile and the corresponding major orbital contributions are presented in Table S5 (ESI†). The simulated absorption spectra for complexes 1–3 agree well with those measured (Fig. 7). The calculated wavelengths of the LE transitions 421 (1), 412 (2), 424 (3) and 416 nm (7) are slightly blue shifted with respect to the experimental λmax values (437 (1), 426 (2), 445 (3) and 435 nm (7); Table 4). In all complexes, this transition has almost pure HOMO → LUMO character (92–95%). Therefore, the LE bands can be assigned to a mixture of 1LC and 1MLCT (L = C^N). The observed modest dependence of the LE transition on the R substituents is reproduced by the calculations on 1–3, i.e. ΔE increases in the order R = tBu < H < CF3. Comparisons with the data obtained for 4–645 show that the level of theory applied also predicts the red-shifts of the LE band observed on moving from the 3,2′-C^N complexes to their 2,2′-C^N counterparts 1–3 (and for 4 → 7); the calculated λ values are 347 (4), 342 (5) and 348 nm (6).
 | ||
| Fig. 7 M06/Def2-QZVP/Def2-SVP-calculated (blue) UV–vis spectra of (a) 1, (b) 2 and (c) 3, and the corresponding experimental data (green) for the PF6− salts in acetonitrile. The ε-axes refer to the experimental data only and the vertical axes of the calculated data are scaled to match the main experimental absorptions. The oscillator strength axes refer to the individual calculated transitions (red). | ||
For 1–3 and 7, the first computed transition involving mainly the N^N ligand (i.e. a dominant component to LUMO+2; in all cases HOMO−2 → LUMO+2 1MLCT) lies to markedly higher energy when compared with the LE transition. The electronic influence of the R substituents is manifested in the predicted energies. The transition is at 3.90 eV (318 nm) for 1, 3.99 eV (311 nm) for 3 and 3.87 eV (320 nm) for 7, but 3.69 eV (336 nm) for 2 due to the stabilisation of the LUMO+2 by the –CF3 groups.
The nature of the luminescence was addressed by optimising the first triplet excited states T1 of 1–3 and 7. The computed geometric parameters of these states are similar to those found for the corresponding ground states S0 (Table S3, ESI†). The lowest energy emissions calculated in acetonitrile as ΔE(T1 − S0) at 531 (1), 527 (2), 538 (3) and 521 nm (7) are close to the experimental values (526 (1), 519 (2), 530 (3) and 520 nm (7); Table 5). Also, the calculations reproduce the experimental trend in the emission wavelengths (λem2 < 1 < 3). The spin densities for the T1 state for all complexes (Fig. 8) are located mainly on one C^N ligand, together with the Ir atom to a lesser extent. Therefore, the emissions can be assigned as 3LC involving the cyclometalating ligand with some 3MLCT contribution. These results are similar to those obtained for complexes 4 and 6,45 showing that the only complex of the nine studied with a different nature of the emission is 5. In that case, the –CF3 substituents stabilise the 3L′C state, causing efficient inter-ligand energy transfer from the C^N to the emitting N^N fragment. In complex 2, the stabilisation due to the –CF3 groups is insufficient to bring the energy of the 3L′C below the 3LC state.
 | ||
| Fig. 8 M06/Def2-QZVP-SVP-calculated spin density plots for the T1 state of the complexes 1, 2, 3 and 7. In each case, the N^N ligand is pointing upwards. | ||
Conclusions
We have synthesised and characterised a series of new IrIII complexes by using three different isomers of 1-methyl-(2′-pyridyl)pyridinium to generate cyclometalating ligands C^N. Because the latter are charge-neutral, adding an α-diimine ancillary ligand N^N affords species with a 3+ charge and unusually high solubility in water when isolated as Cl− salts. Such enhanced aqueous solubility increases the prospects for applications in bioimaging. The complexes are characterised fully as both their PF6− and Cl− salts, and in four cases, their structures are confirmed by single-crystal X-ray crystallography. Electrochemistry reveals for each complex an irreversible oxidation of the {IrIII(C^N)2}3+ unit and multiple ligand-based one-electron reductions. The reductive behaviour is much better defined for the new 2,2′-C^N and 4,2′-C^N complexes when compared with their 3,2′-C^N counterparts, with up to four reversible waves being observed. Within each series, the redox potentials are affected significantly by varying the R substituents on the N^N ligand. All of the complex salts appear yellow coloured and their UV–vis absorption spectra show low energy bands in the region ca. 350–450 nm. DFT and TD-DFT calculations using the M06 functional confirm that these are attributable to 1MLCT transitions with some 1LC, 1ML′CT and also 1LL′CT contributions. Changing the N^N ligand while keeping C^N constant affects the absorption spectra only slightly. The observed red-shifts of the low energy bands for the 2,2′-C^N and 4,2′-C^N complexes with respect their 3,2′-C^N analogues are reproduced theoretically. In contrast to the 3,2′-C^N complexes that show intense blue or blue-green emission, the new isomeric species are all green emitters. The emission is shown by DFT to originate from 3LC excited states involving the cyclometalating ligand with some 3MLCT contribution. Therefore, changing the N^N ligand has only a minor influence on the luminescence. Neither the absorption nor emission spectra show more than slight solvent dependence. In terms of future prospects, there is great scope for modifying the optical properties of these highly charged complexes, for example by changing the substituent on the quaternised N atom and/or attaching other groups to either the C^N or N^N ligands.Acknowledgements
We thank the EPSRC for support (grant EP/J018635/1), and the BBSRC for a PhD studentship (M. K. P.). N. S. S. is supported by an EPSRC Established Career Fellowship (grant EP/J020192/1) and was funded by a Royal Society Wolfson Merit Award. We thank Dr Sarah L. Heath for solving the X-ray crystal structure of 7P·MeCN·Et2O, and Dr Louise S. Natrajan and Michael B. Andrews for assistance with the luminescence measurements.References
- M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- J. A. G. Williams, A. J. Wilkinson and V. L. Whittle, Dalton Trans., 2008, 2081–2099 RSC.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- Y.-M. You and W.-W. Nam, Chem. Soc. Rev., 2012, 41, 7061–7084 RSC.
- S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2985–3007 CrossRef CAS PubMed.
- K. P. S. Zanoni, R. L. Coppo, R. C. Amaral and N. Y. Murakami Iha, Dalton Trans., 2015, 44, 14559–14573 RSC.
- M. A. Baldo, M. E. Thompson and S. R. Forest, Nature, 2000, 403, 750–753 CrossRef CAS PubMed.
- Y. You and S. Y. Park, J. Am. Chem. Soc., 2005, 127, 12438–12439 CrossRef CAS PubMed.
- C.-L. Ho, H. Li and W.-Y. Wong, J. Organomet. Chem., 2014, 751, 261–285 CrossRef CAS PubMed.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J.-J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS PubMed.
- M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. De Cola, Adv. Funct. Mater., 2010, 20, 1812–1820 CrossRef CAS PubMed.
- T. Hu, L. He, L. Duan and Y. Qiu, J. Mater. Chem., 2012, 22, 4206–4215 RSC.
- K. K.-W. Lo, M.-W. Louie and K. Y. Zhang, Coord. Chem. Rev., 2010, 254, 2603–2622 CrossRef CAS PubMed.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS PubMed.
- Y. You, S. Cho and W. Nam, Inorg. Chem., 2014, 53, 1804–1815 CrossRef CAS PubMed.
- K. K.-W. Lo and S. P.-Y. Li, RSC Adv., 2014, 4, 10560–10585 RSC.
- S. Stimpson, D. R. Jenkinson, A. Sadler, M. Latham, A. Wragg, A. J. H. M. Meijer and J. A. Thomas, Angew. Chem., Int. Ed., 2015, 54, 3000–3003 CrossRef CAS PubMed.
- K. Y. Zhang, S.-J. Liu, Q. Zhao, F. Li and W. Huang, Struct. Bonding, 2015, 165, 131–180 CrossRef.
- S. J. Lee, K.-M. Park, K. Yang and Y. Kang, Inorg. Chem., 2009, 48, 1030–1037 CrossRef CAS PubMed.
- C.-H. Yang, M. Mauro, F. Polo, S. Watanabe, I. Muenster, R. Fröhlich and L. De Cola, Chem. Mater., 2012, 24, 3684–3695 CrossRef CAS.
- C.-H. Chang, Z.-J. Wu, C.-H. Chiu, Y.-H. Liang, Y.-S. Tsai, J.-L. Liao, Y. Chi, H.-Y. Hsieh, T.-Y. Kuo, G.-H. Lee, H.-A. Pan, P.-T. Chou, J.-S. Lin and M.-R. Tseng, ACS Appl. Mater. Interfaces, 2013, 5, 7341–7351 CAS.
- Y. Kang, Y.-L. Chang, J.-S. Lu, S.-B. Ko, Y. Rao, M. Varlan, Z.-H. Lu and S. Wang, J. Mater. Chem. C, 2013, 1, 441–450 RSC.
- F. Kessler, Y. Watanabe, H. Sasabe, H. Katagiri, M. K. Nazeeruddin, M. Grätzel and J. Kido, J. Mater. Chem. C, 2013, 1, 1070–1075 RSC.
- J. Frey, B. F. E. Curchod, R. Scopelliti, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and E. Baranoff, Dalton Trans., 2014, 43, 5667–5679 RSC.
- N. Darmawan, C.-H. Yang, M. Mauro, R. Fröhlich, L. De Cola, C.-H. Chang, Z.-J. Wu and C.-W. Tai, J. Mater. Chem. C, 2014, 2, 2569–2582 RSC.
- J. Lee, H. Oh, J. Kim, K.-M. Park, K. S. Yook, J. Y. Lee and Y. Kang, J. Mater. Chem. C, 2014, 2, 6040–6047 RSC.
- J.-B. Kim, S.-H. Han, K. Yang, S.-K. Kwon, J.-J. Kim and Y.-H. Kim, Chem. Commun., 2015, 51, 58–61 RSC.
- K. P. S. Zanoni, A. Ito and N. Y. Murakami Iha, New J. Chem., 2015, 39, 6367–6376 RSC.
- J. Feldman, G. D. Vo, C. D. McLaren, T. C. Gehret, K.-H. Park, J. S. Meth, W. J. Marshall, J. Buriak, L. M. Bryman, K. D. Dobbs, T. H. Scholz and S. G. Zane, Organometallics, 2015, 34, 3665–3669 CrossRef CAS.
- T. Duan, T.-K. Chang, Y. Chi, J.-Y. Wang, Z.-N. Chen, W.-Y. Hung, C.-H. Chen and G.-H. Lee, Dalton Trans., 2015, 44, 14613–14624 RSC.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal Jr., G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2010, 49, 5625–5641 CrossRef CAS PubMed.
- S. B. Meier, W. Sarfert, J. M. Junquera-Hernández, M. Delgado, D. Tordera, E. Ortí, H. J. Bolink, F. Kessler, R. Scopelliti, M. Grätzel, M. K. Nazeeruddin and E. Baranoff, J. Mater. Chem. C, 2013, 1, 58–68 RSC.
- E. C. Constable, C. E. Housecroft, P. Kopecky, C. J. Martin, I. A. Wright, J. A. Zampese, H. J. Bolink and A. Pertegas, Dalton Trans., 2013, 42, 8086–8103 RSC.
- S. Evariste, M. Sandroni, T. W. Rees, C. Roldán-Carmona, L. Gil-Escrig, H. J. Bolink, E. Baranoff and E. Zysman-Colman, J. Mater. Chem. C, 2014, 2, 5793–5804 RSC.
- A. F. Henwood, S. Evariste, A. M. Z. Slawin and E. Zysman-Colman, Faraday Discuss., 2014, 174, 165–182 CAS.
- G. E. Schneider, A. Pertegás, E. C. Constable, C. E. Housecroft, N. Hostettler, C. D. Morris, J. A. Zampese, H. J. Bolink, J. M. Junquera-Hernández, E. Ortí and M. Sessolo, J. Mater. Chem. C, 2014, 2, 7047–7055 RSC.
- K. Hasan, A. K. Pal, T. Auvray, E. Zysman-Colman and G. S. Hanan, Chem. Commun., 2015, 51, 14060–14063 RSC.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- K. K.-W. Lo, K. Y. Zhang, C.-K. Chung and K. Y. Kwok, Chem. – Eur. J., 2007, 13, 7110–7120 CrossRef CAS PubMed.
- C. Dragonetti, S. Righetto, D. Roberto, R. Ugo, A. Valore, F. Demartin, F. De Angelis, A. Sgamellotti and S. Fantacci, Inorg. Chim. Acta, 2008, 361, 4070–4076 CrossRef CAS PubMed.
- M. S. Lowry, W. R. Hudson, R. A. Pascal Jr. and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS PubMed.
- B. J. Coe, M. Helliwell, S. Sánchez, M. K. Peers and N. S. Scrutton, Dalton Trans., 2015, 44, 15420–15423 RSC.
- G. Song, Y. Zhang, Y. Su, W. Deng, K. Han and X. Li, Organometallics, 2008, 27, 6193–6201 CrossRef CAS.
- (a) S. Dholakia, R. D. Gillard and F. L. Wimmer, Inorg. Chim. Acta, 1983, 69, 179–181 CrossRef CAS; (b) F. L. Wimmer and S. Wimmer, Polyhedron, 1985, 4, 1665–1666 CrossRef CAS; (c) F. L. Wimmer and S. Wimmer, Transition Met. Chem., 1985, 10, 238–240 CrossRef CAS; (d) P. Castan, F. Dahan, S. Wimmer and F. L. Wimmer, J. Chem. Soc., Dalton Trans., 1990, 2971–2977 RSC; (e) P. S. Braterman, J.-I. Song, F. L. Wimmer and S. Wimmer, Inorg. Chim. Acta, 1991, 189, 7–9 CrossRef CAS; (f) P. Castan, J. Jaud, S. Wimmer and F. L. Wimmer, J. Chem. Soc., Dalton Trans., 1991, 1155–1158 RSC; (g) F. L. Wimmer, S. Wimmer and P. Castan, J. Organomet. Chem., 1992, 424, 99–104 CrossRef CAS; (h) M. R. Snow and E. R. T. Tiekink, Z. Kristallogr., 1992, 198, 148–150 CrossRef CAS; (i) P. S. Braterman, J.-I. Song, F. M. Wimmer, S. Wimmer, W. Kaim, A. Klein and R. D. Peacock, Inorg. Chem., 1992, 31, 5084–5088 CrossRef CAS; (j) P. Castan, B. Labiad, D. Villemin, F. L. Wimmer and S. Wimmer, J. Organomet. Chem., 1994, 479, 153–157 CrossRef CAS; (k) S. Wimmer and F. L. Wimmer, J. Chem. Soc., Dalton Trans., 1994, 879–884 RSC; (l) L. Yang, F. L. Wimmer, S. Wimmer, J. Zhao and P. S. Braterman, J. Organomet. Chem., 1996, 525, 1–8 CrossRef CAS.
- T. Koizumi, T. Tomon and K. Tanaka, J. Organomet. Chem., 2005, 690, 1258–1264 CrossRef CAS PubMed.
- (a) Y. Ma, S.-J. Liu, H.-R. Yang, Y.-Q. Wu, C.-J. Yang, X.-M. Liu, Q. Zhao, H.-Z. Wu, J.-C. Liang, F.-Y. Li and W. Huang, J. Mater. Chem., 2011, 21, 18974–18982 RSC; (b) Y.-Y. Zhou, J.-L. Jia, W.-F. Li, H. Fei and M. Zhou, Chem. Commun., 2013, 49, 3230–3232 RSC; (c) S.-J. Liu, H. Liang, K. Y. Zhang, Q. Zhao, X.-B. Zhou, W.-J. Xu and W. Huang, Chem. Commun., 2015, 51, 7943–7946 RSC.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 32, 381–388 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, J. C. A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision B.01), Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- F. Weigend, F. Furche and R. Ahlrichs, J. Chem. Phys., 2003, 119, 12753–12762 CrossRef CAS PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 7992–8003 CrossRef CAS PubMed.
- K.-Y. Lu, H.-H. Chou, C.-H. Hsieh, Y.-H. O. Yang, H.-R. Tsai, H.-Y. Tsai, L.-C. Hsu, C.-Y. Chen, I.-C. Chen and C.-H. Cheng, Adv. Mater., 2011, 23, 4933–4937 CrossRef CAS PubMed.
- B. D. Stringer, L. M. Quan, P. J. Barnard, D. J. D. Wilson and C. F. Hogan, Organometallics, 2014, 33, 4860–4872 CrossRef CAS.
- F. Monti, M. G. I. La Placa, N. Armaroli, R. Scopelliti, M. Grätzel, M. K. Nazeeruddin and F. Kessler, Inorg. Chem., 2015, 54, 3031–3042 CrossRef CAS PubMed.
- N. Zhao, Y.-H. Wu, L.-X. Shi, Q.-P. Lin and Z.-N. Chen, Dalton Trans., 2010, 39, 8288–8295 RSC.
- Selected examples: (a) K. A. McGee and K. R. Mann, Inorg. Chem., 2007, 46, 7800 CrossRef CAS PubMed; (b) E. S. Andreiadis, D. Imbert, J. Pécaut, A. Calborean, I. Ciofini, C. Adamo, R. Demadrille and M. Mazzanti, Inorg. Chem., 2011, 50, 8197 CrossRef CAS PubMed; (c) M. Y. Wong, G.-H. Xie, C. Tourbillon, M. Sandroni, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Dalton Trans., 2015, 44, 8419 RSC.
- F.-F. Chen, Z.-Q. Bian and C.-H. Huang, Chin. J. Inorg. Chem., 2008, 24, 1219 CrossRef CAS PubMed.
- (a) L.-G. Yang, A. von Zelewsky, H. P. Nguyen, G. Muller, G. Labat and H. Stoeckli-Evans, Inorg. Chim. Acta, 2009, 362, 3853 CrossRef CAS PubMed; (b) F. Blasberg, J. W. Bats, M. Wagner and H.-W. Lerner, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1837 CAS.
- H. J. Bolink, L. Cappelli, E. Coronado, A. Parham and P. Stössel, Chem. Mater., 2006, 18, 2778–2780 CrossRef CAS.
- N. M. Shavaleev, G.-H. Xie, S. Varghese, D. B. Cordes, A. M. Z. Slawin, C. Momblona, E. Ortí, H. J. Bolink, I. D. W. Samuel and E. Zysman-Colman, Inorg. Chem., 2015, 54, 5907–5914 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation and theoretical studies. CCDC 1427669–1427674. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03753k |
| This journal is © The Royal Society of Chemistry 2015 |