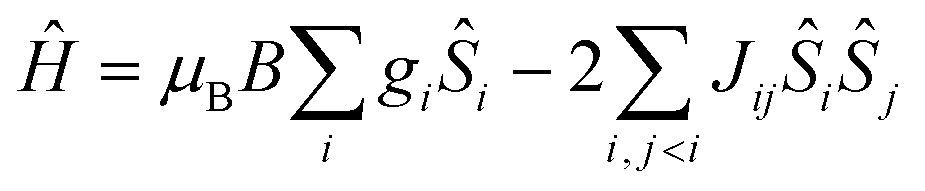DOI:
10.1039/C5DT03615A
(Paper)
Dalton Trans., 2015,
44, 19805-19811
Switching the orientation of Jahn–Teller axes in oxime-based MnIII dimers and its effect upon magnetic exchange: a combined experimental and theoretical study†
Received
15th September 2015
, Accepted 7th October 2015
First published on 12th October 2015
Abstract
A family of MnIII dimers of general formula [MnIII2(R-sao)2(dpa)2](ClO4)2 (1–5) has been synthesised using derivatised phenolic oximes (R-saoH2, where R = H, Me, Et, Ph) in combination with di-(2-picolyl)-amine (dpa). Their structures reveal a double-oxime bridged [MnIII(NO)]2 magnetic core in which the Jahn–Teller axes lie perpendicular to the bridging plane, in contrast to two previously reported family members (6, 7). The switch in the orientation of the Jahn–Teller axes is enforced through the use of the chelating ligand which is present in 1–5 and absent in 6–7. Dc magnetic susceptibility measurements reveal that the exchange interactions between the MnIII metal centres in 1–5 are antiferromagnetic in contrast to that observed for 6 and 7 which are ferromagnetic. DFT calculations performed on complexes 1–6 reproduce both the sign and strength of the J values found experimentally. Molecular orbital analysis unlocks a common mechanism of magnetic coupling based upon the orientation of the Jahn–Teller axis, with the magneto-structural correlation also dependent upon the Mn–N–O–Mn angles – with ferromagnetic interactions at smaller dihedral angles.
Introduction
Molecular magnets – molecules containing exchange-coupled paramagnetic metal ions – are an enormously important class of material with potential application across a diverse range of fields from information storage, quantum computation and molecular spintronics to cryogenic refrigeration and biomedical imaging.1–5 Such species combine the macroscopic properties of magnets with the quantum mechanical properties of molecules, allowing detailed study of quantum size effects in monodisperse, reproducible, orientable, chemically tuneable molecules.6 Recent years have witnessed a plethora of important scientific breakthroughs including the observation of quantum tunnelling of the magnetization, quantum phase interference, memory effects at T = 14 K, single-molecule magnetic detection, the construction of molecular spintronic devices, molecules and coordination polymers displaying enormously enhanced magnetocaloric effects, and the development of protocols for molecule-based quantum information processing.7–20 The discovery of such phenomena, and any potential exploitation thereof, is based upon a detailed understanding of, and subsequent control over, the structure–magnetism relationship. This in turn is derived from a systematic synthetic study of the effects of ligand design, metal identity and reaction conditions upon symmetry and structure, in tandem with detailed physical characterisation and theoretical analysis.21
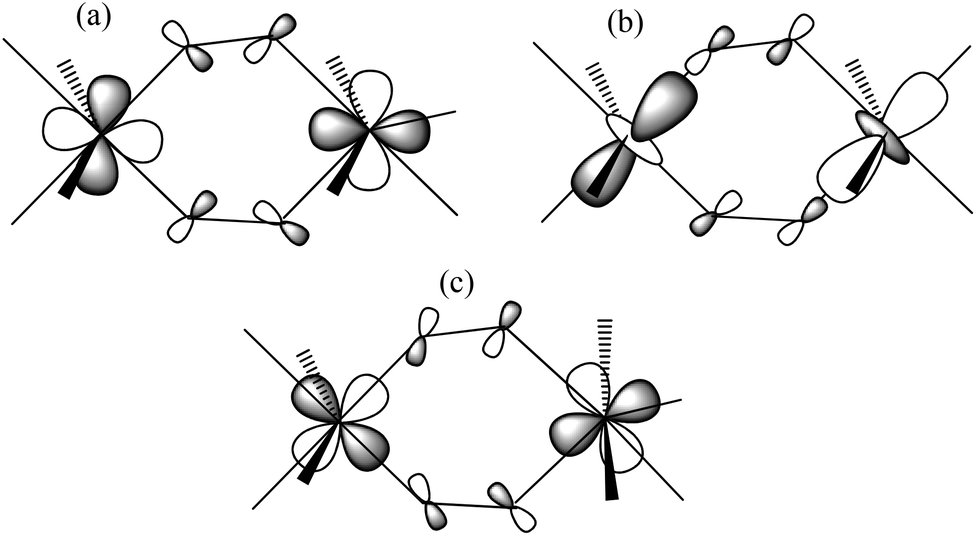
Manganese is often a good choice for constructing such molecules: it commonly exists in the II+, III+ and IV+ oxidation states, resulting in the majority of clusters being mixed-valent, which, in turn, decreases the likelihood of antiferromagnetically coupled cages possessing diamagnetic spin ground states.6 The Jahn–Teller distorted MnIII ion also provides a source magnetic anisotropy that has often been employed in the construction of Single-Molecule Magnets (SMMs),6 whilst the isotropic MnII ion finds application in clusters displaying an enhanced magnetocaloric effect (MCE), a phenomenon that can be exploited for low temperature cooling.17 Application first requires the development of magneto-structural correlations, but this can often be a difficult task due to prohibitively large nuclearities and/or complex exchange interaction patterns. One way to overcome such issues is to construct families of low nuclearity cages (e.g. dimers, trimers, tetramers) which mimic the building blocks of the bigger cages, and investigate how small structural perturbations affect magnetic exchange. Detailed studies on MnIII dimers, however, remain relatively scarce.22 A recent experimental and theoretical study of alkoxide-bridged [MnIII(OR)]2 dimers revealed that the dominant structural feature controlling magnetic exchange (J) was the relative orientations of the Jahn–Teller axes.22 A parallel orientation, perpendicular to the bridging plane of the molecule (Type I, Fig. 1) resulted in weak antiferromagnetic exchange; a parallel orientation within the bridging plane (Type II, Fig. 1) led to borderline cases in which the exchange could be either weakly ferromagnetic or weakly antiferromagnetic; whilst a perpendicular orientation (Type III, Fig. 1) produced ferromagnetic exchange. For the oxime/oxo bridged species [MnIII2(O)(NO)] (Fig. 1, A) the Mn–N–O–Mn torsion angle dominates, with a linear correlation found between the magnitude of J and the torsion angle.22 DFT studies on double-oxime bridged dimers [MnIII(NO)]2 (Fig. 1, B) also revealed a pronounced dependence of the exchange coupling on the relative twisting of the oxime moiety, as proposed previously in more complicated [MnIII3] and [MnIII6] clusters,23 resulting from an accidental orthogonality between the Mn–N–O plane of the first MnIII ion and the Jahn–Teller axis of the second MnIII ion.22
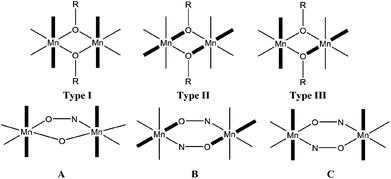 |
| | Fig. 1 Top: alkoxide bridged dimers [MnIII(OR)]2 with Type I–III structures differing in the relative orientations of the Jahn–Teller axes, which are highlighted in bold. Bottom: oxime/oxo dimers [MnIII2(O)(NO)] (A), and the double oxime bridged dimers [MnIII(NO)]2 in which the Jahn–Teller axes lie in the bridging plane (B), and perpendicular to the bridging plane (C). | |
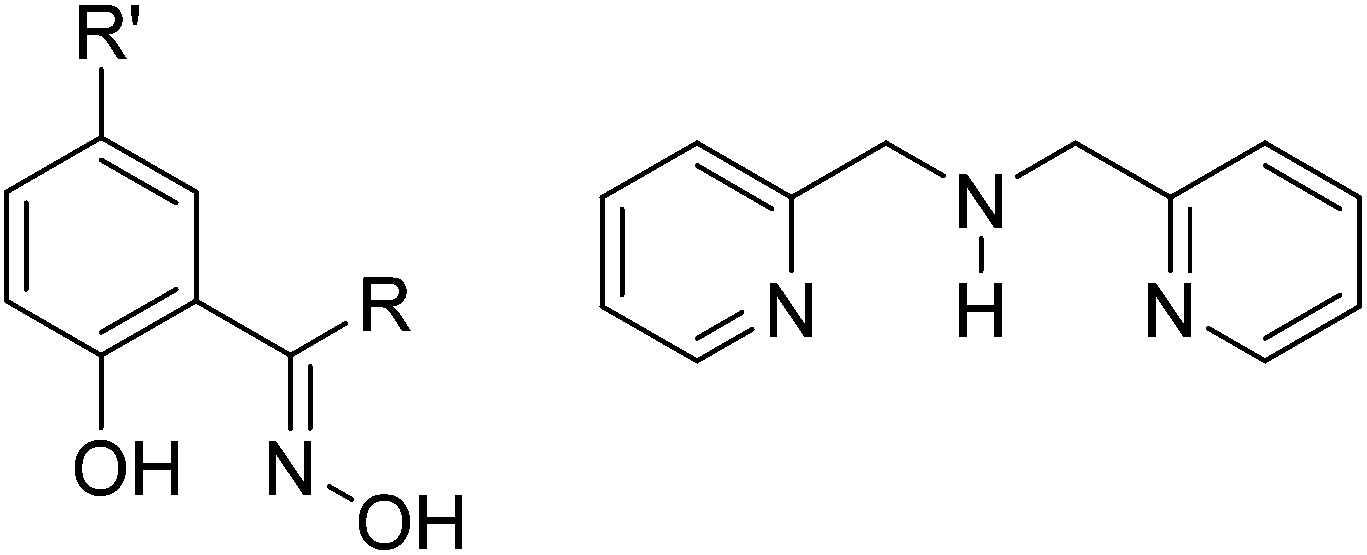
In order to extend the family of double-oxime bridged MnIII dimers – which has just two members – we herein report a series of double-oxime bridged [MnIII(NO)]2 species in which employment of the chelating ligand di-(2-picolyl)-amine, dpa (Fig. 2), switches the orientation of the Jahn–Teller axes from being in the bridging plane (structure type B, Fig. 1) to being perpendicular to the bridging plane (structure type C, Fig. 1) and present a combined experimental and theoretical analyses of the effect of this Jahn–Teller switching upon the magnetic exchange between the two MnIII ions.
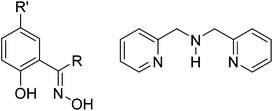 |
| | Fig. 2 Left: generic structure of a phenolic oxime. saoH2, R = R′ = H; Me-saoH2, R = Me, R′ = H, Et-saoH2, R = Et, R′ = H; Ph-saoH2, R = Ph, R′ = H; Me2-saoH2, R = R′ = Me. Right: di-(2-picolyl)-amine, dpa. | |
Experimental
Materials and physical measurements
All synthetic procedures were performed under aerobic conditions using chemicals as received (reagent grade). Caution! Although no problems were encountered here, care should be taken when handling the potentially explosive perchlorate anion. The substituted phenolic oximes were synthesised using the appropriate precursor ketones, hydroxylamine hydrochloride and sodium acetate in EtOH, as described in the literature.24 Variable temperature, solid state magnetic susceptibility data down to 5 K were collected on a Quantum Design MPMS XL SQUID magnetometer equipped with a 7 T dc magnet. Diamagnetic corrections were applied to the observed paramagnetic susceptibilities using Pascal's constants.
Synthesis
General procedure for the synthesis of [MnIII2(R-sao)2(dpa)2](ClO4)2: Mn(ClO4)2·6H2O (0.4 mmol) and the appropriate phenolic oxime (0.4 mmol) were dissolved in MeOH (10 mL). NH4OH (1.6 mmol) was then added to the solution and the reaction left to stir for 40 minutes at room temperature. dpa (0.4 mmol) in EtOH (10 mL) was added to the reaction mixture, which was then filtered after 5 minutes of additional stirring. The black mother liquor was left to evaporate over a period of 4 days, during which time black block-like crystals grew in varying yields. See the ESI† for full synthetic details.
X-Ray crystallography
Diffraction data for samples 1 and 3–5 (CCDC numbers 1044300, 1044302–1044304) were collected on an Oxford Diffraction SuperNova diffractometer using Mo or Cu Kα radiation. The crystal temperature was maintained at 120 K using an Oxford Cryosystems Cryostream 700+ low temperature device. The structures were solved by direct methods and refined by full-matrix least-squares techniques on F2 using the programs SHELXL25a and Olex2.25b X-ray data for complex 2 (CCDC 1044301) were collected at 100 K (Oxford Cryosystems Cobra) on a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum rotating anode generator with VHF Varimax optics (70 m focus). The crystal structure was solved by charge flipping methods in SUPERFLIP25c and the full-matrix least-squares refinement on Fo2 was carried out using SHELXL.25a All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were added at calculated positions and refined using a riding model with isotropic displacement parameters based on the equivalent isotropic displacement parameter (Ueq) of the parent atom. Table S1 in the ESI† contains a summary of the experimental crystallographic parameters.
Computational details
Calculations were performed on the full crystal structures of 1–6. A hybrid B3LYP26 functional with TZV basis set27 was employed for calculating the exchange constants as implemented in the G0928 suite of programs. The high spin states (E-HS) and low spin states (E-BS) were estimated using single determinant wave functions and the broken symmetry approach,29 respectively, and the corresponding J values were computed from the difference between E-HS and E-BS. Further details about the computational methodology are discussed elsewhere.30
Results and discussion
Reacting Mn(ClO4)2·6H2O with the appropriate phenolic oxime (R-saoH2, Fig. 2) and the tridentate ligand dpa in alcohol and in the presence of an appropriate base, produces black crystals of the complexes [MnIII2(Et-sao)2(dpa)2](ClO4)2 (1), [MnIII2(Me-sao)2(dpa)2](ClO4)2 (2), [MnIII2(Me2-sao)2(dpa)2](ClO4)2 (3), [MnIII2(sao)2(dpa)2](ClO4)2 (4), and [MnIII2(Ph-sao)2(dpa)2](ClO4)2 (5) in varying yields over 4 days.
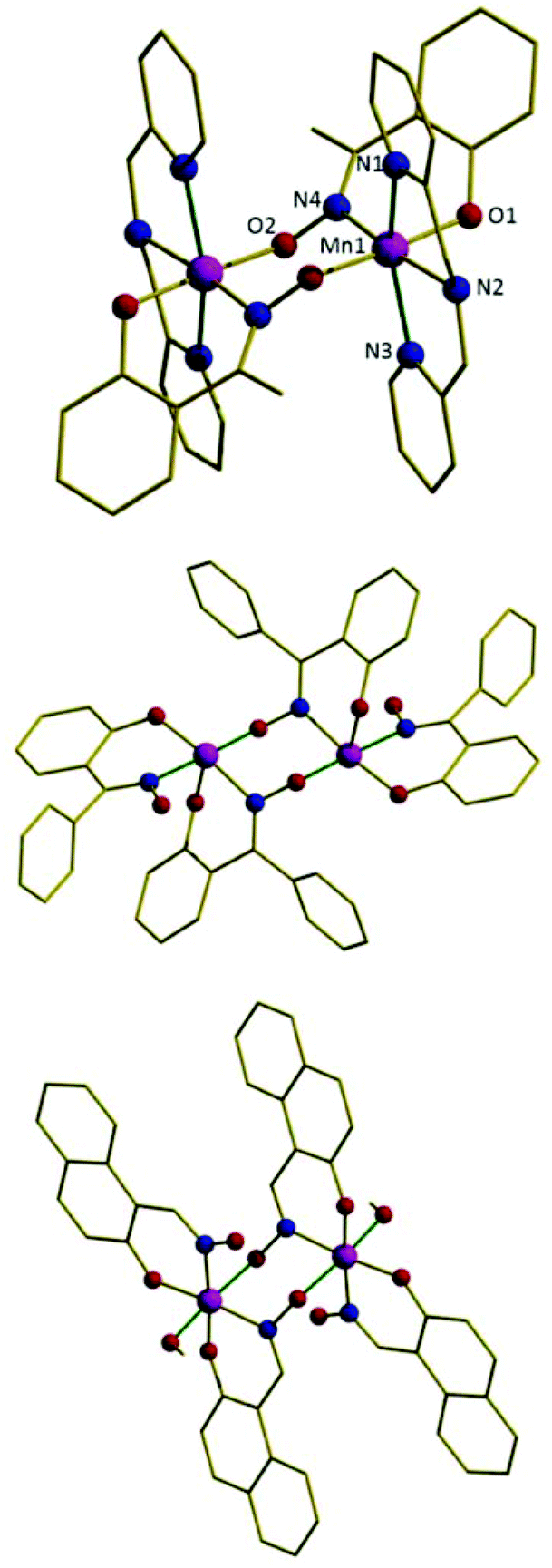
Complexes 1–5 (Fig. 3 shows representative example 2, Table S2† pertinent bond lengths and Table 1 magneto-structural data) describe simple, double-oxime bridged [MnIII(NO)]2 dimers with Mn–O–N–Mn torsion angles of approximately 79° (1), 62° (2), 75° (3), 51° (4) and 63° (5). The remaining coordination sites on each Mn ion are occupied by the three N-atoms of the chelating dpa ligand, and a terminally bonded phenolic O-atom. The metal is thus six coordinate and in a distorted octahedral geometry with the Jahn–Teller axis being the N(pyridine)–Mn–N(pyridine) vector, which lies approximately perpendicular to the bridging [Mn(NO)]2 plane (structure type C, Fig. 1). The two Jahn–Teller axes are not strictly co-parallel but lie at dihedral angles (JT–Mn–Mn–JT) of approximately 26° (1), 24° (2), 21° (3), 23° (4) and 16° (5). The cluster is a 2+ cation with charge balance being maintained through the presence of two ClO4− anions (the packing of the molecules in 1–5 is described in the ESI, Fig. S1–S5†). Complexes 1–5 therefore represent an extension to the family of two previously reported [Mn(NO)]2 dimers, namely the complexes [MnIII2ZnII2(Ph-sao)2(Ph-saoH)4(hmp)2] (6) and [MnIII2(Naphth-sao)2(Naphth-saoH)2(MeOH)2] (7) which are shown in Fig. 3 for comparison.22 Although the bridging [Mn(NO)]2 moiety remains the same in all seven complexes, complexes 6 and 7 differ in that they have their MnIII Jahn–Teller axes co-parallel and in the bridging plane (structure type B, Fig. 1).
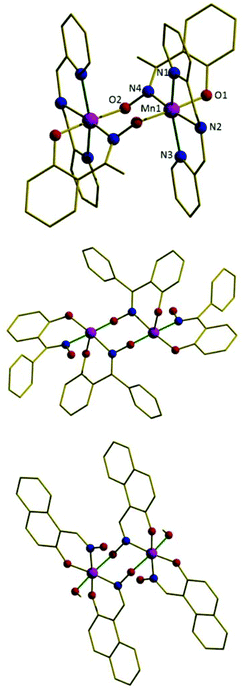 |
| | Fig. 3 The molecular structure of the cation of complex 2 (top), the magnetic core of complex 6 (middle), and complex 7 (bottom). The Jahn–Teller axes of the MnIII ions are highlighted in dark green. Colour code: Mn = purple, O = red, N = blue, C = black. H-atoms are omitted for clarity. | |
Table 1 Summary of pertinent experimentally derived magneto-structural parameters for the [Mn(NO)]2 family of complexes. Theoretically calculated J values for 1–6 are given in parantheses
| Structure |
Type |
Mn–N–O–Mn torsion angle (°) |
J (cm−1) |
|
1
|
C |
78.75 |
−5.73 (−5.76) |
|
2
|
C |
64.59 |
−3.63 (−2.16) |
|
3
|
C |
74.90 |
−5.63 (−5.15) |
|
5
|
C |
63.00 |
−2.05 (−2.99) |
|
6
|
B |
75.21 |
+2.20 (+2.65) |
|
7
|
B |
80.28 |
+1.24 |
SQUID magnetometry
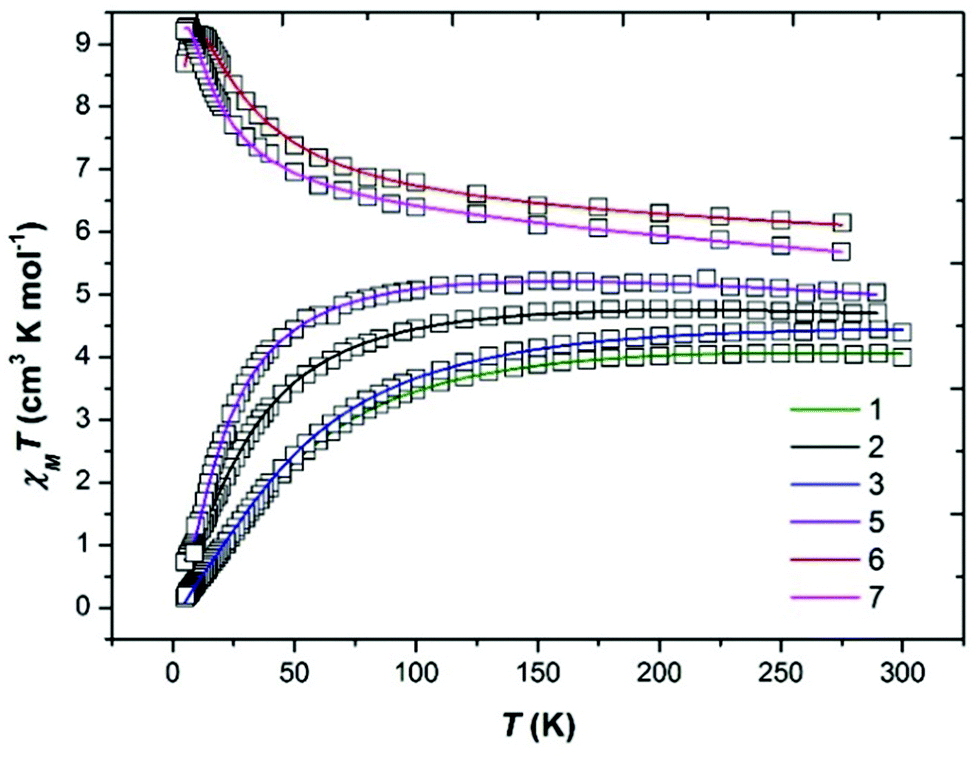
Dc magnetic susceptibility measurements were performed on polycrystalline samples of 1–3 and 5 in the temperature range T = 5–300 K in an applied magnetic field of B = 0.1 T. The poor yield of complex 4 precluded measurement. The experimental results are shown in Fig. 4 in the form of χMT products vs. T, where χM is the molar magnetic susceptibility, along with the data for complexes 6 and 7.22 At the highest temperatures measured the χMT values for 1–3 and 5 are ∼4.1, 4.7, 4.8 and 5.0 cm3 K mol−1, respectively, all somewhat lower than that expected for two non-interacting, high spin d4 ions with g = 2 (6 cm3 K mol−1). Upon cooling the χMT products of each sample behave very similarly, slowly decreasing with decreasing temperature reaching values of ∼0.2 (1), 0.7 (2), 0.2 (3) and 1.1 cm3 K mol−1 (5) at T = 5 K, indicative of the presence of weak antiferromagnetic exchange between the two MnIII ions. This is in stark contrast to that observed for complexes 6 and 7 which display weak ferromagnetic exchange, as discussed in the articles highlighted in ref. 22.| | 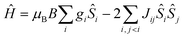 | (1) |
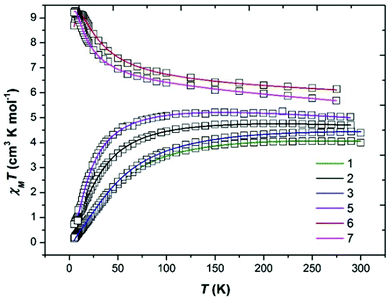 |
| | Fig. 4 Plot of the χMT product versus T for complexes 1–3, 5–7. The solid lines are a fit of the experimental data to isotropic spin-Hamiltonian (1). See text for details of the best fit parameters. Green = 1, black = 2, blue = 3, purple = 5, red = 6 and pink = 7. | |
The data can be fit (Fig. 4, Table 1) to the model described by isotropic spin-Hamiltonian (1), where the indices i and j refer to the two MnIII centres, J is the isotropic exchange interaction parameter, Ŝ is a spin operator, μB is the Bohr magneton, B is the applied magnetic field vector and g = 1.98 is the g-factor of the MnIII ions. The best-fit parameters are J = −5.73 cm−1 (1), −3.63 cm−1 (2), −5.63 cm−1 (3) and −2.05 cm−1 (5). The ground spin-state of all four complexes is a spin singlet (S = 0; energy versus spin state plots are given in the ESI, Fig. S6–S9†). This can be compared to the S = 4 ground states observed for complexes 6 and 7 resulting from exchange interactions of +2.20 and +1.24 cm−1, respectively.22 The difference between the magnetic behaviour of 1–3, 5 and 6–7 is intriguing, particularly given they have the same [Mn(NO)]2 magnetic core, and thus it would appear that the orientation of the Jahn–Teller axes is an important structural parameter. In order to examine this in more detail we have therefore turned to Density Functional Theory.
Theoretical studies
Theoretical studies have been carried out on complexes 1–6 in order to evaluate the exchange parameters and to reveal the mechanism of exchange. The DFT computed J values for complexes 1–6 are −5.76 cm−1, −2.16 cm−1, −5.15 cm−1, −3.66 cm−1, −2.99 cm−1 and +2.65 cm−1, respectively (Table 1). Complexes 1–5 are found to exhibit weak antiferromagnetic exchange interactions, while complex 6 exhibits ferromagnetic exchange. Calculations reproduce not only the sign of J accurately but also their magnitude and the trend in magnitudes compared to that observed experimentally. To understand the electronic reasons behind the variation in the nature and magnitude of exchange and to analyse the effect of the relative orientation of the Jahn–Teller axes (structure types B and C), molecular orbitals (MO) and overlap integrals have been analysed for complexes 1–6. The net exchange interaction in the dinuclear MnIII moiety has two contributions: (i) an antiferromagnetic JAF contribution arising solely from overlap between the singly occupied MOs (SOMOs) of the MnIII ions, and (ii) a ferromagnetic JF contribution arising from the orthogonality of the SOMOs (negligible overlap) and from the cross-interaction22b,c between the occupied and the empty d-orbitals (here between the dx2−y2 and all the other MnIII d-orbitals). Amongst the structures studied, the major contributor to the JAF term is the overlap between the dxy orbitals via the oxime bridge. This contribution is significant for complexes 1–5 possessing type C structures, as indicated in Fig. 1 and Fig. 5a. The second dominant contribution to the JAF term is the dxz–dxz overlap, also routed through the Mn–N–O–Mn bridge. Thus for complexes 1–5 the Mn–N–O–Mn torsion angle clearly dictates the strength of orbital overlap and thus the J value. These two overlaps are expected to be minimal in complex 6, resulting in a very small JAF term. However as the Jahn–Teller axes are now oriented along the oxime bridges, this allows for efficient dz2–dz2 overlap (see Fig. 5b). In this scenario this latter overlap is dominant and this contributes to the JAF term. As shown in Fig. 5b, this overlap is also strongly dependent upon the Mn–N–O–Mn torsion angle, with negligible overlap expected when the torsion angle approaches zero. Apart from these three prominent interactions, other orbital overlap values are small suggesting orthogonality between the SOMOs and a contribution to the JF term. Along with moderate cross-interactions (dx2−y2–dxz) this leads to a significant JF term for all complexes 1–6. The sign of J in each case is dictated by the dominant factor; for 1–5 the two strong overlaps overwhelmingly dominate leading to net antiferromagnetic exchange, while the prominent dz2–dz2 overlap observed in complex 6 is overshadowed by the JF contributions leading to a net ferromagnetic interaction.
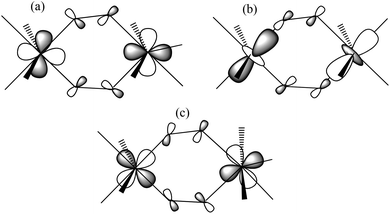 |
| | Fig. 5 Schematic representation of the (a) dxy–dxy overlap in complexes 1–5, (b) dz2–dz2 overlap in complex 6, (c) cross-interaction between dx2−y2 and dxz in complexes 1–6. | |
Spin density plots were computed in order to understand the origin of the electronic differences in complexes 1–6. Unpaired electrons in t2g orbitals usually favour a spin polarisation mechanism, whereas those in the eg orbitals tend to facilitate a spin delocalisation mechanism. In the case of the MnIII ion a mixture of these two mechanisms is found to operate since the ion possesses three unpaired electrons in the t2g orbitals and one unpaired electron in the eg orbital. The spin density of the MnIII ions in 1–6 is found to be <3.8 which shows that it is centred on the metal ion with a dominant spin delocalisation mechanism. The bridging N and O spin densities are different for the two structure types studied; for complex 1 all the bridging N and O atoms possess negative spin density indicating spin polarisation, while complex 6 possesses positive spin density on the O atom due to spin delocalistion as it lies along the Jahn–Teller axis (see Table S8 in ESI† and Fig. 6) and negative spin density on the N atom due to spin polarisation.
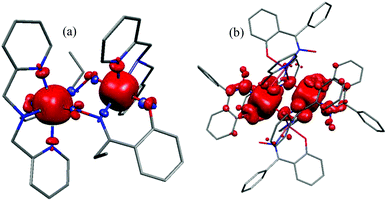 |
| | Fig. 6 Computed spin density HS plot for complex 1 (a) and complex 6 (b). | |
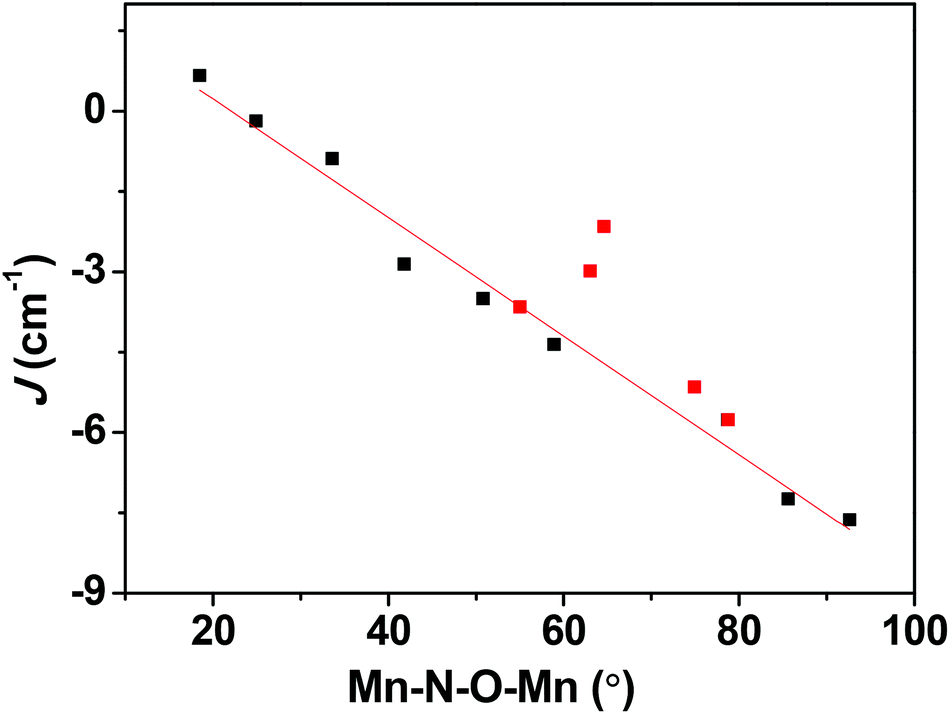
Apart from the orientation of the Jahn–Teller axis, the Mn–N–O–Mn torsion angle also plays a role in dictating the sign and magnitude of the J values. To see if it is possible to obtain ferromagnetic coupling in type C structures, we have developed magneto-structural correlations by varying the Mn–N–O–Mn dihedral angle from 18.5 to 92.8° in complex 1. A plot of J versus Mn–N–O–Mn dihedral angle is shown in Fig. 7. Our correlation reveals that J switches its sign from antiferromagnetic to ferromagnetic with decreasing dihedral angle. As the torsion angle decreases, the cross-interaction between dx2−y2 and dxz/dxy strengthens, leading to a larger JF contribution which in turn results in ferromagnetic exchange. Our calculations predict that a more planar Mn–N–O–Mn moiety of structure type C will exhibit ferromagnetic exchange coupling. Synthetic efforts to produce such a molecule are underway in our laboratory.
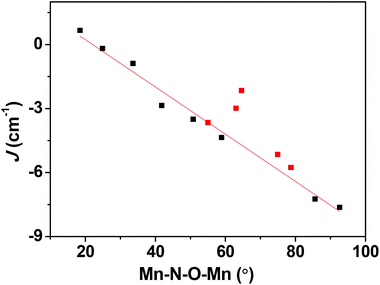 |
| | Fig. 7 Magneto-structural correlations developed by varying the Mn–N–O–Mn dihedral angle in 1. The red squares are experimental J values obtained for complexes 1–5. | |
Conclusions
The simple reaction between a Mn salt, a phenolic oxime and the tridentate chelate dpa affords a small family of double-oxime bridged MnIII dimers, whose structures differ from previous examples in the orientation of their Jahn–Teller axes. Previous family members conform to structure type B with the JT axes co-parallel and in the bridging plane between the MnIII ions, resulting in ferromagnetic exchange. The addition of a chelating ligand results in the novel complexes 1–5 whose JT axes have now switched orientation giving structure type C, where they lie perpendicular to the [Mn(ON)]2 bridging plane. The result is that the magnetic exchange between the metal centres becomes weakly antiferromagnetic. The employed DFT methodology is able to reproduce both the sign and magnitude of the exchange interaction in complexes 1–6. MO analysis reveals two dominant overlaps between the dxy–dxy and dxz–dxz orbitals are found to control the sign and magnitude of exchange in complexes 1–5. In the case of complex 6, strong dz2–dz2 overlap is overcome by cross-interactions and orbital orthogonality resulting in a weak ferromagnetic interaction. The general mechanism of exchange proposed can, in principle, be extended to any double oxime bridged MnIII dimer. Our developed magneto-structural correlation suggests a switch in the sign of the exchange interaction from antiferromagnetic to ferromagnetic upon decreasing the Mn–N–O–Mn torsion angle to approximately 20°.
Acknowledgements
GR thanks the DST (EMR/2014/000247), DST Nanomission and INSA for funding. TR thanks the CSIR for a SRF fellowship.
Notes and references
-
D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, U.K., 2006 Search PubMed.
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464 RSC.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS PubMed.
-
M. Evangelisti, in Molecular Magnets, NanoScience and Technology, ed. J. Bartolomé, F. Luis and J. F. Fernández, Springer-Verlag, Berlin, Heidelberg, 2014, pp. 365–387 Search PubMed.
-
(a) G. Aromí and E. K. Brechin, Struct. Bonding, 2006, 122, 1 CrossRef;
(b) C. J. Milios and R. E. P. Winpenny, Struct. Bonding, 2015, 164, 1 CrossRef.
- W. Wernsdorfer and R. Sessoli, Science, 1999, 284, 133 CrossRef CAS.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135 CrossRef CAS PubMed.
- M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Nat. Nanotechnol., 2013, 8, 165 CrossRef CAS PubMed.
- M. Urdampilleta, S. Klyatskaya, J.-P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502 CrossRef CAS PubMed.
- J.-P. Cleuziou, W. Wernsdorfer, V. Bouchiat, T. Ondarçuhu and M. Monthioux, Nat. Nanotechnol., 2006, 1, 53 CrossRef CAS PubMed.
- M. Baker, T. Guidi, S. Carretta, J. Ollivier, H. Mutka, H.-U. Güdel, G. A. Timco, E. J. L. McInnes, G. Amoretti and R. E. P. Winpenny, Nat. Phys., 2012, 8, 906 CrossRef CAS PubMed.
- S. Hill, R. S. Edwards, N. Aliaga-Alcalde and G. Christou, Science, 2003, 302, 1015 CrossRef CAS PubMed.
- W. Wernsdorfer, N. Aliaga-Alcalde, D. N. Hendrickson and G. Christou, Nature, 2002, 416, 406 CrossRef PubMed.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236 CrossRef CAS PubMed.
- S. Loth, S. Baumann, C. P. Lutz, D. M. Eigler and A. J. Heinrich, Science, 2012, 335, 196 CrossRef CAS PubMed.
-
(a) M. Evangelisti, A. Candini, A. Ghirri, M. Affronte, E. K. Brechin and E. J. L. McInnes, Appl. Phys. Lett., 2005, 87, 072504 CrossRef PubMed;
(b) M. Evangelisti and E. K. Brechin, Dalton Trans., 2010, 39, 4672 RSC;
(c) K. R. Vignesh, S. K. Langley, K. S. Murray and G. Rajaraman, Chem. – Eur. J., 2015, 21, 2881 CrossRef CAS PubMed.
- M. Mannini, F. Pineider, C. Danielli, F. Totti, L. Sorace, P. Sainctavit, M.-A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417 CrossRef CAS PubMed.
- J. Lehmann, A. Gaita-Ariño, E. Coronado and D. Loss, Nat. Nanotechnol., 2007, 2, 312 CrossRef CAS PubMed.
-
(a) J. Liu and S. Hill, Polyhedron, 2013, 66, 147 CrossRef CAS PubMed;
(b) S. Hill, S. Datta, J. Liu, R. Inglis, C. J. Milios, P. L. Feng, J. J. Henderson, E. del Barco, E. K. Brechin and D. N. Hendrickson, Dalton Trans., 2010, 39, 4693 RSC.
-
D. Gatteschi, O. Kahn and R. D. Willet, Magneto-Structural Correlations in Exchange-Coupled Systems, D. Reidel, Dordrecht, 1985 Search PubMed.
-
(a) D. A. Pantazis, V. Krewald, M. Orio and F. Neese, Dalton Trans., 2010, 39, 4959 RSC;
(b) N. Berg, T. Rajeshkumar, S. M. Taylor, E. K. Brechin, G. Rajaraman and L. F. Jones, Chem. – Eur. J., 2012, 18, 5906 CrossRef CAS PubMed;
(c) W. P. Barros, R. Inglis, G. S. Nichol, T. Rajeshkumar, G. Rajaraman, S. Piligkos, H. O. Stumpf and E. K. Brechin, Dalton Trans., 2013, 42, 16510 RSC;
(d) R. Inglis, E. Houton, J. Liu, A. Prescimone, J. Cano, S. Piligkos, S. Hill, L. F. Jones and E. K. Brechin, Dalton Trans., 2011, 40, 9999 RSC;
(e) E. Houton, S. M. Taylor, C. C. Beedle, J. Cano, S. Piligkos, S. Hill, A. G. Ryder, E. K. Brechin and L. F. Jones, Dalton Trans., 2012, 41, 8340 RSC.
- C. J. Milios, R. Inglis, L. F. Jones, S. Piligkos and E. K. Brechin, Chem. Commun., 2012, 48, 181 RSC; C. J. Milios, S. Piligkos and E. K. Brechin, Dalton Trans., 2008, 1809 RSC.
- W. R. Dunstan and T. A. Henry, J. Chem Soc., Trans., 1899, 75, 66 RSC.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed;
(b) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS;
(c) L. Palatinus and G. Chapuis, Superflip - a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
-
(a) A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef PubMed;
(b) A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737 CrossRef CAS PubMed.
-
(a) E. Ruiz, S. Alvarez, J. Cano and P. Alemany, J. Comput. Chem., 1999, 20, 1391 CrossRef CAS;
(b) E. Ruiz, A. Rodriguez-Fortea, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 2003, 24, 982 CrossRef CAS PubMed;
(c) G. Rajaraman, J. Cano, E. K. Brechin and E. J. L. McInnes, Chem. Commun., 2004, 1476 RSC;
(d) P. Christian, G. Rajaraman, A. Harrison, J. J. W. McDouall, J. Raftery and R. E. P. Winpenny, Dalton Trans., 2004, 2550 RSC;
(e) G. Rajaraman, M. Murugesu, E. C. Sanudo, M. Soler, W. Wernsdorfer, M. Helliwell, C. Muryn, J. Raftery, S. J. Teat, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2004, 126, 15445 CrossRef CAS PubMed;
(f) S. Sasmal, S. Hazra, P. Kundu, S. Dutta, G. Rajaraman, E. C. Sañudo and S. Mohanta, Inorg. Chem., 2011, 50, 7257 CrossRef CAS PubMed;
(g) L. F. Jones, G. Rajaraman, J. Brockman, M. Murugesu, J. Raftrey, S. J. Teat, W. Wernsdorfer, G. Christou, E. K. Brechin and D. Collison, Chem. – Eur. J., 2004, 10, 5180 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic and crystallographic details; packing diagrams, additional magnetic data and details of the DFT calculations. CCDC 1044300–1044304. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03615a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Gopalan
Rajaraman
*b and
Euan K.
Brechin
*a
c,
Gopalan
Rajaraman
*b and
Euan K.
Brechin
*a