
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity towards nitriles, cyanamides, and carbodiimides of palladium complexes derived from benzyl alcohol. Synthesis of a mixed Pd2Ag complex†
María-José
Fernández-Rodríguez
a,
Eloísa
Martínez-Viviente
*a,
José
Vicente
a and
Peter G.
Jones
b
aGrupo de Química Organometálica, Dpto. de Química Inorgánica, Facultad de Química, Universidad de Murcia, Apdo. 4021, 30071, Murcia, Spain. E-mail: jvs1@um.es; eloisamv@um.es; Web: http://www.um.es/gqo/
bInstitut für Anorganische und Analytische Chemie der Technischen Universität Braunschweig, Postfach 3329, 38023, Braunschweig, Germany. E-mail: p.jones@tu-bs.de
First published on 25th November 2015
Abstract
The chelate complex [Pd(κ2-C,O-C6H4CH2O-2)(bpy)] (II) reacts with acetonitrile, cyanamides, or carbodiimides, in the presence of AgOTf (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1 molar ratio) and residual water, to form complexes [Pd{κ2-C,N-C6H4{CH2OC(
:5:1 molar ratio) and residual water, to form complexes [Pd{κ2-C,N-C6H4{CH2OC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NX)Y}-2}(bpy)](OTf), where X = H, Y = Me (1), NMe2 (2a), NEt2 (2b), X = R, Y = NHR (R = iPr (3a), Tol (3b)), as a result of the insertion of the unsaturated reagent into the O–Pd bond of II and the protonation of one of the N atoms. In the absence of AgOTf the reaction of II with TolNCNTol (Tol = p-Tolyl) results in the formation of the neutral complex [Pd{κ2-C,N-C6H4{CH2OC(NTol)NTol}-2}(bpy)] (4). Complexes 3b and 4 can be interconverted by deprotonation (3b + KOtBu) or protonation (4 + KOTf + HOTf) reactions. When the reaction of II with TolNCNTol in the presence of AgOTf is carried out in a 1:1:1 stoichiometric ratio, or for a short period of time, a mixture of 3b and a mixed heterometallic Ag2Pd complex 5 is obtained (5 = [Ag(N-4)2](OTf)). Complex 5 is the major product when the AgOTf is added before the carbodiimide, and the reaction is stopped immediately. 5 can also be obtained by reaction of 4 with 0.5 equiv. of AgOTf. When complex [PdI(C6H4CH2OH-2)(bpy)] (I) reacts with iPrNCNiPr in the presence of TlOTf, instead of AgOTf, a ca. 1:1 mixture of 3a and [Pd{κ2-O,N-OCH2{C6H4{C(NHiPr)NiPr}-2}}(bpy)](OTf) (6) forms. Complex 6 is the result of the insertion of the carbodiimide into the C–Pd bond. Complexes 1–6 have been extensively characterized by NMR spectroscopy, and the crystal structures of 2a, 3a, and 5·2.5CHCl3·0.5Et2O have been determined by X-ray diffraction studies.
NX)Y}-2}(bpy)](OTf), where X = H, Y = Me (1), NMe2 (2a), NEt2 (2b), X = R, Y = NHR (R = iPr (3a), Tol (3b)), as a result of the insertion of the unsaturated reagent into the O–Pd bond of II and the protonation of one of the N atoms. In the absence of AgOTf the reaction of II with TolNCNTol (Tol = p-Tolyl) results in the formation of the neutral complex [Pd{κ2-C,N-C6H4{CH2OC(NTol)NTol}-2}(bpy)] (4). Complexes 3b and 4 can be interconverted by deprotonation (3b + KOtBu) or protonation (4 + KOTf + HOTf) reactions. When the reaction of II with TolNCNTol in the presence of AgOTf is carried out in a 1:1:1 stoichiometric ratio, or for a short period of time, a mixture of 3b and a mixed heterometallic Ag2Pd complex 5 is obtained (5 = [Ag(N-4)2](OTf)). Complex 5 is the major product when the AgOTf is added before the carbodiimide, and the reaction is stopped immediately. 5 can also be obtained by reaction of 4 with 0.5 equiv. of AgOTf. When complex [PdI(C6H4CH2OH-2)(bpy)] (I) reacts with iPrNCNiPr in the presence of TlOTf, instead of AgOTf, a ca. 1:1 mixture of 3a and [Pd{κ2-O,N-OCH2{C6H4{C(NHiPr)NiPr}-2}}(bpy)](OTf) (6) forms. Complex 6 is the result of the insertion of the carbodiimide into the C–Pd bond. Complexes 1–6 have been extensively characterized by NMR spectroscopy, and the crystal structures of 2a, 3a, and 5·2.5CHCl3·0.5Et2O have been determined by X-ray diffraction studies.
Introduction
The importance of Pd(II) aryl complexes in organometallic chemistry derives mainly from their involvement in carbon–carbon and carbon–heteroatom bond-forming reactions.1 Their reactivity with unsaturated molecules often results in the insertion of these molecules into the aryl–Pd bonds, forming new ligands or, after decomposition reactions, organic compounds.2 A valuable synthetic tool that we have extensively explored is the incorporation of a substituent at the ortho position of the aryl group,3–7 as this substituent can become involved in the reactivity with the Pd centre and the organic substrate in many interesting ways.3–6,8–12 Very often, the ortho-substitution also results in the formation of cyclopalladated complexes.3,4,7,9,10–12Following this line of research, our group has previously investigated the reactivity of ortho-palladated phenol derivatives.6,10–13 Their reactions with CO, isocyanides, alkenes, alkynes, and allenes did not involve the OH group in the ortho position.6,13 In contrast, the electron-donating ability of this group played a crucial role in the reactivity towards nitriles,10,12 carbodiimides,11,12 cyanamides,12 and isothiocyanates,12 which afforded the first examples of the insertion10–12 of these molecules into a C–M bond of a late transition metal. These insertion reactions occurred together with the deprotonation and coordination of the hydroxyl oxygen to Pd, forming 6-membered chelate rings (Chart 1).10–12 With carbodiimides,11 the addition of the O–H group to one of the CN bonds of the substrate, together with the coordination of the other N to the Pd atom, was an alternative reaction to the insertion.11
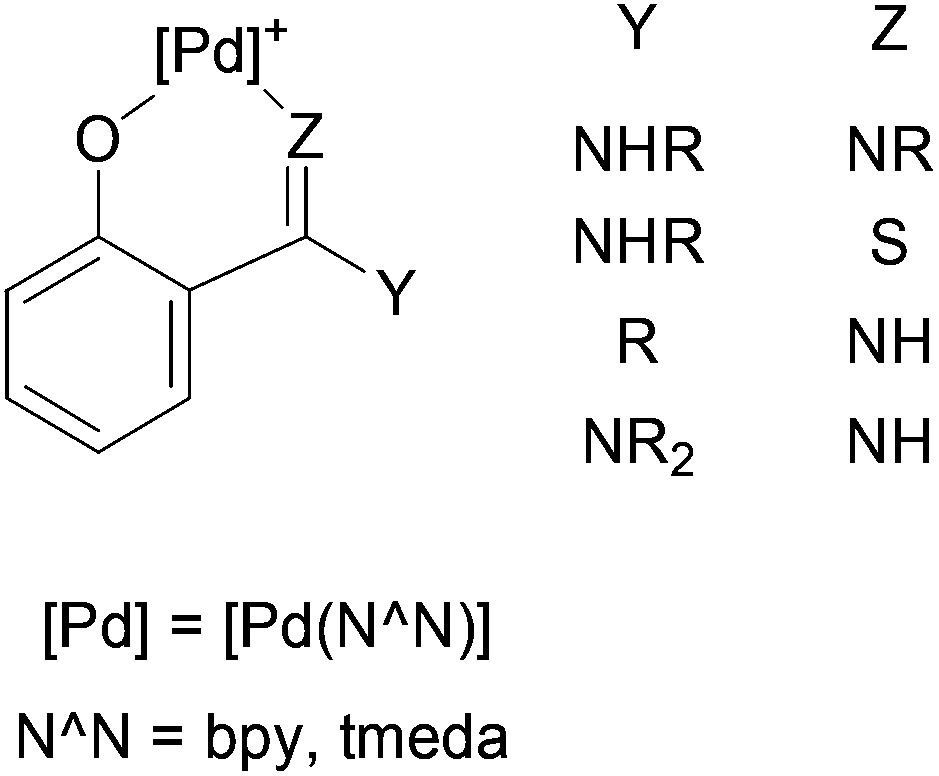 | ||
| Chart 1 | ||
We have recently extended this research to ortho-palladated hydroxymethylphenyl complexes,14 where the methylene link in the alcoholic substituent might significantly influence the reactivity towards unsaturated molecules. There are very few reports of 2-hydroxymethylphenyl palladium complexes15 or oxapalladacycles derived from them.16 These compounds have been used as precatalysts in Heck and cross-coupling reactions,17 but their reactivity towards unsaturated molecules had not been systematically investigated, with a single report on a reaction of a palladacycle with tert-butyl isocyanide, yielding an imidate, while the reaction of the same compound with maleic anhydride resulted in the coordination of the olefin to the Pd(II) centre.16 In our recent work14 we synthesized the complex [PdI(C6H4CH2OH-2)(bpy)] (I) and investigated its reactivity towards alkynes, alkenes, nitriles, cyanamides, allenes, and carbon monoxide, which did not result in clean insertion (C–Pd bond) or addition (O–H bond) reactions.14 Only the reaction of I with XyNC gave a clean insertion product, trans-[PdI{C(NXy)C6H4CH2OH-2}(CNXy)2].14 By deprotonation of complex I we prepared the chelate complex [Pd{κ2-C,O-C6H4(CH2O)-2}(bpy)] (II), which displayed an interesting reactivity towards primary alkyl halides, via a nucleophilic attack of the coordinated oxygen at the alkyl group of the halide.14 We have now extended our research to the chelate complex II, which, in the presence of AgOTf, reacts with acetonitrile, cyanamides, and carbodiimides to give novel complexes containing a [Pd{κ2-C,N-C6H4{CH2OC(NX)Y}-2}] chelate ring, resulting, unexpectedly, from insertion reactions of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N or CN bonds into the O–Pd bond of II. We have not found examples for such a chelate structure with any metal. The Ag+ cations play a key role in these reactions, which is also an interesting observation. An insertion reaction of a carbodiimide into the aryl–Pd bond of I is also described, as is a mixed-metal Pd2Ag complex, which has been characterized by X-ray crystallography. Other heterometallic Pd2Ag18 or Pd2Ag219 complexes have been described in the literature, but their structures differ greatly from the one reported in this work. Thus, the reactivity that we report in this paper differs greatly from the reactions described for complexes I and II in our previous work.14
N or CN bonds into the O–Pd bond of II. We have not found examples for such a chelate structure with any metal. The Ag+ cations play a key role in these reactions, which is also an interesting observation. An insertion reaction of a carbodiimide into the aryl–Pd bond of I is also described, as is a mixed-metal Pd2Ag complex, which has been characterized by X-ray crystallography. Other heterometallic Pd2Ag18 or Pd2Ag219 complexes have been described in the literature, but their structures differ greatly from the one reported in this work. Thus, the reactivity that we report in this paper differs greatly from the reactions described for complexes I and II in our previous work.14
Results and discussion
Reactions with nitriles and cyanamides
The chelate complex [Pd{κ2-C,O-C6H4CH2O-2}(bpy)] (II) reacts with acetonitrile and AgOTf (1:5:1 molar ratio, CH2Cl2), in the presence of residual water, to form [Pd{κ2-C,N-C6H4{CH2OC(NH)Me}-2}(bpy)](OTf) (1, Scheme 1), the result of the insertion of the nitrile into the O–Pd bond of II, and the protonation of the N by the residual water. Complex II reacts similarly with the cyanamides R2NCN (R = Me, Et) and AgOTf to form [Pd{κ2-C,N-C6H4{CH2OC(NH)NR2}-2}(bpy)](OTf) (R = Me (2a), Et (2b), Scheme 1). The presence of Ag+ is a requirement in these reactions (otherwise there is no reaction or, with TlOTf, mixtures of compounds are obtained). There are two possible explanations for this: either the Ag+ forms in situ a complex with the ligands, increasing the electrophilicity of the unsaturated carbon atom, and thus favouring the nucleophilic attack of the O atom of II; or the Ag+ coordinates to the O atom, activating the O–Pd bond in II and favouring the coordination of the ligands to Pd, which would in turn activate them towards the nucleophilic attack of the O. In any case, this influence of added Ag+ on the reactivity of an arylpalladium complex (without halide or pseudohalide ligands), towards unsaturated molecules, controlling the site of the insertion reaction, can be a very useful synthetic tool. Although nucleophilic reactions at coordinated nitriles have been thoroughly investigated,20 the reactions reported here show the interesting feature that the initially uncoordinated nitrile or cyanamide is activated in situ by one of the two proposed mechanisms (coordination to Ag+ or to the Pd centre) and then attacked by an (initially) chelating O atom. Seven-membered C3–Pd–NC–O chelate rings such as those resulting in these reactions (in 1 and 2a,b) have not been described before for any metal.
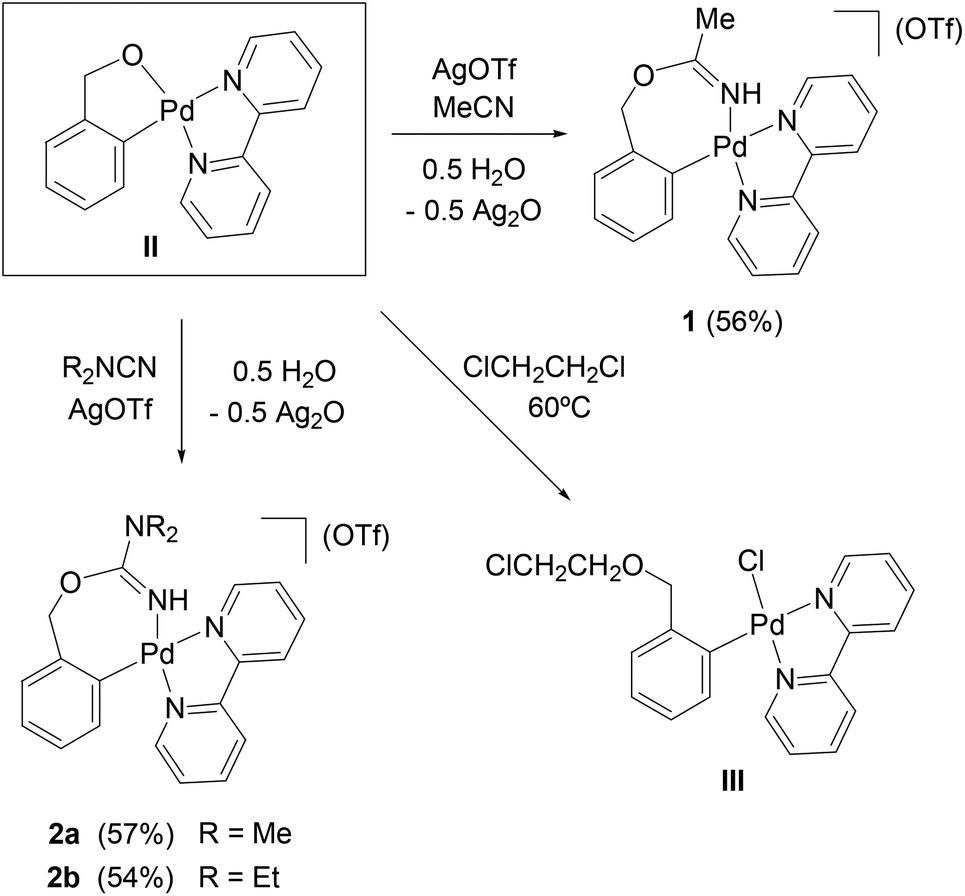 | ||
| Scheme 1 | ||
We have not been able to achieve the insertion of nitriles or cyanamides into the C–Pd bond of complexes I or II. This negative result contrasts with the successful insertion reactions that we observed with the related complexes [PdI(C6H4Y-2)(tmeda)] (Y = OH, NH2, N^N = tmeda, bpy, tbbpy), and a wide variety of nitriles10,12 and cyanamides.12 In those reactions we proposed that the electron-donating OH or NH2 group in ortho position would play a key role in the mechanism, via delocalization of a negative charge on the aryl ipso carbon.10,12 That mechanistic proposal would be now supported by the failure of these insertion reactions with the complexes I and II, for which the CH2 link between the OH function and the aryl ring prevents the delocalization of electron density.
In one of our attempts to react complex II with nitriles, we used 1,2-dichloroethane as solvent and heated to 60 °C. We obtained then the complex [PdCl{C6H4(CH2OCH2CH2Cl)-2}(bpy)] (III), which is the result of the nucleophilic attack of the oxygen in II at a CH2 group of the 1,2-dichloroethane solvent. Complex III has been characterized by X-ray diffraction studies (see the ESI†), but we have not been able to purify and fully characterize it. We have described similar reactions of II with alkyl halides (bromides and iodides) in a previous paper.14
Reactions with carbodiimides
Complex II reacts with the carbodiimides RNCNR (R = iPr, Tol) and AgOTf (1:5:1 molar ratio, CH2Cl2) in the presence of residual water, to form [Pd{κ2-C,N-C6H4{CH2OC(NR)NHR}-2}(bpy)](OTf) (R = iPr (3a), Tol (3b), Scheme 2) which, similarly to 1 and 2a,b, are the result of the insertion of the organic products into the O–Pd bond of II.
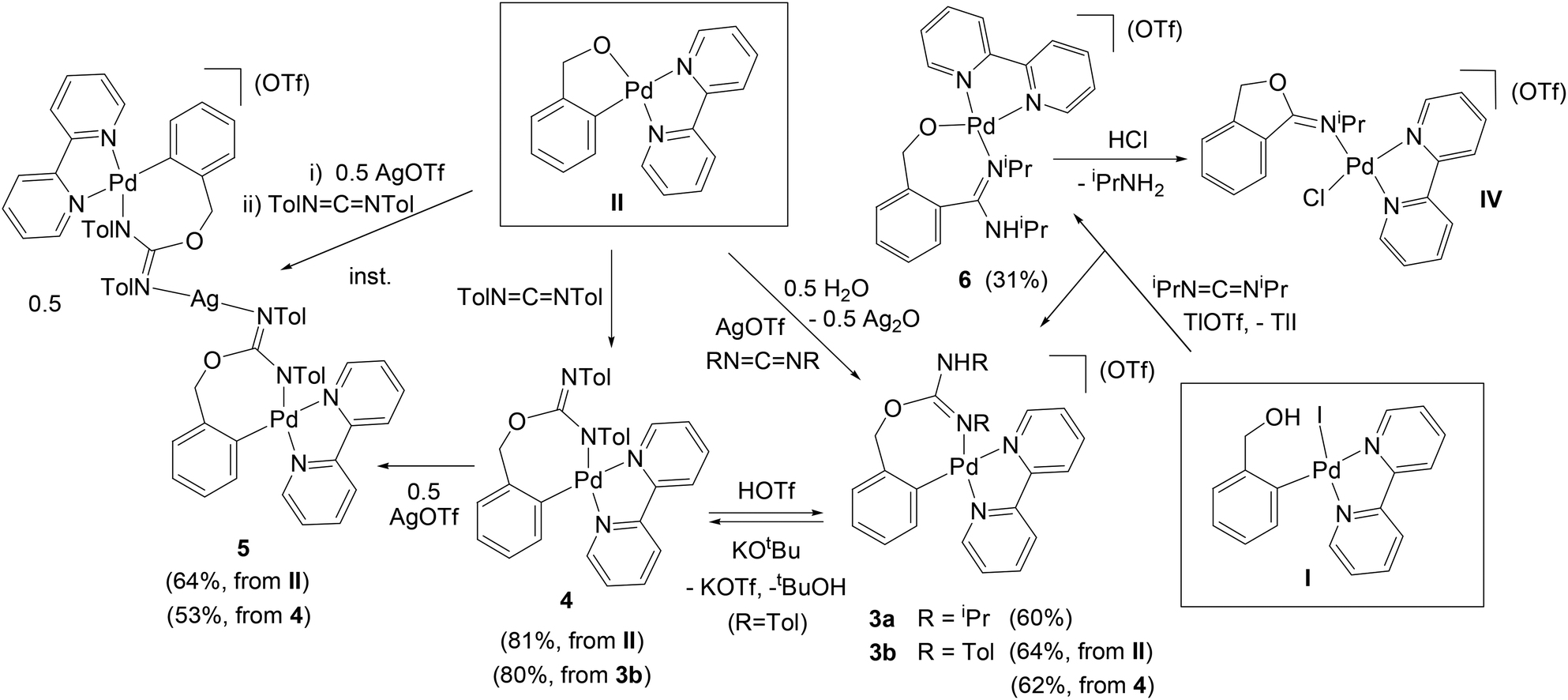 | ||
| Scheme 2 | ||
When these reactions were performed in the absence of AgOTf, however, the results differed for the two carbodiimides investigated. With iPrNCNiPr there was no reaction, whereas with TolNCNTol the reaction in the absence of AgOTf resulted in the formation of [Pd{κ2-C,N-C6H4{CH2OC(NTol)NTol}-2}(bpy)] (4, Scheme 2), which is the conjugate base of 3b. These results suggest that the TolNCNTol is the only reactant investigated in this work that is electrophilic enough to undergo nucleophilic attack by the O atom in II, without the need of activation by Ag+. Complex 4 has a characteristic red color, and it forms after only 5 min in the reaction with either one equivalent or excess of the carbodiimide. It is partially soluble in Et2O.
By deprotonation of the ionic complex 3b with KOtBu, it is possible to obtain the neutral complex 4 and, vice versa, by reaction of 4 with KOTf and HOTf complex 3b is obtained. In this reaction it is necessary to add the KOTf first and then the HOTf after a few minutes, as otherwise a different product forms, which could not be characterized. Thus, the K+ ion seems to stabilize the reaction intermediate, probably by coordinating to the O atom. The deprotonation of the ionic complex 3a (R = iPr) with KOtBu gives a red neutral complex similar to 4, but it re-protonates very easily, so that it could not be characterized. Clearly, the Tol groups in 4 play a very important role in the stability of this complex, most probably through resonance effects.
Curiously, when the reaction of II with AgOTf and a 5-fold excess of TolNCNTol was stopped after only 2 hours, or when it was performed in a ca. 1:1:1 stoichiometric ratio (overnight), a mixture of 3b and a different product formed (in ca. 1.9:1 or 1.4:1 ratio, respectively). This product was identified by X-ray crystallography (see below) as an ionic trinuclear complex consisting of two molecules of 4 coordinated through N to one atom of Ag (complex 5 = [Ag(N-4)2](OTf), Scheme 2). The structure of 5 differs greatly from other heterometallic Pd2Ag18 or Pd2Ag219 complexes found in the literature. With the carbodiimide iPrNCNiPr we did not observe a similar reactivity. The formation of complex 5 is favoured by a shorter reaction time and a smaller amount of carbodiimide, and we have also observed that it is strongly influenced by the order of addition of the reactants. Thus, in the reactions of II with one equivalent of TolNCNTol and AgOTf, if the carbodiimide is added first and then the AgOTf, the major product is 3b (even if the reaction is stopped immediately), although it forms together with a variable amount of 5 (between ca. 5–10%). In contrast, if AgOTf is added first, followed by one equivalent of TolNCNTol, and the reaction is stopped immediately, the trinuclear complex 5 is the major product, with only ca. 20% of 3b (this amount increases if a longer reaction time is allowed). Complex 5 can then be separated from 3b by exploiting differences in solubility (see the Experimental section). From these observations we suggest that the trinuclear complex 5 forms by the nucleophilic attack of II on a [Ag(TolNCNTol)2]+ intermediate, and then it reacts with residual water, losing the Ag atom and forming two molecules of 3b. This “decomposition” to 3b would be favoured by an excess of carbodiimide, which would coordinate to the Ag facilitating the rupture of 5 (in an overnight reaction with a 5-fold excess of TolNCNTol, only 3b is detected, while the same reaction with only one equivalent of TolNCNTol gives a mixture of 3b and 5 in ca. 1:0.8 ratio). In contrast, when the carbodiimide is added before the AgOTf, it would immediately react with II, forming, presumably, first the neutral complex 4 and then, upon addition of the AgOTf, the ionic complex 3b, so that 5 would only be a minor product. We have tried to obtain complex 5 by reaction of 4 with 0.5 equivalents of AgOTf and, after 2 hours in CH2Cl2, the major product of this reaction was indeed the trinuclear complex 5, together with ca. 20% of 3b. Thus, it seems that complex 4 can be transformed in the presence of AgOTf to both 3b or 5, and the favoured product is determined by the reaction conditions. To summarize: to obtain 3b the best method is the reaction of II with AgOTf and TolNCNTol in a 1:1:5 ratio overnight (the order of addition of the reactants is not important), while for 5 the best method is to carry out the reaction in a 1:0.8:1 stoichiometric ratio (better than the theoretical 1:0.5:1 ratio), adding the AgOTf (to a solution of II) before the carbodiimide, and stopping the reaction immediately by evaporation of the solvent. 5 then needs to be separated from 3b by solubility difference (see the Experimental section).
Complexes 3a,b also form in the reaction of the 2-hydroxymethylphenyl Pd complex [PdI(C6H4CH2OH-2)(bpy)] (I) with the corresponding carbodiimides and AgOTf, but with a much lower yield and purity, so that these reactions have not been pursued. Additionally, when complex I reacts with iPrNCNiPr in the presence of TlOTf, instead of AgOTf, a ca. 1:1 mixture of two complexes forms: one is again 3a (which is now the result of the addition of the OH group to the carbodiimide and the coordination of one of the N atoms to Pd) and the other is [Pd{κ2-O,N-OCH2{C6H4{C(NiPr)NHiPr}-2}}(bpy)](OTf) (6, Scheme 2), which is the result of the insertion of the carbodiimide into the C–Pd bond. We have not been able to obtain complex 6 independently of 3a, even by varying the amount of carbodiimide or the reaction time, but we have been able to separate it from 3a by preparative TLC on alumina (see the Experimental section). Additionally, from a CDCl3 solution of 6 we obtained single crystals, the X-ray structure of which showed them to be the unexpected complex IV, apparently formed by reaction of 6 with the residual HCl of the deuterated solvent (the attack of HCl on 6 would promote the intramolecular attack of the O on the CN group of the inserted carbodiimide, the breaking of the C–N and Pd–N bonds and the formation of a new Pd–N bond). Unfortunately, despite much effort we have not been able to reproduce the synthesis of this complex, but we include the X-ray data in the ESI.† Finally, the reaction of I with TolNCNTol and TlOTf instead of AgOTf resulted in the formation of a complex that is probably an insertion product similar to 6 but that was not pure enough to be characterized. The (relatively) cleaner reactivity of the carbodiimides with I and TlOTf, compared to the similar reactions with acetonitrile and cyanamides, which gave intractable mixtures, is probably attributable to a combination of electronic and steric effects. The greater steric hindrance in the carbodiimides, together with their appreciable dipole moments,21 would favour one (or two) major reaction pathways while hindering other secondary reactions.
NMR and IR data
All the complexes reported in this paper have been extensively studied by NMR spectroscopy (1D and 2D experiments), allowing an almost full assignment of the 1H and 13C resonances. To facilitate comparison, the data are collected in Table S.1 in the ESI,† together with a more extended discussion.For the complexes 1–5, the insertion of the organic molecules (MeCN, R2NCN, or RNCNR) into the O–Pd bond, and not the C–Pd bond, is confirmed by the three-bond correlation between the inserted iminic CN carbon and the methylenic CH2OH protons, observed in the 1H,13C-HMBC spectra. For complex 6, in contrast, a three-bond correlation between the iminic CN carbon and the o-H of the aryl ligand is observed. Other NOE and correlation data confirm these structures and allow the assignment of the different groups within the molecules (see the ESI†). For 3a,b and 6, the position of the proton at the uncoordinated N is also confirmed by the 1H-NOESY and 1H,13C-HMBC spectra, and is similar to that observed in the related complexes resulting from the reaction of carbodiimides with ortho-palladated phenol derivatives.11,12 The CNH proton in complex 1 resonates at much higher frequency (δ 8.45 ppm) than in 2a,b (δ 4.81 and 4.76 ppm), for which the partial release of the lone pair from the NR2 group results in a resonance form with a negative charge at the NH group. This electronic delocalization along the R2N–CNH bonds in 2a,b is confirmed by the X-ray diffraction study of 2a, which shows a shortening of the single N–C bond and a lengthening of the double CN bond, relative to other values (see below). In the complexes derived from cyanamides, the three CNH 1H NMR chemical shifts are rather similar: δ 5.57 ppm for 3a, 6.49 ppm for 3b and 6.39 ppm for 6.
The neutral complex 4 shows a fluxional behaviour within the chelate ring, which results in the broadening of one of the methylenic 1H resonances, and also of the 1H resonances of the more external tolyl group. These resonances sharpen at low temperature, but the 1H chemical shifts do not change significantly, so that the room temperature data are given in Table S.1† and in the Experimental section. The 13C NMR data, however, are given for 213 K, because at room temperature the S/N ratio of some resonances is too low.
In the mixed trinuclear Pd2Ag complex 5, the two halves of the molecule are equivalent in solution (not in the solid state, see below), as only one set of 1H and 13C NMR resonances is observed. One of the tolyl groups (the one closer to the Ag) again shows strongly broadened 1H and 13C resonances, indicating that the rotation around that Tol-N bond is hindered by the steric crowding in the molecule.
The IR bands of the CN bonds in 1, 2a,b, 3a,b, and 6 all appear in the range 1599–1635 cm−1. For complex 4, where the CN bond is uncoordinated, the corresponding IR band appears at higher frequency, 1660 cm−1. In the related complex 5 (5 = [Ag(N-4)2](OTf)), however, the coordination of the CN bond to Ag shifts the IR band again to lower frequency, 1600 cm−1. The IR bands of the N–H bonds in 1, 2a,b, 3a,b, and 6 are observed in the range 3213–3401 cm−1.
X-ray structure determinations
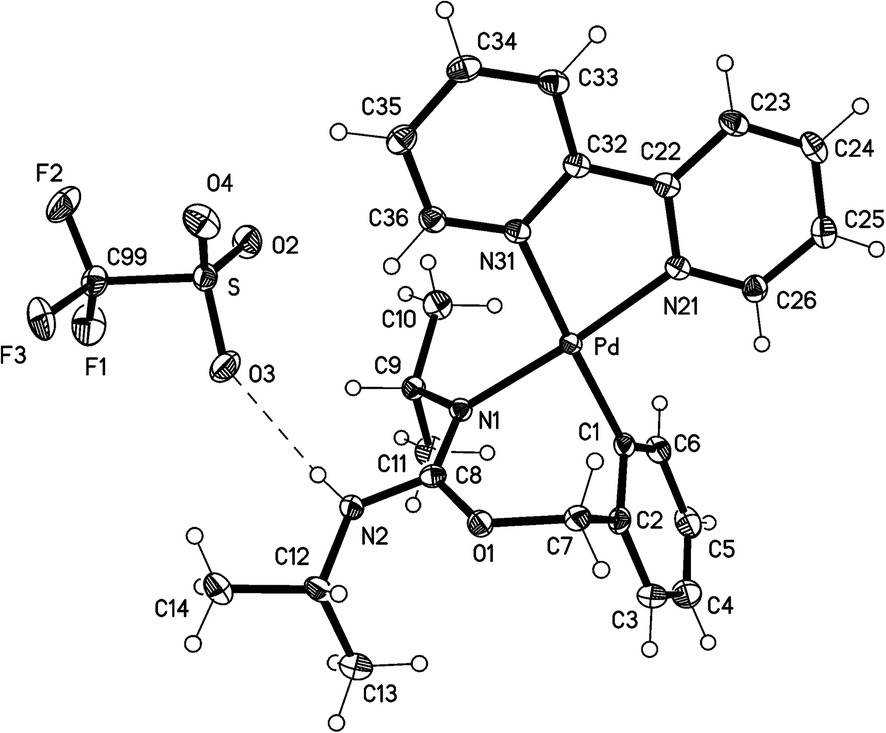
The crystal structures of the complexes 2a (Fig. 1), 3a (Fig. 2), and 5 (Fig. 3; only one of the two independent cations is shown), have been determined by X-ray diffraction studies (see Table S.2† for Experimental details). The crystal structures of III and IV are described in the ESI,† together with details of disordered solvent and anions.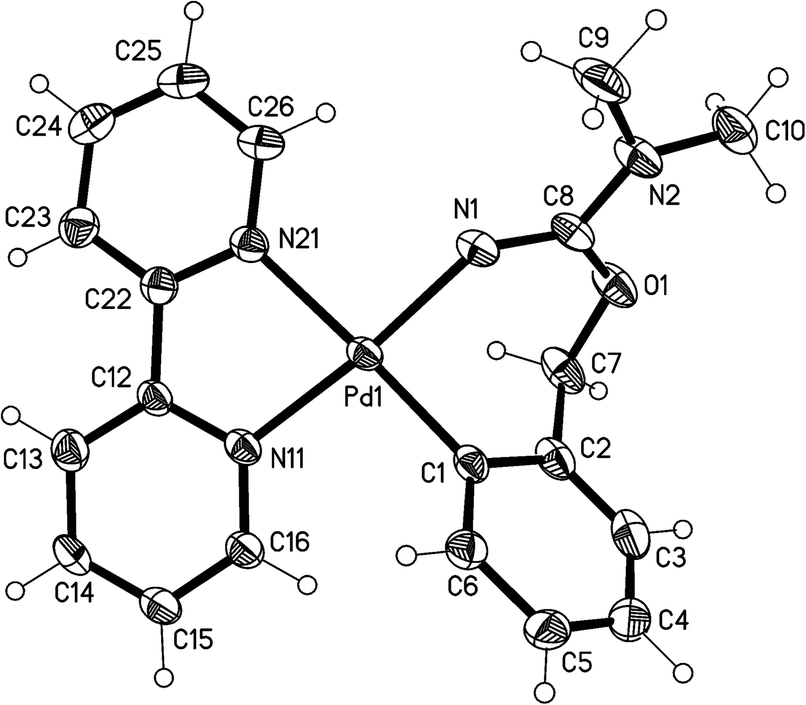 | ||
| Fig. 1 Thermal ellipsoid plot (50% probability level) of the cation of 2a. Selected bond lengths (Å) and angles (°): Pd(1)–C(1) = 1.981(3), Pd(1)–N(1) = 2.026(2), Pd(1)–N(11) = 2.051(2), Pd(1)–N(21) = 2.118(2), O(1)–C(7) = 1.462(4), O(1)–C(8) = 1.334(4), C(8)–N(1) = 1.305(4), C(8)–N(2) = 1.343(4), C(9)–N(2) = 1.450(4), C(10)–N(2) = 1.462(4); C(1)–Pd(1)–N(1) = 87.22(11), C(1)–Pd(1)–N(11) = 97.84(10), N(1)–Pd(1)–N(21) = 95.85(10), N(11)–Pd(1)–N(21) = 79.20(10), N(1)–Pd(1)–N(11) = 174.20(9), C(1)–Pd(1)–N(21) = 176.21(9), C(7)–O(1)–C(8) = 120.0(2), O(1)–C(8)–N(1) = 124.7(3), O(1)–C(8)–N(2) = 112.2(3), N(1)–C(8)–N(2) = 123.1(3), C(8)–N(1)–Pd(1) = 136.3(2). | ||
 | ||
| Fig. 2 Thermal ellipsoid plot (50% probability level) of 3a. Selected bond lengths (Å) and angles (°): Pd–C(1) = 1.9716(15), Pd–N(1) = 2.0313(13), Pd–N(21) = 2.0331(13), Pd–N(31) = 2.1157(12), O(1)–C(7) = 1.4567(19), O(1)–C(8) = 1.3350(19), C(8)–N(1) = 1.3054(18), C(8)–N(2) = 1.346(2), C(9)–N(1) = 1.494(2), C(12)–N(2) = 1.4658(19); C(1)–Pd–N(1) = 84.69(6), C(1)–Pd–N(21) = 98.38(6), N(1)–Pd–N(31) = 97.75(5), N(21)–Pd–N(31) = 79.53(5), N(1)–Pd–N(21) = 175.14(5), C(1)–Pd–N(31) = 174.44(6), C(7)–O(1)–C(8) = 123.79(12), O(1)–C(8)–N(1) = 125.21(14), O(1)–C(8)–N(2) = 111.20(13), N(1)–C(8)–N(2) = 123.58(16), C(8)–N(1)–Pd = 125.84(12). | ||
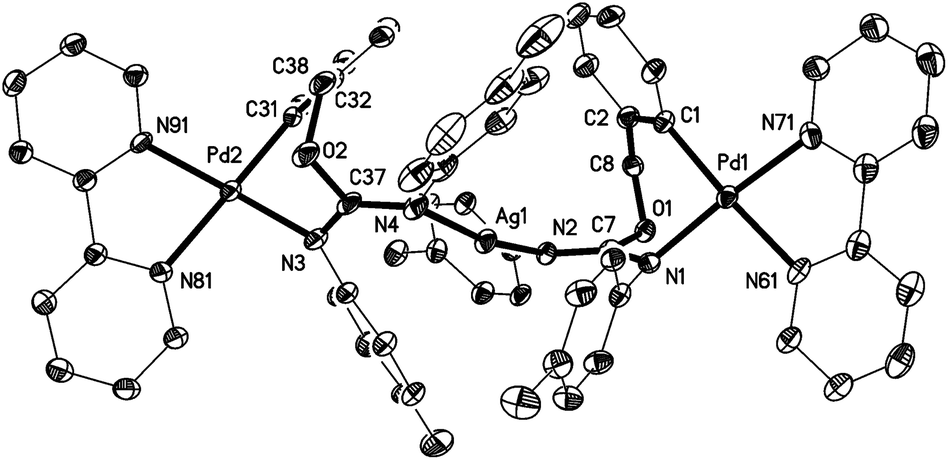 | ||
| Fig. 3 Thermal ellipsoid plot (50% probability level) of one of the two independent cations of 5. Selected bond lengths (Å) and angles (°): Ag(1)–N(4) = 2.121(5), Ag(1)–N(2) = 2.128(4), Pd(1)–C(1) = 2.014(7), Pd(1)–N(1) = 2.044(5), Pd(1)–N(71) = 2.070(5), Pd(1)–N(61) = 2.115(5), Pd(2)–C(31) = 2.009(6), Pd(2)–N(91) = 2.045(5), Pd(2)–N(3) = 2.054(6), Pd(2)–N(81) = 2.117(6), O(1)–C(8) = 1.439(8), O(1)–C(7) = 1.368(7), N(1)–C(7) = 1.341(8), N(1)–C(11) = 1.422(8), N(2)–C(7) = 1.292(8), N(2)–C(21) = 1.418(8), O(2)–C(38) = 1.456(9), O(2)–C(37) = 1.390(7), N(3)–C(37) = 1.322(10), N(3)–C(41) = 1.416(8), N(4)–C(37) = 1.310(9), N(4)–C(51) = 1.415(10); N(4)–Ag(1)–N(2) = 167.5(2), C(1)–Pd(1)–N(1) = 88.7(2), C(1)–Pd(1)–N(71) = 96.5(3), N(1)–Pd(1)–N(61) = 95.7(2), N(71)–Pd(1)–N(61) = 78.5(2), N(1)–Pd(1)–N(71) = 171.9(2), C(1)–Pd(1)–N(61) = 173.1(2), C(31)–Pd(2)–N(91) = 97.9(2), C(31)–Pd(2)–N(3) = 86.4(3), N(91)–Pd(2)–N(81) = 79.7(2), N(3)–Pd(2)–N(81) = 97.5(2), N(91)–Pd(2)–N(3) = 168.8(2), C(31)–Pd(2)–N(81) = 171.3(3), C(7)–O(1)–C(8) = 112.9(4), O(1)–C(7)–N(1) = 111.4(5), O(1)–C(7)–N(2) = 122.1(6), N(1)–C(7)–N(2) = 126.4(6), C(7)–N(2)–Ag(1) = 120.8(4), C(37)–O(2)–C(38) = 112.0(5), O(2)–C(37)–N(3) = 112.2(6), O(2)–C(37)–N(4) = 119.7(7), N(3)–C(37)–N(4) = 128.1(6), C(37)–N(4)–Ag(1) = 120.1(4). | ||
The structures of 2a, 3a, and 5 show somewhat distorted square planar coordination around the Pd atoms. Mean deviations from the best plane through Pd and the four donor atoms are 0.01 Å for 2a, 0.02 Å for 3a, and 0.03 (Pd1), 0.14 (Pd2), 0.04 (Pd1′), and 0.01 (Pd2′) Å for 5. The PdN2C2 chelate rings are all essentially planar and also coplanar with the Pd coordination planes (maximum interplanar angle 8°). The seven-membered rings in 2a and 3a have similar conformations, with the five atoms, Pd, N1, O1, C7, and C8 approximately coplanar, and C1 and C2 lying out of the plane to the same side. For 5, however, all four seven-membered rings have a different form in which C1, C2, N1, and C7 are coplanar, with Pd, O1, and C8 lying out of the plane to the same side.
The Pd–C bond distances for 2a and 3a are 1.981(3) Å and 1.9716(15) Å, respectively, both in the range expected for aryl ligands trans to N (ca. 1.97–2.00 Å).4,11,14,22 The two Pd–C bond distances for 5 are slightly longer, 2.014(7) Å for Pd(1)–C(1) and 2.009 Å for Pd(2)–C(31). The Pd–N (trans to aryl) bond distances are very similar for the three complexes (between 2.115(5) and 2.118(2) Å), and they are longer than the Pd–N (trans to N) bond distances (in the range 2.026(2)–2.070(5) Å), as expected for the stronger trans influence of the aryl ligand with respect to N-donor ligands.
The X-ray diffraction study of 5 (Fig. 3) confirms the structure proposed for this compound, consisting of two molecules of 4 coordinated via nitrogen to a silver atom. The two Ag–N bond lengths are 2.121(5) and 2.128(4) Å, similar to other Ag–N bond distances reported in the literature for compounds with a N–Ag–N moiety.23 The N(2)–Ag(1)–N(4) angle of 167.5(2)° departs significantly from linearity, but is still close to those found in the literature (between 168 and 179°).23
For the three structures we can suggest electronic delocalization along the N–CN group, as a shortening of the single N–C bond and a lengthening of the double CN bond is observed when compared with other bonds in the same or other molecules. Thus, the “single” bonds Me2N(2)–C(8) (1.343(4) Å, 2a), iPrN(2)–C(8) (1.346(2) Å, 3a), and TolN(1)–C(7), TolN(3)–C(37) (1.341(8) and 1.322(10) Å, 5) are much shorter than the C–N bonds (Me–N, iPr–N and Tol–N) in the same complexes, which measure between 1.415(10) and 1.494(2) Å. The corresponding “double” bonds C(8)N(1) (1.305(4) Å, 2a; 1.3054(18) Å, 3a), and C(7)N(2), C(37)N(4) (1.292(8), and 1.310(9) Å, 5) are longer than the mean value in imines (1.279 Å).24 This CN bond lengthening can be attributed to both the electronic delocalization along the N–C–N bonds and the coordination of the iminic nitrogen to Pd (in 2a, 3a) or Ag (in 5) (although it is interesting to note that the coordination of N(1) and N(3) to Pd in 5 does not cause a significant lengthening of the corresponding C–N single bonds (1.341(8) and 1.322(10) Å) with respect to the values for the (uncoordinated) C(8)–N(2) bonds in 2a (1.343(4) Å) and 3a (1.346(2) Å)). Our group has previously observed a similar electronic delocalization along the N–C–N bonds for complexes resulting from the insertion of carbodiimides and cyanamides into the C–Pd bond, or the addition of carbodiimides to the O–H bond, of ortho-palladated phenol derivatives.12 It is also interesting to note that in the trinuclear complex 5 the electronic delocalization in one of the N–CN moieties is much greater than in the other (bond lengths in N(1)–C(7)N(2) are 1.292(8) and 1.341(8) Å while for N(3)–C(37)–N(4) the two bond lengths are more similar, 1.310(9) and 1.322(10) Å).
The structure of 3a shows a classical hydrogen bond between the NH proton of the complex and an oxygen atom of the triflate, with an O(3)⋯H–N(2) distance of 2.22(2) Å.
Conclusions
We have investigated the reactivity of two Pd complexes derived from benzyl alcohol (one of them a κ2-C,O chelate) towards nitriles, cyanamides and carbodiimides. With the chelate complex we have obtained novel neutral or ionic complexes containing a 7-membered κ2-C,N chelate ring, resulting from the insertion of the organic molecules into the O–Pd bond. The presence of AgOTf was necessary for most of these reactions. A novel heterometallic bis-chelate Pd2Ag complex has also been synthesized. Starting from the non-chelate complex, we have achieved the insertion of a carbodiimide into the aryl–Pd bond. All the new compounds have been extensively characterized by NMR spectroscopy, and three of them, including the mixed-metal complex, by X-ray crystallography.Experimental
The 1H and 13C resonances were assigned with the help of 2D NMR experiments measured in Bruker Avance 400 and 600 MHz spectrometers (see Chart 2 for the numbering system). Molar conductivities were measured for ca. 5 × 10−4 M solutions in acetone, using a CRISON micro CM2200 conductivity meter. Infrared spectra were recorded using a Perkin Elmer Spectrum 100 spectrophotometer, and C, H, N, and S elemental analyses were carried out with a Carlo Erba 1106 microanalyzer. Melting points were determined on a Reichert apparatus and are uncorrected. Unless otherwise specified, all experiments were conducted under N2 atmosphere using Schlenk techniques. CH2Cl2 and Et2O were distilled before use. [PdI(C6H4CH2OH-2)(bpy)] (I),14 [Pd(κ2-C,O-C6H4CH2O-2)(bpy)] (II),14 and [Pd(dba)2]25 were prepared according to literature procedures. Other products were obtained from commercial sources and used without further purifications. | ||
| Chart 2 | ||
Synthesis of [Pd{κ2-C,N-C6H4{CH2OC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) NH)Me}-2}(bpy)](OTf) (1)
NH)Me}-2}(bpy)](OTf) (1)
Acetonitrile (55 mg, 1.35 mmol) and AgOTf (69 mg, 0.27 mmol) were added to a solution of [Pd(κ2-C,O-C6H4CH2O-2)(bpy)] (II)14 (100 mg, 0.27 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred in the dark for 16 h at room temperature (the colour darkened and a precipitate formed). It was then filtered over Celite and the resulting yellow solution was concentrated in vacuo to a volume of ca. 1 mL. Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give 1 as a yellow solid, which is soluble in CH2Cl2, CHCl3, and acetone. Yield: 85 mg (56%). Mp: 99 °C. ΛM (acetone): 115 Ω−1 cm2 mol−1. IR (cm−1): ν(SO) 1029, 1279, ν(CN) 1635, ν(NH) 3213. 1H NMR (400 MHz, CDCl3): 8.89 (ddd, 3JHH = 5 Hz, 4JHH = 2 Hz, 5JHH = 1 Hz, 1H, H16′ bpy), 8.45 (br s, 1H, NH), 8.37 (ddd, 3JHH = 6 Hz, 4JHH = 2 Hz, 5JHH = 1, 1H, H16 bpy), 8.25 (d, 3JHH = 8 Hz, 1H, H13 bpy), 8.17 (d, 3JHH = 8 Hz, 1H, H13′ bpy), 8.11 (td, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, H14′ bpy), 8.07 (td, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, H14 bpy), 7.82 (ddd, 3JHH = 8 Hz, 3JHH = 5 Hz, 4JHH = 1 Hz, 1H, H15′ bpy), 7.43 (ddd, 3JHH = 8 Hz, 3JHH = 6 Hz, 4JHH = 1 Hz, 1H, H15 bpy), 7.31–7.27 (m, 1H, H5 aryl), 7.22 (d, 3JHH = 7 Hz, 1H, H6 aryl), 7.15–7.11 (m, 2H, H3,4 aryl), 6.60 and 5.05 (AB system, 2JHH = 11 Hz, 2H, CH2), 2.30 (s, 3H, Me). 13C{1H} NMR (100.6 MHz, CDCl3): 175.1 (CNH), 157.0 (C12 bpy), 152.6 (C12′ bpy), 152.0 (C1 aryl), 151.9 (CH16 bpy), 151.0 (CH16′ bpy), 140.09 and 140.06 (CH14, 14′ bpy), 139.4 (C2 aryl), 134.7 (CH6 aryl), 130.6 (CH5 aryl), 128.3 (CH15′ bpy), 127.6 (CH3 aryl), 126.7 (CH15 bpy), 125.4 (CH4 aryl), 123.0 (CH13 bpy), 122.1 (CH13′ bpy), 121.0 (q, 1JCF = 320 Hz, OTf), 72.2 (CH2), 23.6 (Me). Anal. Calcd for C20H18F3N3O4PdS: C, 42.91; H, 3.24; N, 7.51; S, 5.73. Found: C, 42.58; H, 2.92; N, 7.19; S, 5.42.
Synthesis of [Pd{κ2-C,N-C6H4{CH2OC(NH)NMe2}-2}(bpy)](OTf) (2a)
Dimethylcyanamide (95 mg, 1.35 mmol) and AgOTf (69 mg, 0.27 mmol) were added to a solution of II14 (100 mg, 0.27 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred in the dark for 16 h at room temperature (the color darkened and a precipitate formed). It was then filtered over Celite and the resulting pale yellow solution was concentrated in vacuo to a volume of ca. 1 mL. Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give 2a as a pale yellow solid, which is soluble in CH2Cl2, CHCl3, and acetone. Yield: 91 mg (57%). Mp: 182 °C (dec). ΛM (acetone): 125 Ω−1 cm2 mol−1. IR (cm−1): ν(SO) 1029, 1275, ν(CN) 1602, ν(NH) 3306. 1H NMR (600 MHz, CDCl3): 8.65 (ddd, 3JHH = 5 Hz, 4JHH = 2 Hz, 5JHH = 1 Hz, 1H, H16′ bpy), 8.42 (d, 3JHH = 8 Hz, 1H, H13 bpy), 8.35 (d, 3JHH = 8 Hz, 1H, H13′ bpy), 8.32 (ddd, 3JHH = 6 Hz, 4JHH = 2 Hz, 5JHH = 1 Hz, 1H, H16 bpy), 8.14 (td, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, H14 bpy), 8.08 (td, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, H14′ bpy), 7.79 (ddd, 3JHH = 8 Hz, 3JHH = 5 Hz, 4JHH = 1 Hz, 1H, H15′ bpy), 7.39 (ddd, 3JHH = 8 Hz, 3JHH = 6 Hz, 4JHH = 1 Hz, 1H, H15 bpy), 7.29–7.26 (m, 2H, H5,6 aryl), 7.16–7.10 (m, 2H, H3,4 aryl), 6.62 and 5.10 (AB system, 2JHH = 11 Hz, 2H, CH2), 4.81 (s, 1H, NH), 3.00 (s, 6H, Me). 13C{1H} NMR (150.9 MHz, CDCl3): 161.3 (CNH), 157.0 (C12 bpy), 153.13 and 153.11 (C12′ bpy and C1 aryl), 151.6 (CH16 bpy), 149.1 (CH16′ bpy), 140.5 (CH14′ bpy), 140.3 (CH14 bpy), 139.3 (C2 aryl), 134.9 (CH6 aryl), 130.3 (CH5 aryl), 127.9 (CH15′ bpy), 127.6 (CH3 aryl), 126.7 (CH15 bpy), 125.1 (CH4 aryl), 123.8 (CH13 bpy), 123.0 (CH13′ bpy), 121.0 (q, 1JCF = 320 Hz, OTf), 73.2 (CH2), 38.0 (Me). Anal. Calcd for C21H21F3N4O4PdS: C, 42.83; H, 3.59; N, 9.51; S, 5.44. Found: C, 42.81; H, 3.60; N, 9.43; S, 5.12. Single crystals of 2a were grown by liquid diffusion of Et2O into a solution of 2a in CH2Cl2.
Synthesis of [Pd{κ2-C,N-C6H4{CH2OC(NH)NEt2}-2}(bpy)](OTf) (2b)
Diethylcyanamide (133 mg, 1.35 mmol) and AgOTf (69 mg, 0.27 mmol) were added to a solution of II14 (100 mg, 0.27 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred in the dark for 16 h at room temperature (the color darkened and a precipitate formed). It was then filtered over Celite and the resulting pale yellow solution was concentrated in vacuo to a volume of ca. 1 mL. Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give 2b as a pale yellow solid, which is soluble in CH2Cl2, CHCl3, and acetone. Yield: 100 mg (54%). Mp: 104 °C. ΛM (acetone): 123 Ω−1 cm2 mol−1. IR (cm−1): ν(SO) 1030, 1277, ν(CN) 1599, ν(NH) 3321. 1H NMR (400 MHz, CDCl3): 8.58 (dd, 3JHH = 5 Hz, 4JHH = 1 Hz, 1H, H16′ bpy), 8.44 (d, 3JHH = 8 Hz, 1H, H13 bpy), 8.39 (d, 3JHH = 8 Hz, 1H, H13′ bpy), 8.32 (dd, 3JHH = 6 Hz, 4JHH = 1 Hz, 1H, H16 bpy), 8.15 (td, 3JHH = 8 Hz, 4JHH = 1 Hz, 1H, H14 bpy), 8.13 (td, 3JHH = 8 Hz, 4JHH = 1 Hz, 1H, H14′ bpy), 7.78 (ddd, 3JHH = 8 Hz, 3JHH = 5 Hz, 4JHH = 1 Hz, 1H, H15′ bpy), 7.39 (ddd, 3JHH = 8 Hz, 3JHH = 6 Hz, 4JHH = 1 Hz, 1H, H15 bpy), 7.29–7.26 (m, 2H, H5,6 aryl), 7.14–7.12 (m, 2H, H3,4 aryl), 6.66 and 5.11 (AB system, 2JHH = 11 Hz, 2H, CH2O), 4.76 (s, 1H, NH), 3.33 (m, 4H, CH2CH3), 1.13 (t, 3JHH = 7 Hz, 6H, CH2CH3). 13C{1H} NMR (100.6 MHz, CDCl3): 160.2 (CNH), 157.0 (C12 bpy), 153.2 (C12′ bpy), 153.0 (C1 aryl), 151.6 (CH16 bpy), 148.9 (CH16′ bpy), 140.6 (CH14′ bpy), 140.3 (CH14 bpy), 139.1 (C2 aryl), 134.9 (CH6 aryl), 130.2 (CH5 aryl), 127.7 (CH15′ bpy), 127.4 (CH3 aryl), 126.7 (CH15 bpy), 125.0 (CH4 aryl), 123.9 (CH13 bpy), 123.2 (CH13′ bpy), 121.0 (q, 1JCF = 321 Hz, OTf), 73.1 (CH2O), 43.3 (CH2CH3), 13.6 (CH2CH3). Anal. Calcd for C23H25F3N4O4PdS: C, 44.78; H, 4.08; N, 9.08; S, 5.20. Found: C, 44.95; H, 4.22; N, 8.85; S, 4.89.
Synthesis of [Pd{κ2-C,N-C6H4{CH2OC(NiPr)NHiPr}-2}(bpy)](OTf) (3a)
1,3-Diisopropylcarbodiimide (170 mg, 1.35 mmol) and AgOTf (69 mg, 0.27 mmol) were added to a solution of II14 (100 mg, 0.27 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred in the dark for 16 h at room temperature. It was then filtered over Celite and the resulting yellow solution was concentrated in vacuo to a volume of ca. 1 mL. Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give 3a as a pale yellow solid, which is soluble in CH2Cl2, CHCl3, and acetone. Yield: 104 mg (60%). Mp: 195 °C. ΛM (acetone): 140 Ω−1 cm2 mol−1. IR (cm−1): ν(SO) 1028, 1276, ν(CN) 1611, ν(NH) 3354. 1H NMR (400 MHz, CDCl3): 8.68 (dd, 3JHH = 5 Hz, 4JHH = 1 Hz, 1H, H16′ bpy), 8.52 (dd, 3JHH = 5 Hz, 4JHH = 1 Hz, 1H, H16 bpy), 8.40 (d, 3JHH = 8 Hz, 1H, H13 bpy), 8.35 (d, 3JHH = 8 Hz, 1H, H13′ bpy), 8.16 (td, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, H14 bpy), 8.15 (td, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, H14′ bpy), 7.82 (ddd, 3JHH = 8 Hz, 3JHH = 5 Hz, 4JHH = 1, 1H, H15′ bpy), 7.44 (ddd, 3JHH = 8 Hz, 3JHH = 5 Hz, 4JHH = 1, 1H, H15 bpy), 7.39 (dd, 3JHH = 7 Hz, 4JHH = 1 Hz, 1H, H6 aryl), 7.26 (td, 3JHH = 7 Hz, 4JHH = 2 Hz, 1H, H5 aryl), 7.07 (td, 3JHH = 7 Hz, 4JHH = 1 Hz, 1H, H4 aryl), 7.03 (dd, 3JHH = 7 Hz, 4JHH = 2 Hz, 1H, H3 aryl), 6.65 (A part of AB system, 2JHH = 11 Hz, 1H, CH2), 5.57 (d, 3JHH = 7 Hz, 1H, NH), 5.12 (B part of AB system, 2JHH = 11 Hz, 1H, CH2), 3.89 (dsept, 3JHH = 7 Hz, 3JHH = 6 Hz, 1H, CH iPrA), 3.78 (sept, 3JHH = 6 Hz, 1H, CH iPrB), 1.55 (d, 3JHH = 6 Hz, 3H, Me iPrB), 1.28 (d, 3JHH = 6 Hz, 3H, Me iPrA), 1.14 (d, 3JHH = 6 Hz, 3H, Me iPrA), 0.70 (d, 3JHH = 6 Hz, 3H, Me of iPrB). 13C{1H} NMR (100.6 MHz, CDCl3): 156.9 (C12 bpy), 156.3 (CN), 153.6 (C1 aryl), 153.1 (C12′ bpy), 151.7 (CH16 bpy), 150.8 (CH16′ bpy), 140.4 (CH14′ bpy), 140.3 (CH14 bpy), 138.3 (C2 aryl), 134.4 (CH6 aryl), 129.8 (CH5 aryl), 128.2 (CH15′ bpy), 127.4 (CH3 aryl), 126.9 (CH15 bpy), 124.7 (CH4 aryl), 123.5 (CH13 bpy), 122.7 (CH13′ bpy), 121.1 (q, 1JCF = 320 Hz, OTf), 74.3 (CH2), 51.0 (CH iPrB), 45.4 (CH iPrA), 26.0 (Me iPrB), 23.7 (Me iPrA), 23.1 (Me iPrA), 22.0 (1C, Me iPrB). Anal. Calcd for C25H29F3N4O4PdS: C, 46.55; H, 4.53; N, 8.69; S, 4.97. Found: C, 46.67; H, 4.40; N, 8.37; S, 4.59. Single crystals of 3a were grown by liquid diffusion of Et2O into a solution of 3a in CH2Cl2.
Synthesis of [Pd{κ2-C,N-C6H4{CH2OC(NTol)NHTol}-2}(bpy)](OTf) (3b)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 14.
1,3-Di-p-tolylcarbodiimide (300 mg, 1.35 mmol) and AgOTf (69 mg, 0.27 mmol) were added to a solution of II14 (100 mg, 0.27 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred in the dark for 16 h at room temperature. It was then filtered over Celite and the resulting yellow solution was concentrated in vacuo to a volume of ca. 1 mL. Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give 3b as a yellow solid, which is soluble in CH2Cl2, CHCl3, and acetone. Yield: 128 mg (64%). Starting from 4; KOTf (190 mg, 1.0 mmol) was added to a solution of 4 (60 mg, 0.10 mmol) in commercial CH2Cl2 (20 mL) and in an open flask. The mixture was stirred for 20 min at room temperature, with no change in the yellow color. Then a solution of HOTf in commercial CH2Cl2 (15 mg, 0.10 mmol, in 2 mL) was added dropwise (whereupon the color changed from yellow to red). After the addition the mixture was filtered over Celite and the resulting yellow solution was concentrated in vacuo to a volume of ca. 1 mL. Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give 3b as a yellow solid. Yield: 46 mg (62%). Mp: 182 °C. ΛM (acetone): 125 Ω−1 cm2 mol−1. IR (cm−1): ν(SO) 1030, 1259, ν(CN) 1600, ν(NH) 3401. 1H NMR (600 MHz, CDCl3): 8.61 (d, 3JHH = 5 Hz, 1H, H16′ bpy), 8.54 (d, 3JHH = 8 Hz, 1H, H13 bpy), 8.50 (d, 3JHH = 8 Hz, 1H, H13′ bpy), 8.31 (d, 3JHH = 5 Hz, 1H, H16 bpy), 8.18 (t, 3JHH = 8 Hz, 1H, H14 bpy), 8.14 (t, 3JHH = 8 Hz, 1H, H14′ bpy), 7.69 (dd, 3JHH = 8 Hz, 3JHH = 5, 1H, H15′ bpy), 7.50 (d, 3JHH = 7 Hz, 1H, H6 aryl), 7.44 (td, 3JHH = 7 Hz, 4JHH = 2 Hz, 1H, H5 aryl), 7.39 (dd, 3JHH = 8 Hz, 3JHH = 5 Hz, 1H, H15 bpy), 7.29 (part A of AB system, 3JHH = 8 Hz, 2H, o-H TolB), 7.22 (part A of AB system, 2JHH = 11 Hz, 1H, CH2), 7.20 (part B of AB system, 3JHH = 8 Hz, 2H, m-H TolB), 7.22–7.18 (m, 2, H3,4 aryl), 7.09 (part A of AB system, 3JHH = 8 Hz, 2H, m-H TolA), 6.85 (part B of AB system, 3JHH = 8 Hz, 2H, o-H TolA), 6.49 (s, 1H, NH), 5.27 (part B of AB system, 2JHH = 11 Hz, 1H, CH2), 2.31 (s, 3H, Me TolA), 2.29 (s, 3H, Me TolB). 13C{1H} NMR (150.9 MHz, CDCl3): 156.9 (C12 bpy), 156.5 (CN), 153.7 (C1 aryl), 153.4 (C12′ bpy), 151.6 (CH16 bpy), 149.3 (CH16′ bpy), 141.8 (i-C TolB), 141.0 (2C, CH14, 14′ bpy), 137.9 (C2 aryl), 137.4 (p-C TolB), 136.2 (p-C TolA), 134.2 (CH6 aryl), 133.4 (i-C TolA), 131.3 (2C, m-CH TolB), 131.2 (CH5 aryl), 129.9 (2C, m-CH TolA), 127.9 (CH3 aryl), 127.5 (CH15′ bpy), 126.9 (CH15 bpy), 125.9 (2C, o-CH TolB), 125.6 (CH4 aryl), 124.5 (CH13 bpy), 123.9 (2C, o-CH TolA), 123.8 (CH13′ bpy), 74.2 (CH2), 21.14 (Me TolB), 21.11 (Me TolA). The OTf carbon is not observed. Anal. Calcd for C33H29F3N4O4PdS: C, 53.48; H, 3.94; N, 7.56; S, 4.33. Found: C, 53.12; H, 3.64; N, 7.52; S, 4.09.
14.
1,3-Di-p-tolylcarbodiimide (300 mg, 1.35 mmol) and AgOTf (69 mg, 0.27 mmol) were added to a solution of II14 (100 mg, 0.27 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred in the dark for 16 h at room temperature. It was then filtered over Celite and the resulting yellow solution was concentrated in vacuo to a volume of ca. 1 mL. Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give 3b as a yellow solid, which is soluble in CH2Cl2, CHCl3, and acetone. Yield: 128 mg (64%). Starting from 4; KOTf (190 mg, 1.0 mmol) was added to a solution of 4 (60 mg, 0.10 mmol) in commercial CH2Cl2 (20 mL) and in an open flask. The mixture was stirred for 20 min at room temperature, with no change in the yellow color. Then a solution of HOTf in commercial CH2Cl2 (15 mg, 0.10 mmol, in 2 mL) was added dropwise (whereupon the color changed from yellow to red). After the addition the mixture was filtered over Celite and the resulting yellow solution was concentrated in vacuo to a volume of ca. 1 mL. Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give 3b as a yellow solid. Yield: 46 mg (62%). Mp: 182 °C. ΛM (acetone): 125 Ω−1 cm2 mol−1. IR (cm−1): ν(SO) 1030, 1259, ν(CN) 1600, ν(NH) 3401. 1H NMR (600 MHz, CDCl3): 8.61 (d, 3JHH = 5 Hz, 1H, H16′ bpy), 8.54 (d, 3JHH = 8 Hz, 1H, H13 bpy), 8.50 (d, 3JHH = 8 Hz, 1H, H13′ bpy), 8.31 (d, 3JHH = 5 Hz, 1H, H16 bpy), 8.18 (t, 3JHH = 8 Hz, 1H, H14 bpy), 8.14 (t, 3JHH = 8 Hz, 1H, H14′ bpy), 7.69 (dd, 3JHH = 8 Hz, 3JHH = 5, 1H, H15′ bpy), 7.50 (d, 3JHH = 7 Hz, 1H, H6 aryl), 7.44 (td, 3JHH = 7 Hz, 4JHH = 2 Hz, 1H, H5 aryl), 7.39 (dd, 3JHH = 8 Hz, 3JHH = 5 Hz, 1H, H15 bpy), 7.29 (part A of AB system, 3JHH = 8 Hz, 2H, o-H TolB), 7.22 (part A of AB system, 2JHH = 11 Hz, 1H, CH2), 7.20 (part B of AB system, 3JHH = 8 Hz, 2H, m-H TolB), 7.22–7.18 (m, 2, H3,4 aryl), 7.09 (part A of AB system, 3JHH = 8 Hz, 2H, m-H TolA), 6.85 (part B of AB system, 3JHH = 8 Hz, 2H, o-H TolA), 6.49 (s, 1H, NH), 5.27 (part B of AB system, 2JHH = 11 Hz, 1H, CH2), 2.31 (s, 3H, Me TolA), 2.29 (s, 3H, Me TolB). 13C{1H} NMR (150.9 MHz, CDCl3): 156.9 (C12 bpy), 156.5 (CN), 153.7 (C1 aryl), 153.4 (C12′ bpy), 151.6 (CH16 bpy), 149.3 (CH16′ bpy), 141.8 (i-C TolB), 141.0 (2C, CH14, 14′ bpy), 137.9 (C2 aryl), 137.4 (p-C TolB), 136.2 (p-C TolA), 134.2 (CH6 aryl), 133.4 (i-C TolA), 131.3 (2C, m-CH TolB), 131.2 (CH5 aryl), 129.9 (2C, m-CH TolA), 127.9 (CH3 aryl), 127.5 (CH15′ bpy), 126.9 (CH15 bpy), 125.9 (2C, o-CH TolB), 125.6 (CH4 aryl), 124.5 (CH13 bpy), 123.9 (2C, o-CH TolA), 123.8 (CH13′ bpy), 74.2 (CH2), 21.14 (Me TolB), 21.11 (Me TolA). The OTf carbon is not observed. Anal. Calcd for C33H29F3N4O4PdS: C, 53.48; H, 3.94; N, 7.56; S, 4.33. Found: C, 53.12; H, 3.64; N, 7.52; S, 4.09.
Synthesis of [Pd{κ2-C,N-C6H4{CH2OC(NTol)NTol}-2}(bpy)] (4)
14.
1,3-Di-p-tolylcarbodiimide (60 mg, 0.27 mmol) was added to a solution of II14 (100 mg, 0.27 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred for 5 min at room temperature, with a change in color from yellow to red. Then it was filtered over Celite and the resulting red solution was concentrated to dryness in vacuo. Cold Et2O (15 mL) was added to precipitate a solid, which was filtered off, washed with cold Et2O (3 × 5 mL), and dried in vacuo to give 4 as a red solid, which is soluble in CH2Cl2, CHCl3, and acetone, and partially soluble in Et2O. Yield: 130 mg (81%). Starting from 3b; KOtBu (27 mg, 0.24 mmol) was added to a solution of 3b (60 mg, 0.08 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred for 10 min at room temperature, with a change in color from yellow to red. Work-up as in the previous reaction gave 4 as a red solid. Yield: 38 mg, 80%. Mp: 96 °C. IR (cm−1): ν(CN) 1660. 1H NMR (400 MHz, CDCl3): 8.45 (d, 3JHH = 5 Hz, 1H, H16 bpy), 8.39 (d, 3JHH = 5 Hz, 1H, H16′ bpy), 8.06 (d, 3JHH = 8 Hz, 1H, H13 bpy), 8.02 (d, 3JHH = 8 Hz, 1H, H13′ bpy), 7.96 (t, 3JHH = 8 Hz, 1H, H14 bpy), 7.91 (t, 3JHH = 8 Hz, 1H, H14′ bpy), 7.9 (A part of AB system, 3JHH = 8 Hz, 2H, o-H TolB), 7.70 (d, 3JHH = 7 Hz, 1H, H6 aryl), 7.39–7.30 (m, 2H, H15, 15′ bpy), 7.03 (t, 3JHH = 7 Hz, 1H, H5 aryl), 7.00–6.95 (br m, 4H, o,m-H TolA), 6.93 (t, 3JHH = 7 Hz, 1H, H4 aryl), 6.85 (B part of AB system, 3JHH = 8 Hz, 2H, m-H TolB), 6.70 (d, 3JHH = 7 Hz, 1H, H3), 5.02 and 4.73 (br) (AB system, 2JHH = 14 Hz, 2H, CH2), 2.27 (s, 3H, Me TolA), 2.16 (s, 3H, Me TolB), 1.54 (s, 2H, H2O). 13C{1H} NMR (150.9 MHz, CDCl3, 213 K): 155.7 (CN), 155.2 (C12 bpy), 153.1 (C12′ bpy), 152.1 (CH16 bpy), 149.6 (C1 aryl), 149.3 (CH16′ bpy), 147.9 (i-C TolA), 146.4 (i-C TolB), 140.7 (C2 aryl), 139.1 (CH14′ bpy), 138.8 (CH14 bpy), 136.2 (CH6 aryl), 129.5 (p-C TolB), 128.97 (2C, m-CH TolA), 128.92 (p-C TolA), 128.91 (2C, m-CH TolB), 127.0 (CH15′ bpy), 126.9 (CH5 aryl), 126.7 (CH15 bpy), 124.9 (CH3 aryl), 124.6 (2C, o-CH TolB), 123.5 (CH4 aryl), 122.7 (2C, o-CH TolA), 122.1 (CH13 bpy), 121.6 (CH13′ bpy), 71.2 (CH2), 21.1 (Me TolA), 21.0 (Me TolB). Anal. Calcd for C32H30N4O2Pd (4·H2O): C, 63.11; H, 4.96; N, 9.20. Found: C, 63.25; H, 4.68; N, 9.27.
Synthesis of [Ag(N-4)2](OTf) (5)
14.
AgOTf (57 mg, 0.22 mmol) was added to a solution of II14 (100 mg, 0.27 mmol) in CH2Cl2 (20 mL) under N2, followed by 1,3-di-p-tolylcarbodiimide (60 mg, 0.27 mmol). The solvent was immediately evaporated in vacuo and Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give a mixture of 5 and 3b in ca. 1:0.2 ratio. Yield, 192 mg. This solid was dissolved in CH2Cl2 (20 mL) and the resulting solution was filtered over Celite. The yellow solution was then concentrated in vacuo to a volume of ca. 1 mL. A small amount of Et2O (7 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give pure 5 as a yellow solid, which is soluble in CH2Cl2, CHCl3, and acetone. Yield: 124 mg (64%). Starting from 4; AgOTf (13 mg, 0.05 mmol) was added to a solution of 4 (60 mg, 0.10 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred for 2 h at room temperature, with a change in color from red to yellow. Then it was filtered over Celite, and the resulting yellow solution was evaporated to dryness in vacuo. Et2O (15 mL) was added to precipitate a solid which was filtered off, washed with Et2O (3 × 5 mL), and dried in vacuo to give a mixture of 5 and 3b in ca. 1:0.2 ratio. Yield: 71 mg. This solid was dissolved in CH2Cl2 (20 mL) and the resulting solution was filtered over Celite. The yellow solution was then concentrated in vacuo to a volume of ca. 0.5 mL. A small amount of Et2O (3 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give pure 5 as a yellow solid. Yield: 38 mg (53%). Mp: 159 °C. ΛM (acetone): 148 Ω−1 cm2 mol−1. IR (cm−1): ν(SO) 1030, 1272, ν(CN) 1600. 1H NMR (600 MHz, CDCl3): 8.35 (d, 3JHH = 5 Hz, 2H, H16 bpy), 8.33 (d, 3JHH = 8 Hz, 2H, H13 bpy), 8.28 (d, 3JHH = 8 Hz, 2H, H13′ bpy), 8.13 (d, 3JHH = 5 Hz, 2H, H16′ bpy), 8.10 (td, 3JHH = 8 Hz, 4JHH = 1 Hz, 2H, H14 bpy), 8.00 (td, 3JHH = 8 Hz, 4JHH = 1 Hz, 2H, H14′ bpy), 7.84 (d, 3JHH = 7 Hz, 2H, H6 aryl), 7.39–7.33 (m, 4H, H15 bpy, H5 aryl), 7.33–7.30 (m, 1H, H15′ bpy), 7.17 (t, 3JHH = 7 Hz, 2H, H4 aryl), 7.00 (A part of AB system, 3JHH = 7 Hz, 4H, m-H TolB), 6.95–6.75 (br, 4H, o-H TolA), 6.66 (d, 3JHH = 7 Hz, 2H, H3 aryl), 6.13 (B part of AB system br, 3JHH = 7 Hz, 4H, o-H TolB), 4.88 and 4.19 (AB system, 2JHH = 12 Hz, 2H, CH2), 2.37 (s, 6H, Me TolB), 2.04 (s, 6H, Me TolA). The m-H TolA protons are not observed. 13C{1H} NMR (100.6 MHz, CDCl3): 162.2 (2C, CN), 156.5 (2C, C12 bpy), 153.7 (2C, C12′ bpy), 152.4 (2C, CH16 bpy), 150.0 (2C, C1 aryl), 149.0 (2C, CH16′ bpy), 146.4 (2C, i-C TolB), 143.6 (2C, i-C TolA), 139.9 (2C, CH14′ bpy), 139.8 (2C, CH14 bpy), 138.9 (2C, C2 aryl), 136.1 (2C, CH6 aryl), 132.5 (2C, p-C TolA), 131.3 (2C, p-C TolB), 129.7 (4C, br, m-CH TolA), 128.7 (4C, m-CH TolB), 127.2 (2C, CH5 aryl), 126.9 (2C, CH15′ bpy), 126.8 (2C, CH15 bpy), 126.6 (2C, CH3 aryl), 124.2 (4C, o-CH TolB), 124.1 (2C, CH4 aryl), 123.4 (2C, CH13 bpy), 122.7 (2C, CH13′ bpy), 119.9 (q, 1JCF = 321 Hz, OTf), 72.9 (2C, CH2), 21.2 (2C, Me TolB), 21.0 (2C, Me TolA). The o-CH TolA and OTf carbons are not observed. Anal. Calcd for C65H56AgF3N8O5Pd2S: C, 54.25; H, 3.92; N, 7.79; S, 2.23. Found: C, 54.11; H, 3.81; N, 7.86; S, 2.07. Single crystals of 5·2.5CHCl3·0.5Et2O were grown by liquid diffusion of Et2O into a solution of 5 in CHCl3.
Synthesis of [Pd{κ2-O,N-OCH2{C6H4{C(NiPr)NHiPr}-2}}(bpy)](OTf) (6)
1,3-Diisopropylcarbodiimide (252 mg, 2.0 mmol) and TlOTf (70 mg, 0.20 mmol) were added to a solution of [PdI(C6H4CH2OH-2)(bpy)] (I)14 (100 mg, 0.20 mmol) in CH2Cl2 (20 mL) under N2. The mixture was stirred for 16 h at room temperature. It was then filtered over Celite and the resulting yellow solution was concentrated in vacuo to a volume of ca. 1 mL. Et2O (15 mL) was added to precipitate a solid, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give a mixture of 3a and 6 in a 1:1.3 ratio. Yield: 97 mg. The products were separated by preparative TLC on alumina using acetone as eluent. The band with Rf = 0.48 was collected, and the product was extracted with acetone (30 mL). Evaporation of the acetone and addition of Et2O (15 mL) resulted in the formation of a precipitate, which was filtered off, thoroughly washed with Et2O (3 × 5 mL), and dried in vacuo to give 6 as a yellow solid, which is soluble in CH2Cl2, CHCl3, and acetone. Yield: 54 mg (31%). Mp: 177 °C. ΛM (acetone): 122 Ω1 cm2 mol−1. IR (cm−1): ν(SO) 1032, 1262, ν(CN) 1609, ν(NH) 3318. 1H NMR (600 MHz, CDCl3): 8.85 (d, 3JHH = 5 Hz, 1H, H16′ bpy), 8.52 (d, 3JHH = 5 Hz, 1H, H16 bpy), 8.10–8.06 (m, 2H, H13′, 14′ bpy), 8.04 (d, 3JHH = 8, 1H, H13 bpy), 8.00 (td, 3JHH = 8 Hz, 4JHH = 1 Hz, 1H, H14 bpy), 7.71 (td, 3JHH = 6 Hz, 4JHH = 1 Hz, 1H, H15 bpy), 7.57–7.53 (m, 2H, H15′ bpy, H6 aryl), 7.43 (d, 3JHH = 8 Hz, 1H, H3 aryl), 7.40 (td, 3JHH = 8 Hz, 4JHH = 1 Hz, 1H, H4 aryl), 7.32 (td, 3JHH = 8 Hz, 4JHH = 1 Hz, 1H, H5 aryl), 6.39 (d, 3JHH = 9 Hz, 1H, NH), 4.57 and 3.84 (AB system, 2JHH = 10 Hz, 2H, CH2), 4.25 (sept, 3JHH = 6 Hz, 1H, CH iPrB), 3.55 (dsept, 3JHH = 9 Hz, 3JHH = 6 Hz, 1H, CH iPrA), 1.65 and 1.54 (d, 3JHH = 6 Hz, 3H, Me iPrB), 1.40 and 1.04 (d, 3JHH = 6 Hz, 3H, Me iPrA). 13C{1H} NMR (150.9 MHz, CDCl3): 162.0 (CN), 155.7 (C12′ bpy), 154.8 (C12 bpy), 152.2 (CH16 bpy), 148.1 (CH16′ bpy), 146.0 (C2 aryl), 140.6 (CH14′ bpy), 140.1 (CH14 bpy), 134.2 (C1 aryl), 131.3 (CH4 aryl), 130.8 (CH3 aryl), 128.4 (CH15 bpy), 128.0 (CH6 aryl), 127.9 (CH5 aryl), 126.1 (CH15′ bpy), 122.8 (CH13 bpy), 122.4 (CH13′ bpy), 121.2 (q, 1JCF = 321 Hz, OTf), 69.9 (CH2), 50.7 (CH iPrB), 48.9 (CH iPrA), 25.2 (Me iPrA), 24.9 (Me iPrB), 23.1 (Me iPrA), 22.4 (1C, Me iPrB). Anal. Calcd for C25H29F3N4O4PdS: C, 46.55; H, 4.53; N, 8.69; S, 4.97. Found: C, 46.38; H, 4.80; N, 8.54; S, 4.98.
Acknowledgements
We thank the Ministerio de Educación y Ciencia (Spain), FEDER (Project CTQ2011-24016) and Fundación Séneca (04539/GERM/06) for financial support. M. J. F.-R. is grateful to the Ministerio de Educación y Ciencia (Spain) for a grant.References
- J. Tsuji, Palladium Reagents and Catalysis: Innovations in Organic Synthesis, John Wiley & Sons, Chichester, UK, 1995 CrossRef CAS; N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2047–2067 Search PubMed; J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef; A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef; A. R. Muci and S. L. Buchwald, Top. Curr. Chem., 2002, 219, 131–209 CrossRef; G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285–2309 CrossRef PubMed; R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 2004, 2283–2321 CrossRef; R. C. Larock and G. Zeni, Chem. Rev., 2006, 106, 4644–4680 CrossRef PubMed; S. L. Buchwald, C. Mauger, G. Mignani and U. Scholz, Adv. Synth. Catal., 2006, 348, 23–39 CrossRef; J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef PubMed; R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef PubMed; M. A. Fernández-Rodríguez and J. F. Hartwig, J. Org. Chem., 2009, 74, 1663–1672 CrossRef PubMed; N. Selander and K. J. Szabo, Chem. Rev., 2011, 111, 2048–2076 CrossRef PubMed; J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef PubMed; D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC; C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef PubMed.
- J. Spencer, M. Pfeffer, N. Kyritsakas and J. Fischer, Organometallics, 1995, 14, 2214–2224 CrossRef CAS; M. Catellani, E. Motti and S. Ghelli, Chem. Commun., 2000, 2003–2004 RSC; K. R. Reddy, K. Surekha, G.-H. Lee, S.-M. Peng and S.-T. Liu, Organometallics, 2001, 20, 5557–5563 CrossRef; C. Sirlin, J. Chengebroyen, R. Konrath, G. Ebeling, I. Raad, J. Dupont, M. Paschaki, F. KotzybaHibert, C. HarfMonteil and M. Pfeffer, Eur. J. Org. Chem., 2004, 1724–1731 CrossRef; L. Canovese, F. Visentin, C. Santo, C. Levi and A. Dolmella, Organometallics, 2007, 26, 5590–5601 CrossRef; K. J. Cavell and D. S. McGuinnes, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and M. P. Mingos, Pergamon Press, Oxford, UK, 2007, vol. 8, pp. 197–268 CrossRef; T. Bai, L. Q. Xue, P. Xue, J. Zhu, H. H. Y. Sung, S. M. Ma, I. D. Wiliams, Z. Y. Lin and G. C. Jia, Organometallics, 2008, 27, 2614–2626 CrossRef; Y. Suzaki, M. Shirokawa, T. Yagyu and K. Osakada, Eur. J. Inorg. Chem., 2015, 2015, 421–429 CrossRef.
- J. Vicente, J. A. Abad, K. F. Shaw, J. Gil-Rubio, M. C. Ramírez de Arellano and P. G. Jones, Organometallics, 1997, 16, 4557–4566 CrossRef CAS; J. Vicente, J.-A. Abad, J. López-Serrano, P. G. Jones, C. Nájera and L. Botella-Segura, Organometallics, 2005, 24, 5044–5057 CrossRef; J. Vicente, M. T. Chicote, A. J. Martínez-Martínez, P. G. Jones and D. Bautista, Organometallics, 2008, 27, 3254–3271 CrossRef; J. Vicente, M. T. Chicote, A. J. Martínez-Martínez and D. Bautista, Organometallics, 2009, 28, 5915–5924 CrossRef; J. Vicente, P. González-Herrero, R. Frutos-Pedreño, M. T. Chicote, P. G. Jones and D. Bautista, Organometallics, 2011, 30, 1079–1093 CrossRef; J. Vicente, J. A. Abad, R. M. López-Nicolás and P. G. Jones, Organometallics, 2011, 30, 4983–4998 CrossRef; R. Frutos-Pedreño, P. González-Herrero, J. Vicente and P. G. Jones, Organometallics, 2013, 32, 4664–4676 CrossRef; R. Frutos-Pedreño, P. González-Herrero, J. Vicente and P. G. Jones, Organometallics, 2013, 32, 1892–1904 CrossRef.
- J. Vicente, J. A. Abad, E. Martínez-Viviente, M. C. Ramírez de Arellano and P. G. Jones, Organometallics, 2000, 19, 752–760 CrossRef CAS.
- J. Vicente, J. A. Abad, B. López-Peláez and E. Martínez-Viviente, Organometallics, 2002, 21, 58–67 CrossRef CAS; J. Vicente, J. A. Abad, J. López-Serrano and P. G. Jones, Organometallics, 2004, 23, 4711–4722 CrossRef.
- J. Vicente, J. A. Abad, M. J. López-Sáez, W. Förtsch and P. G. Jones, Organometallics, 2004, 23, 4414–4429 CrossRef CAS.
- J. Vicente and I. Saura-Llamas, Comments Inorg. Chem., 2007, 28, 39–72 CrossRef CAS.
- J. Vicente, J. A. Abad, R. Bergs, M. C. Ramirez de Arellano, E. Martínez-Viviente and P. G. Jones, Organometallics, 2000, 19, 5597–5607 CrossRef CAS; J. Vicente, J. A. Abad, E. Martinez-Viviente and P. G. Jones, Organometallics, 2002, 21, 4454–4467 CrossRef; J. Vicente, J. A. Abad, E. Martínez-Viviente and P. G. Jones, Organometallics, 2003, 22, 1967–1978 CrossRef.
- J. Vicente, I. Saura-Llamas, C. Grünwald, C. Alcaraz, P. G. Jones and D. Bautista, Organometallics, 2002, 21, 3587–3595 CrossRef CAS; M. J. Oliva-Madrid, J.-A. García-López, I. Saura-Llamas, D. Bautista and J. Vicente, Organometallics, 2014, 33, 6420–6430 CrossRef.
- J. Vicente, J. A. Abad, M. J. López-Sáez and P. G. Jones, Angew. Chem., Int. Ed., 2005, 44, 6001–6004 CrossRef CAS PubMed.
- J. Vicente, J. A. Abad, M. J. López-Sáez and P. G. Jones, Organometallics, 2006, 25, 1851–1853 CrossRef CAS.
- J. Vicente, J. A. Abad, M. J. López-Sáez, P. G. Jones and D. Bautista, Chem. – Eur. J., 2010, 16, 661–676 CrossRef CAS PubMed.
- J. Vicente, J. A. Abad, M. J. López-Sáez and P. G. Jones, Organometallics, 2010, 29, 409–416 CrossRef CAS.
- M. J. Fernández-Rodríguez, E. Martínez-Viviente, J. Vicente and P. G. Jones, Organometallics, 2015, 34, 3282–3291 CrossRef.
- M. Shiotsuki, A. Nakagawa, J. Rodriguez Castañon, N. Onishi, T. Kobayashi, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5549–5556 CrossRef CAS; J. Rodriguez Castañon, K. Kuwata, M. Shiotsuki and F. Sanda, Chem. – Eur. J., 2012, 18, 14085–14093 CrossRef PubMed.
- C. Fernández-Rivas, D. J. Cárdenas, B. Martín-Matute, A. Monge, E. Gutiérrez-Puebla and A. M. Echavarren, Organometallics, 2001, 20, 2998–3006 CrossRef.
- M. P. Muñoz, B. Martín-Matute, C. Fernández-Rivas, D. J. Cárdenas and A. M. Echavarren, Adv. Synth. Catal., 2001, 343, 338–342 CrossRef.
- I. Meana, P. Espinet and A. C. Albéniz, Organometallics, 2014, 33, 1–7 CrossRef CAS; T. Nakajima, M. Tsuji, N. Hamada, Y. Fukushima, B. Kure and T. Tanase, J. Organomet. Chem., 2014, 768, 61–67 CrossRef.
- K. Umakoshi, Y. Yamauchi, K. Nakamiya, T. Kojima, M. Yamasaki, H. Kawano and M. Onishi, Inorg. Chem., 2003, 42, 3907–3916 CrossRef CAS PubMed; J. Forniés, A. Martín, V. Sicilia and M. Martín, Chem. – Eur. J., 2003, 9, 3427–3435 CrossRef PubMed; I. Ara, J. Forniés, R. Lasheras, A. Martín and V. Sicilia, Eur. J. Inorg. Chem., 2006, 948–657 CrossRef; K. Umakoshi, T. Kojima, Y. Arikawa and M. Onishi, Chem. – Eur. J., 2006, 12, 5094–5104 CrossRef PubMed.
- R. A. Michelin, M. Mozzon and R. Bertani, Coord. Chem. Rev., 1996, 147, 299–338 CrossRef CAS; V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef PubMed.
- P. H. Mogul, R. N. Kniseley and V. A. Fassel, Spectrosc. Lett., 1977, 10, 959–970 CrossRef CAS.
- J. Vicente, R. V. Shenoy, E. Martínez-Viviente and P. G. Jones, Organometallics, 2009, 28, 6101–6108 CrossRef CAS.
- P. Majumdar, K. K. Kamar, A. Castineiras and S. Goswami, Chem. Commun., 2001, 1292–1293 RSC; J. Dinda, S. Jasimuddin, G. Mostafa, C.-H. Hung and C. Shinha, Polyhedron, 2004, 23, 793–800 CrossRef CAS; C. L. Chen, H. Y. Tan, J. H. Yao, Y. Q. Wan and C. Y. Su, Inorg. Chem., 2005, 44, 8510–8520 CrossRef PubMed; F. Bélanger-Gariépy and A. L. Beauchamp, J. Am. Chem. Soc., 1980, 102, 3461–3464 CrossRef.
- F. H. Allen, O. Kennard, D. G. Watson, A. G. Orpen, L. Brammer and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- Y. Takahashi, S. Ito, S. Sakai and Y. Ishii, J. Chem. Soc., Chem. Commun., 1970, 1065–1066 RSC; R. F. Heck, Palladium Reagents in Organic Synthesis, Academic Press, New York, 1985 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data table of complexes 1–6, extended comments on the NMR data, X-ray crystallographic data, structure refinement details, CIF files for compounds 2a, 3a, 5, III, and IV. CCDC 1424207–1424211. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03597j |
| This journal is © The Royal Society of Chemistry 2016 |