
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalysed homo- and copolymerisation of propylene: steric influence of bis(imino)pyridine ligands†
Takafumi
Kawakami
,
Shingo
Ito
and
Kyoko
Nozaki
*
Department of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: nozaki@chembio.t.u-tokyo.ac.jp
First published on 16th November 2015
Abstract
A series of iron complexes bearing a bis(imino)pyridine ligand were synthesised and examined as precatalysts for homopolymerisation of propylene. The alkyl substituents attached to the aryl group on the imine nitrogen atoms significantly affected the catalytic activity and molecular weight of the obtained polypropylenes. Copolymerisation of propylene and various allyl comonomers catalysed by iron/bis(imino)pyridine was also investigated.
Introduction
Polypropylene is one of the most widely used polyolefins owing to its excellent bulk properties, easy processability, and low manufacturing costs; thus, tens of millions of tons per year are produced worldwide.1 Despite its inherent advantages, the nonpolar nature of polypropylene often leads to poor surface and material properties due to the absence of any polar functional group in its structure. To improve these properties, post-polymerisation functionalisation by radical or plasma methods2 is generally used to produce functional polypropylenes bearing polar functional groups. However, the inert nature of polypropylene requires harsh conditions for functionalisation, makes it difficult to control the polymer composition and structures, and limits the variety of functional groups introduced.An ideal process for synthesising functionalised polypropylenes would be the copolymerisation of propylene and polar olefins bearing functional groups. Although industrial production of polypropylene is usually accomplished using early-transition-metal catalysts, these catalysts are easily deactivated in the presence of polar functional groups. During the last two decades, therefore, group-10-metal catalysts bearing an α-diimine3 or a phosphine–sulfonate ligand4 have attracted significant attention because their high functional group tolerance enables successful copolymerisation of ethylene and polar monomers.5 When these group-10-metal catalysts are applied to propylene polymerisation, however, it is usually difficult to control the regiochemistry in the propylene insertion step. Propylene polymerisation by late-transition-metal catalysts generally affords amorphous polymers, mainly because of “chain-straightening” (red in Scheme 1) caused by formal 3,1-insertion through the sequence of 2,1-insertion of propylene, β-hydride elimination, and subsequent re-insertion in the opposite direction.3,6 Another potential problem is propylene insertion with the opposite regioselectivity, producing a head-to-head or tail-to-tail irregular sequence (blue in Scheme 1). In that sense, the significant advances in group-10-metal catalysts are the development of the C2-symmetric α-diimine nickel complex, reported by Cherian et al., which produces highly isotactic polypropylene (iPP) at −78 °C,7 and the α-keto-β-diimine nickel complex, reported by Azoulay et al., which produces iPP at −60 °C.8 They required extremely low temperature to suppress formal 3,1-insertion and attain high isotactic selectivity. Recently, our group developed regioregular propylene polymerisation and propylene/polar monomer copolymerisation using palladium/imidazo[1,5-a]quinolin-9-olate-1-ylidene (IzQO) complexes,9 in which formal 3,1-insertion was suppressed even at high temperature. However, stereoregularity (tacticity) was not controlled to give atatic polypropylenes.
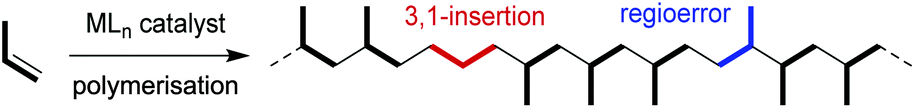 | ||
| Scheme 1 Structural errors of polypropylene in metal-catalysed propylene polymerisation. | ||
In this context, iron(II)/bis(imino)pyridine complexes10 are an important class of catalysts that successfully promote olefin polymerisation without formal 3,1-insertion of propylene. Small and Brookhart11 and Pellecchia, et al.12 independently reported that the iron complexes show unique catalytic activity for propylene polymerisation to produce iPPs with molecular weights of 0.6–8.8 × 103 and isotactic selectivity of up to mmmm = 67.11 After these seminal studies, a range of bis(imino)pyridine ligands and their iron complexes were designed and synthesised to investigate the effect of changing the substituents, especially those attached to the aniline moiety, on polymerisation.13 Most of the studies have focused on the oligo- and polymerisation of ethylene, and much less attention has been paid to those of propylene and higher olefins.14,15 We previously reported that, in ethylene polymerisation by palladium/phosphine–sulfonate catalysts, the introduction of bulky substituents on the phosphorus moiety dramatically increased the molecular weight.16 A plausible explanation for the increase in molecular weight is the steric congestion in the axial positions of the metal center, which could suppress chain transfer reactions.16,17 Motivated by these results, we hypothesized that the introduction of bulky substituents on bis(imino)pyridine ligands18 may affect the molecular weight as well as the tacticity of obtained polypropylene.
Another important requirement for improved utility of late-transition-metal polymerisation catalysts is copolymerisability with other comonomers. However, iron-catalysed copolymerisation of propylene with other olefinic comonomers has never been achieved. Boone et al. provided an important clue by investigating the copolymerisation of ethylene with vinyl chloride or vinyl acetate by iron(II)/bis(imino)pyridine catalysts.19 The attempted copolymerisation with deuterium-labeled vinyl chloride (CD2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CDCl) or vinyl acetate (CD2CDOAc) resulted in the formation of polyethylene bearing a –CDCD2 group as the terminating group in the polyethylene chain. These observations suggested that polymerisation was terminated via β-FG elimination (FG = functional group) after the insertion of comonomers. Hence, it would be necessary to use appropriate comonomers that resist β-FG elimination. We focused on allyl monomers, CH2CHCH2FG, because the presence of a methylene group between the carbon–carbon double bond and the functional group may be effective for suppressing chain termination by functional group elimination.9,16,20
CDCl) or vinyl acetate (CD2CDOAc) resulted in the formation of polyethylene bearing a –CDCD2 group as the terminating group in the polyethylene chain. These observations suggested that polymerisation was terminated via β-FG elimination (FG = functional group) after the insertion of comonomers. Hence, it would be necessary to use appropriate comonomers that resist β-FG elimination. We focused on allyl monomers, CH2CHCH2FG, because the presence of a methylene group between the carbon–carbon double bond and the functional group may be effective for suppressing chain termination by functional group elimination.9,16,20
Herein, we report the synthesis of a series of iron complexes bearing a bis(imino)pyridine ligand with various aryl groups and their catalytic performance in the polymerisation of propylene. We discuss mainly the effect of elongated alkyl substituents on the catalytic activity, molecular weight, and tacticity. We also report our investigation of iron-catalysed copolymerisation of propylene with various allyl monomers.
Results and discussion
A series of bis(imino)pyridine ligands bearing different aryl substituents on the imine nitrogen atoms (Fig. 1, 1a–i) were synthesised by a condensation reaction of 2,6-diacetylpyridine and the corresponding aniline21 using a typical procedure. Subsequent complexation of these ligands with iron(II) chloride in tetrahydrofuran afforded the corresponding iron complexes 2, and their structures were confirmed by elemental analysis, electrospray-ionization (ESI) mass analysis, and X-ray crystallographic analysis:22 Ligands 1a–d and 1f–i were readily transformed to the iron complexes 2a–d and 2f–i, respectively, in good yields (80–96%), whereas the complexation of 1e did not afford complex 2e even at a higher temperature of 60 °C. The reason could be that the 2,6-dimethylheptan-4-yl (dmhep) groups around the coordination site of the ligand are sterically too bulky. The introduced longer alkyl chains increased the solubility of complexes 2a–d and made them less crystalline. The structure of 2c was confirmed by X-ray crystallographic analysis of blue single crystals obtained by recrystallisation from dichloromethane/pentane (Fig. 2A). The iron center of 2c has a distorted square pyramidal geometry, like reported complexes such as 2b (Table 1).10 The recrystallisation of 2d under the same conditions afforded three types of single crystals, which were blue, yellow, or red. The blue single crystals were found to be dichloride 2d, and the red ones were found to be the Fe(II)ClOH complex linked to an FeCl3 moiety via a bridging OH group (2d′), presumably generated by disproportionation of 2d.11 The formation of 2d′ could be explained by reaction with the solvent and/or moisture during recrystallisation. Considering the mass balance of the iron components, the yellow crystals were assumed to be ligand 1d. The crystal structure of 2d′ (Fig. 2B) is closely related to those of 2b and 2c except for the difference in the lengths of the Fe–Cl and Fe–O bonds. Regarding the bis(imino)pyridine–Fe moiety of 2d′, the bond length of Fe–N1(pyridine) is 2.054(6) Å, and those of Fe–N2 and Fe–N3(imine) are 2.225–2.236 Å, which are within the common range of related compounds.10,11,23 The phenyl planes on the imine nitrogen atoms are nearly orthogonal to the plane of the pyridine ring, with dihedral angles of 71° and 86°. These values are slightly smaller than those of the reported Fe(II)ClOH complex bearing isopropyl and methyl groups on the phenyl ring (83° and 89°),11 presumably owing to steric repulsion between the heptan-4-yl groups and the Fe(OH)FeCl3 moiety. Although the long alkyl substituents are placed as it surrounds the iron center, no intramolecular H–Cl bonding interaction was observed. The structure around the iron center of 2d′ is similar to that of the reported Fe(II)ClOH complex:11 For example, the N1–Fe1–Cl1, N1–Fe1–O1, and N2–Fe1–N3 angles of 2d′ are 145.41(17), 97.3(2), and 144.6(2)°, respectively, whereas those of the reported Fe(II)ClOH complex are 147.4(3), 99.0(3), and 144.0(3)°, respectively. In the case of 2g (Fig. 2C), which has unsymmetrically substituted aryl groups on both imine nitrogen atoms, complexation with iron(II) chloride could form two possible isomeric complexes that have C2 or CS symmetry, but the CS symmetric configuration was confirmed by X-ray analysis for complex 2g.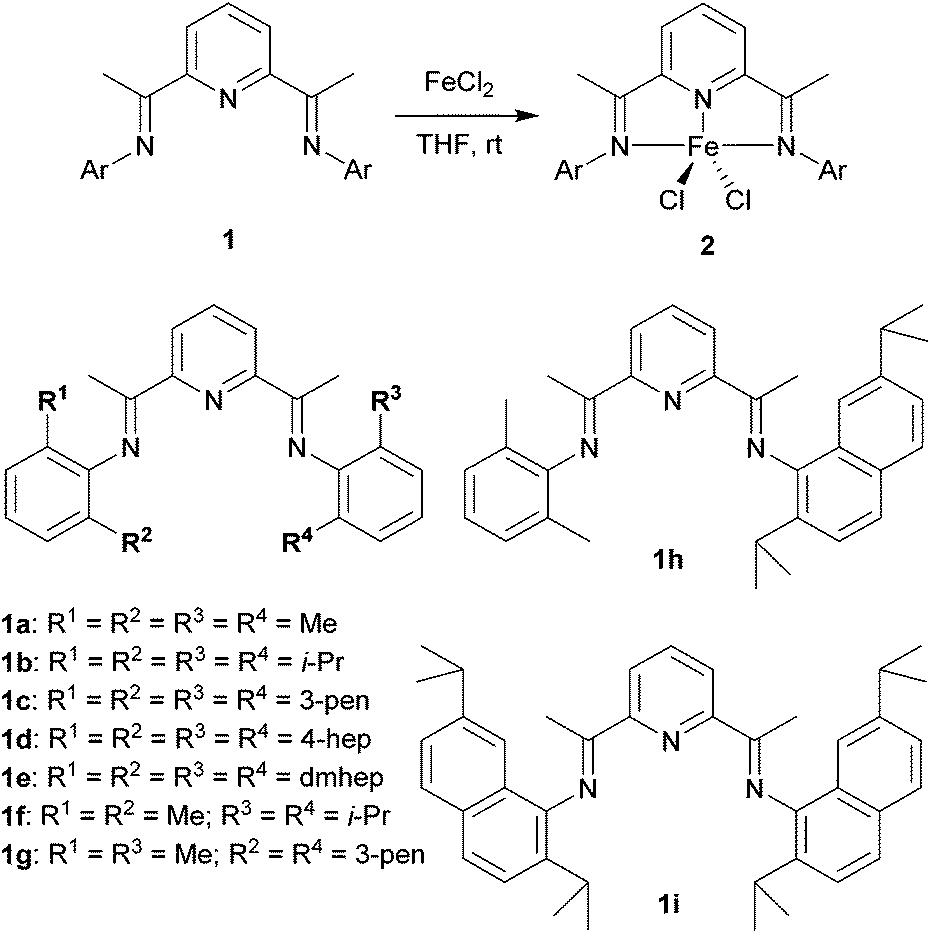 | ||
| Fig. 1 Synthesis of iron complexes 2 bearing a bis(imino)pyridine ligand 1. Abbreviations: Me = methyl; i-Pr = isopropyl; 3-pen = pentan-3-yl; 4-hep = heptan-4-yl; dmhep = 2,6-dimethylheptan-4-yl. | ||
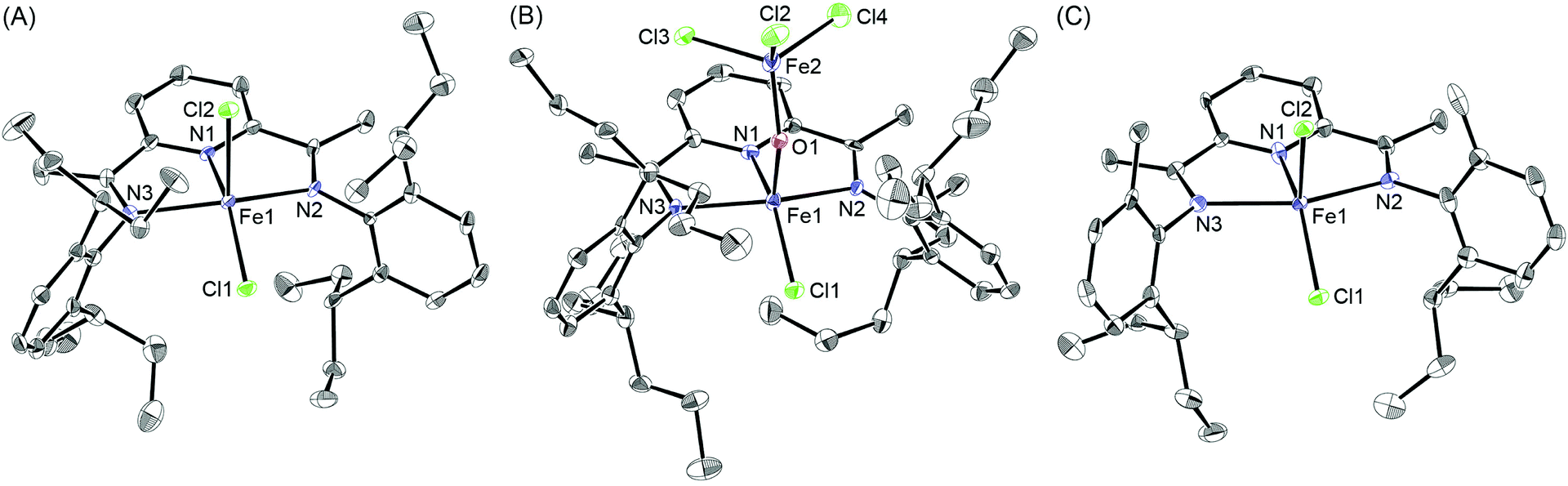 | ||
| Fig. 2 X-ray structures of iron/2,6-bis(imino)pyridine complexes (A) 2c, (B) 2d′, and (C) 2g with 50% probability of thermal ellipsoids. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
| 2b 10b | 2c | 2d′ | (NNN)FeCl{OH(FeCl3)}11 | 2g | |
|---|---|---|---|---|---|
| Fe1–Cl1 | 2.2655(15) | 2.2630(18) | 2.213(5) | 2.179(3) | 2.305(3) |
| Fe1–Cl2 | 2.311(2) | 2.3428(19) | — | — | 2.362(3) |
| Fe1–O1 | — | — | 1.803(5) | 1.769(8) | — |
| Fe1–N1 | 2.088(4) | 2.080(5) | 2.054(6) | 2.076(7) | 2.126(11) |
| Fe1–N2 | 2.250(4) | 2.217(5) | 2.236(6) | 2.197(7) | 2.292(10) |
| Fe1–N3 | 2.238(4) | 2.220(5) | 2.225(6) | 2.163(7) | 2.275(10) |
| N1–Fe1–N2 | 72.88(14) | 74.05(19) | 74.1(2) | 74.4(3) | 73.5(4) |
| N1–Fe1–N3 | 73.22(14) | 73.15(19) | 74.3(2) | — | 72.4(4) |
| N2–Fe1–N3 | 140.08(14) | 141.67(19) | 144.6(2) | 144.0(3) | 145.2(4) |
| Cl1–Fe1–Cl2 | 117.48(7) | 120.67(7) | — | — | 111.14(12) |
| Cl1–Fe1–O1 | — | — | 117.24(17) | 113.58(24) | — |
| N1–Fe1–Cl1 | 147.89(12) | 151.91(15) | 145.41(17) | 147.4(3) | 145.4(3) |
| N1–Fe1–Cl2 | 94.62(12) | 87.42(15) | — | — | 103.4(3) |
| N1–Fe1–O1 | — | — | 97.3(2) | 99.0(3) | — |
| N2–Fe1–Cl1 | 98.94(10) | 100.60(14) | 96.82(16) | 98.89(23) | 101.9(3) |
Homopolymerisation of propylene was investigated using iron complexes 2 activated by modified methylaluminoxane (MMAO) (Table 2). Because the lifetime of the iron catalyst is significantly affected by reaction temperature,11 we performed propylene polymerisation at −20 °C. First, complexes 2a and 2b, originally reported by Brookhart and his coworkers,11 were examined for propylene polymerisation and found to give catalytic activities of 72 and 200 g mmol−1 h−1, respectively (entries 1 and 2). Of these two complexes, 2b, bearing bulkier isopropyl substituents, afforded polypropylene with a higher molecular weight (Mn = 4.7 × 103) than that of 2a, bearing methyl groups (Mn = 1.6 × 103).11 Thus, we hypothesised that the longer alkyl chain could block the axial position of the iron center and inhibit β-hydride elimination to afford high-molecular-weight polypropylenes.16,17 Next, we examined catalysts 2c and 2d, which have longer alkyl substituents (entries 3 and 4), and found that these catalysts were less active by 2 orders of magnitude than less bulky catalysts such as 2b. This suggests that the substituents also severely inhibited the coordination and insertion of propylene. Notably, among the catalysts examined in this study, catalyst 2c yielded the highest isotacticity of 60% (entry 3).24 Next, we focused on ligands bearing unsymmetrically substituted aryl groups. Polymerisation in the presence of complex 2f, bearing a 2,6-diisopropylphenyl group on the nitrogen atom of one imine and a 2,6-dimethylphenyl group on the nitrogen atom of the other imine (entry 5), afforded polypropylene with higher catalytic activity than those obtained using symmetrically substituted groups.11 In the polymerisation by complex 2g, bearing two 2-methyl-6-(pentan-3-yl)phenyl groups (entry 6), the catalytic activity was higher than those of four 3-pentyl-substituted complex 2c (entry 3), but lower than those of four methyl-substituted complex 2a (entry 1) and unsymmetrically substituted complex 2f (entry 5), It should be noted that 2g produced the highest molecular weights of the polypropylenes. We also tried polymerisation by complexes 2h and 2i (entries 7 and 8), bearing 2,7-diisopropylnaphthyl groups on the imine nitrogen atoms. Polymerisation by 2h gave the highest catalytic activity (entry 7), whereas that with 2i gave lower activity (entry 8). The molecular weights of the polymers remained modest, presumably because the 2,7-diisopropylnaphthyl group is assumed to be smaller than the 2,6-diisopropylphenyl group in a proximal position.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Yieldb (g) | Activity (g mmol−1 h−1) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c/103 c/103 |
M
w/Mnc |
[mmmm]d |
| a A mixture of catalyst 2 (10 μmol), MMAO (8 mmol) and propylene in toluene/hexane (15 mL) was stirred in a 50 mL autoclave for 2 h at −20 °C. b Isolated yields after washing with MeOH/1 M HCl aq. mixture and acetone. c Molecular weights determined by size exclusion chromatography (SEC) using polystyrene standards. Molecular weights in parenthesis are determined by 1H NMR analysis. d Ratio of meso pentad determined by 13C NMR analysis.25 e 40 μmol of catalyst was used. f Reaction was carried out for 1 h with 5 μmol of catalyst because of stirring difficulty. | ||||||
| 1 | 2a | 1.4 | 72 | 1.6 | 2.1 | 39 |
| 2f | 2b | 1.0 | 200 | 4.7(9.0) | 2.5 | 54 |
| 3e | 2c | 0.065 | 0.82 | 3.4 | 2.4 | 60 |
| 4e | 2d | 0.041 | 0.76 | 4.5 | 2.1 | 53 |
| 5f | 2f | 1.5 | 290 | 4.1 | 2.3 | 57 |
| 6 | 2g | 0.68 | 34 | 6.6(9.5) | 2.9 | 54 |
| 7f | 2h | 1.8 | 350 | 3.2 | 2.6 | 52 |
| 8 | 2i | 1.3 | 67 | 3.2 | 4.1 | 59 |
The microstructures of the obtained polypropylenes were investigated by nuclear magnetic resonance (NMR) analyses. In 13C NMR spectra of the polypropylenes obtained using 2b (entry 2; Fig. S4 and 5†) and 2g (entry 6; Fig. S13†), the signals corresponding to methine (CH), methylene (CH2), and methyl (CH3) carbons were observed as 1:1:1, respectively. The signals generated by formal 3,1-insertion of propylene, which are supposed to appear at 27.5–27.6 ppm26 or 25.2–25.4 ppm,27 were not observed. On the basis of these results, we concluded that the iron complexes produce polypropylene without observable 3,1-insertion of propylene. Regarding the head-to-tail regioselectivity, the signals of regioerror structures caused by head-to-head and tail-to-tail insertion of propylene (15–18 ppm, 34–38 ppm, 40–44 ppm)11,25,26,28,29 were observed in the case of less sterically hindered catalysts 2a and 2h (Fig. S2 and S15†), which afforded low-molecular-weight polymers, whereas the regioerror was not observed in the case of bulky catalysts 2b–2d, 2f, 2g, and 2i. 13C NMR analyses revealed that all the catalysts examined in this study produced isotactic polypropylene with up to 60% meso pentad content. The low content of the rr diad sequence and almost 1-to-1 ratio of the mmmr and mmrm pentad sequences suggest that the chain-end control mechanism is predominant in this propylene polymerisation, as previously reported.11 These results indicate that the elongation and/or desymmetrisation of substituents do not significantly influence the polypropylene microstructure.
The chain-end structures of the obtained polymers were also elucidated. The major olefinic signals observed in the 13C NMR (around 138 and 116 ppm) and 1H NMR (around 5.9 and 5.1 ppm)30 spectra corresponded to the presence of an allyl group (CH2CHCH2–), which could be generated by 2,1-insertion of propylene followed by β-hydride elimination.29a The molecular weights of polypropylene determined from the 1H NMR spectra assuming the allyl group is the only termination chain end were found to be 9.0 × 103 (entry 2) and 9.5 × 103 (entry 6), which are significantly higher than those determined using gel permeation chromatography analysis, 4.7 × 103 and 6.6 × 103, respectively. This discrepancy could be explained by the presence of saturated alkyl groups as the termination chain end, which could be produced by chain transfer to the aluminum center of the cocatalyst and subsequent hydrolysis during polymerisation. As to the initiation process, there are six types of possible initiating groups generated by 1,2- or 2,1-insertion of propylene into Fe–H, Fe–Me, or Fe–i-Bu bonds. In the 13C NMR spectra (Fig. S5†), we observed only signals representing n-butyl and 3-methylbutyl groups generated by successive 1,2- and 2,1-insertion of propylene into Fe–H and 2,1-insertion of propylene into Fe–i-Bu bonds, respectively, as the dominant initiation chain-end structures.
Copolymerisation of propylene with a range of allyl monomers was examined using catalyst 2b (Table 3).31 First, we investigated copolymerisation with polar monomers bearing CH2CHCH2OR structures, such as allyl acetate (entries 1 and 2), allyl ether (entry 3), and allyl silyl ethers (entries 4 and 5), because the introduction of hydroxy groups into polyolefins is of significant importance.32 In all the attempts, however, the corresponding functionality was not incorporated. In the case of allyl acetate (entry 1), copolymerisation in the presence of 0.20 mL (1.9 mmol) of allyl acetate produced only polypropylene without any incorporation of allyl acetate. When an increased amount of allyl acetate (0.80 mL; 7.4 mmol) was used (entry 2), the production of polymer was completely suppressed. Note that the homopolymerisation of propylene was also significantly suppressed in the presence of ethyl acetate (4.1 mmol).33 Thus, we concluded that the intermolecular interaction between the ester functional group in the acetates and the iron center and/or cocatalyst, MMAO, retarded polymerisation. The attempted copolymerisation with allyl methyl ether and allyl silyl ethers promoted only the homopolymerisation of propylene, and the comonomers were not incorporated at all. Given that oxygen atoms in allyl ethers are known to coordinate to an aluminum cocatalyst,5a a possible explanation of the lack of incorporation of the comonomers is that bulky aluminum or silyl protecting groups retard the coordination of these comonomers than propylene.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R in comonomer | Comonomer (mL) | Propylene (g) | T (°C) | Yieldb (g) | Activity (g mmol–1 h–1) | M n/103c (g mol−1) |
M
w/Mnc |
Incorp.d (%) |
| a A mixture of catalyst 2b, propylene and a comonomer in toluene (15 mL) in a 50 mL autoclave was stirred for 16 hours at an indicated temperature. b Isolated yields after washing with MeOH/1 M HCl aq. mixture and acetone. c Molecular weights determined by SEC analysis using polystyrene standards. d Molar incorporation ratios of comonomers determined by 1H NMR analysis. e Isolated yield after Soxhlet extraction with toluene. | |||||||||
| 1 | R = OAc | 0.20 | 9.4 | 30 | 2.7 | 8.5 | 3.3 | 6.9 | 0 |
| 2 | R = OAc | 0.80 | 9.5 | 30 | 0.0 | — | — | — | — |
| 3 | R = OMe | 0.40 | 9.2 | 30 | 0.37 | 1.1 | 3.7 | 2.1 | 0 |
| 4 | R = OSitBuMe2 | 0.40 | 6.9 | 30 | 1.9 | 5.9 | 3.4 | 2.0 | 0 |
| 5 | R = OSiiPr3 | 0.40 | 9.2 | 30 | 2.7 | 8.4 | 2.4 | 2.0 | 0 |
| 6 | R = Ph | 0.40 | 5.7 | 30 | 2.3 | 7.2 | 0.97 | 2.1 | 1.4 |
| 7 | R = SiMe3 | 0.40 | 7.0 | 30 | 1.4 | 4.5 | 1.3 | 7.8 | 2.1 |
| 8 | R = SiMe2Ph | 0.40 | 7.3 | 30 | 0.67 | 2.1 | 2.0 | 1.9 | 1.7 |
| 9 | R = Si(OEt)3 | 0.40 | 11 | 0 | 0.44e | 1.3 | 3.2 | 2.4 | 0.30 |
In contrast, allylbenzene (entry 6)34–36 and allylsilanes (entries 7–9)37–39 could be incorporated into the polypropylene chain. In the case of allylbenzene, copolymerisation of propylene with 0.40 mL (19 mmol) of allylbenzene afforded the corresponding copolymer with 1.4% comonomer incorporation. 1H and 13C NMR analyses revealed that all the phenyl groups introduced were incorporated into the chain ends of the obtained polypropylene (Fig. 3). This result could be explained by the following mechanism, which is shown in Scheme 2: propylene polymerisation proceeds via the 2,1-insertion mechanism, as proposed in the literature.11 Termination occurs by β-hydride elimination after 2,1-insertion of propylene to form the allyl (Hc2CCHdCH2–) group, which was observed as one of the major chain ends in the 1H NMR spectrum (Fig. 3). Termination also occurs after 2,1-insertion of allylbenzene: the elimination of a benzylic β-hydride forms a terminal styryl (PhCHaCHb–) group, and the elimination of an internal β-hydride forms the PhCHe2CHfCHg– group. These observations suggested that not only propylene but also allylbenzene underwent 2,1-insertion into the polymer chain.
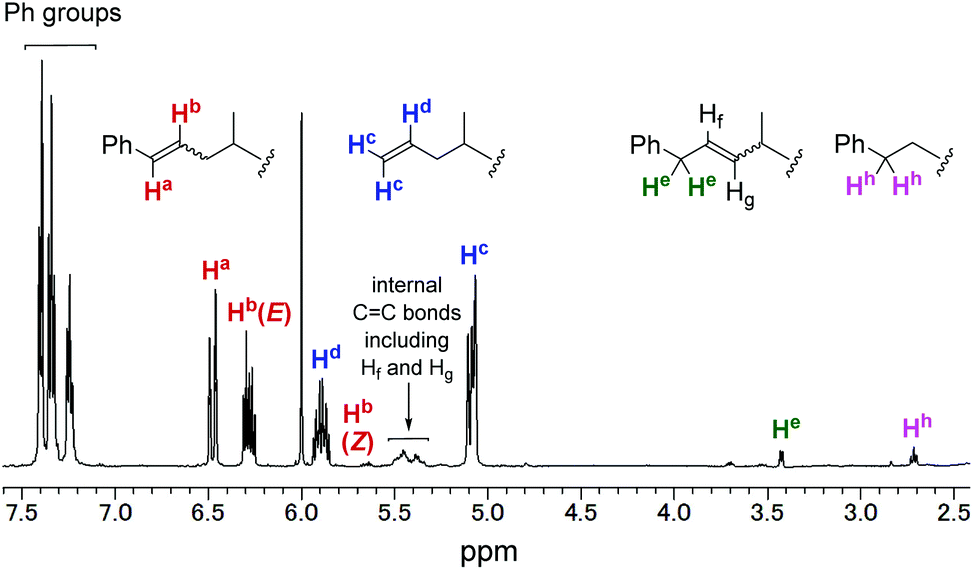 | ||
| Fig. 3 1H NMR spectrum of propylene/allylbenzene copolymer (entry 6 in Table 2). | ||
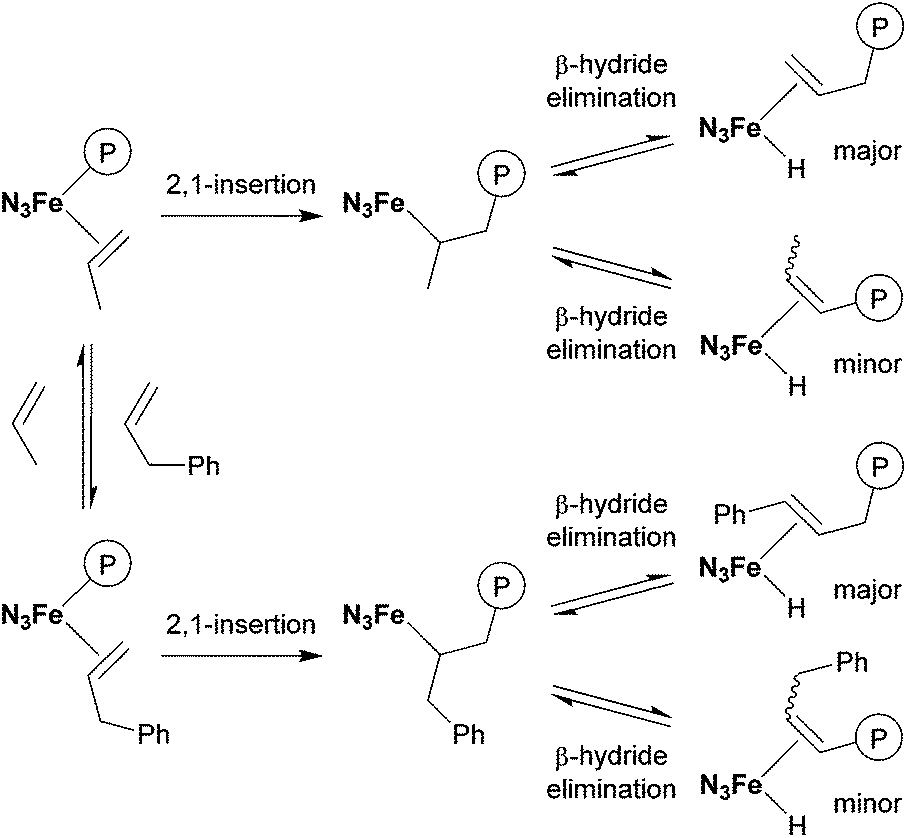 | ||
| Scheme 2 Termination mechanism of the copolymerisation of propylene with allylbenzene. | ||
Copolymerisation of propylene with allylsilanes was also investigated. The copolymerisation reactions in the presence of 0.40 mL of allyl(trimethyl)silane (entry 7), allyl(dimethyl)(phenyl)silane (entry 8), and allyl(triethoxy)silane (entry 9) afforded the corresponding copolymers with comonomer incorporations of 2.1%, 1.7%, and 0.30%, respectively. In the 1H and 13C NMR spectra (Fig. 4) of the propylene/allyl(trimethyl)silane copolymer (entry 7), olefinic signals that correspond to terminal and internal carbon–carbon double bonds were observed. The terminal CC bond is derived from the presence of an allyl group (c and d in Fig. 4). We assigned the internal CC bond to a Me3SiCH2CHCH– group generated by β-hydride elimination after insertion of allyl(trimethyl)silane. This assignment is based on the fact that the integral ratio of the internal olefinic carbons and the predominant signal of the trimethylsilyl group was found to be ca. 1:1 on the basis of quantitative 13C NMR analysis (Fig. S25†) and that the methylene proton next to the CC bond was observed at an unusually high field in the 1H NMR spectrum (Fig. S23†). Regarding the methyl group on the trimethylsilyl group, three other minor signals were observed around −1.5 ppm (SiMe3 shown as red). Although we could not assign all the signals, partly because they overlapped, we assumed that the three signals corresponding to the trimethylsilyl group are derived from in-chain incorporation of allyl(trimethyl)silane.
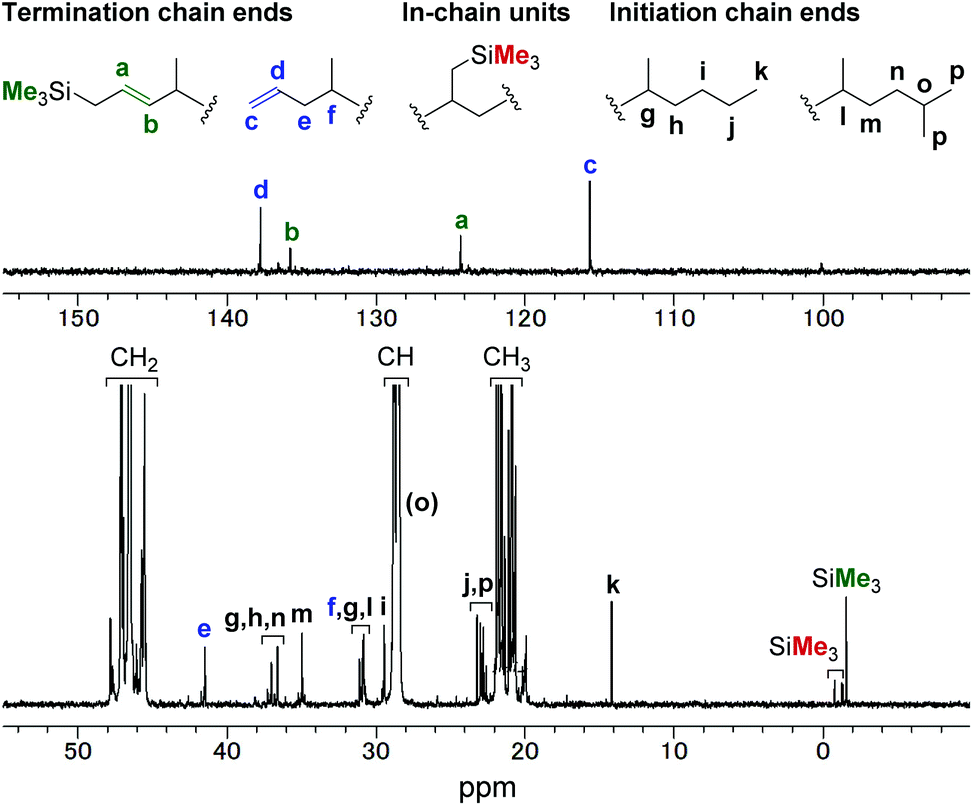 | ||
| Fig. 4 13C NMR spectrum of propylene/allyl(trimethyl)silane copolymer (entry 7 in Table 2). | ||
The structure of the propylene/allyl(dimethyl)(phenyl)silane copolymer was similar to that of the propylene/allyl(trimethyl) silane copolymer. In the 1H NMR (Fig. S27†) and 13C NMR (Fig. S28 and S29†) spectra, the signals of the phenyl and methyl groups of (dimethyl)(phenyl)silyl groups were clearly observed, suggesting that the allylsilane was successfully incorporated into the polymer. Olefinic signals assignable to an allyl group, i.e., a Me2PhSiCH2CHCH– group, were observed (Fig. S29†). As methyl carbons of the (dimethyl)(phenyl)silyl group, six signals were observed around −2.8 ppm in the 13C NMR spectrum, indicating the presence of different types of incorporated structure. The five signals other than that of the Me2PhSiCH2CHCH– group possibly correspond to in-chain incorporation and saturated initiating chain-end structure.
Finally, in the case of allyl(triethoxy)silane, only a terminal allyl group was observed as a major chain end group in the 13C NMR spectrum (Fig. S32†). Although the signals of the ethoxy moiety were observed in NMR analysis, the low content of the silyl groups made it difficult to fully determine the microstructure of the copolymer.
In these copolymerisations, both the catalytic activity and Mn value of the obtained copolymers were lower than those obtained by propylene homopolymerisation. This may be due in part to the role of these comonomers as chain transfer reagents that induce β-hydride elimination caused by retarding the next monomer insertion after incorporation of bulky functional groups into the polymer chain.
Conclusions
In summary, we synthesised a series of bis(imino)pyridine ligands and their iron complexes, and investigated their catalytic activity for propylene polymerisation and propylene/allyl monomer copolymerisation. In these polymerisations, the molecular weights are to some extent correlated with the steric factors of the imine substituents on the bis(imino)pyridine ligands. Thus, the highest molecular weight of polypropylene of Mn = 6.6 kDa mol−1 was achieved by using 2g bearing pentan-3-yl substituted aryl groups. Once the substituent becomes too bulky, however, the catalytic activity was severely decreased. Copolymerisation of various ally monomers with propylene were examined to find that the copolymerisation with allylbenzene and allylsilanes afforded the corresponding copolymers. Although the allyl monomers were incorporated mainly into the terminal chain-end position of the copolymer, this report represents the first example of iron/bis(imino)pyridine-catalysed copolymerisation of propylene with allyl monomers.Conflict of interest
The authors declare no competing financial interests.Acknowledgements
This work was partially supported by JST and CREST. S. I. thanks financial support from the TonenGeneral Sekiyu Foundation.Notes and references
- (a) M. Gahleitner and C. Paulik, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2014, DOI:10.1002/14356007.o21_o04.pub2; (b) Polypropylene Handbook, ed. N. Pasquini, Carl Hanser Verlag, Munich, 2nd edn, 2005 Search PubMed.
- (a) M. Rätzsch, M. Arnold, E. Borsig, H. Bucka and N. Reichelt, Prog. Polym. Sci., 2002, 27, 1195–1282 CrossRef; (b) R. P. Singh, Prog. Polym. Sci., 1992, 17, 251–281 CrossRef CAS; (c) J. Jagur-Grodzinski, Prog. Polym. Sci., 1992, 17, 361–415 CrossRef CAS.
- (a) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS; (b) L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267–268 CrossRef CAS; (c) S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888–899 CrossRef CAS.
- E. Drent, R. van Dijk, R. van Ginkel, B. van Oort and R. I. Pugh, Chem. Commun., 2002, 744–745 RSC.
- (a) L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479–1494 CrossRef CAS PubMed; (b) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS PubMed; (c) A. Berkefeld and S. Mecking, Angew. Chem., Int. Ed., 2008, 47, 2538–2542 CrossRef CAS PubMed; (d) A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215–5244 CrossRef CAS PubMed; (e) S. Ito and K. Nozaki, Chem. Rec., 2010, 10, 315–325 CrossRef CAS PubMed; (f) A. Nakamura, T. M. J. Anselment, J. Claverie, B. Goodall, R. F. Jordan, S. Mecking, B. Rieger, A. Sen, P. W. N. M. van Leeuwen and K. Nozaki, Acc. Chem. Res., 2013, 46, 1438–1449 CrossRef CAS PubMed; (g) B. P. Carrow and K. Nozaki, Macromolecules, 2014, 47, 2541–2555 CrossRef CAS.
- Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059–2062 CrossRef CAS PubMed.
- A. E. Cherian, J. M. Rose, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 13770–13771 CrossRef CAS PubMed.
- J. D. Azoulay, H. Gao, Z. A. Koretz, G. Kehr, G. Erker, F. Shimizu, G. B. Galland and G. C. Bazan, Macromolecules, 2012, 45, 4487–4493 CrossRef CAS.
- R. Nakano and K. Nozaki, J. Am. Chem. Soc., 2015, 137, 10934–10937 CrossRef CAS PubMed.
- (a) B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef CAS; (b) G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef CAS.
- B. L. Small and M. Brookhart, Macromolecules, 1999, 32, 2120–2130 CrossRef CAS.
- C. Pellecchia, M. Mazzeo and D. Pappalardo, Macromol. Rapid Commun., 1998, 19, 651–655 CrossRef CAS.
- (a) B. L. Small, Acc. Chem. Res., 2015, 48, 2599–2611 CrossRef CAS PubMed; (b) W. Zhang, W.-H. Sun and C. Redshaw, Dalton Trans., 2013, 42, 8988–8997 RSC; (c) V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745–1776 CrossRef CAS PubMed; (d) C. Bianchini, G. Giambastiani, I. G. Rios, G. Mantovani, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2006, 250, 1391–1418 CrossRef CAS; (e) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS PubMed; (f) G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef CAS.
- For experimental studies, see: (a) S. T. Babik and G. Fink, J. Mol. Catal. A: Chem., 2002, 188, 245–253 CrossRef CAS; (b) S. T. Babik and G. Fink, J. Organomet. Chem., 2003, 683, 209–219 CrossRef CAS; (c) B. L. Small and R. Schmidt, Chem. – Eur. J., 2004, 10, 1014–1020 CrossRef CAS PubMed; (d) G. Fink and S. T. Babik, Kinet. Catal., 2006, 47, 198–206 CrossRef CAS; (e) A. Rodríguez-Delgado, J. Cámpora, A. M. Naz, P. Palma and M. L. Reyes, Chem. Commun., 2008, 5230–5232 RSC; (f) A. Behr, N. Rentmeister, D. Möller, J. Vosberg, S. Peitz and D. Maschmeyer, Catal. Commun., 2014, 55, 38–42 CrossRef CAS.
- For theoretical studies, see: (a) L. Deng, P. Margl and T. Ziegler, J. Am. Chem. Soc., 1999, 121, 6479–6487 CrossRef CAS; (b) D. V. Khoroshun, D. G. Musaev, T. Vreven and K. Morokuma, Organometallics, 2001, 20, 2007–2026 CrossRef CAS; (c) R. Raucoules, T. de Bruin, P. Raybaud and C. Adamo, Organometallics, 2009, 28, 5358–5367 CrossRef CAS.
- Y. Ota, S. Ito, J. Kuroda, Y. Okumura and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 11898–11901 CrossRef CAS PubMed.
- For a review, see: D. H. Camacho and Z. Guan, Chem. Commun., 2010, 46, 7879–7893 RSC.
- (a) J. E. Steves, M. D. Kennedy, K. P. Chiang, W. S. Kassel, W. G. Dougherty, T. J. Dudley and D. L. Zubris, Dalton Trans., 2009, 1214–1222 RSC; (b) F. Pelascini, F. Peruch, P. J. Lutz, M. Wesolek and J. Kress, Eur. Polym. J., 2005, 41, 1288–1295 CrossRef CAS; (c) L.-H. Guo, H.-Y. Gao, L. Zhang, F.-M. Zhu and Q. Wu, Organometallics, 2010, 29, 2118–2125 CrossRef CAS; (d) J. Yu, H. Liu, W. Zhang, X. Hao and W.-H. Sun, Chem. Commun., 2011, 47, 3257–3259 RSC.
- H. W. Boone, P. S. Athey, M. J. Mullins, D. Philipp, R. Muller and W. A. Goddard, J. Am. Chem. Soc., 2002, 124, 8790–8791 CrossRef CAS PubMed.
- (a) S. Ito, M. Kanazawa, K. Munakata, J. Kuroda, Y. Okumura and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 1232–1235 CrossRef CAS PubMed; (b) S. Ito, Y. Ota and K. Nozaki, Dalton Trans., 2012, 41, 13807–13809 RSC.
- S. Meiries, G. Le Duc, A. Chartoire, A. Collado, K. Speck, K. S. A. Arachchige, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2013, 19, 17358–17368 CrossRef CAS PubMed.
- CCDC 1416774 (2c), 1416775 (2d′), 1416776 (2g) contain the supplementary crystallographic data for this paper.
- (a) L. Guo, H. Gao, L. Zhang, F. Zhu and Q. Wu, Organometallics, 2010, 29, 2118–2125 CrossRef CAS; (b) Y. Chen, R. Chen, C. Qian, X. Dong and J. Sun, Organometallics, 2003, 22, 4312–4321 CrossRef CAS.
- Small and Brookhart achieved the highest value of 67% using ligand bearing R1 = R3 = Me and R2 = R4 = iPr. See ref. 11.
- V. Busico, R. Cipullo, G. Monaco, M. Vacatello and A. L. Segre, Macromolecules, 1997, 30, 6251–6263 CrossRef CAS.
- (a) S. Lipponen, M. Lahelin and J. Seppälä, Eur. Polym. J., 2009, 45, 1179–1189 CrossRef CAS; (b) C. Ruiz-Orta, J. P. Fernandez-Blazquez, A. M. Anderson-Wile, G. W. Coates and R. G. Alamo, Macromolecules, 2011, 44, 3436–3451 CrossRef CAS.
- A. Grassi, A. Zambelli, L. Resconi, E. Albizzati and R. Mazzocchi, Macromolecules, 1988, 21, 617–622 CrossRef CAS.
- M. Mitani, R. Furuyama, J. Mohri, J. Saito, S. Ishii, H. Terao, T. Nakano, H. Tanaka and T. Fujita, J. Am. Chem. Soc., 2003, 125, 4293–4305 CrossRef CAS PubMed.
- (a) P. D. Hustad, J. Tian and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 3614–3621 CrossRef CAS PubMed; (b) L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1345 CrossRef CAS PubMed.
- Although the coupling constants could not be accurately determined due to broadening of signals, the coupling pattern is also consistent with the assignments.
- Some polymerizations were performed using 2a as a catalyst. See ESI† for the data.
- (a) K. W. Doak, in Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. M. Bikales, C. G. Overberger and G. Menges, Wiley, New York, 2nd edn, 1986, vol. 6, pp. 386–429 Search PubMed; (b) D. Jeremic, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2014, DOI:10.1002/14356007.a21_487.pub3.
- See ESI† for details.
- For homopolymerisation of allylbenzene, see: (a) G. F. D'alelio, A. B. Finestone, L. Taft and T. J. Miranda, J. Polym. Sci., 1960, 45, 83–89 CrossRef; (b) D.-J. Byun, D.-K. Shin, J. Jiu and S. Y. Kim, Polym. Bull., 1999, 42, 265–272 CrossRef CAS; (c) D.-J. Byun, D.-K. Shin and S. Y. Kim, Polym. Bull., 1999, 42, 301–307 CrossRef CAS.
- For copolymerisation of allylbenzene with ethylene, see: (a) L.-M. Tang, Y.-G. Li, W.-P. Ye and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5846–5854 CrossRef CAS; (b) L.-M. Tang, T. Hu, L. Pan and Y.-S. Li, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6323–6330 CrossRef CAS; (c) K. R. Kumar and S. Sivaram, Polym. Int., 2001, 50, 367–374 CrossRef CAS; (d) D.-J. Byun and S. Y. Kim, Macromolecules, 2000, 33, 1921–1923 CrossRef CAS; (e) D.-J. Byun, D.-K. Shin and S. Y. Kim, Macromol. Rapid Commun., 1999, 20, 419–422 CrossRef CAS.
- For copolymerisation of allylbenzene with propylene, see: I. Hayashi, Kogyo Kagaku Zasshi, 1967, 70, 735–738 CrossRef CAS.
- For homopolymerisation of allylsilane, see: (a) G. Natta, G. Mazzanti, P. Longi and F. Bernardini, J. Polym. Sci., 1958, 31, 181–183 CrossRef; (b) R. Ziegler, L. Resconi, G. Balbontin, G. Guerra, V. Venditto and C. De Rosa, Polymer, 1994, 35, 4648–4655 CrossRef; (c) S. Habaue, H. Baraki and Y. Okamoto, Macromol. Chem. Phys., 1998, 199, 2211–2215 CrossRef CAS; (d) J.-Y. Lee, M.-C. Shiao, F.-Y. Tzeng, C.-H. Chang, C.-K. Tsai and J.-C. Tsai, Macromolecules, 2012, 45, 2720–2730 CrossRef CAS.
- For copolymerisation of allylsilanes with ethylene, see: (a) S. H. Lipponen and J. V. Seppälä, Organometallics, 2011, 30, 528–533 CrossRef CAS; (b) J. Liu and K. Nomura, Macromolecules, 2008, 41, 1070–1072 CrossRef CAS; (c) S. B. Amin and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 2938–2953 CrossRef CAS PubMed; (d) N. Naga, Macromol. Chem. Phys., 2005, 206, 1959–1966 CrossRef CAS.
- For copolymerisation of allylsilanes with propylene, see: (a) N. Naga, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6083–6093 CrossRef CAS; (b) P. Longi, F. Greco and U. Rossi, Macromol. Chem. Phys., 1968, 116, 113–121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; characterization of ligands, complexes, and polymers; crystallographic data for complexes. CCDC 1416774–1416776. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03551a |
| This journal is © The Royal Society of Chemistry 2015 |