
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-heterocyclic carbene copper(I) catalysed N-methylation of amines using CO2†
Orlando
Santoro
a,
Faïma
Lazreg
a,
Yury
Minenkov
b,
Luigi
Cavallo
b and
Catherine S. J.
Cazin
*a
aEaStCHEM School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK. E-mail: cc111@st-andrews.ac.uk
bPhysical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Kaust Catalysis Center, Thuwal 23955-6900, Saudi Arabia
First published on 30th September 2015
Abstract
The N-methylation of amines using CO2 and PhSiH3 as source of CH3 was efficiently performed using a N-heterocyclic carbene copper(I) complex. The methodology was found compatible with aromatic and aliphatic primary and secondary amines. Synthetic and computational studies have been carried out to support the proposed reaction mechanism for this transformation.
Introduction
The simple methylation of amines is a well-known transformation in organic chemistry leading to compounds having applications in the synthesis of dyes, natural products and fine chemicals.1 Several synthetic protocols exist to achieve this transformation and include the nucleophilic substitution of electrophilic methylating reagents (e.g., methyl iodide) and the methylation with formaldehyde in the presence of reducing reagents (e.g., formic acid and metal hydride).2 However, important drawbacks are associated with these procedures such as reagent toxicity or limitation in the substrate scope. Recently, alternative methodologies have been reported based on the use of CO2 as a methylating reagent. CO2 is an inexpensive, non-toxic and the most abundant carbon source. It is potentially the most attractive C1 feedstock to introduce carbon into molecules.3 In 2013, Cantat and co-workers reported an interesting methodology based on zinc using [Zn(Cl)2(IPr)] (IPr = N,N′-bis-[2,6-(di-iso-propyl)phenyl] imidazol-2-ylidene) as catalyst. Phenylsilane was used as the reducing reagent (Scheme 1).4 Almost concomitantly, Beller and co-workers disclosed a ruthenium-based methodology.5 For the latter, excess silane was required under 30 bar of carbon dioxide. Shortly after, the same group reported a variant of this transformation replacing the silane with hydrogen gas. However, high pressure of H2 (60 bar) and CO2 (20 bar) were required as well as long reaction times (24 h to 48 h) and elevated temperature (140 °C). Independently, Leitner and co-workers reported that an alternative ruthenium based-system could enable this transformation.6 Recently, a nickel/phosphine system able to promote this transformation was investigated.7 Organocatalysts have also been shown as efficient promoters of this transformation.8 Finally, Beller and Cantat also showed that formic acid can be used as C1 source or as both C1 and hydrogen source.9,10 Despite their elegance, these protocols suffer from the use of relatively expensive metals and ligands and, in some cases, high pressures and temperatures. In addition, mechanistic details for this reaction have not been explored yet. Recently, it has been shown that economical copper complexes can promote direct carboxylation of N–H and C–H bonds using CO2.11,12 Indeed, [Cu(OH)(IPr)] (1) as catalyst allowed CO2 insertion into a C–H and N–H bonds leading to the formation of carboxylic and carbamic acid derivatives, respectively.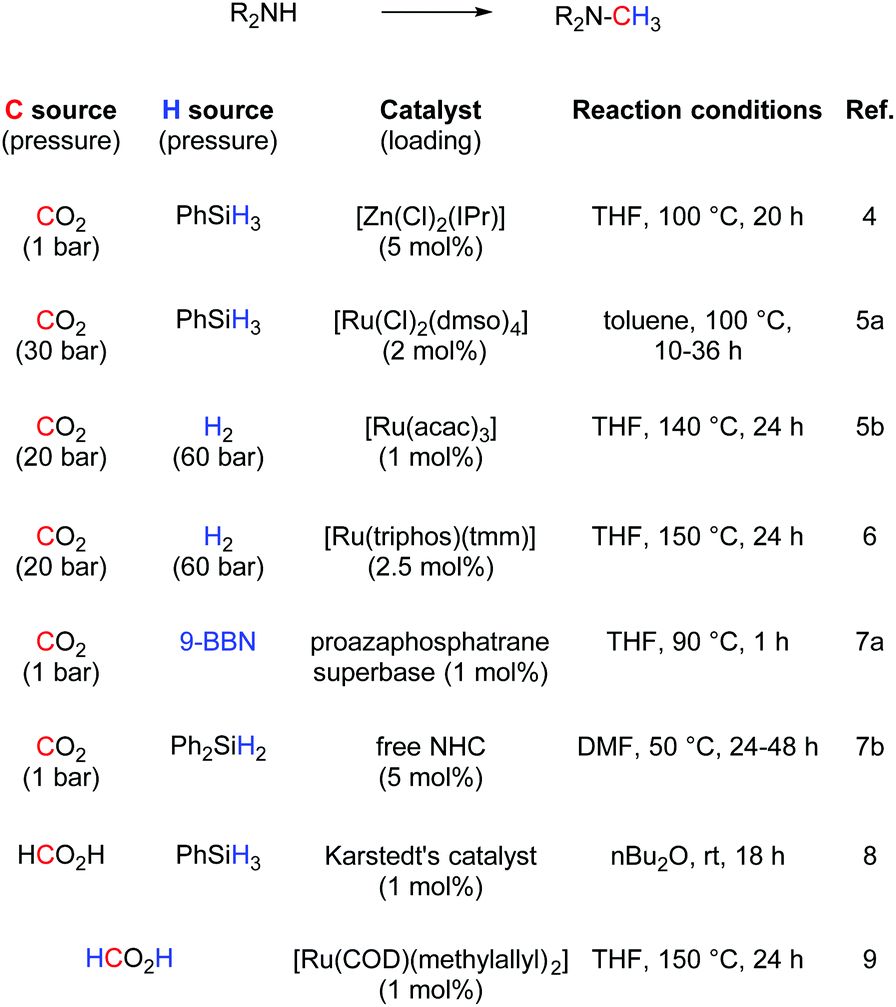 | ||
| Scheme 1 State-of-the-art for the methylation of amines with CO2. | ||
Moreover, copper-based complexes are well-established catalysts for the hydrosilylation of CO2.13,14 Based on these observations, it was hypothesised that the reactivity of NHC–copper complexes could be extended to the methylation of N–H bonds. In particular, this could be performed in two steps: a carboxylation followed by a reduction. Herein, we reported the first Cu-catalysed methylation of amines with mechanistic insight into the fixation of CO2 with amines.
Results and discussion
In the initial optimisation studies, N-methylaniline 5a was chosen as the benchmark substrate (Table 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] | Solvent | Time (h) | 5b | 5c |
| a Reaction conditions: amine (0.25 mmol), [Cu] (10 mol%), PhSiH3 (2 equiv.), solvent (2 mL), CO2 (2 bar), 100 °C. b Conversion determined by GC, based on amine, minimum average of two reactions. | |||||
| 1 | 1 | Toluene | 20 | 35% | 65% |
| 2 | 1 | Benzene | 20 | 40% | 60% |
| 3 | 1 | Isopropanol | 20 | — | — |
| 4 | 1 | THF | 20 | 24% | 76% |
| 5 | 2 | Toluene | 20 | 50% | 50% |
| 6 | 2 | THF | 20 | 48% | 52% |
| 7 | 3 | THF | 20 | 22% | 78% |
| 8 | 4 | THF | 20 | 96% | 4% |
| 9 | 4 | Toluene | 20 | >99% | |
| 10 | 4 | Toluene | 2 | 40% | 60% |
| 11 | 4 | Toluene | 4 | 45% | 55% |
| 12 | 4 | Toluene | 6 | 57% | 43% |
| 13 | 4 | Toluene | 8 | 70% | 30% |
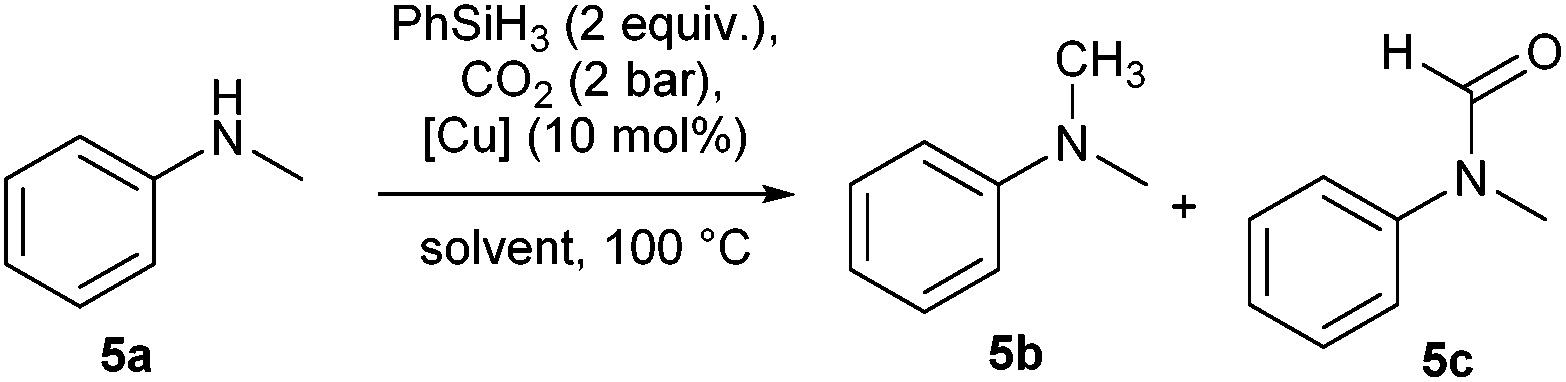
The hydroxide complex 1 (Fig. 1) showed interesting reactivity in solvents such as toluene or benzene. However, no activity was observed in alcohol solvents. By using its tert-butoxide congener, 2, the conversion to the desired compound slightly increased (Table 1, entries 5 and 6). The nature of the NHC ligand was next examined. When the bulkier IPr* was used (3), only 22% conversion to the methylated product was observed. On the other hand, the IMes analogue complex [Cu(OtBu)(IMes)] 4 exhibits improved reactivity. Indeed, in the presence of 4, full conversion into the desired product is observed in toluene after 20 h.
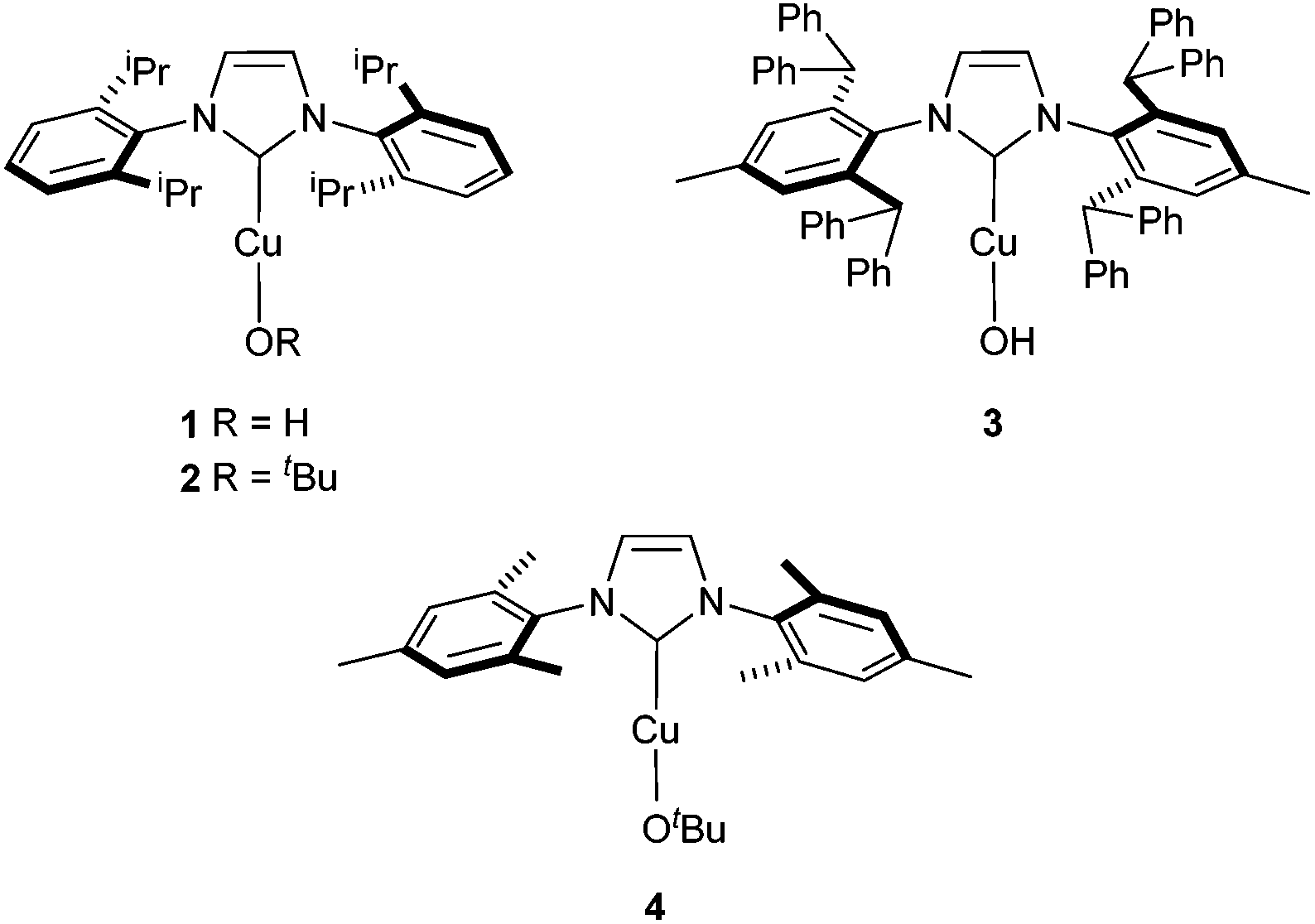 | ||
| Fig. 1 Catalysts studied in the N-methylation. | ||
Based on these promising results, the influence of the reaction time was examined. After 2 hours all starting material was converted into a mixture of amide (60%) and N-methylamine (40%). This highlights that the CO2 insertion occurs in less than 2 hours and that it is not the rate-limiting step in the transformation. As a function of time, the amount of N-methylation increased. After 8 hours, 70% of the desired compound was obtained. Finally, the optimal reaction conditions for the transformation were determined to be [Cu(OtBu)(IMes)] (10 mol%), phenylsilane at 100 °C under 2 bar of CO2 for 20 h.
Using these conditions, the scope of the reaction was investigated, starting with secondary amines (Scheme 2).
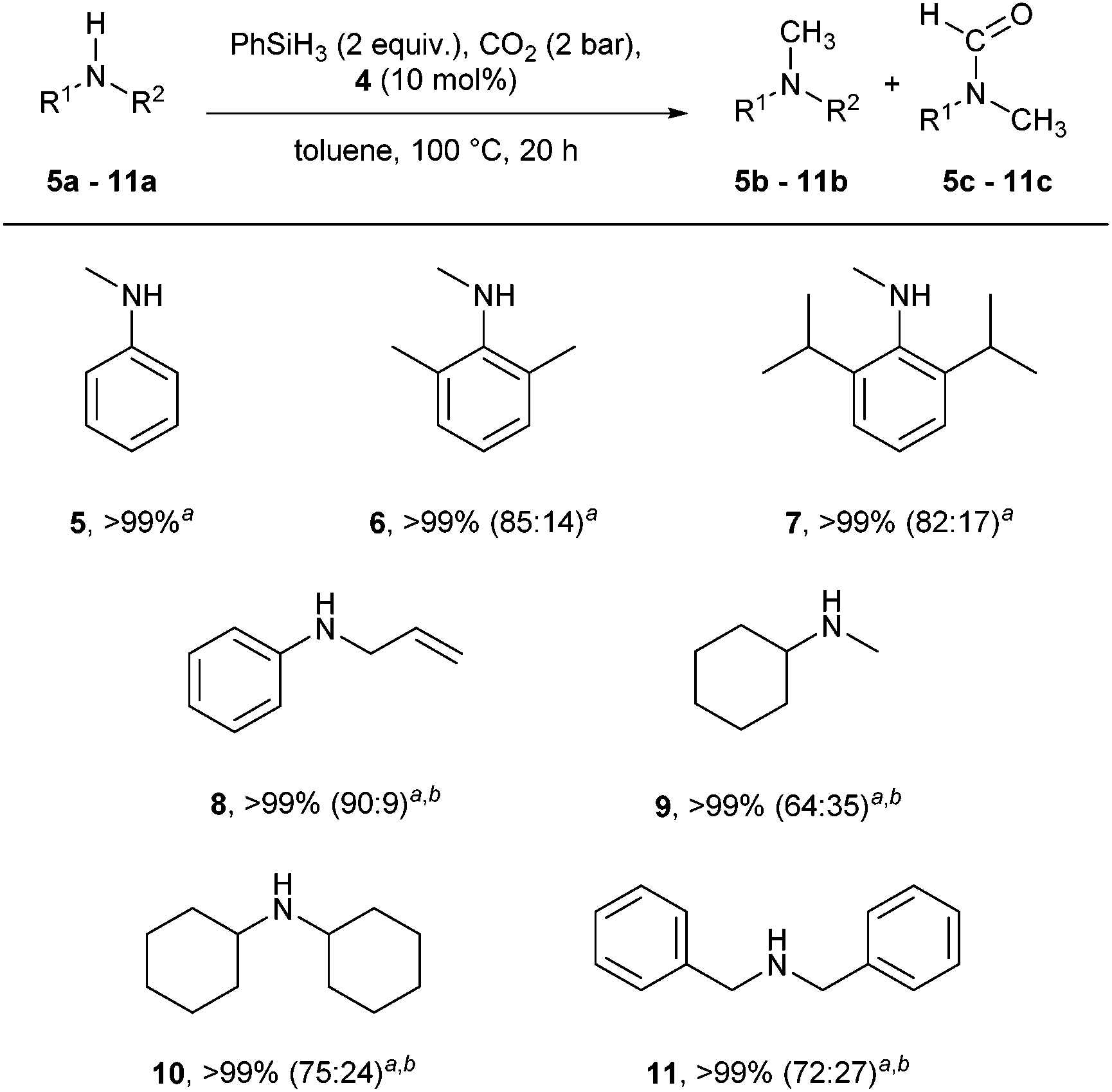 | ||
Scheme 2 Cu-catalysed methylation of secondary amines using CO2 as C1 building block. Reaction conditions: amine (0.25 mmol), 4 (10 mol%), PhSiH3 (2 equiv.), toluene (2 mL), CO2 (2 bar), 100 °C, 20 h. Conversions determined by GC, based on amine, minimum average of two reactions. ab![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :c ratio. bKOtBu (10 mol%), PhSiH3 (4 equiv.). :c ratio. bKOtBu (10 mol%), PhSiH3 (4 equiv.). | ||
N,N-Dimethylaniline 5b was obtained quantitatively and selectively. Sterically congested N-methylanilines such as 6a and 7a were efficiently converted into the desired compounds, the major product being the methylated compound (85% and 82%, respectively). In contrast, while full consumption of starting materials was also observed with compounds 8–11, the major product under these reaction conditions (2 equiv. PhSiH3, no base) was the amide intermediate. In order to overcome this issue, the reaction conditions were further optimised, and the addition of additives was investigated (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Reaction conditions | |||||
| Entry | Substrate | I | II | III | IV |
| Reaction conditions: amine (0.25 mmol), 4 (10 mol%), toluene (2 mL), CO2 (2 bar), 100 °C, 20 h. Conversions determined by GC, based on amine, minimum average of two reactions. I: 2 equiv. PhSiH3, no base. II: 2 equiv. PhSiH3, 10 mol% KOtBu. III: 4 equiv. PhSiH3, no base. IV: 4 equiv. PhSiH3, 10 mol% KOtBu. | |||||
| 1 | 8a | b: 49% | b: 53% | b: 44% | b: 90% |
| c: 50% | c: 46% | c: 55% | c: 9% | ||
| 2 | 9a | b: 14% | b: 40% | b: 33% | b: 64% |
| c: 85% | c: 59% | c: 66% | c: 35% | ||
| 3 | 10a | b: 45% | b: 62% | b: 70% | b: 75% |
| c: 54% | c: 37% | c: 29% | c: 24% | ||
| 4 | 11a | b: 15% | b: 59% | b: 46% | b: 72% |
| c: 84% | c: 40% | c: 53% | c: 27% | ||
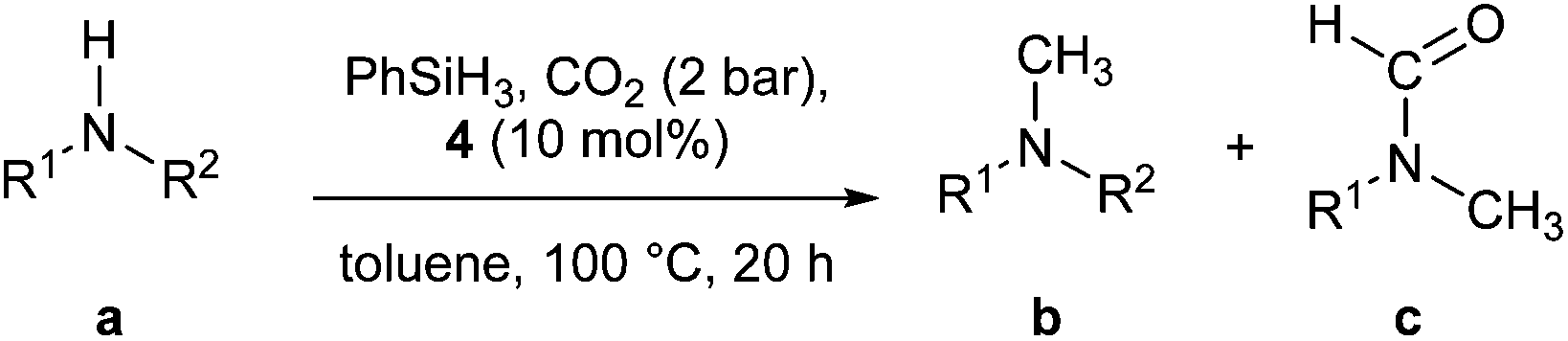
By adding a catalytic amount of KOtBu (10 mol% – II), an important increase in the formation of the methylated compound was observed for substates 9a–11a while only a slight improvement was observed for 8a. The same trend was observed by doubling the amount of silane. Finally, by combining the two variations (4 equiv. of silane and 10 mol% KOtBu), the conversion of all substrates towards the desired compounds was drastically improved. Although KOtBu may be required to promote catalyst regeneration, by doubling its amount no further improvement was observed.
It is interesting to note that, under our standard reaction conditions, i.e. without adding 10 mol% of KOtBu, substrate 9a leads to a 14% conversion of the methylated compound while the more hindered N,N-dicyclohexylamine (10a) is converted in 45%.
Primary amines were next considered (Scheme 3). All substrates investigated were completely converted into a mixture of mono- and di-methylated compounds and their corresponding amide derivatives. Interestingly, the bulkiness of the ortho substituents of substrates 12a and 13a did not affect the outcome of the reaction; both led to the dimethylated product. No amide intermediate was observed with substrate 14a. In the case of methylthio-anilines, the position of the (S–CH3) substituent proved to be crucial. While 2-(methylthio)-aniline (15a) was fully converted into the dimethylated product, only 10% of the corresponding para-substituted product was isolated. Also in this case, the addition of a catalytic amount of KOtBu proved to have a positive effect on the reaction. In fact, the formation of the monomethylated compound 13b was improved while an increase in dimethylated product was observed for substrates 12a and 14a.
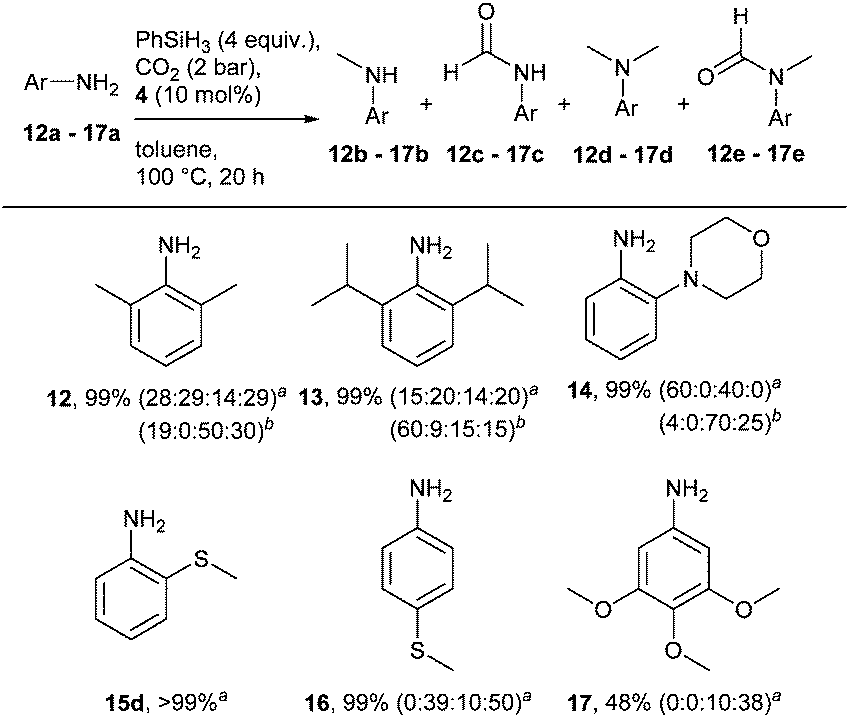 | ||
| Scheme 3 Cu-catalysed methylation of anilines. Reaction conditions: amine (0.25 mmol), 4 (10 mol%), PhSiH3 (2 equiv.), toluene (2 mL), CO2 (2 bar), 100 °C, 20 h. Conversions determined by GC, based on amine, minimum average of two reactions. ab:c:d:e ratio. bKOtBu (10 mol%). | ||
Mechanistic studies
The proposed mechanism for the methylation of amines with CO2 and silane is depicted in Scheme 4. The first steps of this pathway involve the formation of a hydride species followed by the insertion of CO2 into the Cu–H bond affording a Cu-formato species (I). The latter complex undergoes metathesis with the silane to afford the copper hydride species and the siloxane II. The siloxane then reacts with the amine to form the amide derivative (III). The amide would further react with the [Cu(H)(NHC)] to form species IV. This intermediate, in the presence of silane, would regenerate the hydrido complex and release the desired compound.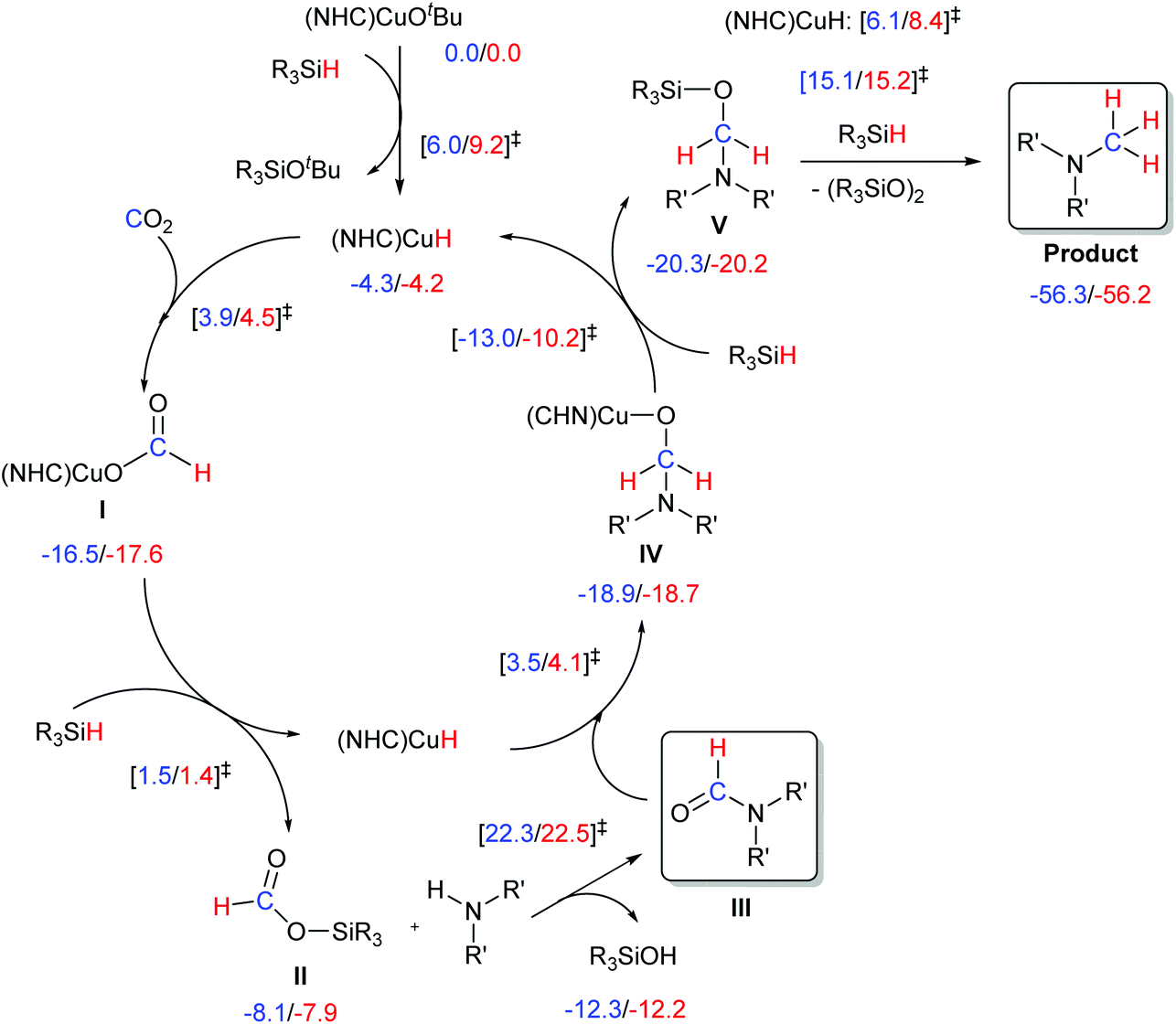 | ||
| Scheme 4 Pathways for methylation via CO2 insertion. DFT calculated ΔG (kcal mol−1) are reported for NHC = IMes and IPr. | ||
In order to obtain a more detailed understanding of the pathway described, we carried out DFT calculations with both IMes- and IPr-based catalysts. The energetics of the transformation are reported in Scheme 4. Calculations indicate that activation of the initial pre-catalyst to the [Cu(H)(NHC)] species, as well as the CO2 activation step by its insertion into the Cu–H bond of [Cu(H)(NHC)], leading to the formato species I, are relatively easy steps, with free energy barriers less than 10 kcal mol−1. The reactivity of I with the silane has a relatively low energy barrier of 18–19 kcal mol−1. This step regenerates [Cu(H)(NHC)], and liberates HC(O)OSiR3 (II), which can react with the amine to generate the amide intermediate III. Although this organic transformation is energetically quite expensive, ca. 30 kcal mol−1, it is consistent with the harsh experimental conditions and previous computational work.15,16 Once formed, the amide can easily react with [Cu(H)(NHC)] to generate [Cu(OCH2NR′2)(NHC)] (IV), which can liberate R3Si–O–CH2–NR′2 (V) through reaction with R3SiH via fairly small activation barrier of 10–13 kcal mol−1. The next step, reduction of V with hydrosilane, despite being highly favorable thermodynamically, occurs via fairly high activation barriers of ∼35.0 kcal mol−1 when the transformation is not catalysed.15
On the other hand, when catalysed by [Cu(H)(NHC)] the barrier of this transformation is reduced to 26–28 kcal mol−1.15
To support these computational findings, synthetic studies were carried out. Phenylsilane was added to a solution of [Cu(OH)(IPr)] and the reaction was monitored by 1H NMR.
The data are in accordance with the in situ formation of [Cu(H)(IPr)] (18) (Scheme 5).17 The subsequent reaction of this species with CO2 (2 bar) led to the formation of [Cu{OC(O)H}(IPr)] (19).13,18 The same outcome was obtained by reacting [Cu(OH)(IPr)] with formic acid in benzene.
 | ||
| Scheme 5 Formation of [Cu{OC(O)H}(IPr)]. | ||
In order to corroborate the other steps of the proposed mechanism, stoichiometric NMR experiments were carried out (Scheme 6). By reacting 19 with N-methylaniline 5a in the presence of PhSiH3, 50% conversion towards N-methylformanilide 5c was observed. Noteworthy, the formation of the dimethylated compound was not observed. This is probably due to the short reaction time (7 hours rather than 20 hours).15 As highlighted in Scheme 6, the presence of silane is crucial for the formylation of N-methylaniline. In fact, the reaction solely promoted by 19 was unsuccessful in the presence or absence of KOtBu.15
 | ||
| Scheme 6 NMR experiments. | ||
Finally, the reduction of 5c promoted by PhSiH3 in the presence of the copper catalyst was carried out. After 7 hours, the reaction led to 64% conversion of the amide into the methylated product 5b confirming the methylation proceeds via N-formylamine intermediates.15
Conclusions
In conclusion, the first NHC-copper-catalysed methylation of N–H bonds using CO2 and silane as reagents was reported. The scope of this transformation showed that secondary amines are converted under relatively mild conditions into the desired methylated products while primary amines provide a distribution of products depending on the nature of the substrate. DFT calculations supported by experimental work were carried out in order to elucidate the mechanism of this transformation.Experimental
General information
All reactions were carried out under argon atmosphere using standard Schlenk and glovebox techniques. Chemicals were used as received unless otherwise noted. Dry toluene was obtained from a PureSolv SPS-400-5 solvent purification system. 1H, and 13C-{1H} Nuclear Magnetic Resonance (NMR) spectra were recorded on a Bruker-400 MHz or 300 MHz spectrometers using the residual solvent peak as reference (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm, CD2Cl2: δH = 5.32 ppm, δC = 53.84 ppm, C7D8: δH = 2.08 ppm, δC = 20.4 ppm) at 298 K. Elemental analyses were performed at London Metropolitan University. HMRS analyses were carried out by the EPSRC National Mass Spectro,metry Service Centre at Swansea University.Synthesis of [Cu(OH)(IPr*)] (3)
In a glovebox, a round bottom flask was charged with [Cu(Cl)(IPr*)] (250 mg, 0.25 mmol), CsOH (74 mg, 2 equiv.) and THF (12.5 ml). The reaction mixture was stirred at room temperature during 15 hours. The suspension was filtered through a plug of celite. The solution was concentrated and pentane (10 mL) was added. The colourless precipitate was collected by filtration and washed with pentane (3 × 5 mL). The product was obtained as a colourless solid in 89% yield (215 mg, 0.22 mmol). 1H NMR (300 MHz, C7D8, 298 K): δ = 1.74 (s, 6H, CH3), 5.58 (s, 2H, H4 and H5), 5.60 (s, 4H, CHPh2), 6.97–7.01 (m, 28H, CHAr), 7.17 (m, 8H, CHAr), 7.41 (m, 8H, CHAr). 13C-{1H} NMR (75 MHz, C7D8, 298 K): δ = 21.1 (s, CH3), 51.7 (s, CHPh2), 123.2 (s, C4 and C5), 126.8 (s, CH Ar), 127.0 (s, CH Ar), 128.3 (s, CH Ar), 128.6 (s, CH Ar), 128.7 (s, CH Ar), 129.1 (s, CH Ar), 129.9 (s, CH Ar), 130.3 (s, CIV Ar), 130.6 (s, CIV Ar), 141.7 (s, CIV Ar), 143.5 (s, CIV Ar), 143.7 (s, CIV Ar). C2 has not been observed. Elem. Anal.: Calcd for C25H33CuN2O: C, 83.40; H, 5.78, N, 2.82. Found: C, 83.46, H, 5.65, N, 2.74.General procedure for the methylation
Under an argon atmosphere, a 3 mL vial was charged with [Cu(OtBu)(IMes)] (4) (11 mg, 10 mol%), KOtBu (2.8 mg, 10 mol%) and toluene (2 mL). The amine substrate (0.25 mmol, 1 equiv.) and PhSiH3 (123 μL, 1.0 mol, 4 equiv.) were added and the vial was sealed with a septum cap. The septum cap was pierced with a syringe needle and placed into a six-slot steal autoclave. The autoclave was sealed, purged twice with CO2 and heated at 100 °C (oil bath) under CO2 atmosphere (2 bar) for 20 hours. The reaction mixture was then allowed to cool and the gas was carefully released. The reaction mixture was analysed by gas chromatography (GC). In the case of isolated products, dichloromethane (5 mL) was added to the crude and the mixture was extracted with HCl 1 M (3 × 10 mL). The aqueous layer was neutralised by addition of K2CO3 (pH = 12) and extracted with diethyl ether (3 × 10 mL). The ether layer was dried over Na2SO4. Removal of the solvent afforded the desired compounds.Synthesis of [Cu{OC(O)H}(IPr)], (19)
Under an argon atmosphere, a vial was charged with [Cu(OH)(IPr)] (1) (200 mg, 0.21 mmol, 1 equiv.), formic acid (16 μL, 0.21 mmol, 1 equiv.) and benzene (2 mL). The mixture was stirred at room temperature for 1 hour, the solution was then concentrated under reduced pressure. The addition of pentane (10 mL) afforded a colourless solid collected by filtration (Yield = 82%). 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.22 (d, 3JH–H = 6.8 Hz, 12H, CH–CH3), 1.29 (d, 3JH–H = 6.8 Hz, 12H, CH–CH3), 2.56 (sept, 3JH–H = 7.0 Hz, 4H, CH–CH3 isopropyl), 7.15 (s, 2H, H4 and H5), 7.29 (d, 3JH–H = 7.8 Hz, 4H, CH phenyl), 7.48 (t, 3JH–H = 7.9 Hz, 2H, CH phenyl), 8.11 (s, 1H, C(O)H formyl) 13C-{1H} NMR (75 MHz, CDCl3, 298 K): δ = 24.0 (s, CH–CH3), 24.8 (s, CH–CH3) 28.8 (s, CH–CH3), 123.3 (s, CIV Ar), 124.3 (s, CH Ar), 130.6 (s, C4 and C5), 134.5 (s, CIV Ar), 145.7 (s, CH Ar), 167.9 (s, OC(O)H), 180.4 (s, C2).Computational details
The default, adaptively generated PRIRODA grid, corresponding to an accuracy of the exchange–correlation energy per atom (1 × 10−8 hartree) was decreased by a factor of 100 for more accurate evaluation of the exchange–correlation energy. Default values were used for the Self–Consistent–Field (SCF) convergence and the maximum gradient for geometry optimisation criterion (1 × 10−4 au), whereas the maximum displacement geometry convergence criterion was decreased to 0.0018 au.
Translational, rotational, and vibrational partition functions for thermal corrections to arrive at total Gibbs free energies were computed within the ideal-gas, rigid-rotor, and harmonic oscillator approximations. The temperature used in the calculations of thermochemical corrections was set to 298.15 K in all the cases.
Acknowledgements
The authors gratefully acknowledge the Royal Society (University Research Fellowship to C. S. J. C.), the EPSRC (DTG011 EP/J500549/1) and the King Abdullah University of Science and Technology for funding, and the EPSRC National Mass Spectrometry Service Centre at Swansea University for HMRS analyses.Notes and references
- (a) J. M. Adams and S. Cory, Nature, 1975, 255, 28 CrossRef CAS PubMed; (b) D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek and G. Rechavi, Nature, 2012, 485, 201 CrossRef CAS PubMed; (c) E. L. Greer and Y. Shi, Nat. Rev. Genet., 2012, 13, 343 CrossRef CAS PubMed; (d) K. D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason and S. R. Jaffrey, Cell, 2012, 149, 1635 CrossRef CAS PubMed; (e) Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, New York, 4th edn, 1992, p. 438 Search PubMed.
- (a) H. T. Clarke, H. B. Gillespie and S. Z. Weisshaus, J. Am. Chem. Soc., 1933, 55, 4571 CrossRef CAS; (b) E. Farkas and C. J. Sunman, J. Org. Chem., 1985, 50, 1110 CrossRef CAS; (c) J. R. Harding, J. R. Jones, S. Y. Lu and R. Wood, Tetrahedron Lett., 2002, 43, 9487 CrossRef CAS; (d) S. Kim, C. H. Oh, J. S. Ko, K. H. Ahn and Y. J. Kim, J. Org. Chem., 1985, 50, 1927 CrossRef CAS; (e) H. Alinezhad, M. Tajbakhsh and R. Zamani, Synth. Commun., 2006, 36, 3609 CrossRef CAS PubMed; (f) H. Alinezhad, M. Tajbakhsh, F. Salehian and K. Fazli, Synth. Commun., 2010, 40, 2415 CrossRef CAS PubMed; (g) M. Tanis and G. Rauniyar, US Pat, 5105013, 1992 Search PubMed; (h) M. Selva, P. Tundo and A. Perosa, J. Org. Chem., 2002, 67, 9238 CrossRef CAS PubMed; (i) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706 CrossRef CAS PubMed; (j) M. Selva, P. Tundo and A. Perosa, J. Org. Chem., 2003, 68, 7374 CrossRef CAS PubMed; (k) M. Selva, P. Tundo and T. J. Foccardi, Org. Chem., 2005, 70, 2476 CrossRef CAS PubMed; (l) M. Selva, A. Perosa, P. Tundo and D. Brunelli, J. Org. Chem., 2006, 71, 5770 CrossRef CAS PubMed; (m) M. Selva and A. Perosa, Green Chem., 2008, 10, 457 RSC; (n) T. Oku, Y. Arita, H. Tsuneki and T. Ikariya, J. Am. Chem. Soc., 2004, 126, 7368 CrossRef CAS PubMed; (o) Y. Zhao, S. W. Foo and S. Saito, Angew. Chem., Int. Ed., 2011, 50, 3006 CrossRef CAS PubMed.
- (a) Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Wienheim, 2010 Search PubMed; (b) M. Aresta, in Activation of Small Molecules, ed. W. B. Tolman, Wiley-VCH, Weinheim, 2006, ch. 1 Search PubMed; (c) D. H. Gibson, in Comprehensive Coordination Chemistry-II, ed. J. A. McCleverty and T. J. Meyer, Elsevier Ltd., Amsterdam, 2003, vol. 1, section 1.30, p. 595 Search PubMed.
- O. Jacquet, X. Frogneux, C. D. N. Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127 RSC.
- (a) Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568 CrossRef CAS PubMed; (b) Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156 CrossRef CAS PubMed.
- K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554 CrossRef CAS PubMed.
- L. González-Sebastián, M. Flores-Alamo and J. J. García, Organometallics, 2015, 34, 763 CrossRef.
- (a) E. Blondiaux, J. Pouessel and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 12186 CrossRef CAS PubMed; (b) S. Das, F. D. Bobbink, G. Laurenczy and P. J. Dyson, Angew. Chem., Int. Ed., 2014, 53, 12876 CrossRef CAS PubMed.
- I. Sorribes, K. Junge and M. Beller, Chem. – Eur. J., 2014, 20, 7878 CrossRef CAS PubMed.
- S. Savourey, G. Lefèvre, J. Berthet and T. Cantat, Chem. Commun., 2014, 50, 14033 RSC.
- I. I. F. Boogaerts, G. C. Fortman, M. R. L. Furst, C. S. J. Cazin and S. P. Nolan, Angew. Chem., Int. Ed., 2010, 49, 8674 CrossRef CAS PubMed.
- G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3966 CrossRef CAS.
- L. Zhang, J. Cheng and Z. Hou, Chem. Commun., 2013, 49, 4782 RSC.
- K. Motokura, D. Kashiwame, N. Takahashi, A. Miyaji and T. Baba, Chem. – Eur. J., 2013, 19, 10030 CrossRef CAS PubMed.
- See ESI† for further information.
- S. Ilieva, B. Galabov, D. G. Musaev, K. Morokuma and H. F. Schaefer, J. Org. Chem., 2003, 68, 1496 CrossRef CAS PubMed.
- (a) N. P. Mankad, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 3369 CrossRef CAS; (b) M. R. Uehling, R. P. Rucker and G. Lalic, J. Am. Chem. Soc., 2014, 136, 8799 CrossRef CAS PubMed; (c) A. M. Whittaker and G. Lalic, Org. Lett., 2013, 15, 1112 CrossRef CAS PubMed.
- C. M. Wyss, B. K. Tate, J. Bacsa, T. G. Gray and J. P. Sadighi, Angew. Chem., Int. Ed., 2013, 52, 12920 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS; J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef.
- D. N. Laikov and Y. A. Ustynyuk, Russ. Chem. Bull., 2005, 54, 820 CrossRef CAS.
- D. N. Laikov, Chem. Phys. Lett., 2005, 416, 116 CrossRef CAS PubMed.
- K. G. Dyall, J. Chem. Phys., 1994, 100, 2118 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 224306 CrossRef PubMed.
- (a) F. Weigend and R. Ahlrichs, PCCP, 2005, 7, 3297 RSC; (b) F. Weigend, PCCP, 2006, 8, 1057 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for the synthesis of the complexes, optimisation and protocol for catalysis, characterisation of reaction products, DFT calculations, stoichiometric reactions assisting in supporting mechanistic proposal. See DOI: 10.1039/c5dt03506f |
| This journal is © The Royal Society of Chemistry 2015 |