
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Group 1 and group 2 metal complexes supported by a bidentate bulky iminopyrrolyl ligand: synthesis, structural diversity, and ε-caprolactone polymerization study†
Ravi K.
Kottalanka
,
A.
Harinath
,
Supriya
Rej
and
Tarun K.
Panda
 *
*
Department of Chemistry, Indian Institute of Technology Hyderabad, Kandi – 502 285, Sangareddy, Telangana, India. E-mail: tpanda@iith.ac.in; Fax: +91 (40)2301 6032; Tel: +91 (40)2301 6036
First published on 13th October 2015
Abstract
We report here a series of alkali and alkaline earth metal complexes, each with a bulky iminopyrrolyl ligand [2-(Ph3CN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)C4H3NH] (1-H) moiety in their coordination sphere, synthesized using either alkane elimination or silylamine elimination methods or the salt metathesis route. The lithium salt of molecular composition [Li(2-(Ph3CNCH)C4H3N)(THF)2] (2) was prepared using the alkane elimination method, and the silylamine elimination method was used to synthesize the dimeric sodium and tetra-nuclear potassium salts of composition [(2-(Ph3CNCH)C4H3N)Na(THF)]2 (3) and [(2-(Ph3CNCH)C4H3N)K(THF)0.5]4 (4) respectively. The magnesium complex of composition [(THF)2Mg(CH2Ph){2-(Ph3CNCH)C4H3N}] (5) was synthesized through the alkane elimination method, in which [Mg(CH2Ph)2(OEt2)2] was treated with the bulky iminopyrrole ligand 1-H in 1
CH)C4H3NH] (1-H) moiety in their coordination sphere, synthesized using either alkane elimination or silylamine elimination methods or the salt metathesis route. The lithium salt of molecular composition [Li(2-(Ph3CNCH)C4H3N)(THF)2] (2) was prepared using the alkane elimination method, and the silylamine elimination method was used to synthesize the dimeric sodium and tetra-nuclear potassium salts of composition [(2-(Ph3CNCH)C4H3N)Na(THF)]2 (3) and [(2-(Ph3CNCH)C4H3N)K(THF)0.5]4 (4) respectively. The magnesium complex of composition [(THF)2Mg(CH2Ph){2-(Ph3CNCH)C4H3N}] (5) was synthesized through the alkane elimination method, in which [Mg(CH2Ph)2(OEt2)2] was treated with the bulky iminopyrrole ligand 1-H in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, whereas the bis(iminopyrrolyl)magnesium complex [(THF)2Mg{2-(Ph3CNCH)C4H3N}2] (6) was isolated using the salt metathesis route. The heavier alkaline earth metal complexes of the general formula {(THF)nM(2-(Ph3CNCH)C4H3N)2} [M = Ca (7), Sr (8), and n = 2; M = Ba (9), n = 3] were prepared in pure form using two synthetic methods: in the first method, the bulky iminopyrrole ligand 1-H was directly treated with the alkaline earth metal precursor [M{N(SiMe3)2}2(THF)n] (where M = Ca, Sr and Ba) in 2:1 molar ratio in THF solvent at ambient temperature. The complexes 7–9 were also obtained using the salt metathesis reaction, which involves the treatment of the potassium salt (4) with the corresponding metal diiodides MI2 (M = Ca, Sr and Ba) in 2:1 molar ratio in THF solvent. The molecular structures of all the metal complexes (1-H, 2–9) in the solid state were established through single-crystal X-ray diffraction analysis. The complexes 5–9 were tested as catalysts for the ring-opening polymerization of ε-caprolactone. High activity was observed in the heavier alkaline earth metal complexes 7–9, with a very narrow polydispersity index in comparison to that of magnesium complexes 5 and 6.
:1 molar ratio, whereas the bis(iminopyrrolyl)magnesium complex [(THF)2Mg{2-(Ph3CNCH)C4H3N}2] (6) was isolated using the salt metathesis route. The heavier alkaline earth metal complexes of the general formula {(THF)nM(2-(Ph3CNCH)C4H3N)2} [M = Ca (7), Sr (8), and n = 2; M = Ba (9), n = 3] were prepared in pure form using two synthetic methods: in the first method, the bulky iminopyrrole ligand 1-H was directly treated with the alkaline earth metal precursor [M{N(SiMe3)2}2(THF)n] (where M = Ca, Sr and Ba) in 2:1 molar ratio in THF solvent at ambient temperature. The complexes 7–9 were also obtained using the salt metathesis reaction, which involves the treatment of the potassium salt (4) with the corresponding metal diiodides MI2 (M = Ca, Sr and Ba) in 2:1 molar ratio in THF solvent. The molecular structures of all the metal complexes (1-H, 2–9) in the solid state were established through single-crystal X-ray diffraction analysis. The complexes 5–9 were tested as catalysts for the ring-opening polymerization of ε-caprolactone. High activity was observed in the heavier alkaline earth metal complexes 7–9, with a very narrow polydispersity index in comparison to that of magnesium complexes 5 and 6.
Introduction
Biodegradable polyesters such as poly(ε-caprolactone) (PCL) and poly(lactic acid) (PLA) have attracted widespread interest from industrial and academic research groups, particularly polylactones, which are widely used in medicinal applications such as drug delivery systems, medical sutures, and as plastic modifiers in industries.1–3 Although they can be obtained by traditional poly-condensation reactions,4 these polymers are best prepared through the ring-opening polymerization (ROP) of cyclic esters. Macromolecular engineering of these polymers is gaining importance for the synthesis of telechelics, block, graft, and star-shaped polymers.5 This, in turn, requires the synthesis of polyesters with predictable molecular weights, low polydispersity indices, and control over end groups. Various metal complexes such as aluminium,6 titanium,7 tin,8 zinc,9 magnesium,10 and rare earth metals11 have been used as initiators for the ROP of ε-caprolactone (ε-CL). In many cases, the molecular weight distribution of PCL became broader after the monomer was completely consumed, which suggested the occurrence of trans-esterification (Chart 1).11b,12 These trans-esterifications occur very often during the polymerization of ε-CL by metal alkoxides, and the rate at which they occur are related to both the nature of the metal ion and the groups surrounding the ion. Subsequently, it was shown that sterically demanding groups attached to a metal ion can prevent PCL chains from coordinating to the ion, and therefore minimize trans-esterification reactions.13 | ||
| Chart 1 Intramolecular and intermolecular trans-esterification. | ||
Thus, the development of very efficient metal initiators with strong control over the initiation, propagation, and termination steps is needed to synthesize polyesters with predictable molecular weights and narrow polydispersity indices. So far, the ROP process initiated by the heavier alkaline earth (Ae) metals calcium, strontium, and barium has been explored less when compared to other metal complexes,6–13 and only a few well-defined neutral heteroleptic and homoleptic Ae ROP catalysts are mentioned in the literature.14,15 This reflects the lack of information in the literature about the synthesis, stability, and reactivity of complexes of these highly electropositive metals. Recurring issues typical of Ca, Sr, and Ba complexes include their kinetic lability with Schlenk-type equilibria in solution.16 However, significant efforts have been made of late in order to make them friendly and exploit the high reactivity of these complexes. Strategies aimed at suppressing solution distribution equilibria through the careful selection of ancillary ligands such as tris(pyrazolyl)borates,17 β-diketiminates,18 aminotrop-(on)iminates,19,20 or bulky nucleophilic substituents have been invented,21 while the range of synthetic precursors is growing steadily.22,23 As a result, single-site Ae-based catalysts have shown an astounding ability for a variety of transformations involving σ-bond metathesis processes.24
Recently, iminopyrrole ligands have been commonly employed in the synthesis of several transition metals and rare earth metal compounds.25 The metal complexes including main group metals, transition metals as well as rare earth metals with 2-iminopyrrolyl in their coordination spheres act as efficient polymerization catalysts.26 The Mashima group reported a series of alkaline earth metal complexes stabilized by the [2-(2,6-iPr2C6H3NCH)C4H3NH] ligand and those Ae-complexes act as efficient catalysts for the ROP of ε-CL. They also investigated the effects of ionic radii of metal ions on the rate of polymerization and concluded that it was rather good for heavier alkaline earth metal complexes.27 However, we have noticed that PCLs obtained by heavier alkaline earth metal complexes supported by the [2-(2,6-iPr2C6H3NCH)C4H3NH] ligand have moderate polydispersity indices (PDI = 2.0 for Ba and 1.9 for Mg complexes). This could be due to intra or intermolecular trans-esterification reactions or other side reactions, which are very common in the ROP of cyclic esters (Chart 1). In our ongoing research into heavier alkaline earth metal chemistry, we have previously reported a series of alkaline earth metal complexes with amidophosphine-chalcogenides/boranes [R2NHPh2P(E)]n (R = C(CH3)3, CHPh2, CPh3, *CH(CH3)(Ph), –CH2–CH2–; E = O, S, Se, and BH3; n = 1 or 2) in their coordination spheres.28 In these complexes the anionic ligands presented novel molecular structural characteristics via coordination from the amido-nitrogen atom and coordination from the chalcogenide atom or borane group through the hydrogen atoms either in a η1 or η2 fashion. A few such complexes have presented excellent catalytic activity towards the ROP of ε-CL.28d,f
In this context, we introduce another sterically demanding ancillary ligand [2-(Ph3CNCH)–C4H3NH] (1-H) into alkaline earth metal coordination chemistry. We envisage that the sterically demanding bulky substituent on the imine nitrogen atom will completely shield the metal ion, preventing trans-esterification or other side reactions to afford narrower polydispersity indices for PCL (Chart 2).
 | ||
| Chart 2 Control of trans-esterification in ROP by changing substituent on imine nitrogen of iminopyrrolyl ligand. | ||
Here, we describe the detailed synthesis and structural studies of a bidentate rigid bulky-iminopyrrolyl ligand [2-(Ph3CNCH)–C4H3NH] (1-H) and its corresponding alkali metal complexes [Li{2-(Ph3CNCH)C4H3N}(THF)2] (2), [{2-(Ph3CNCH)–C4H3N}-Na(THF)]2 (3), and [{2-(Ph3CNCH)C4H3N}-K(THF)0.5]4 (4), and alkaline earth metal complexes [(THF)2Mg(CH2Ph){2-(Ph3CNCH)C4H3N}] (5), [(THF)2Mg-{2-(Ph3CNCH)C4H3N}2] (6), and [(THF)nM{2-(Ph3CNCH)C4H3N}2] (M = Ca (7), Sr (8) and n = 2; M = Ba (9), n = 3). We also report in detail the ROP study of ε-CL using newly synthesized alkaline earth metal complexes (5–9) as catalysts with different monomer/catalyst ratios.
Results and discussion
Ligand synthesis
The (E)-N-((1H-pyrrol-2-yl)methylene)-1,1,1-triphenylmethanamine ligand [2-(Ph3CNCH)C4H3NH] (1-H) was prepared in good yield and high purity by the condensation reaction of pyrrol-2-carboxyaldehyde with 1 equiv. of tritylamine in the presence of a catalytic amount of glacial acetic acid in methanol solvent (Scheme 1). The ligand 1-H was fully characterized using standard spectroscopic/analytical techniques and its solid-state structure was established using single-crystal X-ray diffraction analysis.
 | ||
| Scheme 1 Synthesis of ligand 1-H and corresponding alkali metal complexes 2–4. | ||
A strong absorption band observed at 1629 cm−1 in FT-IR spectra indicates a CN bond in the ligand 1-H. This value is within the range reported in the literature.29,30 The 1H NMR spectrum of ligand 1-H showed a broad resonance signal at δ 9.56 ppm for the N–H proton of the pyrrole moiety. The singlet resonance signal at δ 7.71 ppm can be assigned to the imine NC–H proton. In addition, the singlet at 6.96 ppm, doublet at 6.45 ppm, and multiplets centered at 6.29 ppm in the 1H NMR spectrum clearly represent the resonance of pyrrole ring protons. In the 13C{1H} NMR spectrum, we observed a strong resonance signal at δ 150.2 ppm for the imine carbon atom –CN, which is in good agreement for the compound [2-(2,6-iPr2C6H3NCH)–C4H3NH] (δ 153.2 ppm) and for the compound [2-(2-Ph2PC6H4NCH)–C4H3NH] (δ 148.7 ppm) reported in the literature.8 The resonance signal at δ 77.8 ppm corresponds to the tertiary carbon atom of the CPh3 group.
The bulky iminopyrrolyl ligand 1-H readily crystallizes in CH2Cl2 at room temperature and therefore, the solid-state structure was established using single-crystal X-ray diffraction analysis. The molecular structure of ligand 1-H is shown in Fig. 1 and details of the structural parameters are given in Table TS1 in the ESI.† Ligand 1-H crystallizes in the monoclinic space group P21/c with two independent molecules in the asymmetric unit. The bond distance of 1.357(6) Å observed for C1–N1 is in good agreement with the value reported for the compound [2-(2,6-iPr2C6H3NCH)–C4H3NH]31 [1.354(4) Å] and for the [2-(2,6-iPr2C6H3NCMe)–C4H3NH] [1.3604(17) Å].32 The C5–N2 bond distance was 1.260(6) Å, which is slightly shorter than the C2–N5 distance of 1.2835(19) Å for the compound [2-(2,6-iPr2C6H3NCMe)–C4H3NH].32
 | ||
| Fig. 1 Solid-state structure of ligand 1-H. All hydrogen atoms (except H1) are omitted for clarity. Selected bond lengths (Å) and bond angles (°): C1–N1 1.357(6), C1–C2 1.364(8), C2–C3 1.410(7), C3–C4 1.372(7), C4–C5 1.433(6), N2–C5 1.260(6), N2–C6 1.485(6), C6–C7 1.544(7), C6–C13 1.552(6), C6–C19 1.530(6); C1–N1–C4 109.3(4), C4–C5–N2 122.6(5), C5–N2–C6 120.6(4), N2–C6–C7 104.2(4). | ||
Synthesis and characterization of alkali metal complexes
Treatment of LiCH2SiMe3 with 1 equiv. of the bulky iminopyrrolyl ligand 1-H in THF solvent resulted in the corresponding lithium complex of the molecular formula [Li{2-(Ph3CNCH)C4H3N}(THF)2] (2). Similarly, treatment of 1 equiv. of either sodium or potassium bis(trimethylsilyl)amide with 1-H in THF solvent resulted in the corresponding dimeric sodium complex [(2-(Ph3CNCH)C4H3N)Na(THF)]2 (3) and tetra-nuclear potassium complex of molecular composition [(2-(Ph3CNCH)C4H3N)K(THF)0.5]4 (4) in very good yield (Scheme 1).33 The alkali metal complexes 2–4 were fully characterized using standard spectroscopic/analytical techniques and their solid-state structures were established using single-crystal X-ray diffraction analysis. The 1H NMR spectra of compounds 2–4 showed a singlet resonance signal at δ 8.04 (for 2), 8.03 (for 3), and 8.17 (for 4) indicating the presence of the imine proton in each complex. The 13C{1H} NMR spectra also supported the presence of the imine carbon atom in each complex, showing resonance signals at δ 147.9 (for 2), 147.8 (for 3), and 147.9 (for 4). The other pyrrole ring protons and aromatic protons showed resonance signals in each complex at expected regions. Both complexes 3 and 4 displayed only one set of signals in solution, which indicates their dynamic behavior in the solution state. In the solid state, lithium complex 2 crystallized in the orthorhombic space group Pbca with 16 molecules in the unit cell. The details of the structural parameters are given in the ESI.† The solid-state structure confirmed the κ2-NN ligation of ligand 1 with two THF molecules. Complex 2 is shown in Fig. 2. The iminopyrrolyl ligand 1 acts as a bidentate chelating ligand and coordinates to the lithium center through the pyrrolide nitrogen and imine nitrogen atoms. Therefore, the geometry around the lithium ion in 2 can be best described as distorted tetrahedral with bond angles of 87.7(3)° for N1–Li1–N2, 114.0(4)° for O1–Li1–N1, 110.9(3)° for O2–Li1–N1, 115.5(4)° for N2–Li1–O1, 120.5(4)° for N2–Li1–O2, and 107.20(18)° for O1–Li1–O2. The Li–N bond lengths of 1.993(8) and 2.097(7) Å observed in compound 2 are in good agreement with the Li–N bond lengths found in the reported molecules. For example, Li–N bond lengths of 2.068(3) and 2.085(3) Å were observed in the complex {[η2:η1-2-(2,6-Me2C6H3NCH)C4H3N]Li(THF)}2 and 2.105(4) and 2.088(4) Å were observed in the lithium complex {[η2:η1-2-(2,6-iPr2C6H3NCH)C4H3N]Li(THF)}2.34 The C1–N1 bond distance of 1.345(4) Å and C5–N2 bond distance of 1.289(5) Å of the anionic ligand moiety are in the range similar to that of the free ligand 1-H [1.357(6) Å for C1–N1 and 1.260(6) Å for C5–N2] upon coordination to the lithium ion. The Li–O bond distances of 1.944(7) and 1.946(9) Å are within the range of Li–O bond distances reported in the literature. Therefore, in the lithium complex 2 a five-membered metallacycle Li1–N1–C4–C5–N2 was formed with a bite angle of 87.7(3)°.
 | ||
| Fig. 2 Solid-state structure of lithium complex 2. All hydrogen atoms (except H10) are omitted for clarity. Selected bond lengths (Å) and bond angles (°): Li1–N1 1.993(8), Li1–N2 2.097(7), Li1–O1 1.944(7), Li–O2 1.946(9), N1–C1 1.345(4), C1–C2 1.397(6), C2–C3 1.401(5), C3–C4 1.408(5), N1–C4 1.366(4), C4–C5 1.431(4), N2–C5 1.289(5), N2–C6 1.492(4), C6–C13 1.550(5); N1–Li–N2 87.7(3), N1–Li1–O1 114.0(4), N1–Li1–O2 110.9(3), N2–Li1–O1 115.5(4), N2–Li1–O2 120.5(4), O1–Li1–O2 107.2(3), C4–C5–N2 122.4(3), N1–C4–C5 121.3(3), N1–C4–C3 111.4(3), N1–C1–C2 112.3(3), Li1–N1–C4 104.6(3), Li1–N2–C5 103.6(3), Li1–N2–C6 138.4(3), N2–C6–C13 107.3(2). | ||
The sodium complex 3 crystallizes in the monoclinic space group P21/n, with two molecules in the unit cell. The details of the structural parameters are given in TS1 in the ESI.† The solid-state structure of complex 3 confirms its dimeric structure owing to the larger size of the sodium ion than the lithium ion when compared to the monomeric lithium complex 2. The solid-state structure and selected bond lengths and bond angles are shown in Fig. 3. In the dimeric sodium complex 3, each sodium ion is surrounded by one anionic ligand moiety in a bidentate (κ2) fashion and one THF molecule. Each sodium ion further has π-interactions with pyrrole ring carbons in η3 mode in the dimeric sodium complex 3. Therefore, each ligand in the dimeric sodium complex 3 bonds to the sodium ions in (σ + π) mode. The geometry of each sodium ion is best described as distorted tetrahedral, formed due to the coordination between two nitrogen atoms of the iminopyrrolyl ligand, one oxygen atom of the THF molecule, and η3-coordination from the pyrrolyl ring of the dimer fragment. The bite angles of 95.46(5)° for N1–Na1–N2, 75.44(5)° for N1i–Na1–N2, and 85.56(6)° for N1–Na1–N1i were observed for each of the iminopyrrolyls chelated to the sodium atoms. The Na–N bond distances of 2.3586(17) and 2.4641(16) Å were in the range similar to the Na–N distances observed in the compound [μ2:κ2-2-(2,6-Me2C6H3NCH)C4H3NNa(OEt2)]2 [2.405(3) and 2.4285(3) Å].31 The distance between the sodium ion and the pyrrole ring atoms (C1, N1, and C4 or C1i, N1i, and C4i) were found to be 2.790(2), 2.6998(17), and 2.8670(19) Å respectively. These distances are somewhat longer when compared to the Na-pyrrolyl centroid distances of 2.447(3) Å and 2.494(3) Å found in the polymeric sodium compounds of the type [{Na(μ2:κ2-N,N′-iminopyrrolyl)}2n(OEt2)2x] (n ≥ 1; x = 0 or 1), (aryl = C6H5 or 2,6-Me2C6H3),31 indicating that moderate π-interactions exist between the sodium ions and pyrrolyl rings in the dimeric sodium complex 3. The bond distances of 1.349(2), 1.436(3), and 1.289(2) Å for C1–N1, C4–C5, and C5–N2 respectively were almost unchanged compared to that of the free ligand [C1–N1: 1.357(6), C4–C5: 1.433(6), and C5–N2: 1.260(6) Å] upon coordination to the sodium ion. Therefore, each bidentate iminopyrrolyl ligand forms a five-membered metallacycle Na1–N1–C4–C5–N2 or Na1i–N1i–C4i–C5i–N2i with the sodium ion, where the sodium ions are slightly deviated from the planarity. Each sodium ion in the dimeric complex 3 is further stabilized by the coordination from one THF molecule. The Na–O bond distance of 2.3315(17) Å fits well with reports in the literature. A short contact Na⋯H between the sodium ion and one of the phenyl protons (Na1⋯H13 2.707 Å) is observed, which can be characterized as a remote or secondary M⋯H interaction.35 However, in solution all phenyl protons appear equivalent, as observed in the 1H NMR study, presumably due to the dynamic behavior of the complex.
 | ||
| Fig. 3 Solid-state structure of sodium complex (3). All hydrogen atoms (except H13) are omitted for clarity. Selected bond lengths (Å) and bond angles (°): Na1–N1i 2.3586(17), Na1–N2 2.4641(16), Na1–O1 2.3315(17), Na1–N1 2.6998(17), Na1–C1 2.8670(19), Na1–C2 3.080(2), Na1–C3 3.034(2), Na1–C4 2.790(2), Na1–C5 3.0870(19), C4–C5i 1.436(3), C5–N2 1.289(2), N2–C6 1.493(2), C1–N1 1.382(2), C1–C2 1.403(3), C2–C3 1.396(3), C3–C4 1.396(3); N2–Na1–N1i 75.44(5), N1–Na1–N2 95.46(5), N1–Na1–O1 147.46(6), N2–Na1–O1 117.06(6), N1i–Na1–O1 102.83(6), N1–Na1–N1i 85.56(6), C1–Na1–O1 144.32(7), C2–Na1–O1 117.02(6), C3–Na1–O1 105.33(6), C4–Na1–O1 118.92(6). | ||
In contrast to sodium complex 3, the potassium complex 4 crystallizes in the monoclinic space group P21/c with two molecules in the unit cell. The solid-state structure and selected bond lengths and bond angles are shown in Fig. 4. The asymmetric unit of potassium complex 4 contains two iminopyrrolyls, two potassium ions, and one coordinated THF molecule. It must be noted that the coordination spheres of both the potassium ions are different. The ion K1 is chelated by two iminopyrrolyl ligands in a bidentate fashion, and one THF molecule, whereas the second ion, K2, is chelated by two nitrogens from a iminopyrrolyl ligand in a κ2 fashion and π interaction (η5-mode) from one pyrrole ring of the adjacent iminopyrrolyl ligand. Therefore, in the grown structure, two potassium ions (K2 and K2i) are observed as sandwiched between two pyrrolyl ring π-electron densities in a η5-fashion and further chelated by imine-nitrogen atoms of the iminopyrrolyls. The other two potassium atoms (K1 and K1i) are surrounded by iminopyrrolyl moieties in a bidentate fashion and one THF molecule. Further, K1 has weak interactions with the aromatic ring hydrogen atoms (K1⋯H24 and K1⋯H48). Therefore, the geometry of K1 is best described as distorted trigonal-bipyramidal, while that of K2 as distorted tetrahedral. The K2–N1pyrrolyl and K2–N3pyrrolyl bond distances, 3.155(3) and 3.041(3) Å respectively, fit well with the K–N distances observed in [K(THF)2{Ph2P(Se)N(CMe3)}]n (3.047(3) Å).28e The K1–N1pyrrolyl and K1–N3pyrrolyl bond distances of 2.971(3) and 2.667(3) Å respectively, which are in good agreement with K–N distances, were observed in the complexes [{Ph2P(Se)NCHPh2}K(THF)2]2 [(2.725(3) Å] and [{Ph2P(BH3)NCHPh2}K(THF)2]2 [(2.691(2) Å] previously reported by our group.28 The K2–N2imine and K1–N4imine bond distances of 2.946(3) and 3.013(3) Å respectively were also observed. An average distance of 2.913 Å for the potassium-pyrrolyl ring centroid was observed in the potassium complex 4, which indicates that highly electropositive and larger potassium atoms have considerable interactions with pyrrolyl π-electron density. In addition, K1 has very weak interactions with the aromatic ring hydrogen atoms (K1⋯H24 3.047 Å and K1⋯H48 2.891 Å), reducing their coordination unsaturation. The observed K1–O1 bond distance is 2.668(3) Å, which is in a range similar to that reported in the literature. To the best of our knowledge this is the only example observed of a μ2-(η1-η5) – binding mode between a pyrrole ring and potassium atoms when considering different binding modes such as μ2-(η1–ηn)-reported many times in the literature.36
 | ||
| Fig. 4 Solid-state structure of potassium complex 4. All hydrogen atoms (except H24 and H48) are omitted for clarity. Selected bond lengths (Å) and bond angles (°): K1–N1 2.971(3), K1–N2 3.005(3), K1–N3 2.667(3), K1–N4 3.013(3), K1–O1 2.668(3), K1–C5 3.419(4), K2–N1 3.155(3), K2–N2 2.946(3), K2–N3 3.041(3), K2–C1 3.082(4), K2–C2 3.063(4), K2–C3 3.095(4), K2–C4 3.128(4), C1–N1 1.355(5), C4–N1 1.396(4), C5–N2 1.292(4), C6–N2 1.488(5), C25–N3 1.364(5), C28–N3 1.384(4), C29–N4 1.282(4), C30–N4 1.487(5); N3–K1–O1 114.95(9), N3–K1–N1i 77.85(9), O1–K1–N1i 97.13(9), N3–K1–N2 104.62(8), O1–K1–N2 127.32(8), N1i–K1–N2 58.07(8), N3–K1–N4 60.28(9), O1–K1–N4 112.87(8), N1i–K1–N4 135.55(8), N2–K1–N4 116.53(8), N2–K1–C48 133.36(9), N4–K1–C48 50.33(9), N1i–K2–N2 60.09(8), N1i–K2–N3 74.12(8), N2–K2–N3 97.26(8), N1i–K2–N1 92.85(8), N2–K2–N1 102.47(8), N3–K2–N1 146.71(9). | ||
Synthesis and characterization of alkaline earth metal complexes
We have synthesized various alkaline earth metal complexes of the bulky iminopyrrolyl ligand using alkane elimination, silylamine elimination methods, and salt metathesis routes. The heteroleptic magnesium complex of composition [(THF)2Mg(CH2Ph){2-(Ph3CNCH)C4H3N}] (5) was synthesized through the alkane elimination method, in which [Mg(CH2Ph)2(OEt2)2] was treated with the bulky iminopyrrole ligand 1-H in 1:1 molar ratio in toluene at ambient temperature (Scheme 2). Re-crystallization from a THF/n-pentane mixture afforded the magnesium complex 5 in good yield. However, the homoleptic bis(iminopyrrolyl)magnesium complex of composition [(THF)2Mg{2-(Ph3CNCH)C4H3N}2] (6) was synthesized in 90% yield through the salt metathesis route, where the potassium complex 4 was charged with anhydrous MgI2 in 2:1 molar ratio in THF solvent (Scheme 2).
 | ||
| Scheme 2 Synthesis of heteroleptic (5) and homoleptic (6) magnesium complexes. | ||
The two magnesium complexes 5 and 6 were fully characterized using spectroscopic and analytical techniques. The molecular structures of complexes 5 and 6 were established by single-crystal X-ray diffraction analysis. In the 1H NMR spectra of 5 and 6 recorded in C6D6, the resonance of the imine proton was observed as a singlet at δ 7.91 ppm (5) and 7.66 ppm (6). The resonances of the two benzyl protons of the –CH2Ph group were obtained as singlets at δ 1.73 ppm for complex 5. In the 13C{1H} NMR spectra, resonance at δ 146.3 ppm (for 5) and 145.8 ppm (for 6) can be assigned to the imine carbon (HCN) present in the ligand moiety. However, these values are significantly up-field shifted compared to the free ligand 1-H (150.3 ppm). In addition, a resonance signal at δ 39.3 ppm was observed for the benzylic carbon atom in complex 5.
In the solid state, complexes 5 and 6 crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with four molecules of 5 and one molecule of 6 in the unit cell. The details of the structural parameters are given in TS1 in the ESI.† The solid-state structures of complexes 5 and 6 confirmed the attachment of one (for 5) and two (for 6) iminopyrrolyl ligands to the magnesium ion through the κ2-NN mode. Fig. 5 and 6 show the molecular structures of complexes 5 and 6 respectively. The central magnesium ion in complex 5 is chelated via two nitrogen atoms of the iminopyrrolyl moiety, one benzyl carbon of the –CH2Ph group, and two oxygen atoms from two THF molecules. Thus, the geometry of the magnesium ion in this complex can be best described as distorted trigonal bipyramidal, with two oxygen atoms in the apical position, and two nitrogen atoms and one carbon atom in the basal plane. In contrast, the central magnesium atom in complex 6 is coordinated by two iminopyrrolyl moieties and two THF molecules to adopt a distorted octahedral geometry around the magnesium ion. Both complexes 5 and 6 display two sets of Mg–N distances: one short and one long. The short bond distances Mg–Npyr, 2.070(2) (for 5) and 2.0813(14) Å (for 6), indicate the Mg–N covalent bond. Mg–Npyr bond distances observed in complexes 5 and 6 are in agreement with reported values; for example, the Mg–N bond distance reported as 1.970(3) Å for [{(LiPr)2Mg(THF)2}·(THF)], 2.094(3) Å for [{(LiPr)2Mg}·(THF)] (where LiPr = [(2,6-iPr2C6H3)NC(Me)]2), and 2.051(2) Å for [(LMes)2Mg(THF)3] and 2.070(2) Å for [(LMes)2Mg] (where LMes = [(2,4,6 Me3C6H2)-NC(Me)]2).37 Recently, our group also synthesized magnesium complexes of the type [Mg{C2H4(NPh2P(Se))2}-(THF)3] in which we observed Mg–N distances of 2.066(3) and 2.083(3) Å, which are in good agreement with the observed values of 2.070(2) and 2.0813(14) Å for the complexes 5 and 6 respectively.28e
, with four molecules of 5 and one molecule of 6 in the unit cell. The details of the structural parameters are given in TS1 in the ESI.† The solid-state structures of complexes 5 and 6 confirmed the attachment of one (for 5) and two (for 6) iminopyrrolyl ligands to the magnesium ion through the κ2-NN mode. Fig. 5 and 6 show the molecular structures of complexes 5 and 6 respectively. The central magnesium ion in complex 5 is chelated via two nitrogen atoms of the iminopyrrolyl moiety, one benzyl carbon of the –CH2Ph group, and two oxygen atoms from two THF molecules. Thus, the geometry of the magnesium ion in this complex can be best described as distorted trigonal bipyramidal, with two oxygen atoms in the apical position, and two nitrogen atoms and one carbon atom in the basal plane. In contrast, the central magnesium atom in complex 6 is coordinated by two iminopyrrolyl moieties and two THF molecules to adopt a distorted octahedral geometry around the magnesium ion. Both complexes 5 and 6 display two sets of Mg–N distances: one short and one long. The short bond distances Mg–Npyr, 2.070(2) (for 5) and 2.0813(14) Å (for 6), indicate the Mg–N covalent bond. Mg–Npyr bond distances observed in complexes 5 and 6 are in agreement with reported values; for example, the Mg–N bond distance reported as 1.970(3) Å for [{(LiPr)2Mg(THF)2}·(THF)], 2.094(3) Å for [{(LiPr)2Mg}·(THF)] (where LiPr = [(2,6-iPr2C6H3)NC(Me)]2), and 2.051(2) Å for [(LMes)2Mg(THF)3] and 2.070(2) Å for [(LMes)2Mg] (where LMes = [(2,4,6 Me3C6H2)-NC(Me)]2).37 Recently, our group also synthesized magnesium complexes of the type [Mg{C2H4(NPh2P(Se))2}-(THF)3] in which we observed Mg–N distances of 2.066(3) and 2.083(3) Å, which are in good agreement with the observed values of 2.070(2) and 2.0813(14) Å for the complexes 5 and 6 respectively.28e
 | ||
| Fig. 5 Solid-state structure of heteroleptic magnesium complex 5. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°): molecule 1: Mg1–N1 2.070(2), Mg1–N2 2.200(2), Mg1–C25 2.185(3), Mg1–O1 2.2040(19), Mg1–O2 2.225(2), C1–N1 1.353(3), C1–C2 1.392(4), C2–C3 1.399(3), C3–C4 1.399(3), C4–C5 1.426(3), C4–N1 1.385(3), C5–N2 1.303(3), N2–C6 1.494(3), C6–C7 1.541(3), C25–C26 1.464(4); N1–Mg1–N2 80.98(8), O1–Mg1–O2 173.46(7), N1–Mg1–O1 89.79(8), N1–Mg1–O2 92.71(8), N2–Mg1–O1 84.74(7), N2–Mg1–O2 89.68(7), N1–Mg1–C25 123.72(10), O1–Mg1–C25 92.51(10), O2–Mg1–C25 91.13(10), N2–Mg1–C25 155.19(10), C1–N1–C4 105.8(2), N1–C4–C5 119.0(2), C4–C5–N2 121.2(2), C5–N2–C6 120.02(19), C4–N1–Mg1 110.34(15), C5–N2–Mg1 108.06(15). | ||
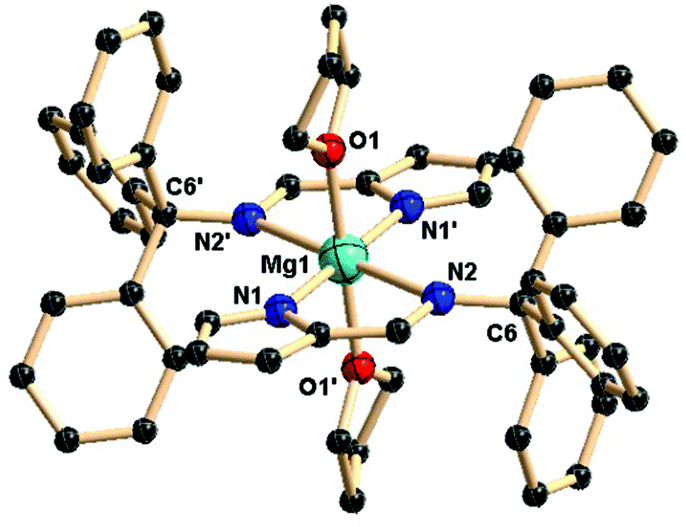 | ||
| Fig. 6 Solid-state structure of homoleptic magnesium complex 6. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°): Mg1–N1 2.0813(14), Mg1–N2 2.5422(14), Mg1–O1 2.1517(12), C1–N1 1.343(2), N1–C4 1.383(2), C4–C5 1.428(2), C5–N2 1.295(2), N2–C6 1.5030(19); N1–Mg1–N1i 180.0, N2–Mg1–N2i 180.0, O1–Mg1–O1i 180.0, N1–Mg1–O1 89.87(5), N2–Mg1–O1 90.80(4), N1–Mg1–N2 75.72(5), N1–Mg1–N2i 104.28(5), C1–N1–C4 104.92(13), N1–C4–C5 121.60(14), C4–C5–N2 122.89(15), C5–N2–Mg1 103.64(10). | ||
The slightly elongated Mg1–N2 distance of 2.200(2) Å in complex 5 represents the coordination bond between the imine nitrogen and the magnesium ion. The Mg–N bond distances also agree well with the Mg–Nimine bond distances [2.194(16) Å for [{C4H3N(2-CH2NMe2)}Mg{N(SiMe3)2}]2; 2.225(10) Å for [{C4H3N(2-CH2NEt2)}Mg{N(SiMe3)2}]2 reported by Ting-Yu Lee et al.38 In contrast, the Mg–Nimine bond length of 2.5422(14) Å in complex 6 is longer than the Mg–Nimine distance of 2.200(2) Å observed in complex 5 and literature reports.28e,37–39 The elongated Mg–Nimine bond length in complex 6 can be explained by the steric congestion–created moiety around the magnesium ion by the presence of two bulky triphenyl groups attached to the imine nitrogen atoms of the bulky iminopyrrolyl. In complex 5, the coordinating nitrogen atoms N1 and N2 formed a bite angle of N1–Mg1–N2 80.98(8)° with the magnesium ion. However, the bite angle of N1–Mg1–N2 75.72(5)° is slightly reduced in complex 6. In complex 5, the Mg1–C25 bond distance of 2.185(3) Å is in good agreement with the Mg–C bond distances of 2.1697(17) Å in [(tmeda)Mg(CH2Ph)2] and 2.1325(18) Å in [η2-HC{C(CH3)NAr′}2Mg(CH2Ph)(thf)] (Ar′ = 2,6-diisopropylphenyl) observed and reported by P. J. Bailey et al.40 In complexes 5 and 6, the Mg–O bond distances of 2.204 and 2.225 Å (for 5) and 2.1517(12) Å (for 6) fit well with the values we previously reported.28e,38,39 For complex 5, a five-membered magnesium metallacycle N1–C4–C5–N2–Mg1 and for complex 6, two five-membered metallacycles, N1–C4–C5––N2–Mg1 and N1i–C4i–C5i–N2i–Mg1, are observed due to the κ2-NN coordination of the iminopyrrolyl ligand 1.
The heavier alkaline earth metal complexes of composition [M(2-(Ph3CNCH)C4H3N)2(THF)n] [M = Ca (7), Sr (8), and n = 2; M = Ba (9), n = 3] were synthesized using two methods. In the first method, the bulky iminopyrrolyl ligand 1-H was directly charged with the corresponding alkaline earth metal bis(trimethylsilyl)amides [M{N(SiMe3)2}2(THF)n] (where M = Ca, Sr, and Ba) in 2:1 molar ratio in THF solvent at ambient temperature. The same alkaline earth metal complexes 7–9 were also obtained using the salt metathesis reaction involving the treatment of potassium salt 4 with the corresponding alkaline earth metal diiodides MI2 (M = Ca, Sr and Ba) in 2:1 molar ratio in THF solvent (Scheme 3).33
 | ||
| Scheme 3 Synthesis of heavier alkaline earth metal complexes 7–9. | ||
In the 1H NMR spectra, each of the complexes 7–9 shows a sharp singlet resonance at δ 7.95 (for 7), 8.04 (for 8), and 7.89 (for 9) ppm, indicating the presence of the imine –C–H proton in the metal complexes, which is slightly downfield shifted compared to the free ligand (7.67 ppm). The coordinated THF molecules can be easily recognized by the 1H NMR spectra as two multiplets centered at 3.61 and 1.76 ppm (for 7), 3.38 and 1.17 ppm (for 8), and 3.56 and 1.40 ppm (for 9). One set of resonance signals was observed for the aromatic ring protons in each metal complex, indicating dynamic behavior in the solution state. The solid-state structures of complexes 7–9 were established through single-crystal X-ray diffraction analysis. The centro-symmetric calcium complex 7 crystallizes in the monoclinic space group P21/n, with two molecules in the unit cell. In contrast, both the strontium and barium complexes 8 and 9 crystallize in the non-centro-symmetric triclinic space group P, with two molecules each in their respective unit cells. The details of the structural parameters are given in TS1 in the ESI.† The molecular structures of complexes 7–9 are shown in Fig. 7a–c respectively. The selected bond lengths and bond angles of complexes 7–9 are given in Table 1. The calcium complex 7 is iso-structural to the corresponding magnesium complex 6, in which the central calcium ion is surrounded by two anionic iminopyrrolyl ligands and two THF molecules trans to each other. Each ligand moiety coordinates to the metal center through the Npyrrolyl and Nimine atoms, and forms two five-membered metallacycles, N1–C4–C5–N2–Ca1 and N1i–C4i–C5i–N2i–Ca1, with a bite angle of 71.76(13)°. In complex 8, the strontium ion was ligated by two chelating bulky iminopyrrolyls and two THF molecules. However, the two THF molecules are cis to each other. In both complexes 7 and 8, the geometry around the central metal ion can be best described as distorted octahedral.
 | ||
| Fig. 7 Solid-state structures of complexes 7 (a), 8 (b), and 9 (c). All hydrogen atoms are omitted for clarity. | ||
| Ca (7) | Sr (8) | Ba (9) | |
|---|---|---|---|
| Bond lengths (Å) | |||
| M–N1pyrrolyl | 2.423(4) | 2.570(5) | 2.731(5) |
| 2.546(6) | 2.762(5) | ||
| M–N2imine | 2.567(4) | 2.677(5) | 2.946(5) |
| 2.679(5) | 2.933(5) | ||
| M–O1 | 2.361(4) | 2.621(5) | 2.812(5) |
| M–O2 | 2.361(4) | 2.593(5) | 2.842(4) |
| M–O3 | — | — | 2.830(4) |
| C4–N1pyrrolyl | 1.400(5) | 1.382(8) | 1.348(8) |
| C5–N2imine | 1.291(8) | 1.304(7) | |
| Bond angles (°) | |||
| N1–M–N2 | 71.76(13) | 68.35(15) | 63.78(14) |
| 68.32(16) | 63.14(15) | ||
| N1–M–N3 | — | 154.21(18) | 166.32(15) |
| N1–M–N4 | — | 129.28(16) | 127.40(15) |
| O1–M–O2 | 180.00(12) | 89.05(17) | 134.20(14) |
| 65.90(14) | |||
| 68.76(15) | |||
| O1–M–N1 | 86.72(14) | 84.62(17) | 89.67(16) |
| 80.22(17) | 84.29(16) | ||
| O1–M–N2 | 89.32(15) | 78.967(7) | 75.024(5) |
In complex 7, the Ca–Npyr bond distance of 2.423(4) Å and Ca–Nimin bond distance of 2.567(4) Å are in good agreement with the Ca–N bond distances reported for the complexes of composition [(ImpDipp)2Ca(THF)2] [Ca–N1 2.422(2) Å; Ca–N3 2.393(2) Å and Ca–N2 2.526(2) Å; Ca–N4 2.534(2) Å] and [(ImpDipp)Ca(N(SiMe3)2)(THF)2] [Ca–N1 2.388(2) Å; Ca–N3 2.312(2) Å, Ca–N2 2.467(2) Å], and [(ImpMe)2Ca(THF)2] [Ca–N1 2.398(4) Å; Ca–N2 2.448(3) Å] (where ImpDipp = 2-(2,6-C6H3iPr2CNCH)C4H3N and ImpMe = 2-(2,6-C6H3Me2-CNCH)C4H3N).27 The Ca1–O1 bond distance of 2.361(4) Å is in the range of normal Ca–O bonds.41 In complex 8, the Sr–Npyr bond distances of Sr1–N1 2.570(5) Å and Sr1–N3 2.546(6) Å are relatively longer than the corresponding value [2.423(4) Å] observed for complex 7 due to the larger ion radius of strontium when compared to calcium. The Sr–Nimine bond distances [Sr1–N2 2.677(5) Å and Sr1–N4 2.679(5) Å] are also longer than the value [2.567(4) Å] observed in complex 7. However, these values are in good agreement with the strontium–nitrogen bond distances [2.6512(2) and 2.669(2) Å] reported previously for the strontium complex [(ImpDipp)2Sr(THF)3] (ImpDipp = (2,6-iPr2C6H3NCH)–C4H3N).27 In the strontium complex 8, each monoanionic bidentate chelate ligand forms a five-membered metallacycle with the strontium atom N1–C4–C5–N2–Sr1 with a bite angle of 68.35(15)° and N3–C28–C29–N4–Sr1 with a bite angle of 68.32(16)°. The two planes containing the N1, N2, Sr1 and N3, N4, Sr1 atoms are almost orthogonal to each other with a dihedral angle of 85.02°. The Sr–O bond distances, Sr1–O1 2.621(5) Å and Sr1–O2 2.593(5) Å, are in the range of normal Sr–O bonds.41
In the barium complex 9, the Ba–Npyr bond distances [Ba1–N1 2.731(5) Å; Ba1–N3 2.762(5) Å] are the longest among all the complexes 7–9 due to the largest ionic radius of Ba2+ among three metal ions. However, these values fit well to the Ba–N bond distances reported for the complexes of composition [Ba((Dipp)2DAD)(μ-I)(THF)2]2 [2.720(4) and 2.706(4) Å].21 The Ba–Nimine bond distances of Ba1–N2 2.946(5) Å and Ba1–N4 2.933(5) Å are slightly longer than the Ba–N distances reported for the complexes of composition [(ImpDipp)2Ba(THF)2] [Ba–N1 2.821(5) Å and (Ba–N2 2.823(4) Å] and [Ba((Dipp)2DAD)(μ-I)(THF)2]2 (2.720(4) and 2.706(4) Å] (where ImpDipp = 2-(2,6-C6H3iPr2–CNCH)–C4H3N).27,42 The Ba–N bond distances are also comparable to the barium complexes we reported previously.28 Each ligand moiety is ligated to the barium ion through the Npyr and Nimine atoms to form two five-membered metallacycles N1–C1–C5–N2–Ba1 with a bite angle of 63.78(14)° and N3–C28–C29–N4–Ba1 with a bite angle of 63.14(15)°. A dihedral angle of 87.08° between two planes having N1, N2, and Ba1 and N3, N4, and Ba1 atoms indicates the orthogonal arrangement of two five-membered metallacycles to each other. The Ba–O bond distances of Ba1–O1 2.812(5), Ba1–O2 2.842(4), and Ba1–O3 2.830(4) Å are in the range of normal Ba–O bonds reported in the literature.41
ROP studies on ε-CL
A series of alkaline-earth metal complexes supported by a bulky iminopyrrolyl ligand were studied as initiators for the living ROP of ε-CL. The typical ring-opening polymerization process is depicted in Scheme 4. We mostly concentrated on living ROP and not on immortal ROP of ε-CL in order to understand the influence of steric bulk on rate of polymerization and the influence of the nature of the metal centre. In the living ROP of cyclic esters, the metal complex acts an initiator, that is, each metal center produces only one polymer chain. On the other hand, an immortal ROP is performed upon addition of a large excess of a protic agent (typically an alcohol) acting as an exogenous initiator and a chain transfer agent, and the complex acts as a catalyst: if the transfer between growing and dormant macromolecules is fast and reversible, the number of polymer chains generated per metal center is equal to the [transfer agent]0-to-[metal]0. The selected data obtained with alkaline-earth metal complexes (5–9) as initiators for living ROP of ε-CL are shown in Table 2. The catalytic efficiency of newly synthesized heteroleptic and homoleptic magnesium complexes 5 and 6 to promote ROP of ε-CL was first evaluated (Table 2, entries 1–6). Indeed, although some previously reported studies on similar magnesium complexes having less-bulky iminopyrrolyls in their coordination sphere gave poor results under similar polymerization conditions,27 our preliminary investigations on magnesium complexes 5 and 6 showed that they are active in the ROP of ε-CL at 25 °C in toluene with conversion over 90% within 15 minutes (Table 2, entries 1–6).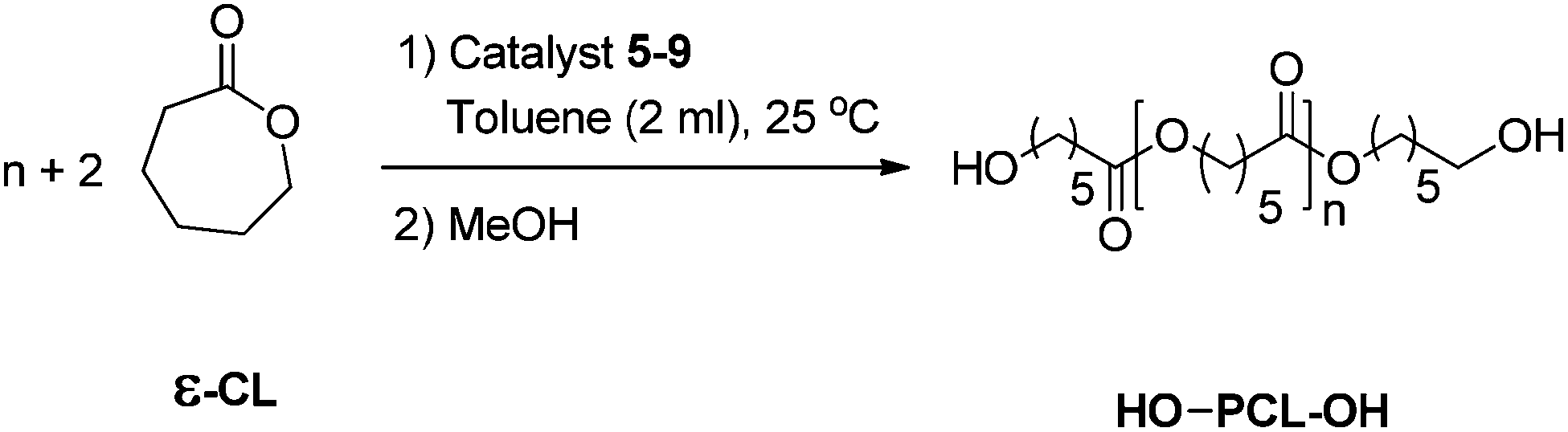 | ||
| Scheme 4 ROP of ε-CL initiated by alkaline earth metal complexes 5, 7–9. | ||
| Entry | Cat. [M] | [εCL]0/[M]0 | Reac. timeb [min] | Conv.c [%] | M n(theo) [g mol−1] | M n(GPC) [g mol−1] | M w(GPC) [g mol−1] |
M
w/Mnf |
|---|---|---|---|---|---|---|---|---|
| a Results are representative of at least two experiments. b Reaction times were not necessarily optimized. c Monomer conversions were determined by 1H NMR spectroscopy. d Theoretical molar mass values calculated from the relation: [monomer]0/[M]0 × monomer conversion where [M]0 = 8.76 × 10−3 mmol and monomer weight of ε-CL = 114 g mol−1. e Experimental molar masses were determined by GPC versus polyethylene glycol standards. f Molar mass distribution was calculated from GPC. g These data have been included for comparison in ROP with the alkyl magnesium43,44 analogues. | ||||||||
| 1 | 5 | 200/1 | 10 | 90 | 19826 |
21141 |
31440 |
1.48 |
| 2 | 5 | 400/1 | 10 | 87 | 34847 |
21147 |
31875 |
1.50 |
| 3 | [Mg(CH2SiMe3)(K2-η5-bpzcp)]g | 5000/1 | 10 | 65 | 2233 | — | 151000 |
1.45 |
| 4 | [Mg(CH2SiMe3)(tbpamd)]g | 500/1 | 1 | 97 | 3320 | — | 52000 |
1.41 |
| 5 | 6 | 200/1 | 15 | 89 | 17824 |
13781 |
21994 |
1.59 |
| 6 | 6 | 400/1 | 15 | 91 | 36449 |
39622 |
72653 |
1.83 |
| 7 | 7 | 150/1 | 5 | 96 | 14419 |
17263 |
26204 |
1.51 |
| 8 | 7 | 200/1 | 5 | 92 | 18425 |
22538 |
37073 |
1.64 |
| 9 | 7 | 300/1 | 10 | 94 | 28238 |
32219 |
50483 |
1.56 |
| 10 | 7 | 400/1 | 10 | 92 | 39613 |
60613 |
97678 |
1.61 |
| 11 | 7 | 500/1 | 15 | 95 | 48065 |
85808 |
138365 |
1.61 |
| 12 | 8 | 100/1 | 5 | 99 | 11896 |
11664 |
12173 |
1.04 |
| 13 | 8 | 200/1 | 5 | 97 | 20397 |
25190 |
34481 |
1.36 |
| 14 | 8 | 300/1 | 5 | 94 | 32944 |
36973 |
55396 |
1.49 |
| 15 | 8 | 400/1 | 10 | 91 | 35087 |
76030 |
93067 |
1.22 |
| 16 | 8 | 500/1 | 10 | 93 | 50288 |
86307 |
137623 |
1.59 |
| 17 | 9 | 100/1 | 5 | 99 | 12887 |
12480 |
13076 |
1.04 |
| 18 | 9 | 200/1 | 5 | 98 | 20608 |
36990 |
46567 |
1.25 |
| 19 | 9 | 300/1 | 5 | 95 | 32343 |
41739 |
55528 |
1.33 |
| 20 | 9 | 400/1 | 10 | 96 | 40374 |
73272 |
105361 |
1.43 |
| 21 | 9 | 600/1 | 10 | 98 | 61824 |
107891 |
139168 |
1.28 |
The molar mass distribution PDI values obtained from GPC analysis are narrow (PDI < 1.8, for entries 1–6) and controlled molecular weight distribution was observed. We noticed that the heteroleptic Mg complex (5) is more active than the homoleptic Mg complex (6). The difference in reactivity could be understood by the initiation steps in both the cases. In the case of complex 5 polymerization follows a nucleophilic route and is initiated by the transfer of an alkyl ligand to the monomer, with cleavage of the acyl–oxygen bond and formation of a metal alkoxide-propagating species.43 A similar mechanism was also suggested by the A. M. Rodrıguez group for the magnesium complex of composition [Mg(CH2SiMe3)(κ2-η5-bpzcp)] (where bpzcp = 2,2-bis(3,5-dimethylpyrazol-1-yl)-1,1-diphenylethyl cyclopentadienyl) as an initiator for the living ROP of ε-CL.44 The results obtained therein (PDI < 1.5 with controlled molecular weights) are comparable to our observations (see Table 2) suggesting that the ligand steric bulk and the nature of the metal centre play a crucial role in the ROP of ε-CL. The calcium complex 7 also showed comparable activity towards the ROP of ε-CL with magnesium analogues (5 and 6) with narrow PDI values and controlled molecular weight distributions (Table 2, entries 7–11). Indeed, the sluggish reactivity of the calcium complexes is very similar to that observed in some previously reported studies using other calcium complexes for ROP of ε-CL,45,46 we have noted living polymerization characteristics at room temperature without using any initiating agent like alcohol (entry 9, PDI = 1.5 and Mw = 50483) indicating that the triphenylmethyl group on the ligand backbone strongly influences the activity of calcium complexes towards the ROP of ε-CL. We anticipated that strontium (8) and barium (9) complexes could be more active than those of magnesium and calcium complexes having bulky iminopyrrolyls due to the larger ionic radii of Sr2+ and Ba2+ ions.47,48 Both strontium and barium analogues showed higher reactivity towards the conversion of ε-caprolactone to poly-caprolactone and up to 600 ε-CL units were successfully converted in high yields (90 to 98%) within 5–10 minutes at 25 °C. The control over the ROP process was rather good, affording PCLs, with controlled molar mass values, as well as very narrow dispersity data (PDI < 1.4, entries 12–21). Therefore, the overall catalytic efficiency of ring-opening polymerization by heavier alkaline-earth metal complexes (Sr2+ and Ba2+) supported by sterically hindered iminopyrrolyl ligands was much better and afforded poly-caprolactone with controlled molecular weights and narrow PDI values. From the 1H NMR spectrum of low-molecular weight PCL by 9 (run 17), we found resonance signals assignable to a terminal iminopyrrolyl group (Fig. S19†), indicating that in the case of amido complexes of alkaline-earth metal complexes (6–9) the initial step of the polymerization was a nucleophilic attack of the pyrrolyl nitrogen atom towards the carbonyl carbon of the monomer followed by acyl-oxygen cleavage.
Experimental
General consideration
All manipulations of air-sensitive materials were performed with the rigorous exclusion of oxygen and moisture in flame-dried Schlenk-type glassware either on a dual manifold Schlenk line, interfaced to a high vacuum (10−4 Torr) line, or in an argon-filled M. Braun glove box. THF was pre-dried over Na wire and distilled under nitrogen from sodium and benzophenone ketyl prior to use. Hydrocarbon solvents (toluene and n-pentane) were distilled under nitrogen from LiAlH4 and stored in the glove box. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Bruker Avance III-400 spectrometer. Bruker Alpha FT-IR was used for FT-IR measurements. Elemental analyses were performed on a Bruker Euro EA at the Indian Institute of Technology Hyderabad and GPC measurements were performed on a Shimadzu LCsolution GPC instrument with polyethylene glycol standards at Graduate School of Engineering Science, Osaka University Japan. Alkaline earth metal diiodides (MgI2, CaI2, SrI2, and BaI2), [NaN(SiMe3)2], [KN(SiMe3)2], pyrrole-2-carboxyaldehyde, tritylamine, and ε-CL were purchased from Sigma Aldrich and used as such. The alkaline earth metal bis(trimethylsilyl)amides [M{N(SiMe3)2}2−(THF)n], (M = Ca, Sr, Ba), [Mg(CH2Ph)2−(OEt2)2] and LiCH2SiMe3 were prepared according to procedures prescribed in the literature.30,49,50 The NMR solvent C6D6 and CDCl3 were purchased from Sigma Aldrich and dried under either Na/K alloy (for C6D6) or a molecular sieve prior to use.Preparation of [2-(Ph3CN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH)C4H3NH] (1-H)
CH)C4H3NH] (1-H)
To a dried methanol solution (10 mL) of pyrrole-2-carboxyaldehyde (2.0 g, 21.0 mmol), methanol solution (10 mL) of tritylamine (5.45 g, 21.0 mmol) and a catalytic amount of glacial acetic acid (0.25 mL) were added under stirring. The reaction mixture was stirred for another 12 h at room temperature. The solution was filtered and the solid was washed with cold methanol (5 mL). The compound was dissolved in n-hexane (5 mL) and the solvent was evaporated under reduced pressure to afford the final product as an off-white powder. Re-crystallization from hot toluene gave the crystalline product in 79% yield (5.62 g). 1H NMR (400 MHz, CDCl3): δ 9.56 (br, 1H, N-H), 7.71 (s, 1H, NC–H), 7.35–7.28 (m, 15H, CPh3), 6.96 (d, 1H, 5-pyr), 6.45 (d, 1H, 3-pyr), 6.29 (m, 1H, 4-pyr) ppm. 13C NMR (100 MHz, CDCl3): δ 150.2 (NCH), 145.9 (ArC), 131.0 (2-pyr), 129.8 (o-ArC), 127.7 (m-ArC), 126.7 (p-ArC), 121.5 (5-pyr), 114.4 (3-pyr), 110.0 (4-pyr), 77.8 (CPh3) ppm. FT-IR (selected frequencies, ν): 3445 (br, N–H), 3025 (w, ArC–H), 1629 (s, CN) cm−1. Elemental Analysis: C24H20N2 (336.42): Calcd C 85.68, H 5.99, N 8.33. Found C 85.42, H 5.62, N 8.19.
Preparation of [{2-(Ph3CNCH)C4H3N}Li(THF)2] (2)
To a THF solution (2 mL) of LiCH2SiMe3 (50 mg, 0.53 mmol), a THF solution (5 mL) of 1 equivalent of ligand 1-H (178.6 mg, 0.53 mmol) was added dropwise at room temperature under continuous stirring. The mixture was then stirred for 3 h. The solution was kept under reduced pressure to remove the volatile SiMe4 and solvent to give a light yellow product. The yellow residue was washed with n-pentane (3 mL) and dried in vacuo to afford complex 2 in 85% yield (220.0 mg). Single crystals suitable for X-ray analysis were grown from the THF/n-pentane mixture (1:2) at −35 °C in one day. 1H NMR (400 MHz, C6D6): δ 8.04 (s, 1H, NC–H), 7.13–7.11 (m, 9H, CPh3), 7.06 (s, 1H, 5-pyr), 6.98–6.94 (m, 9H, CPh3), 6.43 (d, 1H, 3-pyr), 6.23 (d, 1H, 4-pyr), 3.38–3.36 (m, THF), 1.27–1.23 (m, THF) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 147.9 (NCH), 130.4 (ArC), 129.4 (2-pyr), 128.3 (o-ArC), 128.1 (m-ArC), 128.0 (p-ArC), 126.6 (5-pyr), 125.7 (3-pyr), 111.6 (4-pyr), 78.5 (CPh3), 67.8 (THF), 25.4 (THF) ppm. FT-IR (selected frequencies, ν): 3025 (w, ArC–H), 1629 (s, CN) cm−1. Elemental Analysis: C32H35LiN2O2 (486.29): Calcd C 78.99, H 7.25, N 5.76. Found C 78.64, H 6.98, N 5.56.
Preparation of [{2-(Ph3CNCH)C4H3N}Na(THF)]2 (3)
To a THF solution (5 mL) of ligand 1-H (300 mg, 0.89 mmol), a solution of 5 mL THF and one equivalent of sodium bis(trimethylsilyl)amide (163.5 mg, 0.89 mmol) was added dropwise, with stirring, at room temperature. Stirring was continued for another 12 h and the volatile compounds were then removed under reduced pressure. The title compound was obtained as a white solid, which was further purified by washing with n-pentane (5 mL). Single crystals suitable for X-ray diffraction analysis were obtained from the THF/n-pentane mixture (1:2) solvent at −35 °C after one day. 91% Yield (350.0 mg). 1H NMR (400 MHz, C6D6): δ 8.03 (s, 1H, NC-H), 7.17–7.15 (m, 6H, CPh3), 7.02 (s, 1H, 5-pyr), 6.98–6.91 (m, 9H, CPh3), 6.49 (d, 1H, 3-pyr), 6.27 (d, 1H, 4-pyr), 3.27–3.24 (m, THF), 1.21–1.18 (m, THF) ppm. 13C NMR (100 MHz, C6D6): δ 147.8 (NCH), 145.9 (ArC), 130.2 (2-pyr), 128.3 (o-ArC), 128.1 (m-ArC), 127.8 (p-ArC), 126.8 (5-pyr), 119.1 (3-pyr), 111.1 (1C, 4-pyr), 78.3 (CPh3), 67.8 (THF), 25.6 (THF) ppm. FT-IR (selected frequencies, ν): 3025 (w, ArC–H), 1629 (s, CN) cm−1. Elemental Analysis: C56H54N4Na2O2 (861.01): Calcd C 78.12, H 6.32, N 6.51. Found C 77.94, H 5.99, N 6.38.
Preparation of [{2-(Ph3CNCH)C4H3N}K(THF)0.5]4 (4)
To a THF solution (5 mL) of ligand 1-H (300 mg, 0.89 mmol), one equivalent of potassium bis(trimethylsilyl)amide (177.8 mg, 0.89 mmol) was added dropwise, with stirring, at room temperature. Stirring was continued for another 12 h and the volatile compounds were then removed under reduced pressure. The title compound was obtained as a white solid, which was further purified by washing with n-pentane. Single crystals suitable for X-ray diffraction analysis were obtained from the THF/n-pentane mixture (1:2) at −35 °C after one day. 95% Yield (380.5 mg). 1H NMR (400 MHz, C6D6): δ 8.17 (s, 1H, NC–H), 7.18–7.01 (m, 15H, CPh3), 7.11 (s, 1H, 5-pyr), 6.74 (m, 1H, 3-pyr), 6.58 (m, 1H, 4-pyr), 3.22–3.19 (m, THF), 1.22–1.19 (m, THF) ppm. 13C NMR (100 MHz, C6D6): δ 147.9 (NCH), 137.2 (2-pyr), 130.2 (ArC), 128.8 (o-ArC), 128.1 (m-ArC), 127.9 (p-ArC), 126.8 (5-pyr), 122.4 (3-pyr), 111.3 (4-pyr), 79.2 (CPh3), 68.1 (THF), 25.4 (THF) ppm. FT-IR (selected frequencies, ν): 3026 (w, ArC–H), 1630 (s, CN) cm−1. Elemental Analysis: C104H92K4N8O2 (1642.26): Calcd C 76.06, H 5.65, N 6.82. Found C 75.88, H 5.32, N 6.51.
Preparation of [{2-(Ph3CNCH)C4H3N}–{PhCH2}Mg(THF)2] (5)
In a 25 mL of Schlenk flask one equivalent of ligand 1-H (100 mg, 0.297 mmol) was dissolved in 10 mL of toluene. To this solution, one equivalent of [Mg(CH2Ph)2(Et2O)2] (105.4 mg, 0.297 mmol) in toluene (5 mL) was added dropwise at room temperature. The reaction mixture was stirred for 6 h and the solvents were then removed under reduced pressure. The colorless residue was washed with n-pentane twice (5 mL) and crystals suitable for X-ray analysis were grown from the THF/n-pentane (1:2) mixture. Yield 160.5 mg (90%). 1H NMR (400 MHz, C6D6): δ 7.91 (s, 1H, NC–H), 7.18–7.07 (m, 15H, CPh3), 7.02–6.96 (m, 5H, Ar–H), 6.74 (d, 1H, 5-pyr), 6.66 (m, 1H, 3-pyr), 6.56 (m, 1H, 4-pyr), 1.73 (s, 2H, CH2Ph) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 146.3 (NCH), 137.8 (ArC), 136.2 (CH2Ph), 135.3 (2-pyr), 129.1 (o-CH2Ph), 128.7 (o-ArC), 128.1 (m-CH2Ph), 127.2 (m-ArC), 126.1 (p-ArC), 125.4 (p-CH2Ph), 127.4 (5-pyr), 121.8 (3-pyr), 114.2 (4-pyr), 77.7 (CPh3), 39.3 (CH2Ph) ppm. FT-IR (selected frequencies, ν): 3025 (w, ArC–H), 1631 (s, CN) cm−1. Elemental Analysis: C39H42MgN2O2 (595.06): Calcd C 78.72, H 7.11, N 4.71. Found C 78.39, H 6.93, N 4.46.
Preparation of [{2-(Ph3CNCH)C4H3N}2Mg(THF)2] (6)
In a pre-dried 25 mL Schlenk flask potassium salt 4 (200 mg, 0.448 mmol) and MgI2 (62.30 mg, 0.224 mmol) were mixed with THF (10 mL) solvent. The reaction mixture was stirred for 12 h at room temperature and a white precipitate of KI was removed by filtration. The solvent was removed under reduced pressure to leave a white residue. The magnesium complex 6 was re-crystallized from the THF/n-pentane (1:2) mixture. Yield: 175 mg (93%). 1H NMR (400 MHz, C6D6): δ 7.66 (s, 1H, NC–H), 7.15–7.10 (m, 9H, CPh3), 7.03–6.99 (m, 6H, CPh3), 6.76 (m, 1H, 5-pyr), 6.47 (d, 1H, 3-pyr), 5.85 (m, 1H, 4-pyr), 3.56–3.52 (m, THF), 1.39–1.37 (m, THF) ppm. 13C NMR (100 MHz, C6D6): δ 145.8 (NCH), 135.6 (2-pyr), 127.9 (ArC), 126.8 (o-ArC), 126.0 (m-ArC), 124.2 (p-ArC), 119.2 (5-pyr), 115.4 (3-pyr), 114.9 (4-pyr), 77.3 (CPh3), 64.5 (THF), 22.4 (THF) ppm. FT-IR (selected frequencies, ν): 3025 (w, ArC–H), 1629 (s, CN) cm−1. Elemental Analysis: C56H54MgN4O2 (839.34): Calcd C 80.13, H 6.48, N 6.67. Found C 79.71, H 6.27, N 6.31.
Preparation of [{2-(Ph3CNCH)C4H3N}2M(THF)n] [M = Ca (7), Sr (8) and n = 2; M = Ba (9) and n = 3]
Route 1: Ligand 1-H (200 mg, 0.594 mmol) and [Ca{N(SiMe3)2}2(THF)2] (150 mg, 0.297 mmol) were dissolved in THF (5 mL). The reaction mixture was stirred for 6 h at room temperature and all volatiles were removed under reduced pressure. The remaining white solid was washed with n-pentane 95 mL) and dried in vacuo to give the calcium complex 7 as a white powder. Re-crystallization from THF/n-pentane (1:2) gave colorless crystals suitable for X-ray diffraction measurements. Yield: 241 mg (95%).
Route 2: In a pre-dried Schlenk flask potassium salt 4 (200 mg, 0.448 mmol) and CaI2 (65.8 mg, 0.224 mmol) were mixed with THF (10 mL) solvent. The reaction mixture was stirred for 12 h at room temperature and the white precipitate of KI was removed by filtration through a G-4 frit. The solvent was evaporated under reduced pressure to give a white residue. The calcium complex 7 was re-crystallized from the THF/n-pentane (1:2) mixture. Yield: 172 mg (90%). 1H NMR (400 MHz, C6D6): δ 7.95 (s, 1H, NC–H), 7.20–7.17 (m, 15H, CPh3), 6.33 (m, 1H, 5-pyr), 6.13 (s, 1H, 3-pyr), 5.89 (m, 1H, 4-pyr), 3.61–3.57 (m, THF), 1.76–1.72 (m, THF) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 157.7 (NCH), 147.1 (ArC), 136.3(2-pyr), 128.9 (o-ArC), 128.2 (m-ArC), 125.3 (p-ArC), 116.8 (5-pyr), 113.4 (3-pyr), 111.5 (4-pyr), 66.8 (CPh3), 65.4 (THF), 25.1 (THF) ppm. FT-IR (selected frequencies, ν): 3025 (w, ArC–H), 1632 (s, CN) cm−1. Elemental Analysis: C66H70CaN4O4 (999.32): Calcd C 77.46, H, 6.89, N 5.47. Found C 76.98, H 6.42, N 5.28.
Other heavier alkaline earth bis(iminopyrrolyl) complexes 8 and 9 were prepared in a manner similar to complex 7 using two routes.
Complex 8
Route 1: Yield 248 mg (92%) and Route 2: Yield 182 mg (90%). 1H NMR (400 MHz, C6D6): δ 8.04 (s, 1H, NC–H), 7.17–6.94 (m, 15H, CPh3), 6.51 (m, 1H, 5-pyr), 6.15 (m, 1H, 3-pyr), 5.92 (m, 1H, 4-pyr), 3.38–3.36 (m, THF), 1.20–1.17 (m, THF) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 163.9 (NCH), 148.1 (ArC), 137.3 (2-pyr), 130.3 (o-ArC), 128.6 (m-ArC), 127.8 (p-ArC), 122.6 (5-pyr), 116.4 (3-pyr), 111.4 (4-pyr), 79.4 (CPh3), 68.3 (THF), 25.5 (THF) ppm. FT-IR (selected frequencies, ν): 3026 (w, ArC–H), 1629 (s, CN) cm−1. Elemental Analysis: C60H62N4O3Sr (974.76): Calcd C 73.93, H 6.41, N 5.75. Found C 73.42, H 6.22, N 5.43.
Complex 9
Route 1: Yield 283 mg (93%) and Route 2: Yield 200 mg (88%). 1H NMR (400 MHz, C6D6): δ 7.89 (s, 1H, NC–H), 7.39–7.37 (m, 6H, CPh3), 7.16–7.03 (m, 9H, CPh3), 6.43 (m, 1H, 5-pyr), 6.25 (s, 1H, 3-pyr), 6.15 (m, 1H, 4-pyr), 3.58–3.55 (m, THF), 1.42–1.39 (m, THF) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 146.8 (NCH), 130.4 (ArC), 128.3 (o-ArC), 128.0 (m-ArC), 127.9 (2-pyr), 127.8 (p-ArC), 127.0 (5-pyr), 115.4 (3-pyr), 110.2 (4-pyr), 78.3 (CPh3), 67.8 (THF), 25.8 (THF) ppm. FT-IR (selected frequencies, ν): 3025 (w, ArC–H), 1629 (s, CN) cm−1. Elemental Analysis: C68H77BaN4O5 (1167.68): Calcd C 69.94, H 6.65, N 4.80. Found C 69.48, H 6.16, N 4.53.
Typical polymerization experiment
In a glove box under an argon atmosphere, the catalyst was dissolved in appropriate amount (1.0 mL) of dry toluene. ε-CL in 1.0 mL of toluene was then added along with vigorous stirring. The reaction mixture was stirred at room temperature for 5–10 minutes, after which the reaction mixture was quenched by addition of a small amount of (1.0 mL) methanol, to which a slight excess of acidified methanol was then added. The polymer was precipitated in excess methanol and it was filtered and dried under vacuum. The final polymer was then analyzed using NMR and GPC.X-Ray crystallographic studies of complexes 1a, 1d, 2b, 2c, 3a, 4a, 5a
Single crystals of compounds 2–9 were grown from a THF and n-pentane mixture at −35 °C under an inert atmosphere. Single crystals of compound 1-H suitable for X-ray measurements were obtained from CH2Cl2 at room temperature. For compounds 1–9 (except 5 and 7) a crystal of suitable dimensions was mounted on a CryoLoop (Hampton Research Corp.) with a layer of light mineral oil and placed in a nitrogen stream at 150(2) K and all measurements were made on an Agilent Supernova X-calibur Eos CCD detector with graphite-monochromatic Cu-Kα (1.54184 Å) radiation. For compounds 5 and 7, data were collected at 113(2) K and measurements were made on a Rigaku RAXIS RAPID imaging plate area detector or a Rigaku Mercury CCD area detector with graphite-monochromated Mo-Kα (0.71075 Å) radiation. Crystal data and structure refinement parameters are summarized in Table TS1 in the ESI.† The structures were solved by direct methods (SIR92)51 and refined on F2 by full-matrix least-squares methods; using SHELXL-97.52 Non-hydrogen atoms were anisotropically refined. H atoms were included in the refinement in calculated positions riding on their carrier atoms. The function minimized was [∑w(Fo2 − Fc2)2] (w = 1/[σ2(Fo2) + (aP)2 + bP]), where P = (Max(Fo2,0) + 2Fc2)/3 with σ2(Fo2) from counting statistics. The functions R1 and wR2 were (∑||Fo| − |Fc||)/∑|Fo| and [∑w(Fo2 − Fc2)2/∑(wFo4)]1/2 respectively. The Diamond-3 program was used to draw the molecules. Crystallographic data (excluding structural factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1418542–1418550.Conclusion
We have successfully prepared alkali metal complexes of the bulky iminopyrrolyl ligand and their solid-state structures reveal the flexibility and multidentate behavior of the iminopyrrolyl ligand 1. In the case of the sodium complex, we observed a dimeric structure whereas, due to lower charge density of the potassium ion, a tetranuclear structure was found in the solid state. The heteroleptic and homoleptic magnesium complexes 5 and 6 respectively were successfully synthesized and characterized using X-ray crystallography. In the solid state, complex 5 is five-fold coordinated and shows a trigonal bipyramidal geometry around the magnesium ion, whereas the magnesium ion in complex 6 adopts a octahedral arrangement due to the six-fold coordination from ligand 1 and THF molecules. The heavier alkaline earth metal complexes 7–9 were synthesized either by silylamine elimination or salt metathesis routes using [M{N(SiMe3)2}2(THF)n] or MI2 (M = Ca, Sr, and Ba) as starting materials. In the solid state, the effect of the ion radii of the central metal ions as well as the steric bulk of the ligand backbone determined the metal coordination sphere. The calcium complex 7 is centro-symmetric and adopts an octahedral geometry, whereas the strontium and barium complexes (8 and 9), due to their larger size, are non-centro-symmetric and adopt distorted octahedral and distorted pentagonal-bipyramidal geometries respectively. In addition, the M–Npyr and M–Nimine bonds are relatively longer than those of the other amido-metal complexes of barium and strontium. The bis(iminopyrrolyl)complexes of strontium (8) and barium (9) were highly active for the ROP of ε-CL, affording high molecular weight PCLs compared to the polymers produced by the calcium and magnesium complexes.Acknowledgements
This work is supported by the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), India, under project no. (SB/S1/IC/045/2013). We thank Prof. K. Mashima and Dr H. Tsurugi, Division of Chemistry, Graduate School of Engineering Science, Osaka University for providing the facility to initiate this work at Osaka University under the short-term student exchange program and measure the crystals of Mg (5) and Ca (7) compounds.Notes and references
- (a) K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed; (b) R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS; (c) A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed; (d) S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 CrossRef CAS PubMed; (e) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (f) Y. Sarazin, B. Liu, T. Roisnel, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069–9087 CrossRef CAS PubMed.
- (a) Y. Ikada and H. Tsuji, Macromol. Rapid Cummun., 2000, 21, 117–132 CrossRef CAS; (b) K. Sudehsh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503–1555 CrossRef; (c) M. Vert, Biomacromolecules, 2005, 6, 538–546 CrossRef CAS PubMed; (d) L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- (a) G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS; (b) K. M. Stridsberg, M. Ryner and A.-C. Albertsson, Adv. Polym. Sci., 2002, 157, 42–65 CrossRef; (c) A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2002, 3, 1–41 CrossRef; (d) A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- (a) K. Ito and Y. Yamashita, Macromolecules, 1978, 11, 68–72 CrossRef CAS; (b) N. Spassky, Makromol. Chem., Macromol. Symp., 1991, 42/43, 15–49 CrossRef CAS; (c) J. Okuda and I. L. Rushkin, Macromolecules, 1993, 26, 5530–5532 CrossRef CAS; (d) A. Duda and S. Penczek, Macromolecules, 1995, 28, 5981–5992 CrossRef CAS; (e) H. R. Kricheldorf and S. Eggerstedt, Macromolecules, 1997, 30, 5693–5697 CrossRef CAS; (f) A. Nakayama, N. A. Nakayama, N. Kawasaki, S. Aiba, Y. Maeda, I. Arvanitoyannis and N. Yamamoto, Polymer, 1998, 39, 1213–1222 CrossRef CAS.
- (a) T. Ouhadi, A. Hamitou, R. Jerome and P. H. Teyssié, Macromolecules, 1976, 9, 927–931 CrossRef CAS; (b) J. Heuschen, R. Jérôme and P. H. Teyssié, Macromolecules, 1981, 14, 242–246 CrossRef CAS; (c) C. Jacobs, Ph. Dubois, R. Jérôme and P. H. Teyssié, Macromolecules, 1991, 24, 3027–3034 CrossRef CAS; (d) P. Kurcok, P. Dubois, W. Sikorska, Z. Jedlinski and R. Jérôme, Macromolecules, 1997, 30, 5591–5595 CrossRef CAS; (e) D. Tian, Ph. Dubois and R. Jérôme, Macromolecules, 1997, 30, 1947–1954 CrossRef CAS; (f) M. Trollsas, J. L. Hedrick, D. Mecerreyes, Ph. Dubois, R. Jérôme, H. Ihre and A. Hult, Macromolecules, 1998, 31, 2756–2763 CrossRef; (g) H. R. Kricheldorf and S. Eggerstedt, Macromol. Chem. Phys., 1998, 199, 283–290 CAS; (h) M. Trollsas, B. Atthoff, H. Claesson and J. L. Hedrick, Macromolecules, 1998, 31, 3439–3445 CrossRef; (i) H. R. Kricheldorf and K. Hauser, Macromolecules, 1998, 31, 614–620 CrossRef CAS.
- (a) A. Hamitou, T. Ouhadi, R. Jerome and P. H. Teyssié, J. Polym. Sci., 1977, 15, 865–873 CAS; (b) M. Endo, T. Aida and S. Inoue, Macromolecules, 1987, 20, 2982–2988 CrossRef CAS; (c) R. C. Yu, C. H. Hung, J. H. Huang, H. Y. Lee and J. T. Chen, Inorg. Chem., 2002, 41, 6450–6455 CrossRef CAS PubMed; (d) D. Chakraborty and E. Y. X. Chen, Organometallics, 2003, 22, 769–774 CrossRef CAS; (e) C. T. Chen, C. A. Huang and B. H. Huang, Macromolecules, 2004, 37, 7968–7973 CrossRef CAS; (f) P. Hormnirun, E. L. Marshall, V. C. Gibson, J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed; (g) N. Nomura, T. Aoyama, R. Ishii and T. Kondo, Macromolecules, 2005, 38, 5363–5366 CrossRef CAS; (h) W. Ziemkowska, J. Kochanowski and M. K. Cyrański, J. Organomet. Chem., 2010, 695, 1205–1209 CrossRef CAS; (i) X. Pang, R. Duan, X. Li, Z. Sun, H. Zhang, X. Wang and X. Chen, RSC Adv., 2014, 4, 57210–57217 RSC.
- (a) J. Okuda and I. L. Rushkin, Macromolecules, 1993, 26, 5530–5532 CrossRef CAS; (b) Y. Takashima, Y. Nakayama, K. Watanabe, T. Itono, N. Ueyama, A. Nakamura, H. Yasuda and A. Harada, Macromolecules, 2002, 35, 7538–7544 CrossRef CAS; (c) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC.
- (a) H. Yavuz, C. Babac, K. Tuzlakoglu and E. Piskin, Polym. Degrad. Stab., 2002, 75, 431–437 CrossRef CAS; (b) A. Kowalski, J. Libiszowski, T. Biela, M. Cypryk, A. Duda and S. Penczek, Macromolecules, 2005, 38, 8170–8176 CrossRef CAS; (c) G. Jiang, I. A. Jones, C. D. Rudd and G. S. Walker, J. Appl. Polym. Sci., 2009, 114, 658–662 CrossRef CAS.
- (a) M. Vivas and J. Contreras, Eur. Polym. J., 2003, 39, 43–47 CrossRef CAS; (b) H. Y. Chen, B. H. Huang and C. C. Lin, Macromolecules, 2005, 38, 5400–5405 CrossRef CAS; (c) D. J. Darensbourg and O. Karroonnirun, Macromolecules, 2010, 43, 8880–8886 CrossRef CAS.
- (a) E. L. Marshall, V. C. Gibson and H. S. Rzepa, J. Am. Chem. Soc., 2005, 127, 6048–6051 CrossRef CAS PubMed; (b) L. F. Hsueh, N. T. Chuang, C. Y. Lee, A. Datta, J. H. Huang and T. Y. Lee, Eur. J. Inorg. Chem., 2011, 5530–5537 CrossRef CAS; (c) H. J. Fang, P. S. Lai, J. Y. Chen, S. C. N. Hsu, W. D. Peng, S. W. Ou, Y. C. Lai, Y. J. Chen, H. Chung, Y. Chen, T. C. Huang, B. S. Wu and H. Y. Chen, J. Polym. Sci., 2012, 50, 2697–2704 CrossRef CAS.
- (a) Y. Shen, Macromolecules, 1996, 29, 3441–3446 CrossRef CAS; (b) Y. Shen, Z. Shen, Y. Zhang and K. Yao, Macromolecules, 1996, 29, 8289–8295 CrossRef CAS; (c) D. Barbier-Baudry, A. Bouazza, C. H. Brachais, A. Dormond and M. Visseaux, Macromol. Rapid Commun., 2000, 21, 213–217 CrossRef CAS; (d) M. Visseauxa, C. H. Brachaisa, C. Boissonb and T. C. R. Karine, Acad. Sci. Paris, Série IIc, Chimie 3, 2000, 631–638 Search PubMed; (e) Y. Matsuo, K. Mashima and K. Tani, Organometallics, 2001, 20, 3510–3518 CrossRef CAS; (f) L. Zhang, C. Yu and Z. Shen, Polym. Bull., 2003, 51, 47–53 CrossRef CAS; (g) K. Nakano, N. Kosaka, T. Hiyamab and K. Nozaki, Dalton Trans., 2003, 4039–4050 RSC; (h) T. J. Woodman, M. Schormann, D. L. Hughes and M. Bochmann, Organometallics, 2004, 23, 2972–2979 CrossRef CAS; (i) J. Cheng, D. Cui, W. Chen, N. Hu, T. Tang and B. Huang, J. Organomet. Chem., 2004, 689, 2646–2653 CrossRef CAS; (j) H. Zhou, H. Guo, Y. Yao, L. Zhou, H. Sun, H. Sheng, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 958–964 CrossRef CAS PubMed; (k) S. Zhou, S. Wang, G. Yang, Q. Li, L. Zhang, Z. Yao, Z. Zhou and H. Song, Organometallics, 2007, 26, 3755–3761 CrossRef CAS; (l) C. E. Willans, M. A. Sinenkov, G. K. Fukin, K. Sheridan, J. M. Lynam, A. A. Trifonov and F. M. Kerton, Dalton Trans., 2008, 3592–3598 RSC; (m) M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC; (n) G. Du, Y. Wei, W. Zhang, Y. Dong, Z. Lin, H. He, S. Zhang and X. Li, Dalton Trans., 2013, 42, 1278–1286 RSC.
- (a) S. J. Mclain and N. E. Drysdale, Polym. Prepr., 1992, 174 CAS; (b) Z. Q. Shen, Y. Q. Shen, J. Q. Sun, F. Y. Zhang and Y. F. Zhang, Chin. Sci. Bull., 1994, 39(13), 1096 CAS; (c) Y. Q. Shen, Z. Q. Shen, F. Y. Zhang and Y. F. Zhang, Polym. J., 1995, 27, 59–64 CrossRef CAS.
- S. Agarwal, C. Mast, K. Dehnicke and A. Greiner, Macromol. Rapid Commun., 2000, 21, 195–212 CrossRef CAS.
- (a) M. Westerhausen, S. Schneiderbauer, A. N. Kneifel, Y. Söoltl, P. Mayer, H. Nöoth, Z. Zhong, P. J. Dijkstra and J. Feijen, Eur. J. Inorg. Chem., 2003, 3432–3439 CrossRef CAS; (b) M. S. Hill and P. B. Hitchcock, Chem. Commun., 2003, 1758–1759 RSC; (c) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48–49 RSC; (d) M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS PubMed; (e) D. J. Darensbourg, W. Choi and C. P. Richers, Macromolecules, 2007, 40, 3521–3523 CrossRef CAS; (f) D. J. Darensbourg, W. Choi, O. Karroonnirun and N. Bhuvanesh, Macromolecules, 2008, 41, 3493–3502 CrossRef CAS; (g) V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820–9827 RSC; (h) X. Xu, Y. Chen, G. Zou, Z. Mac and G. Li, J. Organomet. Chem., 2010, 695, 1155–1162 CrossRef CAS; (i) Y. Sarazin, D. Ros-ca, V. Poirier, T. Roisnel, A. Silvestru, L. Maron and J.-F. Carpentier, Organometallics, 2010, 29, 6569–6577 CrossRef CAS.
- (a) Z. Zhong, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Macromolecules, 2001, 34, 3863–3868 CrossRef CAS; (b) D. J. Darensbourg, W. Choi, P. Ganguly and C. P. Richers, Macromolecules, 2006, 39, 4374–4379 CrossRef CAS; (c) Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC; (d) M. G. Davidson, C. T. O'Hara, M. D. Jones, C. G. Keir, M. F. Mahon and G. Kociok-Köohn, Inorg. Chem., 2007, 46, 7686–7688 CrossRef CAS PubMed.
- (a) T. P. Hanusa, Chem. Rev., 1993, 93, 1023–1036 CrossRef CAS; (b) T. P. Hanusa, Coord. Chem. Rev., 2000, 210, 329–367 CrossRef CAS; (c) J. S. Alexander and K. Ruhlandt-Senge, Eur. J. Inorg. Chem., 2002, 2761–2774 CrossRef CAS; (d) W. D. Buchanan, D. G. Allis and K. Ruhlandt-Senge, Chem. Commun., 2010, 46, 4449–4465 RSC.
- M. H. Chisholm, Inorg. Chim. Acta, 2009, 362, 4284–4290 CrossRef CAS and references therein.
- (a) M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042–2043 CrossRef CAS PubMed; (b) S. Harder and J. Brettar, Angew. Chem., Int. Ed., 2006, 45, 3474–3478 CrossRef CAS PubMed; (c) M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670–9685 CrossRef CAS PubMed; (d) S. P. Sarish, S. Nembenna, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2011, 44, 157–170 CrossRef CAS PubMed and references therein.
- (a) S. Datta, P. W. Roesky and S. Blechert, Organometallics, 2007, 26, 4392–4394 CrossRef CAS; (b) S. Datta, M. T. Gamer and P. W. Roesky, Organometallics, 2008, 27, 1207–1213 CrossRef CAS.
- (a) M. Arrowsmith, M. S. Hill and G. Kociok-Köohn, Organometallics, 2009, 28, 1730–1738 CrossRef CAS; (b) M. Arrowsmith, A. Heath, M. S. Hill, P. B. Hitchcock and G. Kociok-Köohn, Organometallics, 2009, 28, 4550–4559 CrossRef CAS; (c) J. Jenter, R. Köoppe and P. W. Roesky, Organometallics, 2011, 30, 1404–1413 CrossRef CAS.
- M. Arrowsmith, M. S. Hill and G. Kociok-Köohn, Organometallics, 2011, 30, 1291–1294 CrossRef CAS.
- (a) C. Eaborn, S. A. Hawkes, P. B. Hitchcock and J. D. Smith, Chem. Commun., 1997, 1961–1962 RSC; (b) M. J. Harvey, T. P. Hanusa and V. G. Young Jr., Angew. Chem., Int. Ed., 1999, 38, 217–219 CrossRef CAS; (c) M. R. Crimmin, A. G. M. Barrett, M. S. Hill, D. J. MacDougall, M. F. Mahon and P. A. Procopiou, Chem. – Eur. J., 2008, 14, 11292–11295 CrossRef CAS PubMed; (d) A. M. Johns, S. C. Chmely and T. P. Hanusa, Inorg. Chem., 2009, 48, 1380–1384 CrossRef CAS PubMed; (e) P. Jochmann, T. S. Dols, T. P. Spaniol, L. Perrin, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2009, 48, 5715–5719 CrossRef CAS PubMed; (f) K. Yan, B. M. Upton, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2009, 131, 15110–15111 CrossRef CAS PubMed.
- (a) M. M. Gillett-Kunnath, J. G. MacLellan, C. M. Forsyth, P. C. Andrews, G. B. Deacon and K. Ruhlandt-Senge, Chem. Commun., 2008, 4490–4492 RSC; (b) M. Göartner, H. Göorls and M. Westerhausen, Dalton Trans., 2008, 1574–1582 RSC; (c) C. Glock, H. Göorls and M. Westerhausen, Inorg. Chem., 2009, 48, 394–399 CrossRef CAS PubMed; (d) T. K. Panda, H. Kaneko, K. Pal, H. Tsurugi and K. Mashima, Organometallics, 2010, 29, 2610–2615 CrossRef CAS.
- (a) M. Z. Westerhausen, Anorg. Allg. Chem., 2009, 635, 13–32 CrossRef CAS; (b) A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. London, Ser. A, 2010, 466, 927–963 CrossRef CAS; (c) S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed; (d) S. Kobayashi and Y. Yamashita, Acc. Chem. Res., 2011, 44, 58–71 CrossRef CAS PubMed.
- K. Mashima and H. Tsurugi, J. Organomet. Chem., 2005, 690, 4414–4423 CrossRef CAS and references cited therein.
- (a) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS; (b) G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849–850 RSC; (c) S. Matsui, M. Mitani, J. Saito, Y. Tohi, H. Makio, N. Matsukawa, Y. Takagi, K. Tsuru, M. Nitabaru, T. Nakano, H. Tanaka, N. Kashiwa and T. Fujita, J. Am. Chem. Soc., 2001, 123, 6847–6856 CrossRef CAS and references cited therein; (d) J. Tian, P. D. Hustand and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 5134–5135 CrossRef CAS PubMed and references cited therein; (e) H. Hao, S. Bhandari, Y. Ding, H. W. Roesky, J. Magull, H. G. Schmidt, M. Noltemeyer and C. Cui, Eur. J. Inorg. Chem., 2002, 1060–1065 CrossRef CAS; (f) Y. Matsuo, K. Mashima and K. Tani, Organometallics, 2001, 20, 3510–3518 CrossRef CAS; (g) C. Cui, A. Shafir, C. L. Reeder and J. Arnold, Organometallics, 2003, 22, 3357–3359 CrossRef CAS.
- T. K. Panda, K. Yamamoto, K. Yamamoto, H. Kaneko, Y. Yang, H. Tsurugi and K. Mashima, Organometallics, 2012, 31, 2268–2274 CrossRef CAS.
- (a) R. K. Kottalanka, K. Naktode, S. Anga, H. P. Nayek and T. K. Panda, Dalton Trans., 2013, 42, 4947–4956 RSC; (b) R. K. Kottalanka, S. Anga, K. Naktode, P. Laskar, H. P. Nayek and T. K. Panda, Organometallics, 2013, 32, 4473–4482 CrossRef CAS; (c) R. K. Kottalanka, P. Laskar, K. Naktode, B. S. Mallik and T. K. Panda, J. Mol. Struct., 2013, 1047, 302–309 CrossRef CAS; (d) R. K. Kottalanka, A. Harinath, J. Bhattacharjee, H. V. Babu and T. K. Panda, Dalton Trans., 2014, 8757–8766 RSC; (e) J. Bhattacharjee, R. K. Kottalanka, A. Harinath and T. K. Panda, J. Chem. Sci., 2014, 126, 1463–1475 CrossRef CAS; (f) R. K. Kottalanka, A. Harinath and T. K. Panda, RSC Adv., 2015, 5, 37755–37767 RSC.
- Y. Yang, Bo. Liu, K. Lv, W. Gao, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 4575–4584 CrossRef CAS.
- Y. Yang, S. Li, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 671–678 CrossRef CAS.
- S. B. Clara, D. Gomes, P. T. Suresh, L. F. Gomes, M. T. Veiros, G. N. Teresa and M. C. Oliveira, Dalton Trans., 2010, 39, 736–748 RSC.
- S. A. Carabineiro, L. C. Silva, P. T. Gomes, L. C. J. Pereira, L. F. Veiros, S. I. Pascu, M. T. Duarte, S. Namorado and R. T. Henriques, Inorg. Chem., 2007, 46, 6880–6890 CrossRef CAS PubMed.
- The bonding situation in the drawing of the ligand system is simplified for clarity.
- Q. Li, J. Rong, S. Wang, S. Zhou, L. Zhang, X. Zhu, F. Wang, S. Yang and Y. Wei, Organometallics, 2011, 30, 992–1001 CrossRef CAS.
- (a) W. T. Klooster, L. Brammer, C. J. Schaverien and P. H. M. Budzelaar, J. Am. Chem. Soc., 1999, 121, 1381–1382 CrossRef CAS; (b) W. Scherer and G. S. McGrady, Angew. Chem., Int. Ed., 2004, 43, 1782–1806 CrossRef CAS PubMed.
- (a) S. De Angelis, E. Solan, C. Flonani, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1994, 116, 5702–5713 CrossRef CAS; (b) S. De Angelis, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Chem. Soc., Dalton Trans., 1994, 2467–2469 RSC; (c) J. Jubb, S. Gambarotta, R. Duchateau and J. H. Teuben, J. Chem. Soc., Chem. Commun., 1994, 2641–2642 RSC; (d) D. Jacoby, S. Isoz, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1995, 117, 2805–2816 CrossRef CAS; (e) L. Bonomo, E. Solari, R. Scopelliti, C. Floriani and N. Re, J. Am. Chem. Soc., 2000, 122, 5312–5326 CrossRef CAS.
- J. Gao, Y. Liu, Y. Zhao, X.-J. Yang and Y. Sui, Organometallics, 2011, 30, 6071–6077 CrossRef CAS.
- L.-F. Hsueh, N.-T. Chuang, C.-Y. Lee, A. Datta, J.-H. Huang and T.-Y. Lee, Eur. J. Inorg. Chem., 2011, 5530–5537 CrossRef CAS.
- G. J. Moxey, F. Ortu, L. G. Sidley, H. N. Strandberg, A. J. Blake, W. Lewis and D. L. Kays, Dalton Trans., 2014, 43, 4838–4846 RSC.
- P. J. Bailey, R. A. Coxall, C. M. Dick, S. Fabre, L. C. Henderson, C. Herber, S. T. Liddle, D. Loroño-Gonzalez, A. Parkin and S. Parsons, Chem. – Eur. J., 2003, 9, 4820–4828 CrossRef CAS PubMed.
- (a) S. Harder, S. Müller and E. Hübner, Organometallics, 2004, 23, 178–183 CrossRef CAS; (b) O. Michel, C. Meermann, K. W. Toürnroos and R. Anwander, Organometallics, 2009, 28, 4783–4790 CrossRef CAS.
- T. K. Panda, H. Kaneko, O. Michel, H. Tsurugi, K. Pal, K. W. Toernroos, R. Anwander and K. Mashima, Organometallics, 2012, 31, 3178–3184 CrossRef CAS.
- (a) L. F. Sánchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2006, 25, 1012–1020 CrossRef; (b) L. F. Sánchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2005, 24, 5329–5334 CrossRef; (c) L. F. Sánchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2005, 24, 3792–3799 CrossRef.
- A. Garcés, L. F. Sánchez-Barba, C. Alonso-Moreno, M. Fajardo, J. Fernández-Baeza, A. Otero, A. Lara-Sánchez, I. López-Solera and A. M. Rodríguez, Inorg. Chem., 2010, 49, 2859–2871 CrossRef PubMed.
- M. Kuzdrowska, L. Annunziata, S. Marks, M. Schmid, C. G. Jaffredo, P. W. Roesky, S. M. Guillaume and L. Maron, Dalton Trans., 2013, 42, 9352–9360 RSC.
- M. G. Cushion and P. Mountford, Chem. Commun., 2011, 47, 2276–2278 RSC.
- B. Liu, T. Roisnel, J.-P. Guegan, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 18, 6289–6301 CrossRef CAS PubMed.
- Y. Sarazin, B. Liu, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069–9087 CrossRef CAS PubMed.
- M. Westerhausen, Coord. Chem. Rev., 1998, 176, 157–210 CrossRef CAS.
- D. George, K. Vaughn, K. Alex and J. A. Gladysz, Organometallics, 1986, 5, 936–942 CrossRef.
- A. Altomare, M. C. Burla, G. Camalli, G. Cascarano, C. Giacovazzo, A. Gualiardi and G. Polidori, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1418542–1418550. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03222a |
| This journal is © The Royal Society of Chemistry 2015 |