
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTin(II)-functionalization of the archetypal {P8W48} polyoxotungstate†
N. V.
Izarova
*a,
L.
Klaß
ab,
P.
de Oliveira
c,
I.-M.
Mbomekalle
c,
V.
Peters
b,
F.
Haarmann
b and
P.
Kögerler
 *ab
*ab
aJülich-Aachen Research Alliance (JARA-FIT) and Peter Grünberg Institute – PGI 6, Forschungszentrum Jülich, D-52425 Jülich, Germany. E-mail: n.izarova@fz-juelich.de; paul.koegerler@ac.rwth-aachen.de
bInstitute of Inorganic Chemistry, RWTH Aachen University, D-52074 Aachen, Germany
cEquipe d'Electrochimie et de Photoélectrochimie, Laboratoire de Chimie-Physique, Université Paris-Sud, UMR 8000 CNRS, Orsay F-91405, France
First published on 7th October 2015
Abstract
The synthesis of [K4.5 ⊂ (ClSnII)8P8W48O184]17.5−, featuring Sn(II) ions in trigonal-pyramidal SnO2Cl environment coordinating to the two inner rims of the wheel-shaped {P8W48}-type polyoxotungstate(VI) archetype, showcases how high chloride ligand concentrations as well as the control of the polyanion solubility via electrolytes and evaporation rates are essential to prevent numerous competing reactions that can hamper the Sn(II) functionalization of polyoxometalates.
Introduction
The reactivity of main group metals towards polyoxotungstates (POTs) has received relatively little attention to date in comparison with that of transition metals and lanthanides. Among such main group metals, tin is one of the most investigated elements due to interest in POM functionalization with organotin moieties with Sn in its formal +IV oxidation state.1 At the same time, examples of POTs incorporating SnII ions are still rather elusive, despite the general potential to exploit their structure-directing electron pair effect, in line with the lone pair effects that were e.g. utilized in the synthesis of As(III)-functionalized high-nuclearity POTs.2In the pioneering work in the late 1970s, Knoth explored the possibility to attach organometallic RMSnII moieties (RM = CpFe(CO)2, (OC)3Co, π-C3H5Pd, (C7H8)2Rh, etc.) to monolacunary Keggin-type POTs.3a Several years later, Chorghade and Pope thoroughly characterized a series of α/β-[XW11O39Sn]n− (X = BIII, GaIII, SiIV, GeIV, PV) and α2-[P2W17O61Sn]8− complexes where SnII centers coordinate with monovacant Keggin- and Wells–Dawson-type species.3b A related polyanion with a postulated composition [PMo2W9O39Sn]5− was reported to catalyze the oxidation of various organic sulfides to sulfoxides by H2O2.3c
In the complexes of SnII with α-A-isomers of trilacunary Keggin-type POTs two trivacant polyoxometalate (POM) ligands typically sandwich several tin(II) centers: three in [Sn3(XW9O34)2]n− (X = SiIV, PV, AsV),4 six in [Sn6(XW9O33)2]n− (X = SnII, SbIII),5 or even nine in the polymeric {(H2O)mNa}n[Cl5Sn9(XW9O34)2]n (X = SiIV, GeIV).5b In [{Sn4(SiW9O34)2}2]24− polyanions, which are catalytically active in hydrogen evolution from aqueous media, two {Sn3(SiW9O34)2} fragments are linked via two additional SnII centers into a dimer.6 In the β-B-type trilacunary POTs [Sn1.5(WO2(OH))0.5(WO2)2(XW9O33)2]12− (X = SbIII, BiIII) the SnII ions occupy the outer positions in the inner belt of the sandwich-type structure.7 SnII ions in all the above mentioned species exhibit either trigonal or tetragonal pyramidal coordination environments and possess a lone electron pair, which, if sterically accessible, can be used for further functionalization of the SnII-containing POMs via coordination with organometallic moieties.
Besides acting as an external heterometal, SnII ions can also assume the role of internal heteroatoms composing POT structures as was observed in [H3SnW18O60]7− (ref. 7) and the {SnW9O33} subunit in the above-mentioned [Sn6(SnW9O33)2]8−,5a as well as act as a linker between the intact paratungstate-B structures leading to anionic chains [{H2SnW12O42}n]8n−.8
At the same time the reactivity of SnII towards multilacunary (i.e. more reactive) derivatives of Wells–Dawson-type POTs or POMs with more complex structures remains unstudied. Therefore we decided to investigate the reaction of SnII chloride with the archetypal macrocyclic phosphotungstate [P8W48O184]40− (= {P8W48}), possessing a wheel-shaped structure composed of four identical hexalacunary {P2W12O48} Wells–Dawson fragments connected to each other via oxo-bridges. The large inner cavity of {P8W48} allows for the incorporation of a wide range of hetero-transition metal ions, as exemplified by [Cu20X(OH)24(H2O)12P8W48O184]25− (X = Cl, Br, I),9a,b [Cu20(N3)6(OH)18P8W48O184]24−,9c [K8 ⊂ {P8W48O184}-{VV4VIV2O12(H2O)2}2]24−,10a [Fe16(OH)28(H2O)4P8W48O184]20−,10b [K8 ⊂ {P8W48O184}{MoVIO2}4{MoV4O10(H2O)3}2]24−,10c and others.11 {P8W48} can also assume the role of a secondary building block in the carbon-free 3D coordination framework K18Li6[Mn8(H2O)48P8W48O184]·108H2O, exhibiting rigid cages with an approximate volume of 7.24 nm3.12
Herein we report the first example of a {P8W48} POT incorporating the main group element, namely the polyanion [K4.5 ⊂ (ClSnII)8P8W48O184]17.5− (1) that was synthesized in a one-pot reaction in an aqueous medium, isolated as the hydrated potassium/lithium salt K10Li17.5[K4.5⊂(ClSnII)8P8W48O184]·50H2O (KLi-1), and characterized by single-crystal X-ray, elemental and thermogravimetric analyses; FT-IR/Raman, UV-Vis, solid-state MAS and solution 31P NMR spectroscopy as well as cyclic voltammetry and electrochemical quartz crystal microbalance studies.
Results and discussion
Synthesis
The polyanions 1 have been synthesized by the reaction of [H7P8W48O184]33− with SnCl2 in 4 M aqueous LiCl solution at 50 °C. Alternatively heating at 50 °C for 1 h, the reaction mixture could be stirred at room temperature for 4–5 h. The large bright-orange block-shaped crystals of KLi-1 form within several days by the evaporation of the obtained solutions under a hood. During the reaction the color of the reaction mixture changes from bright-orange to brown and then to dark-green indicating partial reduction of WVI centers of {P8W48} by SnII to WV ions which are then slowly reoxidized by air oxygen. To minimize this undesirable process, lowering the yield of the product, a large excess of SnII ions should be excluded and access of the reaction mixture to air oxygen needs to be provided which is in part achieved by a sufficient surface/volume ratio of the reaction solution (that, in turn, makes difficult upscaling of the reaction volume). Prolonged heating of the reaction mixture as well as heating at higher temperatures (70–90 °C) speed up the WVI reduction process and thus should be prevented. The evaporation rate of the reaction solution also seems to play a crucial role. Thus, evaporation under a fumehood results in the rapid crystallization of KLi-1 within 1–6 days, while slower evaporation of the reaction mixture (e.g. outside a fumehood) leads to gradual decomposition of 1 with the formation of a white viscous gel-like precipitate. This product most likely represents a mixed Sn(II) chloride-hydroxide (Sn(OH)Cl), the formation of which is followed by a visible color change of the reaction mixture, which complicates the isolation of pure KLi-1. A relatively high Cl− concentration is also important as it serves as a source of terminal chloride ligands on the SnII centers in 1. Thus, the polyanion 1 cannot be crystallized from other media (e.g., LiOAc (pH 2–6), NaOAc (pH 2–6), Li2SO4 (pH 2–3) or 2 M LiCl and NaCl) where the reaction products of [H7P8W48O184]33− with SnCl2 turn out to be mixtures of hydrated K+/Li+ salts of polyanions with the general composition [(HOSnII)xP8W48O184](40−x)− with x = 3–6. Representative examples which we were able to structurally characterize are K46.75Li26.25[(HOSnII)4P8W48O184][(HOSnII)3P8W48O184]·nH2O (CSD-430081) and K11Li23[(HOSnII)6P8W48O184]·nH2O (CSD-430082), both of which exhibit correspondingly decreased crystallographic occupancy factors for the Sn positions.Crystal structure analysis
Compound KLi-1 crystallizes in tetragonal symmetry in the space group I4/m. The polyanion 1 consists of a macrocyclic phosphotungstate {P8W48} which is coordinated by eight SnII ions that are located in the inner cavity of {P8W48} at the eight vacant sites between neighboring {P2W12} subunits. Each Sn center binds to two terminal oxygen positions belonging to two adjacent {P2W12} groups (Sn–O 2.140(10)–2.152(11) Å; Fig. 1). Thus, the POM possesses an idealized D4h symmetry. The bond lengths and angles within the phosphotungstate framework are in the usual range (see Table S1†).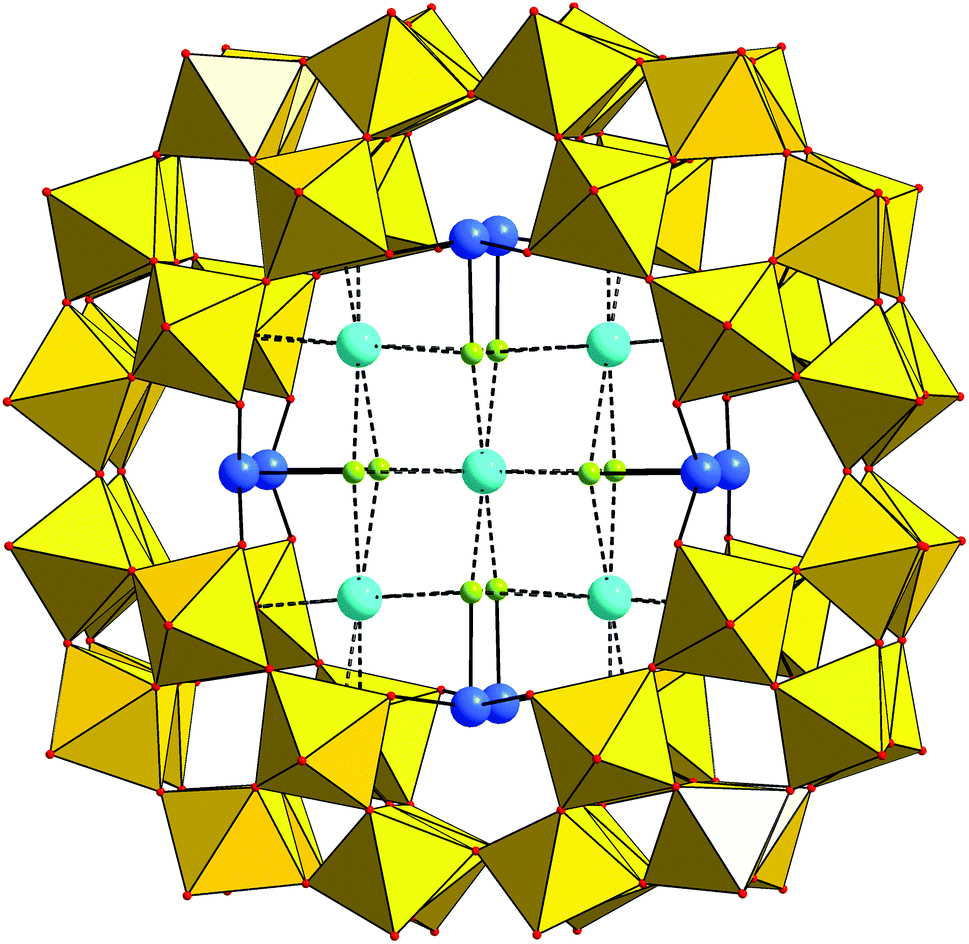 | ||
| Fig. 1 Structure of 1 in combined polyhedral/ball-and-stick representation. Color legend: WO6, yellow octahedra; Sn, blue; K, light blue; Cl, lime green; O, red spheres. The P centers are not visible in this perspective. | ||
Each SnII center resides in a trigonal pyramidal SnO2Cl coordination environment with a terminal chloride ligand (Sn–Cl 2.515(6) Å) directed towards the center of the {P8W48} ring. This trigonal-pyramidal coordination mode is reminiscent of several other anionic Sn(II) complexes such as SnCl3−, that are formed in aqueous solutions at higher halide concentrations, or SnCl2(H2O). The Sn–Cl bond lengths in 1 can be compared with those of 2.523 and 2.595 Å in Cs[SnCl3]13a and [SnCl2(H2O)]·H2O,13b respectively. The Sn–O bond distances in 1 also correlate rather well with the Sn–O bond lengths in [SnCl2(H2O)]·H2O (2.169 Å)13b and other SnII-containing POTs.3–8 The O–Sn–O and O–Sn–Cl angles are in the usual range and constitute 91.5(4)° and 85.8(3)–86.3(3)°, respectively. The lone electron pair on each SnII center is oriented towards the center of the polyanion and thus is sterically inaccessible. The closest Sn⋯Sn distances are 3.956(1) Å between the SnII centers situated on the opposite side of {P8W48} and 7.248(1) Å between the SnII centers located on the same side of the wheel.
The inner cavity of 1 is occupied by four K+ countercations each forming weak electrostatic interactions with four oxygen atoms of two phosphate groups of one of the {P2W12} units (K–O: 2.815(11)–2.828(11) Å) and four chloride ligands on the adjacent SnII centers (K–Cl: 3.278(8)–3.301(8) Å). In addition, half of the polyanions in the solid-state structure of KLi-1 incorporate a central K+ cation, which coordinates eight Cl− ligands of eight SnII ions (K–Cl: 3.155(5) Å).
According to bond valence sum calculations,14 all tungsten centers in 1 are in the oxidation state +VI and all oxygen atoms of {P8W48} are unprotonated (see Table S2†). Thus, the partial reduction of WVI centers in the {P8W48} precursor due to the reaction with SnII might explain the relatively low yield of KLi-1, the final formula of which was determined from crystal structural data, BVS calculations and elemental analysis.
To the best of our knowledge 1 is the first example of a {P8W48}-type polyanion with a main group metal, as only transition metal and lanthanide-functionalized {P8W48} derivatives have been reported to date. The main difference in SnII (vs. transition metal/lanthanide) complexation with {P8W48} is the relatively small coordination number (3) of SnII. This feature, however, limits the long-term solution stability of 1 (vide infra). On the other hand, it leaves rather a significant space inside the {Sn8P8W48} macrocycles that can potentially be used to incorporate other ligands/metals.
In the solid-state structure of KLi-1 the polyanions 1 are interlinked via K+ countercations into a 3D framework with pseudolayers of 1 packed in an ⋯ABABAB⋯ pattern (Fig. S1 and S2†) along the crystallographic c axis. A network of channels along the a and b axes with 5 × 9 Å diameter are filled by crystal water molecules.
FT-IR spectrum
The FT-IR spectrum of KLi-1 (Fig. S3†) exhibits a set of bands in the range of 1135–400 cm−1 typical of the {P8W48} framework. Thus, the positions of the bands characteristic of vibrations of P–O bonds in KLi-1 (1134, 1084 and 1022 cm−1) are almost the same as those attributed to P–O vibrations of a tin-free K/Li salt, KLi-P8W48 (1138, 1085 and 1017 cm−1), despite a small shoulder at 1120 cm−1 for KLi-1. The intense band at 926 cm−1 belongs to the stretching vibrations of the terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. The main differences between KLi-1 and KLi-P8W48 in the 850–400 cm−1 range concern W–O–W, W–O–P and W–O–Sn vibrations (716, 569, 530, 465 and 401 cm−1 for KLi-1 and 807, 686, 573, 527 and 472 cm−1 for KLi-P8W48) and, especially in the 850–550 cm−1 region, reflect the SnII coordination to {P8W48}.
O bonds. The main differences between KLi-1 and KLi-P8W48 in the 850–400 cm−1 range concern W–O–W, W–O–P and W–O–Sn vibrations (716, 569, 530, 465 and 401 cm−1 for KLi-1 and 807, 686, 573, 527 and 472 cm−1 for KLi-P8W48) and, especially in the 850–550 cm−1 region, reflect the SnII coordination to {P8W48}.
Characterization in aqueous solution
KLi-1 is soluble in water, and its solubility is significantly enhanced in the presence of Li+ ions. The addition of K+ salts to solutions of KLi-1 leads to immediate precipitation of the complex. The stability of the polyanions 1 in various Li+-based media has been investigated by 31P NMR and UV-Vis spectroscopy.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 measured in the same medium (Fig. S7†). This result indicates short-term stability of the title compound in 2 M Li2SO4/H2SO4 buffer (pH 3.0). At the same time, after several hours other signals start to appear in the spectrum evident of slow decomposition of 1 in aqueous solution via the gradual release of SnII ions. Thus, the spectrum of the same solution measured for 1 day (Fig. S8†) shows a set of additional, weaker signals at −8.1 ppm (13.6% of the total intensity of all the signals), −8.3 ppm (6.1%), −8.4 ppm (2.7%), −8.7 ppm (2.3%), −8.8 ppm (1.4%) and −10.7 ppm (6.4%) along with the original one at −8.0 ppm (67.5%). The decomposition of the polyanion in 4 M LiCl seems to be even faster, already after one hour the spectrum of KLi-1 redissolved in this medium exhibits a set of badly resolved signals in the range of −6.3 to −7.5 ppm (85%) and two singlets at −8.0 (8.8%) and −10.7 ppm (6.2%). The 31P NMR spectrum of 1 in this medium (Fig. S9†) does not change further significantly within 24 h. The signals at −8.1 and −10.7 ppm in the spectra measured in both 2 M Li2SO4 (pH 3.0) and 4 M LiCl after 24 h suggest the existence of a rather stable decomposition intermediate(s) of the {Sn8P8W48} complex, most likely of composition {SnxP8W48} with x < 8, which form via the release of one or more SnII centers from {Sn8P8W48}.
15 measured in the same medium (Fig. S7†). This result indicates short-term stability of the title compound in 2 M Li2SO4/H2SO4 buffer (pH 3.0). At the same time, after several hours other signals start to appear in the spectrum evident of slow decomposition of 1 in aqueous solution via the gradual release of SnII ions. Thus, the spectrum of the same solution measured for 1 day (Fig. S8†) shows a set of additional, weaker signals at −8.1 ppm (13.6% of the total intensity of all the signals), −8.3 ppm (6.1%), −8.4 ppm (2.7%), −8.7 ppm (2.3%), −8.8 ppm (1.4%) and −10.7 ppm (6.4%) along with the original one at −8.0 ppm (67.5%). The decomposition of the polyanion in 4 M LiCl seems to be even faster, already after one hour the spectrum of KLi-1 redissolved in this medium exhibits a set of badly resolved signals in the range of −6.3 to −7.5 ppm (85%) and two singlets at −8.0 (8.8%) and −10.7 ppm (6.2%). The 31P NMR spectrum of 1 in this medium (Fig. S9†) does not change further significantly within 24 h. The signals at −8.1 and −10.7 ppm in the spectra measured in both 2 M Li2SO4 (pH 3.0) and 4 M LiCl after 24 h suggest the existence of a rather stable decomposition intermediate(s) of the {Sn8P8W48} complex, most likely of composition {SnxP8W48} with x < 8, which form via the release of one or more SnII centers from {Sn8P8W48}.
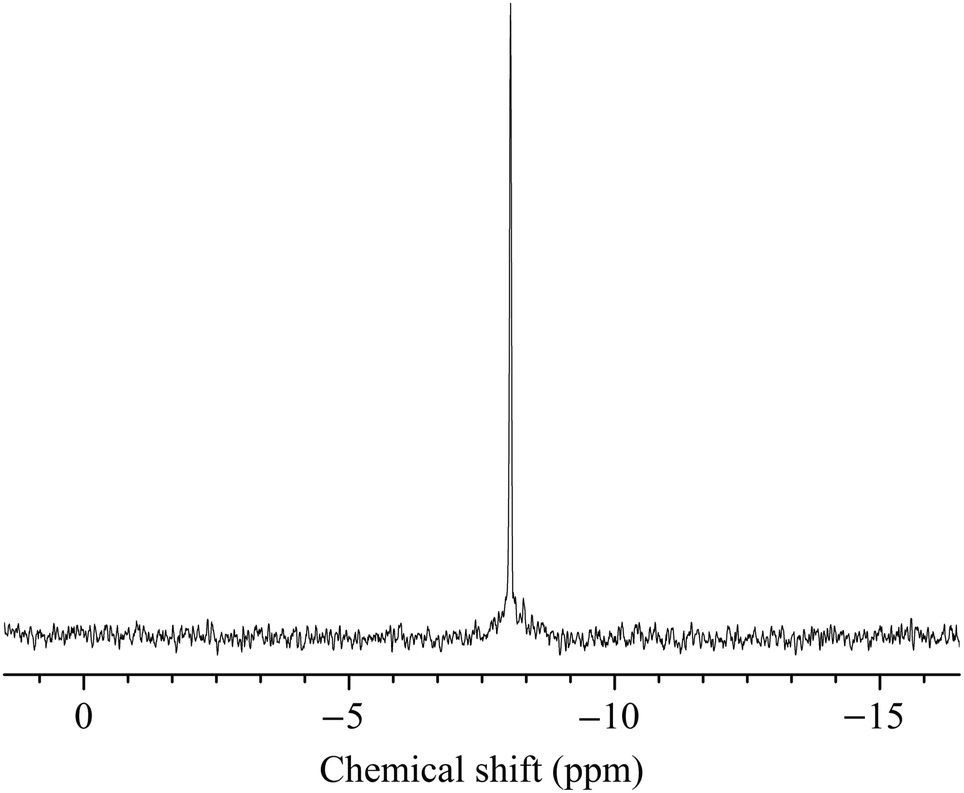 | ||
| Fig. 2 Room temperature liquid-state 31P NMR spectrum of 1 in 2 M Li2SO4/H2SO4 buffer solution (pH 3.0) after 1 h. | ||
800 M−1 cm−1) and 275 nm (ε = 97800 M−1 cm−1) and at 385 nm (ε = 9800 M−1 cm−1), responsible for the orange color of the compound (Fig. S10†). Time-dependent measurements on solutions of KLi-1 in 2 M Li2SO4/H2SO4 buffer (pH 3.0) (c = 1.0 × 10−4 M) show a gradual decrease in the intensity of the maximum at 385 nm starting after 1.5–2 h (Fig. S11 and S12†), which supports the conclusions on slow solution decomposition of the polyanions based on the 31P NMR spectroscopy data. The spectra in some other media show similar characteristics (see the Experimental section and Fig. S13 and S14†).
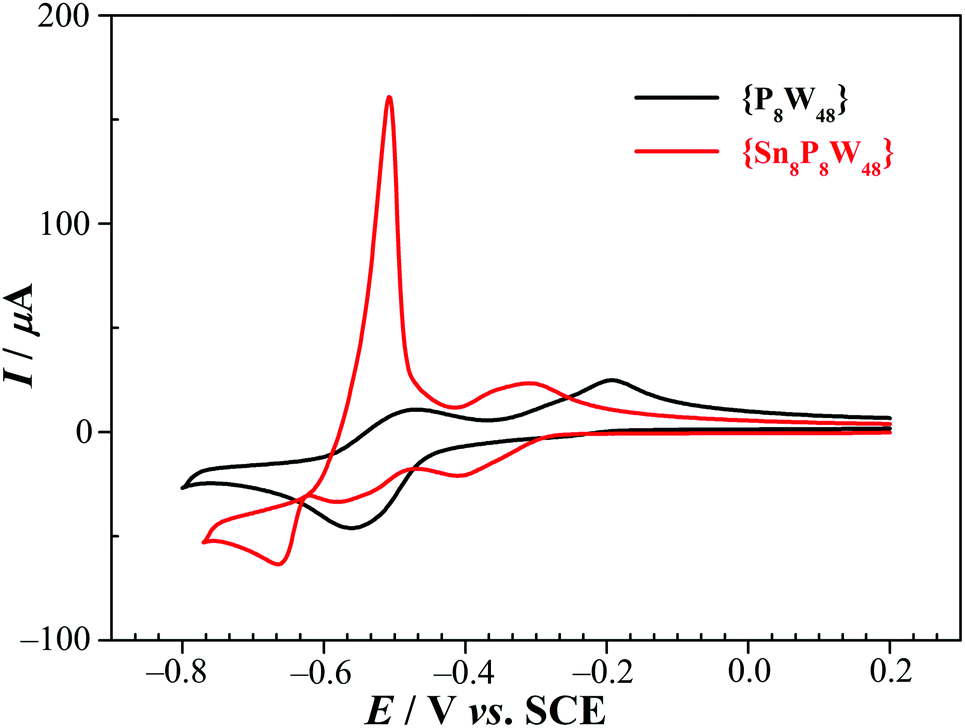 | ||
| Fig. 3 CVs of 1 (red) and {P8W48} (black) in 0.5 M Li2SO4 + H2SO4 (pH 2.0). POM concentration: 0.5 mM. Scan rate: 100 mV s−1; working electrode: glassy carbon; reference electrode: SCE. | ||
In order to prove unambiguously that the potential scan restricted to a range not going below −0.58 V vs. SCE concerns just the W centers, we carried out a study of the dependence of the reduction and oxidation peak currents, Ipc and Ipa, on the scan rate. It revealed that Ip depends on the square root of the scan rate, despite the fact that a relatively narrow set of scan rates was used (10 to 50 mV s−1), this confirms that the mass transport phenomena coupled to the electron transfer are diffusion controlled, excluding the possible involvement of the Sn centers (Fig. S17†).
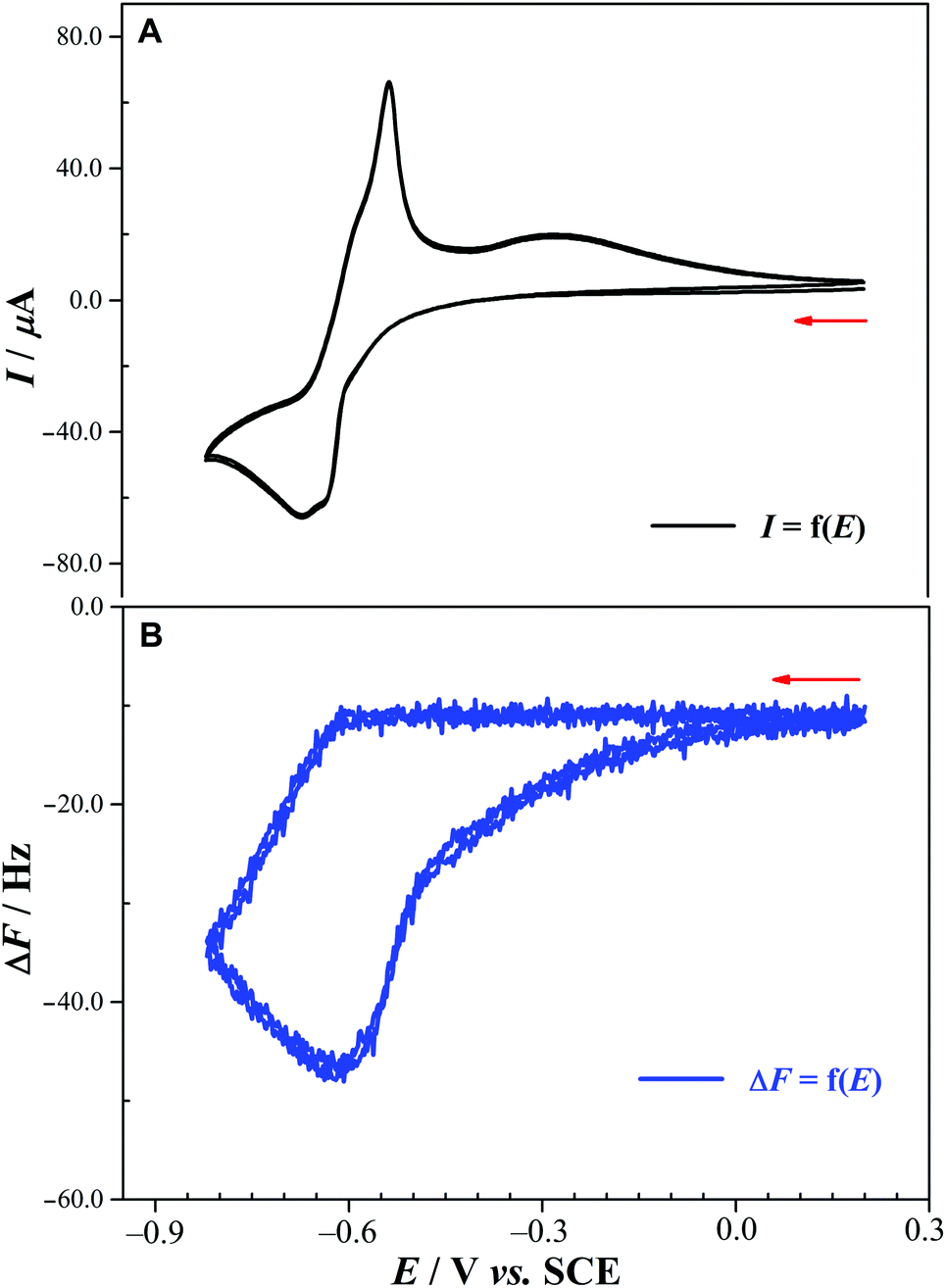 | ||
| Fig. 4 (A) CV of 1 in 0.5 M Li2SO4 + H2SO4 (pH 2.0). Working electrode: glassy carbon-covered quartz crystal. Reference electrode: SCE. Scan rate: 2 mV s−1. POM concentration: 0.5 mM. Potential scan extended to the reduction waves of the SnII centers. (B) Quartz crystal vibration frequency variation, ΔF, as a function of the working electrode potential. | ||
Experimental section
General methods and materials
The reagents were used as purchased without further purification. K28Li5[H7P8W48O184]·92H2O (KLi-P8W48) was obtained according to the reported procedure15 and its identity and purity were confirmed by IR and 31P NMR spectroscopy. Elemental analysis results (ICP-OES) were obtained from Central Institute for Engineering, Electronics and Analytics (ZEA-3), Forschungszentrum Jülich GmbH (D-52425 Jülich, Germany). TGA/DTA measurements were carried out with a Mettler Toledo TGA/SDTA 851 in dry N2 (60 ml min−1) at a heating rate of 5 K min−1. Vibrational spectra were recorded on a Bruker Vertex 70 FT-IR spectrometer coupled with a RAM II FT-Raman module (1064 nm Nd:YAG laser) on KBr disks for the FT-IR and the solid material for the Raman measurements. UV-Vis spectra were recorded using 10 mm quartz cuvettes on an Analytik Jena Specord S600 spectrophotometer. Solution 31P NMR spectra were recorded at room temperature in 5 mm tubes using a Bruker Avance 600 MHz spectrometer equipped with a prodigy probe, operating at 242.95 MHz for 31P. Chemical shifts are reported with respect to 85% H3PO4; all chemical shifts downfield of the reference are reported as positive values. The solid-state 31P NMR measurements were performed at ambient temperature using a Bruker Avance III spectrometer with a 9.4 T magnetic field. The spectrometer was equipped with a Bruker triple resonance probe for rotors of 1.3 mm diameter. The ground sample powder was filled into a ZrO2 rotor for the experiments with and without sample spinning. A rotation frequency of 67 kHz was applied during MAS. An eight-fold cyclisation of the pulse sequences with a cycle delay of 10 s was used. The signal shift is referred to H3PO4.Synthesis of K10Li17.5[K4.5⊂(ClSn)8P8W48O184]·50H2O (KLi-1)
K28Li5[H7P8W48O184]·92H2O (0.10 g; 6.75 μmol) and SnCl2·2H2O (0.013 g; 57.6 μmol) were dissolved in 3 mL of 4 M aqueous solution of LiCl in a 20 mL vial which was then closed with a screw cap. The reaction mixture was stirred at 50 °C for 1 h. During the heating the color of the solution was gradually changing from bright orange (obtained directly after the reagent dissolution) to brown and then to dark-green (originating from partial WVI reduction). After that the reaction mixture was cooled down to room temperature and filtered. The obtained dark-green solution was then evaporated under a hood resulting in large block-shaped orange crystals of the product within 2 to 6 days. Yield: 0.025 g (25% based on {P8W48}).Elemental analysis: calculated for H100Cl8K14.5Li17.5O234P8Sn8W48 (found): Cl, 1.91 (1.96); K, 3.82 (3.75); Li, 0.82 (0.83); P, 1.67 (1.45); Sn, 6.4 (6.1); W, 59.5 (61)%.
IR spectrum (KBr pellets), cm−1: 3411 (s, br); 1620 (m); 1134 (m); 1120 (m); 1084 (m); 1022 (w); 926 (s); 716 (s); 569 (m); 530 (m); 465 (w); 401 (w).
Raman (solid sample, λe = 1064 nm), cm−1: 1084 (w); 964 (s); 870 (w); 852 (w); 783 (m); 658 (m); 542 (w); 534 (w); 482 (w); 415 (w); 390 (w); 366 (w); 324 (w); 287 (w); 260 (m); 228 (m); 206 (m); 154 (m); 112 (m); 64 (s); 59 (s).
31P NMR (H2O/D2O): −8.0 ppm (singlet). 31P MAS NMR (67 kHz): −7.6 ppm (singlet).
UV-Vis (2 M Li2SO4 buffer solution, pH 3.0): λ = 216 nm, ε = 168400 M−1 cm−1; λ = 275 nm, ε = 97800 M−1 cm−1; λ = 385 nm, ε = 9789 M−1 cm−1.
UV-Vis (0.5 M Li2SO4 buffer solution, pH 2.0): λ = 219 nm, ε = 214600 M−1 cm−1; λ = 274.5 nm, ε = 134800 M−1 cm−1; λ = 385 nm, ε = 8330 M−1 cm−1.
X-ray crystallography
Single-crystal X-ray diffraction data for KLi-1 were collected on a SuperNova (Agilent Technologies) diffractometer with Mo Kα radiation (λ = 0.71073 Å) at 120 K. A crystal was mounted on a Hampton cryoloop with Paratone-N oil to prevent water loss. Absorption corrections were applied numerically based on a multifaceted crystal model using CrysAlis software.17 The SHELXTL software package18 was used to solve and refine the structure. The structure was solved by the direct methods and refined by the full-matrix least-squares method against |F|2 with anisotropic thermal parameters for all POM skeleton atoms (Sn, Cl, P, W, O) and potassium countercations and in isotropic approximation for the O atoms of co-crystallized solvent molecules. The Li+ countercations and hydrogen atoms of the crystal water molecules were not located. The relative site occupancy factors for the disordered potassium countercations and solvent oxygens were first refined in an isotropic approximation with Uiso = 0.05 and then fixed at the obtained values and refined without the thermal parameter restrictions.The number of crystal water molecules found by XRD was smaller than that determined by elemental analysis and TGA (41 vs. 50 and 102, respectively) reflecting the high degree of solvent disorder in the solid-state structure of KLi-1. This is also consistent with large solvent-accessible volume remaining in the structure. On the other hand, the sample used for TGA was dried to less than that taken for elemental analysis (2 days vs. 3 to 4 weeks). For the overall consistency the formula in the CIF file corresponds to the composition of the bulk material determined by elemental analysis since all the further studies were performed on the isolated well-dried bulk material of KLi-1.
Additional crystallographic data are summarized in Table 1. Further details on the crystal structure investigation may be obtained from Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany [fax (+49) 7247-808-666; e-mail crysdata@fiz-karlsruhe.de], upon quoting the depository number CSD 429908.
| Empirical formula | H100Cl8K14.5Li17.5O234P8Sn8W48 |
| Formula weight, g mol−1 | 14838.88 |
| Crystal system | Tetragonal |
| Space group | I4/m |
| a, Å | 25.4151(3) |
| c, Å | 21.7748(4) |
| Volume, Å3 | 14065.0(3) |
| Z | 2 |
| D calc, g cm−3 | 3.504 |
| Absorption coefficient, mm−1 | 20.666 |
| F(000) | 13016 |
| Crystal size, mm | 0.19 × 0.28 × 0.35 |
| Theta range for data collection | 4.09°–26.02° |
| Completeness to Θmax | 99.4% |
| Index ranges | −31 ≤ h ≤ 30, |
| −31 ≤ k ≤ 30, | |
| −22 ≤ l ≤ 26 | |
| Reflections collected | 35937 |
| Independent reflections | 7076 |
| R int | 0.1192 |
| Observed (I > 2σ(I)) | 5862 |
| Absorption correction | Empirical using spherical harmonics |
| T min/Tmax | 0.0562/0.1109 |
| Data/restraints/parameters | 7076/36/379 |
| Goodness-of-fit on F2 | 1.092 |
| R 1, wR2 (I > 2σ(I)) | R 1 = 0.0583, |
| wR2 = 0.1563 | |
| R 1, wR2 (all data) | R 1 = 0.0709, |
| wR2 = 0.1691 | |
| Largest diff. peak and hole, e Å−3 | 6.755 and −3.837 |
Electrochemical experiments
Cyclic voltammograms of 1 were recorded in comparison with the heterometal-free {P8W48} salt KLi-P8W48 at room temperature in three different media: (1) on a 0.55 mM solution of KLi-1 in 1 M LiCl/HCl buffer (pH 2.0), (2) 0.57 mM solution of KLi-1 in 0.5 M Li2SO4/H2SO4 buffer (pH 2.0) and (3) 0.52 mM non-buffered KLi-1 solution in 2 M LiCl. The corresponding concentrations of KLi-P8W48 were 0.51 mM, 0.56 mM and 0.53 mM, respectively. Electrochemical data were obtained using an EG&G 273A potentiostat controlled by M270 software. A conventional one-compartment three-electrode electrochemical cell included a glassy carbon working electrode (GC, Mersen, France) with a diameter of 3 mm, a platinum gauze counter electrode with a large surface area and a saturated calomel reference electrode. The source, mounting, and polishing of the glassy carbon electrode has been previously described.19 The reference and counter electrodes were separated from the studied solutions via fritted compartments filled with the same electrolyte. Prior to the experiments the solutions were thoroughly deaerated with pure argon for at least 30 min and then kept under a positive argon pressure during the measurements. The midpoint redox potentials were determined from the average values of the anodic and cathodic peak potentials and are reported vs. the saturated calomel reference electrode (SCE).The electrochemical quartz crystal microbalance (EQCM) measurements were carried out using a QCA 922 (Seiko/EG&G) system with 9 MHz AT-cut crystals. New crystals equipped with carbon electrodes possessing a true surface area of 0.3 cm2 were provided by Seiko. Frequency variations were recorded and used for discussion in this work.
Conclusions
In summary, we have prepared the first complex of macrocyclic phosphotungstate with the main group metal ions, [K4.5⊂(ClSn)8P8W48O184]17.5− (1), which also represents a rather rare example of SnII-containing POMs. In 1 the SnII centers occupy the eight vacant sites of the polyanion resulting in the POT with D4h symmetry. Polyanions 1 have been isolated in the solid state as the hydrated mixed potassium/lithium salt KLi-1 and characterized by single-crystal X-ray diffraction, FT-IR/Raman and 31P MAS NMR spectroscopy as well as by elemental and thermogravimetric analyses. Solution studies by 31P NMR, UV-Vis and electrochemistry showed that the complex is stable in aqueous medium for several hours and then slowly decomposes, releasing SnII. In contrast with transition metal and lanthanide-functionalized POMs, the SnII ions in 1 display a small coordination number of 3 and coordinate only one terminal Cl− ligand each, thus retaining a relatively large void in the inner cavity of the macrocyclic complex. Currently we are investigating the possibility of further functionalization of {Sn8P8W48} via incorporation of various heterometals in this inner core volume.Acknowledgements
We gratefully acknowledge financial support by Forschungszentrum Jülich and the EU ERC Starting Grant MOLSPINTRON, no. 308051 (P.K.) as well as by the Université Paris-Sud and the CNRS (I.M.M and P.d.O.) and by an STSM action attributed by the COST CM1203 “Polyoxometalate Chemistry for Molecular Nanoscience (PoCheMoN)”.Notes and references
- See for example: (a) P. Gouzerh and A. Proust, Chem. Rev., 1998, 98, 77–111 CrossRef CAS PubMed; (b) A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837–1852 RSC; (c) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed; (d) M.-P. Santoni, G. S. Hanan and B. Hasenknopf, Coord. Chem. Rev., 2014, 281, 64–85 CrossRef CAS PubMed , and references therein.
- See for example: (a) S. Duval, M.-A. Pilette, J. Marrot, C. Simonnet-Jégat, M. N. Sokolov and E. Cadot, Chem. – Eur. J., 2008, 14, 3457–3466 CrossRef CAS PubMed; (b) Y. Zhang, L. Li, T. Sun, B. Liu, H. Hu and G. Xue, Inorg. Chem., 2011, 50, 2613–2618 CrossRef CAS PubMed; (c) C. Ritchie, M. Speldrich, R. W. Gable, L. Sorace, P. Kögerler and C. Boskovic, Inorg. Chem., 2011, 50, 7004–7014 CrossRef CAS PubMed; (d) S. Duval, J. Marrot, C. Simonnet-Jégat, I. M. Mbomekalle, M. N. Sokolov and E. Cadot, Dalton Trans., 2012, 41, 3174–3184 RSC.
- (a) W. H. Knoth, J. Am. Chem. Soc., 1979, 101, 2211–2213 CrossRef CAS; (b) G. S. Chorghade and M. T. Pope, J. Am. Chem. Soc., 1987, 109, 5134–5138 CrossRef CAS; (c) E. Rafiee, I. M. Baltork, S. Tangestaninejad, A. Azad and S. Moinee, Z. Naturforsch., B: Chem. Sci., 2006, 61, 601–606 CrossRef CAS.
- (a) F. Xin and M. T. Pope, J. Am. Chem. Soc., 1996, 118, 7731–7736 CrossRef CAS; (b) A. Botar, B. Botar, P. Gili, A. Müller, J. Meyer, H. Bögge and M. Schmidtmann, Z. Anorg. Allg. Chem., 1996, 622, 1435–1440 CrossRef CAS PubMed; (c) R. Khoshnavazi and L. Bahrami, J. Coord. Chem., 2009, 62, 2067–2075 CrossRef CAS PubMed.
- (a) M. N. Sokolov, N. V. Izarova, A. V. Virovets, V. P. Fedin, Z. A. Starikova and M. Yu. Antipin, Dalton Trans., 2003, 4389–4390 RSC; (b) C. Zhao, E. N. Glass, B. Chica, D. G. Musaev, J. M. Sumliner, R. B. Dyer, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 12085–12091 CrossRef CAS PubMed.
- Z. Zhang, Q. Lin, S.-T. Zheng, X. Bu and P. Feng, Chem. Commun., 2011, 47, 3918–3920 RSC.
- B. Krebs, E. Droste, M. Piepenbrink and G. Vollmer, C. R. Acad. Sci., Ser. IIc: Chim., 2000, 3, 205–210 CrossRef CAS.
- M. N. Sokolov, I. V. Kalinina, E. V. Peresypkina, N. K. Moroz, D. Yu. Naumov and V. P. Fedin, Eur. J. Inorg. Chem., 2013, 1772–1779 CrossRef CAS PubMed.
- (a) S. S. Mal and U. Kortz, Angew. Chem., Int. Ed., 2005, 44, 3777–3780 CrossRef CAS PubMed; (b) S. S. Mal, B. S. Bassil, M. Ibrahim, S. Nellutla, J. van Tol, N. S. Dalal, J. A. Fernández, X. López, J. M. Poblet, R. Ngo Biboum, B. Keita and U. Kortz, Inorg. Chem., 2009, 48, 11636–11645 CrossRef CAS PubMed; (c) C. Pichon, P. Mialane, A. Dolbecq, J. Marrot, E. Rivière, B. Keita, L. Nadjo and F. Sécheresse, Inorg. Chem., 2007, 46, 5292–5301 CrossRef CAS PubMed.
- (a) A. Müller, M. T. Pope, A. M. Todea, H. Bögge, J. van Slageren, M. Dressel, P. Gouzerh, R. Thouvenot, B. Tsukerblat and A. Bell, Angew. Chem., Int. Ed., 2007, 46, 4477–4480 CrossRef PubMed; (b) S. S. Mal, M. H. Dickman, U. Kortz, A. M. Todea, A. Merca, H. Bögge, T. Glaser, A. Müller, S. Nellutla, N. Kaur, J. van Tol, N. S. Dalal, B. Keita and L. Nadjo, Chem. – Eur. J., 2008, 14, 1186–1195 CrossRef CAS PubMed; (c) F. L. Sousa, H. Bögge, A. Merca, P. Gouzerh, R. Thouvenot and A. Müller, Chem. Commun., 2009, 7491–7493 RSC.
- (a) M. Zimmermann, N. Belai, R. J. Butcher, M. T. Pope, E. V. Chubarova, M. H. Dickman and U. Kortz, Inorg. Chem., 2007, 46, 1737–1740 CrossRef CAS PubMed; (b) S. S. Mal, N. H. Nsouli, M. H. Dickman and U. Kortz, Dalton Trans., 2007, 2627–2630 RSC; (c) S. G. Mitchell, D. Gabb, C. Ritchie, N. Hazel, D.-L. Long and L. Cronin, CrystEngComm, 2009, 11, 36–39 RSC; (d) B. S. Bassil, M. Ibrahim, S. S. Mal, A. Suchopar, R. Ngo Biboum, B. Keita, L. Nadjo, S. Nellutla, J. van Tol, N. S. Dalal and U. Kortz, Inorg. Chem., 2010, 49, 4949–4959 CrossRef CAS PubMed; (e) S. G. Mitchell, T. Boyd, H. N. Miras, D.-L. Long and L. Cronin, Inorg. Chem., 2011, 50, 136–143 CrossRef CAS PubMed; (f) X. Fang, P. Kögerler, Y. Furukawa, M. Speldrich and M. Luban, Angew. Chem., Int. Ed., 2011, 50, 5212–5216 CrossRef CAS PubMed; (g) S.-W. Chen, K. Boubekeur, P. Gouzerh and A. Proust, J. Mol. Struct., 2011, 994, 104–108 CrossRef CAS PubMed; (h) V. S. Korenev, S. Floquet, J. Marrot, M. Haouas, I.-M. Mbomekallé, F. Taulelle, M. N. Sokolov, V. P. Fedin and E. Cadot, Inorg. Chem., 2012, 51, 2349–2358 CrossRef CAS PubMed; (i) Z.-J. Liu, Z.-M. Zhang, H. Fu, Y.-G. Li, W.-L. Chen, H.-H. Wu and E.-B. Wang, Dalton Trans., 2012, 41, 11700–11705 RSC; (j) L. Huang, L. Cheng, W.-H. Fang, S.-S. Wang and G.-Y. Yang, Eur. J. Inorg. Chem., 2013, 1693–1698 CrossRef CAS PubMed; (k) Y.-Q. Jiao, C. Qin, X.-L. Wang, C.-G. Wang, C.-Y. Sun, H.-N. Wang, K.-Z. Shao and Z.-M. Su, Chem. – Asian J., 2014, 9, 470–478 CrossRef CAS PubMed.
- S. G. Mitchell, C. Streb, H. N. Miras, T. Boyd, D. L. Long and L. Cronin, Nat. Chem., 2010, 2, 308–312 CrossRef CAS PubMed.
- (a) F. R. Poulsen and S. E. Rasmussen, Acta Chem., Scand., 1970, 24, 150–156 CrossRef CAS; (b) B. Kamenar and D. Grdenic, J. Chem. Soc., 1961, 3954–3958 RSC.
- (a) I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef; (b) K. Knížek, Kalvados – Software for crystal structure and powder diffraction; see http://www.fzu.cz/~knizek/kalvados/index.html.
- R. Contant and A. Tézé, Inorg. Chem., 1985, 24, 4610–4614 CrossRef CAS.
- B. Keita, Y. W. Lu, L. Nadjo and R. Contant, Electrochem. Commun., 2000, 2, 720–726 CrossRef CAS.
- CrysAlisPro, Version 1.171.36.21 (release 14-08-2012 CrysAlis171.NET), Agilent Technologies Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- N. Vila, P. A. Aparicio, F. Sécheresse, J. M. Poblet, X. Lopez and I. M. Mbomekallé, Inorg. Chem., 2012, 51, 6129–6138 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data in CIF format, BVS calculation details, crystal packing in KLi-1, IR/Raman, solid-state and time-dependent solution 31P NMR and UV-vis spectra; TGA, CV and EQCM curves. See DOI: 10.1039/c5dt03164h |
| This journal is © The Royal Society of Chemistry 2015 |