DOI:
10.1039/C5DT03119B
(Paper)
Dalton Trans., 2015,
44, 20216-20231
Ring-opening polymerization of rac-lactide mediated by tetrametallic lithium and sodium diamino-bis(phenolate) complexes†
Received
12th August 2015
, Accepted 26th October 2015
First published on 5th November 2015
Abstract
Lithium and sodium compounds supported by tetradentate amino-bis(phenolato) ligands, [Li2(N2O2BuBuPip)] (1), [Na2(N2O2BuBuPip)] (2) (where [N2O2BuBuPip] = 2,2′-N,N’-homopiperazinyl-bis(2-methylene-4,6-tert-butylphenol), and [Li2(N2O2BuMePip)] (3), [Na2(N2O2BuMePip)] (4) (where [N2O2BuMePip] = 2,2′-N,N’-homopiperazinyl-bis(2-methylene-4-methyl-6-tert-butylphenol) were synthesized and characterized by NMR spectroscopy and MALDI-TOF mass spectrometry. Variable temperature NMR experiments were performed to understand solution-phase dynamics. The solid-state structures of 1 and 4 were determined by X-ray diffraction and reveal tetrametallic species. PGSE NMR spectroscopic data suggests that 1 maintains its aggregated structure in CD2Cl2. The complexes exhibit good activity for controlled ring-opening polymerization of rac-lactide (LA) both solvent free and in solution to yield PLA with low dispersities. Stoichiometric reactions suggest that the formation of PLA may proceed by the typical coordination–insertion mechanism. For example, 7Li NMR experiments show growth of a new resonance when 1 is mixed with 1 equiv. LA and 1H NMR data suggests formation of a Li-alkoxide species upon reaction of 1 with BnOH.
Introduction
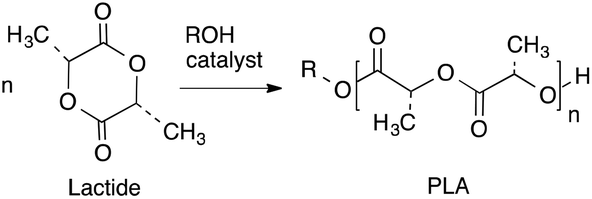
In recent years, considerable attention has been directed toward the design of bio-based and degradable polymers, particularly linear aliphatic polyesters such as polylactide (PLA) and polycaprolactone (PCL).1–3 PLA has received a significant amount of interest industrially as a packing material and in the pharmaceutical and biomedical fields4–14 because it is biodegradable, biocompatible and can be synthesized from renewable feedstocks. An equally important aspect of PLA is its unique physical properties, which make it a viable alternative to more traditional polymers, like polystyrene and polyethylene terephthalate for containers including bottles.7,8,12 In the synthesis of PLA, to achieve a high degree of control (including a high molecular weight with a low dispersity), ring opening polymerization (ROP) of lactide (LA) with an initiator/catalyst is the ideal route (Scheme 1).14
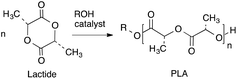 |
| | Scheme 1 Synthesis of polylactide by ROP route. | |
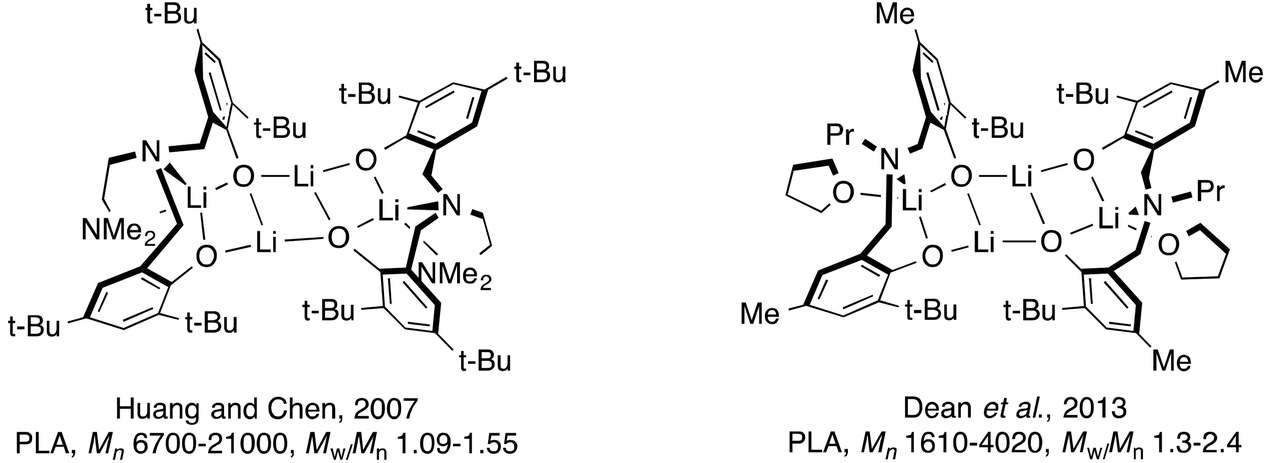
Ligands with N- and O-donor atoms such as amino-phenolates have become an important ligand class in this field due to their ability to coordinate to a wide range of metal centers. Variation of the steric properties of the ligand is also readily achieved by changing either the backbone and/or the phenolate substituent.15 To initiate the polymerization of lactide and ε-caprolactone, main group metals are often employed with these ligands: magnesium,16–27 calcium,16,26,28 barium,29 aluminum30–35 and indium36,37 have all been used. In addition, alkali metals such as lithium26,38–50 and sodium26,50–54 bearing bulky ligands have shown promise in ROP of cyclic esters with few side reactions. Both metals tend to form aggregates in solution and in the crystalline state depending on the steric properties of the ligand or solvent used.38–52,54 These complexes are attractive in this field because of their stability, low cost and toxicity. Of particular note are the lithium complexes studied by Huang and Chen, and Dean et al. (Fig. 1), which were used in ROP of LA.38,44 In these examples, the tetradentate ligand employed in the earlier work led to lithium complexes that were able to better control the polymerization reactions (e.g. lower polymer dispersity) compared with the related tridentate ligand complexes. In later mechanistic studies by Chen and co-workers, using different lithium amino-phenolate complexes, they were able to show that BnOH is activated by the lithium first, followed by insertion of the resulting benzyl alkoxy group to the carbonyl group of LA.43
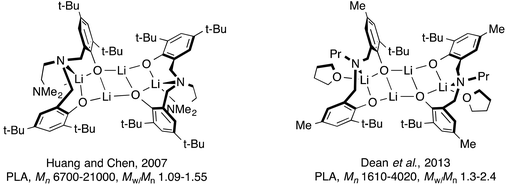 |
| | Fig. 1 Known tetralithium complexes of amino-bis(phenolate) ligands. | |
The current work targets the preparation of a series of multinuclear lithium and sodium complexes and subsequent investigation of their catalytic activity in the ROP of rac-lactide, both solvent free and in solution, in the presence and absence of benzyl alcohol. This will allow valuable direct comparison between lithium- and sodium-derived catalysts in these important reactions.
Results and discussion
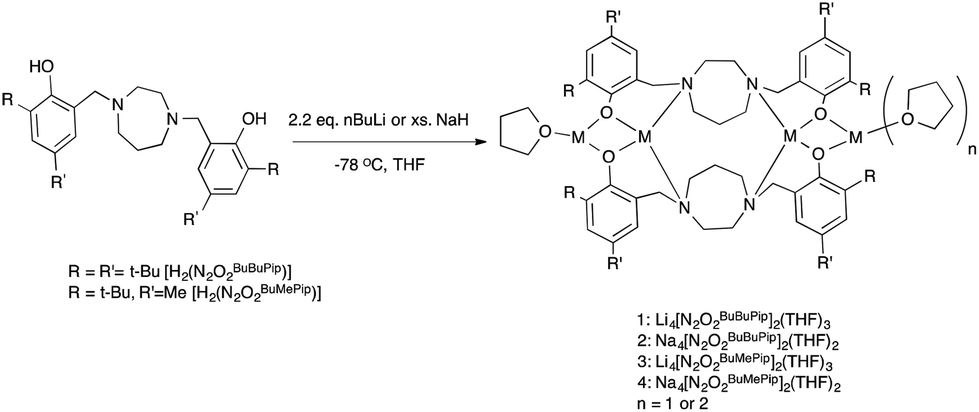
Synthesis and characterization of ligands, lithium and sodium complexes
The tetradentate amine-bis(phenol) ligands were synthesized via a modified Mannich condensation reaction in water.55 These and closely related ligands have previously been used to prepare complexes of Ti,56–59 Mn,60 Al,61 Zr and Hf,62 Mo,63 and Fe.64–66 As shown in Scheme 2, lithium complexes with the formulation (Li2O2N2BuBuPip)2(THF)3 and (Li2O2N2BuMePip)2(THF)3 were synthesized by the reaction of H2[O2N2]BuBuPip and H2[O2N2]BuMePip with 2.2 equiv. n-BuLi in THF. Tetranuclear sodium complexes were produced by reacting the appropriate ligand with an excess of NaH as shown in Scheme 2 but could also be prepared via reaction of ligands with two equiv. of sodium tert-butoxide. The complexes were characterized using MALDI-TOF MS and 1H, 13C and 7Li NMR spectroscopies. For 2 and 4, identical spectroscopic data was obtained irrespective of synthetic route. The structures of 1 and 4 were determined via single crystal X-ray diffraction analysis. Unfortunately, elemental analyses often gave lower than expected carbon values across all samples, which may be indicative of incomplete combustion. Although for the Li complexes, it may also suggest contamination with lithium oxide impurities.
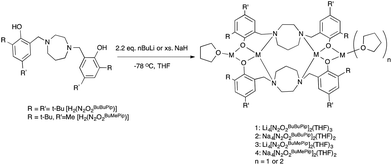 |
| | Scheme 2 Synthesis of lithium and sodium complexes. | |
Crystal structure determination
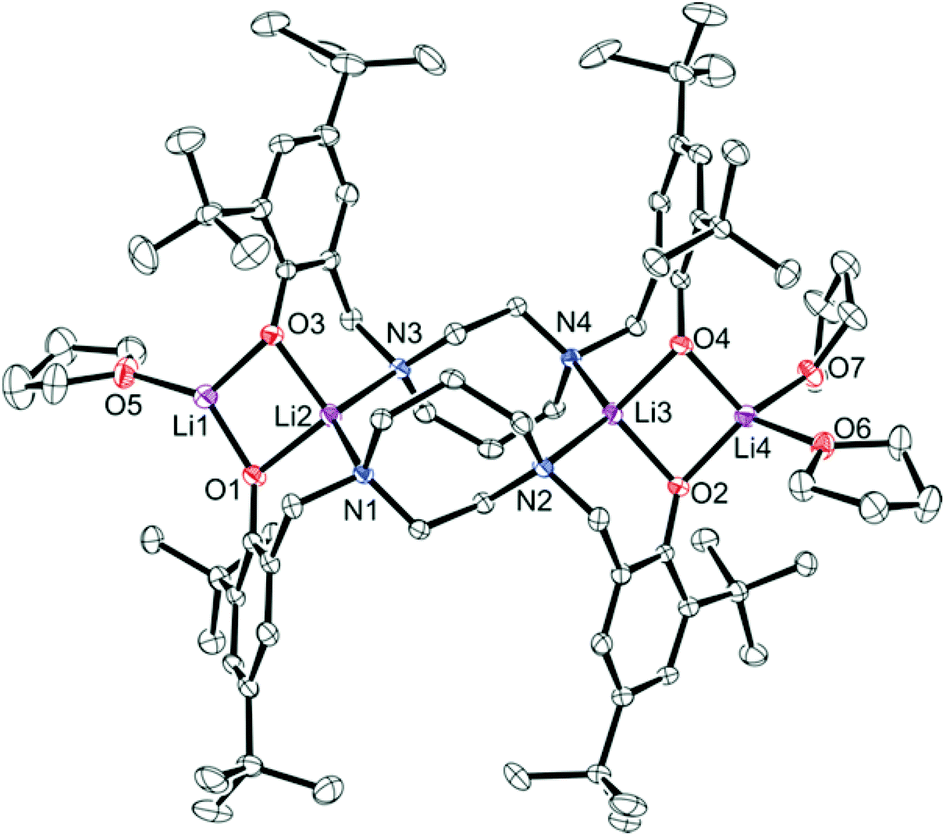
For complex 1, crystals suitable for single crystal X-ray diffraction were formed by slow evaporation of a toluene/pentane solution under an inert atmosphere at −35 °C. The ORTEP structure of complex 1 ({Li2[N2O2BuBuPip]}2·3THF) is shown in Fig. 2 and the crystallographic data are collected in Table S1.† The compound contains four Li atoms, capped by two amino-bis(phenolate) ligands. At its core, only three of the four Li centers are tetracoordinate, with the fourth being tricoordinate. Selected bond lengths (Å) and angles (°) for compound 1 are presented in Table S2.† Two terminal lithium centers Li(1) and Li(4) are bonded to two phenolate oxygen atoms (Li(1)–O(1), 1.804 Å, Li(1)–O(3), 1.848 Å, Li(4)–O(2), 1.899 Å, Li(4)–O(4) 1.922 Å) with distances in agreement to those reported by Chen et al. for a series of tetranuclear ladder-like lithium phenolates,44 and other similar species.38,47 The terminal Li atoms are bound by one or two THF molecules (Li(1)–O(5), 1.934 Å, Li(4)–O(7), 2.085 Å, Li(4)–O(6) 2.009 Å) resulting in pseudo-trigonal planar (Li(1)) and tetrahedral (Li(4)) coordination environments. Each central Li atom is bound by two N atoms and bridging phenolate O atoms. Li–O and Li–N bond lengths are in good agreement with previously observed literature values for similar systems.44,45 Each of these lithium atoms interacts weakly with an adjacent terminal lithium atom (Li(1)–Li(2), 2.493 Å, Li(3)–Li(4), 2.583 Å) and agrees with the values reported by Chen et al. for analogous Li-containing compounds (2.403(7)–2.444(4) Å).38,44
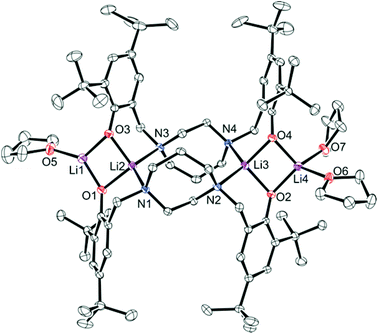 |
| | Fig. 2 Molecular structure (ORTEP) and partial numbering scheme for 1. Ellipsoids are shown at the 50% probability level (H-atoms omitted for clarity). | |
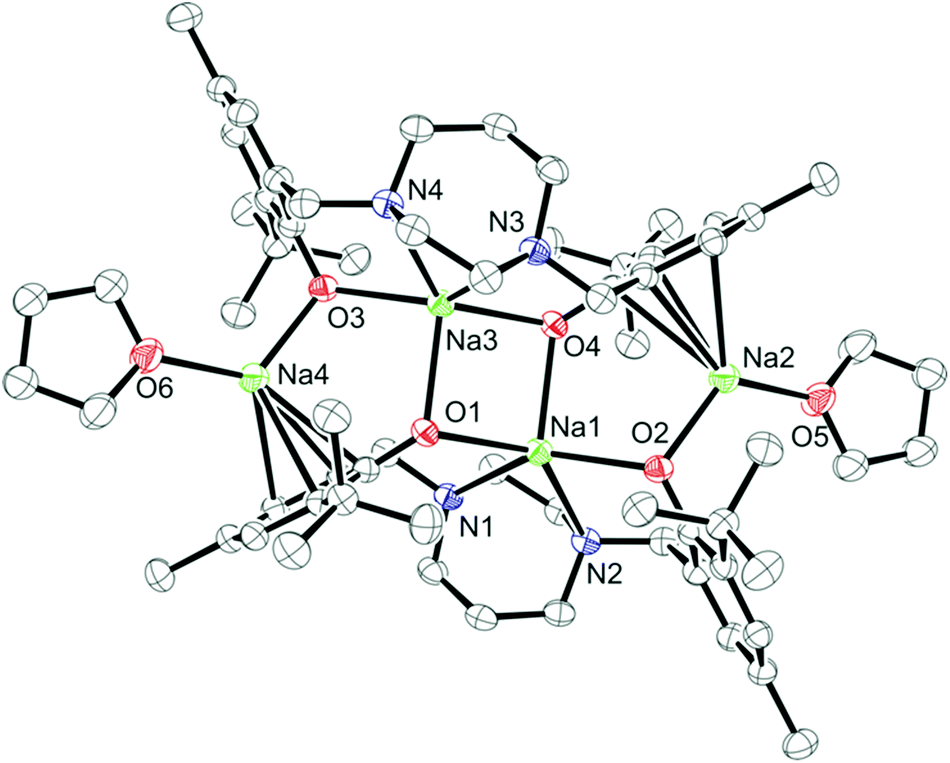
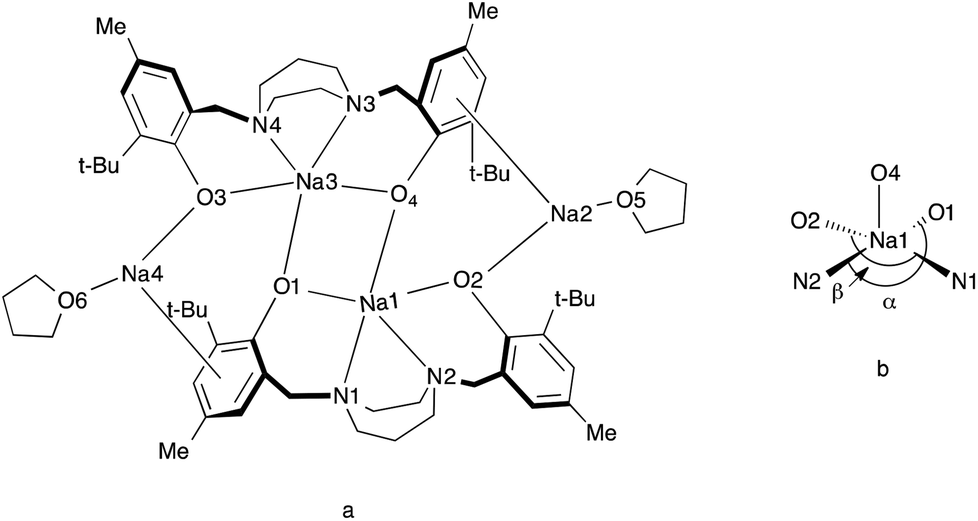
Colorless crystals of complex 4 were collected upon recrystallization in toluene/pentane under an inert atmosphere at −35 °C. The molecular structure of complex 4 ({Na2[N2O2BuMePip]}2·2THF) is shown in Fig. 3 and the crystallographic parameters are given in Table S1.† The arrangement of the metal centers is different to compound 1 due to the larger size of sodium compared to lithium. Complex 4 is dimeric with the sodium atoms forming a tetranuclear node, THF molecules are bonded to the terminal sodium atoms in a symmetric arrangement. A simplified illustration of the bonding in 4 is shown in Fig. 4. Two sodium atoms Na(1) and Na(3) form a rhomboid structure with two bridging phenolate oxygen donors, O(1) and (O4). Atoms Na(1) and Na(3) are each five coordinate and bonded to both the amine nitrogen atoms and the other phenolate oxygen donors of the tetradentate ligand. Selected bond lengths (Å) and angles (°) for compound 4 are presented in Table S2.† According to the calculated tau parameter (τ = (β − α)/60), the geometry around the inner sodium centers can be described as distorted square pyramidal.67–69 The τ value is close to 0, as β the largest angle is 139.96(5)° and α the second largest angle in the coordination sphere is 134.44(5)° as shown in Fig. 4b. A Na(1)–Na(3) interatomic distance of 3.3769 Å is observed which is within the typical range observed for related complexes.52 In comparison to the bonding observed in 1, the bond distances for Na(1)–O(4), Na(1)–O(1), Na(1)–N(1) and Na(1)–N(2) are as expected longer 2.3957(13), 2.3450(14), 2.6761(16) and 2.6392(16) Å. All values are in statistical agreement with previously observed related bond distances in the literature.41,50,51,53,54 The phenyl rings of the ligand display π interactions with the outer sodium atoms and this is attributed to the flexibility of the aromatic rings to bend toward the metal centers. The Na⋯C π bond is supported by the short distances between the sodium and carbon atoms, with Na(2)–C(21), Na(2)–C(26), Na(2)–C(8), Na(2)–C(7) bond distances of 2.6683(18), 2.7093(19), 2.8526(19), 2.8788(19) Å, respectively. These are comparable to those reported in previous studies.41,53,54
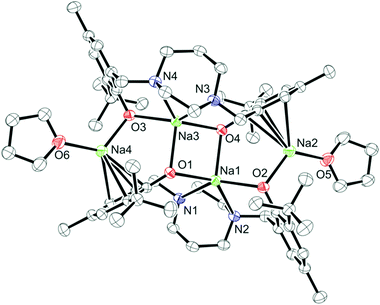 |
| | Fig. 3 Molecular structure (ORTEP) and partial numbering scheme for 4. Ellipsoids are shown at the 50% probability level (H-atoms omitted for clarity). | |
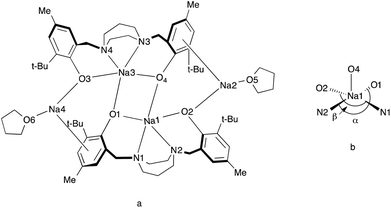 |
| | Fig. 4 [a] Schematic representation of 4. [b] Representation of the five-coordinate environment of Na(1). | |
Solution-state NMR spectroscopy
The solution structures of complexes 1, 2, 3 and 4 in C6D6 and C5D5N were investigated by 1H and, where appropriate, 7Li NMR spectroscopy. For complexes 1 and 2 in C6D6 at 298 K, only one set of ArCH2, t-Bu and homopiperazine (CH2) resonances are observed, which is indicative of a centrosymmetric species (Fig. S1†). We assumed that disaggregation might be occuring as a result of steric crowding/congestion, wherein the structure of the ligand causes the degree of Li–O aggregation to be less in solution than the solid-state. The dissociation behavior of related tetranuclear lithium compounds in C6D6 and C5D5N at 296.2 K has previously been noted by others38,47 in which ladder complexes undergo dissociation in solution rather than remaining intact.
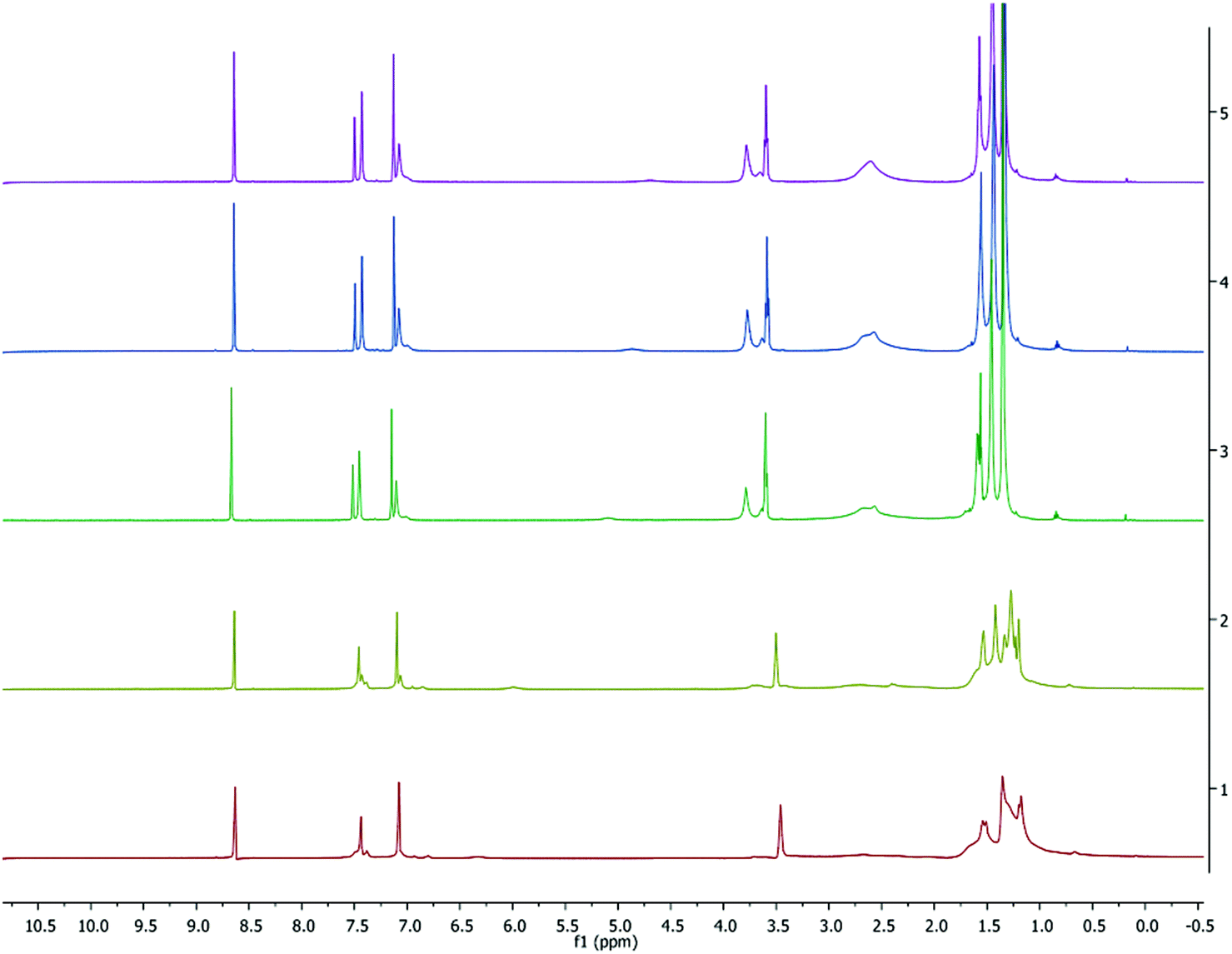
To further study the aggregation behavior of complex 1, variable-temperature (VT) 1H and 7Li NMR spectra were obtained in C5D5N from 233 to 318 K (shown in Fig. 5 and 6, respectively). At room temperature, the proton resonances are noticeably sharper in C5D5N compared to those seen in C6D6 (Fig. S2†), suggesting that the pyridine preferentially coordinates to the metal center and reduces fluxionality in the complexes.
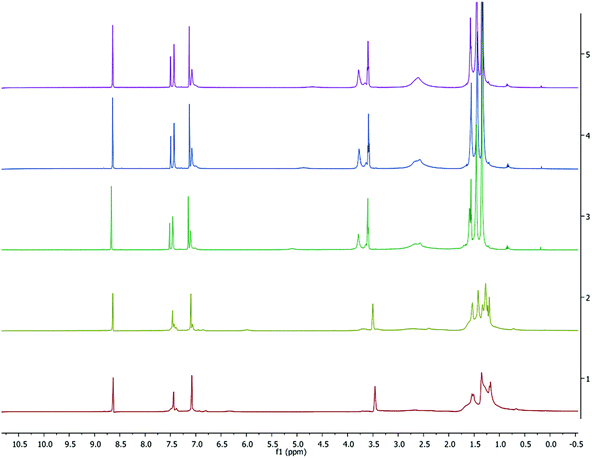 |
| | Fig. 5 VT 1H NMR spectrum (500 MHz, C5D5N) of 1. Temperatures from top to bottom: 318, 308, 298, 253, 233 K. | |
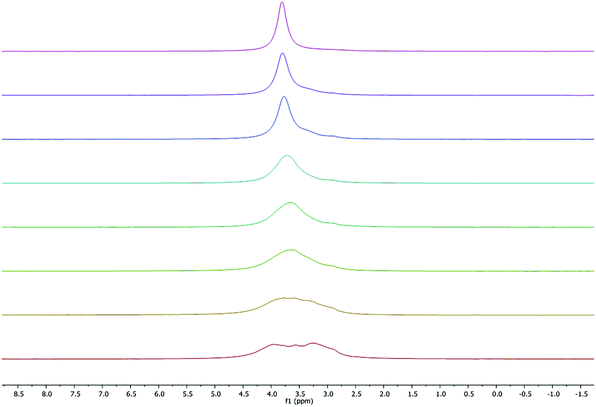 |
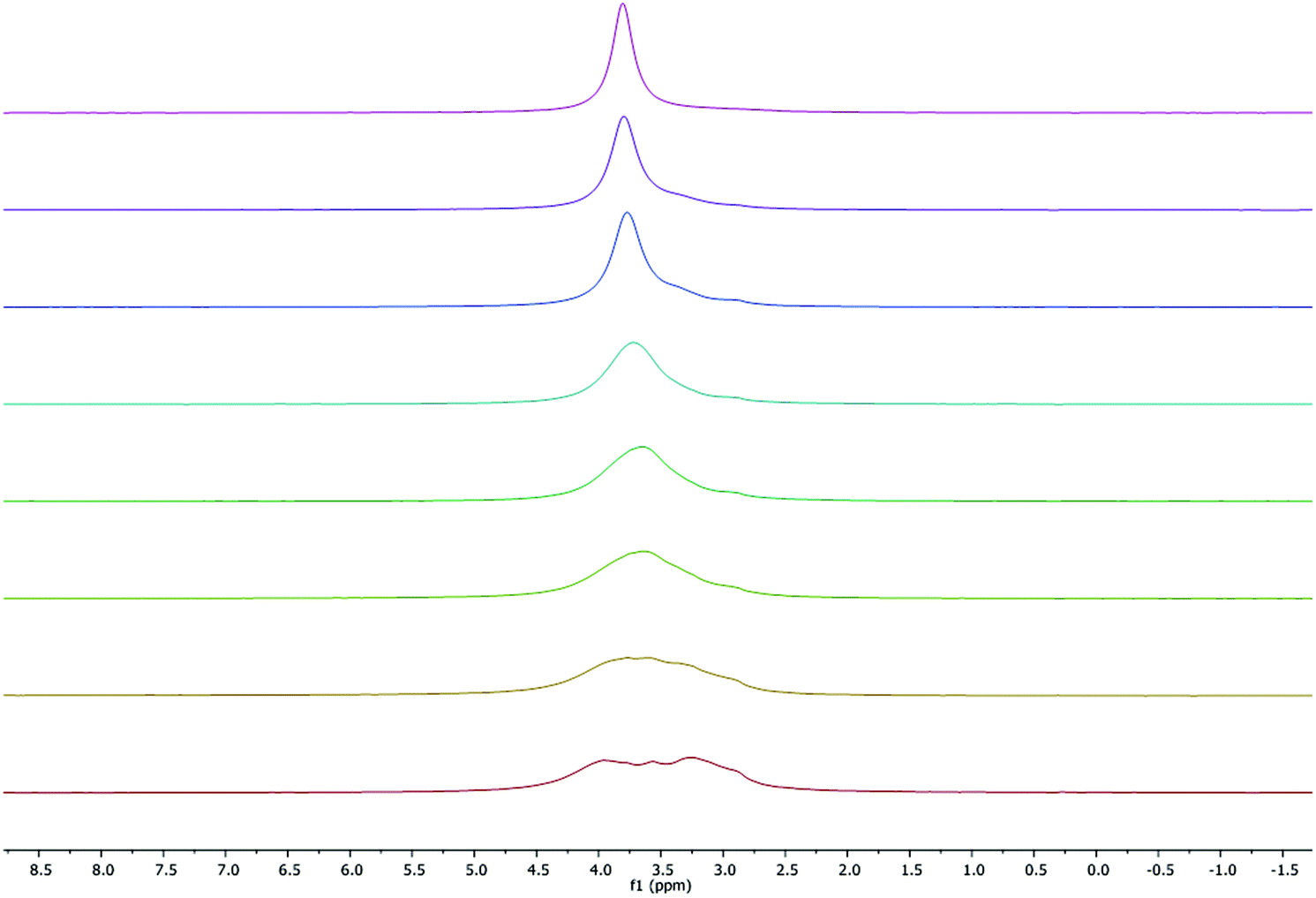
| | Fig. 6 VT 7Li NMR spectrum (300 MHz, C5D5N) of 1 (ω1/2 values were calculated from the line fitting program in MestReNova NMR processing software). Temperatures from top to bottom: 333, 318, 298, 272, 263, 252, 242, 233 K. | |
As seen in Fig. 6, at room temperature and above, the 7Li NMR spectra display a single peak at approximately 3.8 ppm (ω1/2 = 29.6 Hz), corresponding to a single type of lithium environment, which contrasts with the solid-state structure where two environments are present. However, at low temperature (233 K) four Li environments were observed at 4.0 (ω1/2 = 70.6 Hz), 3.8 (ω1/2 = 10.01 Hz), 3.6 (ω1/2 = 27.2 Hz) and 3.3 (ω1/2 = 94.5 Hz) ppm. Our results contrast with the previously reported work by Kozak38 and Chen44 groups, who observed symmetric Li environments for similar complexes. This unique observation is likely because at low temperatures the tetralithium adduct is the dominant species present. On going from 233 K to 333 K, a small change in the chemical shift was observed which is consistent with those reported for other Li-based phenolates.38,447Li NMR spectra were also obtained in the formally non-coordinating solvent deuterated benzene. As expected, a major lithium environment at lower frequency, 1.58 ppm (Fig. S4†).
For complex 4, the spectra in C6D6 solutions showed broad peaks indicative of fluxional behavior (Fig. S5†) and this led us to pursue VT experiments in C5D5N as shown in Fig. 7. The methylene groups of the homopiperazine and those between the nitrogen and the aromatic ring twist and are averaged at high temperatures but at low temperatures (233 and 253 K) the spectrum consists of separate peaks. Therefore, low temperature NMR data are reported for 4 (Fig. S6a†). At room and high temperature, the separate peaks coalesce to yield single broad peaks as shown in Fig. S6b.†
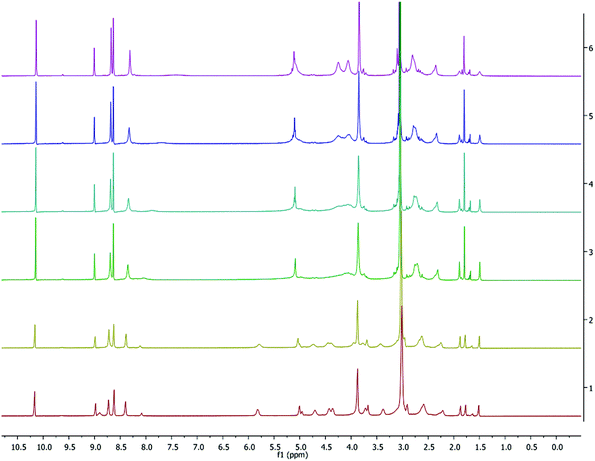 |
| | Fig. 7 VT 1H NMR spectrum (500 MHz, C5D5N) of 4. Temperatures from top to bottom: 333, 318, 308, 298, 253, 233 K. | |
Pulse-gradient spin-echo (PGSE) NMR spectroscopy is a useful way to determine the size of molecules in solution.70 As polymerization reactions were performed in dichloromethane, the nuclearity of 1 in CD2Cl2 was assessed by PGSE NMR spectroscopy. The value of the hydrodynamic radius (rH,PGSE) of 1 was calculated using a previously described method and found to be 15.1 Å.39 This is moderately smaller than rX-ray = 18.76 Å, which was calculated according to rX-ray = (a2b)1/3 where a and b are the major and minor semi-axes of the prolate ellipsoid formed by the complex, as determined from the solid-state structure (a = 19.94 Å, b = 16.61 Å). This indicates that 1 likely retains its tetrametallic structure in this non-coordinating solvent. However, it should be noted that this data reflects the solution-state structure at room temperature and bimetallic species may exist at elevated temperatures. Although many previously reported Li4 phenolate complexes disaggregate in solution, the Kerton group have recently published a closely related Li4 complex that also retains its aggregated state at room temperature in solution.71
Polymerization of rac-lactide
The catalytic behavior of 1, 2, 3 and 4 in the ring-opening polymerization of rac-lactide in the presence and absence of benzyl alcohol as co-initiator was investigated.
Solvent free polymerization
In order to reduce reaction times and achieve higher turnover frequencies,4,72 polymerization reactions were conducted under bulk/melt conditions at high temperatures. The results are summarized in Table 1. ROP reactions were performed with 1, 2, 3 and 4 at 150 °C and no significant differences in reactivity were observed. All complexes were stable and capable of initiating the ROP of rac-lactide, with (entries 5–8) or without benzyl alcohol (BnOH) (entries 1–4). Under these conditions, PLA molecular weights were slightly lower for reactions in the presence of alcohol (see below). Moreover, compared to work done previously with main group metals under the same conditions but with higher monomer loadings (greater than 50), higher conversions could be achieved in shorter reaction times with lower dispersities values using these complexes.27,73,74 To study the effect of the temperature and the co-initiator, compound 3 was further scrutinized (entries 9–12). The complex was able to efficiently polymerize rac-lactide at 130 °C, which is particularly relevant to industry.4 For bulk polymerization, studies indicate a pseudo first-order dependence on the monomer concentration as shown in conversion vs. time plots (Fig. S7†). However, kinetic data cannot be obtained from such graphs because the reaction proceeds too quickly to ascertain initial rates where rac-lactide concentrations will still be high. In addition, as expected it was found that the conversion rates were greater with BnOH than without it (entries 9 and 11). The control of macromolecular features is also much improved when BnOH is used, both in terms of PDIs and agreement between Mncalvs. Mn. All the generated polymers have molecular weights higher than the theoretical values (Mncal), which might be attributed to intermolecular transesterification reactions,75,76 but are less significant in reactions when BnOH is present. Notably, the obtained dispersities (with or without BnOH) were more narrow than previously reported values27,73,74 for polymers prepared in the melt-phase suggesting a more controlled polymerization. It should be noted that the Mn values increase moderately with longer reaction times (entries 11 and 12). In addition, there was no significant difference in the molecular weights between the polymers produced at 150 °C and 130 °C. However, lower molecular weights were observed when polymerizations were run with higher monomer loadings (entries 13 and 14).
Table 1 Polymerization of rac-lactide using 1, 2, 3 and 4 in the presence and absence of BnOH in the melt phase
| Entry |
Complex |
[LA]0/[M]0/[BnOH]0 |
t/min |
T/°C |
Conv.a /% |
M
ncal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b × 103 b × 103 |
M
nc × 103 |
M
w/Mnc |
|
Determined by 1H NMR spectroscopy.
The Mncal value of the polymer was calculated with Mncal = ([LA]0/[M]0) × 144.13 × conv. %/[BnOH] + 108.14.
M
n (g mol−1) determined by triple detection gel permeation chromatography (GPC) in THF using a dn/dc value of 0.049 mL g−1.
|
| 1 |
1
|
50/1/0 |
90 |
150 |
98 |
7.1 |
13.0 |
1.30 |
| 2 |
2
|
50/1/0 |
90 |
150 |
97 |
7.0 |
10.2 |
1.10 |
| 3 |
3
|
50/1/0 |
90 |
150 |
99 |
7.1 |
12.0 |
1.14 |
| 4 |
4
|
50/1/0 |
90 |
150 |
96 |
7.0 |
11.3 |
1.20 |
| 5 |
1
|
50/1/1 |
90 |
150 |
98 |
7.1 |
8.2 |
1.05 |
| 6 |
2
|
50/1/1 |
90 |
150 |
98 |
7.2 |
9.2 |
1.06 |
| 7 |
3
|
50/1/1 |
90 |
150 |
99 |
7.2 |
9.0 |
1.20 |
| 8 |
4
|
50/1/1 |
90 |
150 |
99 |
7.2 |
10.3 |
1.10 |
| 9 |
3
|
50/1/0 |
10 |
130 |
93 |
6.7 |
14.0 |
1.14 |
| 10 |
3
|
50/1/0 |
120 |
130 |
98 |
7.0 |
16.2 |
1.21 |
| 11 |
3
|
50/1/1 |
5 |
130 |
97 |
7.1 |
9.4 |
1.04 |
| 12 |
3
|
50/1/1 |
120 |
130 |
99 |
7.2 |
11.0 |
1.18 |
| 13 |
2
|
250/1/0 |
90 |
150 |
73 |
26.3 |
8.0 |
1. 22 |
| 14 |
2
|
250/1/1 |
90 |
150 |
99 |
35.8 |
6.4 |
1.18 |
Polymerizations in solution
1–4 were examined for ROP of rac-lactide in CH2Cl2 at room temperature. To examine solvent effects that may influence activities in these reactions, polymerization with 3 was further explored in THF and toluene. Representative results are reported in Table 2. Comparison between melt phase and solution polymerization data (Table 1, melt phase, entries 13 and 14, and Table 2, CH2Cl2, entries 7 and 8) shows that although polymer dispersities are similar, the molecular weights of polymers prepared in solution are higher and in closer agreement with theoretically predicted values than those obtained under melt phase conditions. This difference is seen both in the presence and absence of BnOH.
Table 2 Polymerization of rac-lactide using 1, 2, 3 and 4 in the presence and absence of BnOH
| Entry |
Complex |
[LA]0/[M]0/[BnOH]0 |
t/min |
Conv.d /% |
M
ncale × 103 |
M
nf × 103 |
M
w/Mnf |
| All reactions performed at 25 °C. CH2Cl2 (5 mL). Toluene (30 mL). THF (20 mL). Determined by 1H NMR spectroscopy. The Mncal value of the polymer was calculated with Mncal = ([LA]0/[M]0) × 144.13 × conv. %/[BnOH] + 108.14. The Mn (g mol−1) determined by triple detection gel permeation chromatography (GPC) in THF using a dn/dc value of 0.049 mL g−1. Low molecular weight oligomers formed. Data from NMR end group analysis: n = 5, Mn ∼ 384 g mol−1. CH2Cl2 (20 mL). 0.2 mL n-BuLi (0.16 M, 0.02 mmol) was added to CH2Cl2 solution (5 mL) containing rac-lactide (2.95 mmol) and BnOH (0.0118) at 25 °C. NaH (0.16 M, 0.047 mmol) was added to CH2Cl2 solution (5 mL) containing rac-lactide (2.95 mmol) and BnOH (0.0118 mmol) at 25 °C. Pr values for polymers produced by 2 and 3 were typically 0.44–0.47. |
| 1 |
1
|
250/1/0a |
80 |
92 |
33.1 |
28.0 |
1.31 |
| 2 |
1
|
250/1/1a |
80 |
93 |
33.6 |
17.2 |
1.29 |
| 3 |
1
|
250/1/2a |
40 |
92 |
16.7 |
8.00 |
1.15 |
| 4 |
1
|
250/1/2a |
300 |
99 |
17.9 |
12.1 |
1.24 |
| 5 |
1
|
250/1/4a |
30 |
90 |
8.2 |
4.00 |
1.19 |
| 6 |
1
|
250/1/4a |
300 |
95 |
8.7 |
7.80 |
1.15 |
| 7 |
2
|
250/1/0a |
40 |
73 |
26.3 |
24.0 |
1.30 |
| 8 |
2
|
250/1/1a |
7 |
93 |
33.6 |
19.0 |
1.18 |
| 9 |
2
|
250/1/0h |
60 |
53 |
19.1 |
13.0 |
1.11 |
| 10 |
2
|
250/1/0c |
180 |
55 |
19.8 |
21.0 |
1.10 |
| 11 |
3
|
50/1/0a |
5 |
100 |
7.2 |
ND |
— |
| 12 |
3
|
250/1/0a |
80 |
94 |
33.9 |
33.4 |
1.33 |
| 13 |
3
|
250/1/1a |
80 |
91 |
33.0 |
29.6 |
1.32 |
| 14 |
3
|
250/1/0b |
180 |
69 |
24.9 |
NDg |
— |
| 15 |
3
|
250/1/1b |
120 |
93 |
33.6 |
NDg |
— |
| 16 |
3
|
250/1/0c |
240 |
52 |
18.7 |
NDg |
— |
| 17 |
3
|
250/1/1c |
3 |
98 |
35.4 |
NDg |
— |
| 18 |
4
|
250/1/0a |
40 |
91 |
32.8 |
26.3 |
1.20 |
| 19 |
4
|
250/1/1a |
5 |
94 |
34.1 |
22.4 |
1.36 |
| 20 |
4
|
250/1/1a |
300 |
99 |
35.8 |
34.8 |
1.22 |
| 21 |
4
|
250/1/0c |
3 |
90 |
32.4 |
36.2 |
1.41 |
| 22 |
4
|
250/1/1c |
3 |
100 |
36.1 |
20.2 |
1.10 |
| 23i |
n-BuLi |
250/1/1a |
60 |
96 |
34.7 |
11.8 |
1.13 |
| 24j |
NaH |
250/1/1a |
>540 |
65 |
23.5 |
11.7 |
1.47 |
All solution polymerizations showed a first-order dependence on lactide concentration in the form of a linear relationship of ln([LA]0/[LA]t) versus time as shown in Fig. 8–13 and S8–S12.† Polymerization data (Table 2) shows that catalytic activities were strongly influenced by the nature of the solvent and the presence of BnOH (initiator). It should be noted that a large amount of toluene (30 mL) was essential due to the low solubility of LA in toluene at room temperature. Performing the reaction in CH2Cl2 (entry 12) in the absence of BnOH using 3 resulted in a much faster reaction compared to toluene and THF (entries 14 and 16) with high conversion after only 80 min. Furthermore, see below, the molecular weights of the polymers obtained in toluene and THF was very low (∼384 g mol−1) despite moderate conversion levels. Due to the high solubility of both LA and catalysts in CH2Cl2, reactions in this solvent normally contained 2.95 mmol of LA and the desired amount of catalyst in 5 mL. When reactions were performed using 2 under similar conditions (entries 7 and 9) but using different volumes of CH2Cl2, Mn values were lower than theoretically predicted under more dilute reaction conditions whereas good agreement with calculated Mn values was seen when only 5 mL CH2Cl2 was used. This shows that in addition to rate effects, the volume of solvent used can also affect polymerization reactions in terms of polymer properties. Compared to CH2Cl2 and toluene, lower conversions were observed when THF was employed and this could be due to the coordinating nature of THF, which competes with the incoming monomer for coordination at the metal center (entry 16 compared with entries 12 and 14).29,40,74 Moreover, BnOH is able to significantly speed up the polymerizations carried out in THF (entry 17), whereas little difference in activities were observed for both CH2Cl2 and toluene (entries 13 and 15). Therefore, kinetic data for reactions performed in THF in the presence of BnOH could not be accurately obtained, as assumptions regarding the steady-state concentration of rac-lactide could not be made (Fig. S8†). It should be noted that with 3 low molecular weight oligomers were likely obtained when toluene and THF were used (entries 14–17) because no polymer precipitated upon the addition of cold methanol to reaction mixtures and therefore, GPC data were not obtained for these samples. The formation of low molecular weight species was confirmed by end group analysis where the 1H NMR spectra of the polymers. Integration of the –OH end group resonance relative to the CH3 group showed that low molecular weight polymer formed (n = 5384 g mol−1).
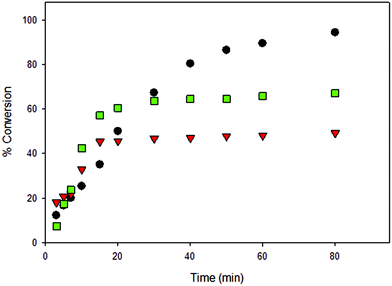 |
| | Fig. 8 Conversion (%) vs. time for the ROP of LA initiated by 3 [conditions: 2.95 mmol LA, 250LA:1Li:0BnOH, 25 °C]. ● CH2Cl2 (5 mL),  toluene (30 mL), toluene (30 mL),  THF (20 mL). THF (20 mL). | |
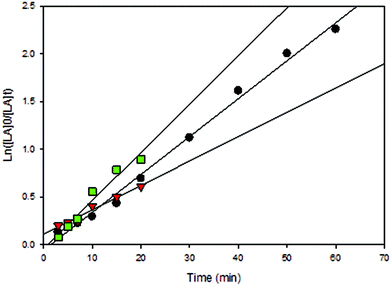 |
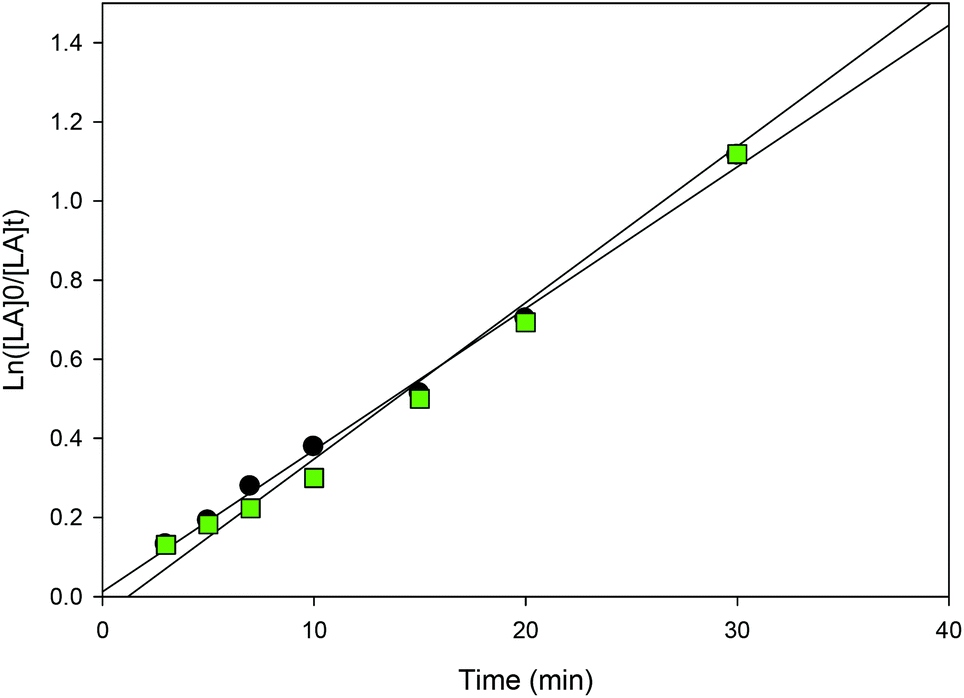
| | Fig. 9 First-order plot of LA consumption initiated by 3. [conditions: 2.95 mmol LA, 250LA:1Li:0BnOH, 25 °C]. ● CH2Cl2 (5 mL, y = 0.0397x − 0.0594, R2 = 0.9933),  toluene (30 mL, 0.0506x − 0.0448, R2 = 0.96), toluene (30 mL, 0.0506x − 0.0448, R2 = 0.96),  THF (20 mL THF (20 mL  toluene (y = 0.0255x + 0.1097, R2 = 0.9742). toluene (y = 0.0255x + 0.1097, R2 = 0.9742). | |
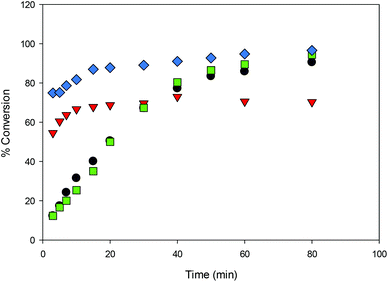 |
| | Fig. 10 Conversion (%) vs. time for the ROP of LA initiated by 1, 2, 3 and 4 in CH2Cl2 (5 mL) [conditions: 2.95 mmol LA, 250LA:1M:0BnOH, 25 °C] ● 1,  2, 2,  3, 3,  4. 4. | |
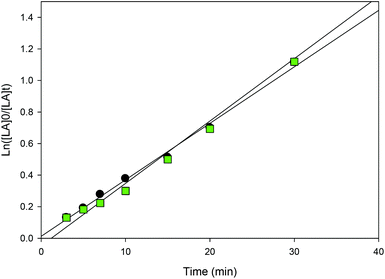 |
| | Fig. 11 First-order plot of LA consumption initiated by 1 and 3 in CH2Cl2 (5 mL) [conditions: 2.95 mmol LA, 250LA:1Li:0BnOH, 25 °C], ● 1 (y = 0.0358x + 0.0123, R2 = 0.9954),  3 (y = 0.0395x − 0.0471, R2 = 0.9951). 3 (y = 0.0395x − 0.0471, R2 = 0.9951). | |
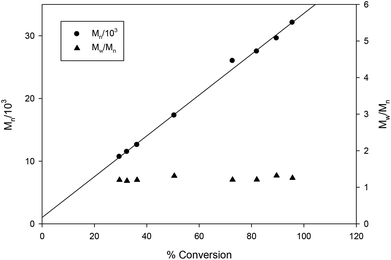 |
| | Fig. 12 Plot of PLA Mn and dispersity (Mw/Mn) as a function of rac-lactide conversion [conditions: CH2Cl2 5 mL, 2.95 mmol LA, 250LA:1Li:1BnOH, 25 °C]. Line shown to indicate the linear trend and proportional increases in Mn as conversion increases. | |
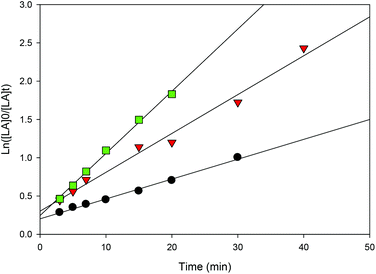 |
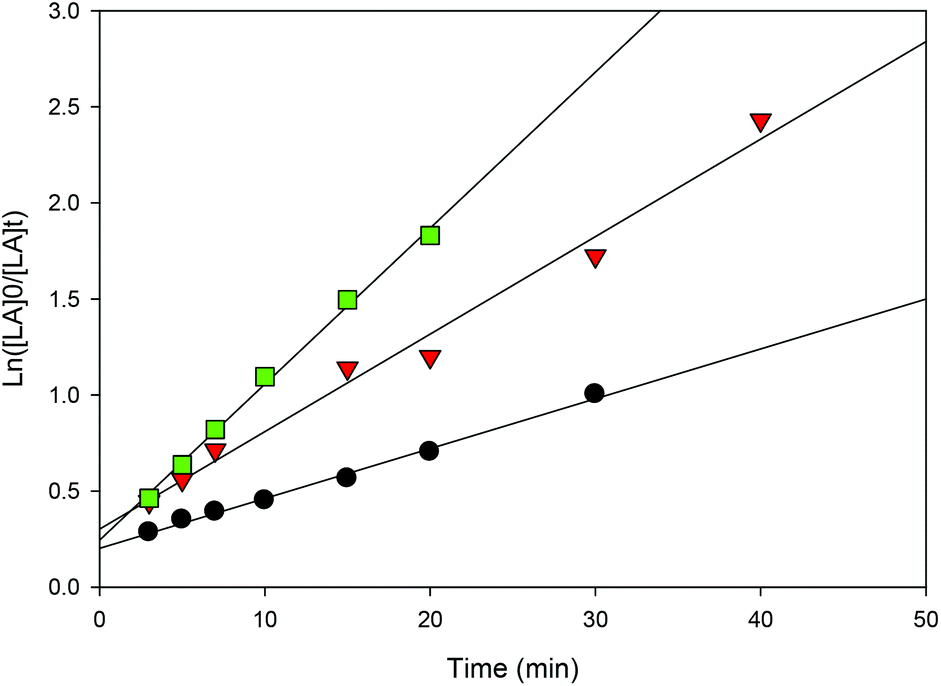
| | Fig. 13 First-order plot of LA consumption initiated by 1 with different quantities of BnOH [conditions: CH2Cl2 (5 mL), 2.95 mmol LA, 250LA:1Li:nBnOH, 25 °C]. ● 1 eq. BnOH (y = 0.0507x + 0.3033, R2 = 0.9858),  2 eq. BnOH (y = 0.0259x + 0.2026, R2 = 0.9944), 2 eq. BnOH (y = 0.0259x + 0.2026, R2 = 0.9944),  4 eq. BnOH (y = 0.0812x + 0.2446, R2 = 0.9963). 4 eq. BnOH (y = 0.0812x + 0.2446, R2 = 0.9963). | |
The steric bulk of substituents on the aromatic rings was found to have less influence on the activity compared to the identity of the metal center (entries 1 and 12). That is, the activity trend decreases in the order 4 > 2 > 3 ≥ 1 and can be partially explained by the larger ionic radius of sodium compared to lithium.26,41,50,51 It is not surprising that the initiator is not required for ROP catalyzed by 1 and 3 as the same observation was reported by Kozak's group for related amino-bis(phenolate) lithium complexes.38 In contrast, the presence of the initiator was necessary to accelerate reactions using sodium complexes (entries 18 and 19) and this has also been reported by others.52,53 The observed Mn values of these solution-phase polymers are in some cases close to the expected molecular weights and the low dispersities obtained (ranging from 1.10–1.36) indicated that the polymerization has characteristics of controlled propagation. The controlled behavior is also demonstrated by the linear relationship between Mn and % conversion, and the narrow dispersity of the polymers throughout their chain growth (Fig. 12).31,39,76
Increasing the amount of BnOH can be used to control both the molecular weight and the polymerization rate. For instance, upon doubling the amount of alcohol to two equiv. per lithium center, the molecular weight of the polymer diminished to half its original value (entries 2 and 3). While it decreased to one quarter with the addition of four equiv. BnOH (entries 2 and 4). In addition, as shown in Fig. 13 and S12,† in both cases the reaction rates are faster compared to performing the reaction with one equiv. of benzyl alcohol. Such observations are an important feature of well-behaved immortal ROP with reversible and fast chain transfer between dormant and growing macroalcohols.39,76 Performing the reaction at longer times gives higher molecular weight compared with short reaction times (e.g. entries 3 and 4) suggesting the occurrence of chain transfer reactions during polymerization.27,39 We propose that once LA conversion is close to completion, the lithium-polymeryl species undergo intermolecular chain transfer (as there is little remaining monomer to react with) and this leads to higher Mn values.
Moreover, a comparison of the catalytic activities of 1–4 and diamino-bis(phenolate)-free systems (i.e. control reactions) has been undertaken under identical ROP conditions. These polymerization systems use the precursor metal reagents as initiators and no phenolic ligand. One equiv. of n-BuLi or four equiv. NaH was added into a CH2Cl2 solution containing 250 equiv. of rac-lactide and one equiv. of BnOH (entries 23 and 24). ROP of lactide using the in situ formed lithium alkoxide was complete within 60 min with a conversion of 96% whereas the sodium alkoxide exhibited lower efficiency with moderate conversion (65%) and a more disperse polymer was produced. All the generated polymers from phenolate-free systems had lower molecular weights than those produced with 1–4.
NMR spectroscopy of polymers in bulk and solvent polymerization
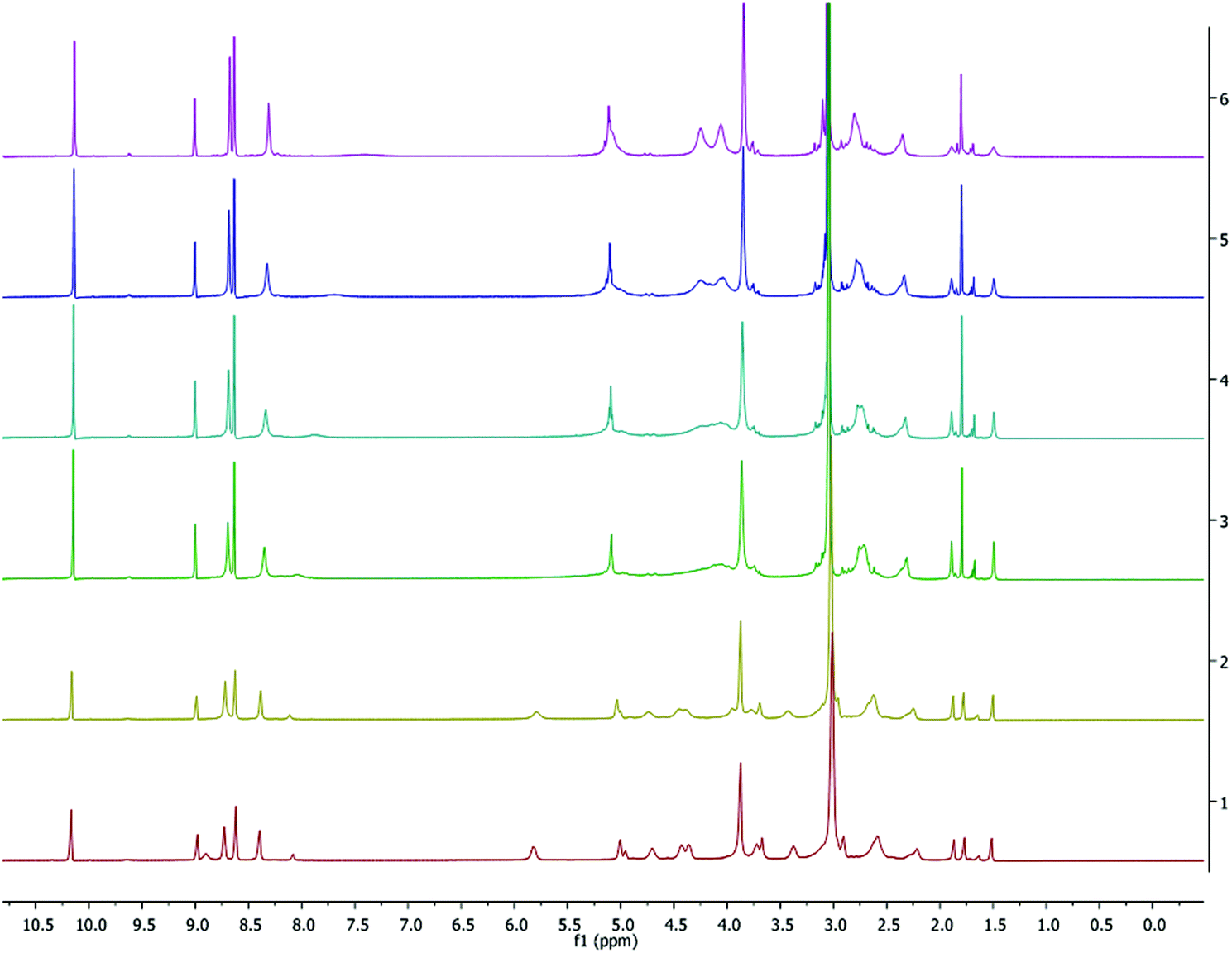
Similar signals were found in 1H NMR spectra for the polymers from reactions performed in both the absence and the presence of one equiv. BnOH: a hydroxyl group (d) at 2.72 ppm, a hydroxymethine group (HOCH–) (c) at 4.35 ppm, methyl groups (CH–CH3) (a) between 1.50 and 1.55 ppm (Fig. S13†).38 No evidence for benzyl ester group formation could be found when one equiv. BnOH was added, suggesting the presence of inter or intratransesterfication reactions. However, a benzyl ester group (OCH2Ph) (e) at 7.32 ppm, hydroxyl group (f) and hydroxymethine group (HOCH–) (c) at 4.33 ppm were observed as end-groups with the addition of two and four equiv. BnOH (Fig. S14†).
Determination of the polymer tacticity was achieved by homodecoupled 1H and 13C NMR experiments (Fig. S15 and S16,† respectively). The probability of racemic enchainment (probability of forming a new racemic diad) was calculated from the deconvoluted homonuclear decoupled 1H NMR spectra.7 The Pr values for polymers produced by 2 and 3 are within the range 0.44–0.47, which indicates a negligible isotactic bias (a value of 0.5 is expected for a perfectly atactic polymer).53 Isotactic polymers have previously been reported in some cases for lithium and sodium phenolates.38,53 In 13C spectra the signal assignments in the methine region show multiple possible tetrad sequences iii, iis, sii and isi which is in good agreement with previously reported ROP of rac-lactide by an achiral catalyst.77 However, the unusual increase in the intensity of the iss, sss and ssi tetrads in Fig. S15† and the presence of weaker peaks at 69.58, 69.47 and 69.25 ppm in Fig. S16† indicate stereorandom transesterification during the course of the polymerization reactions.38,53,78
Mass spectrometry of polymers
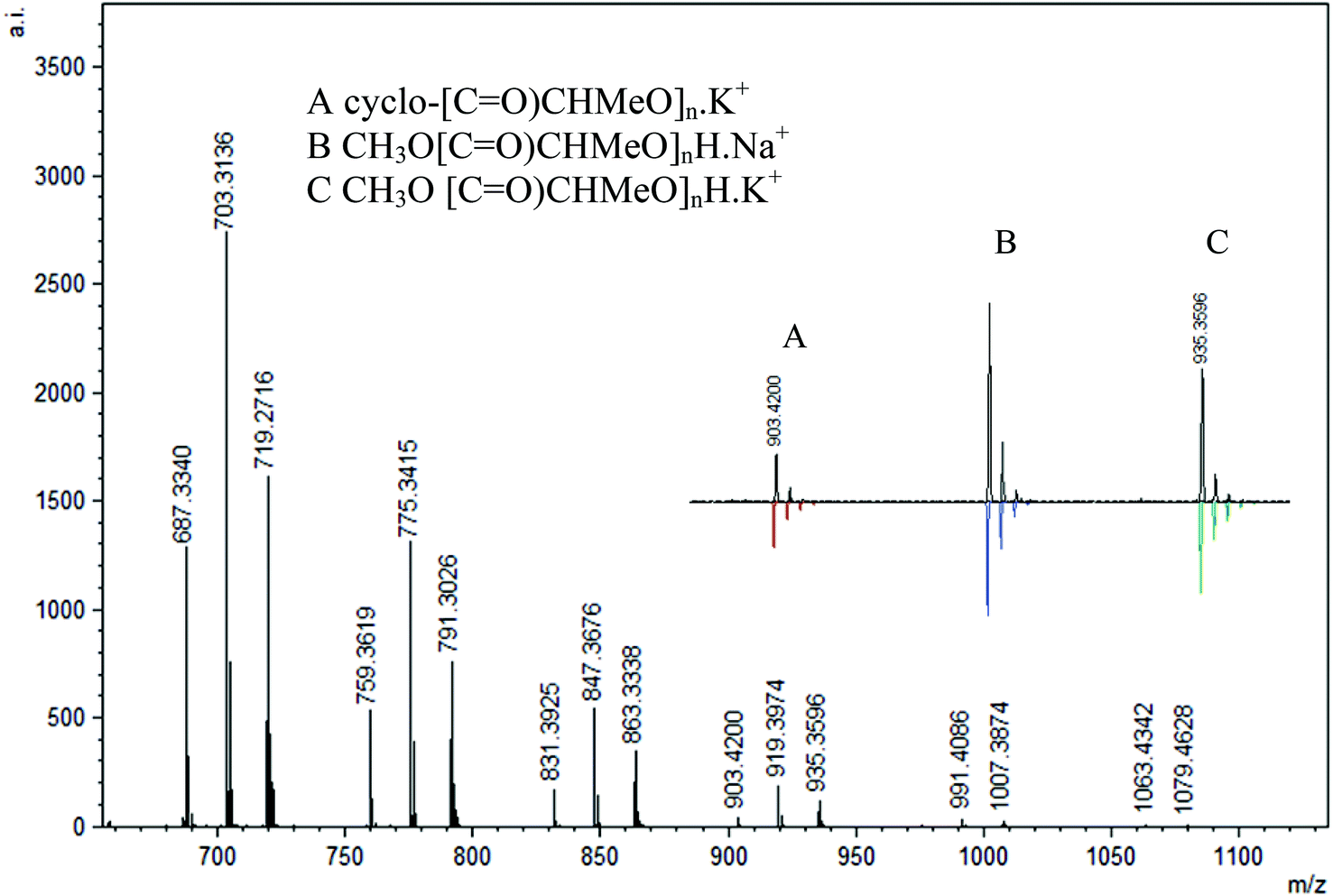
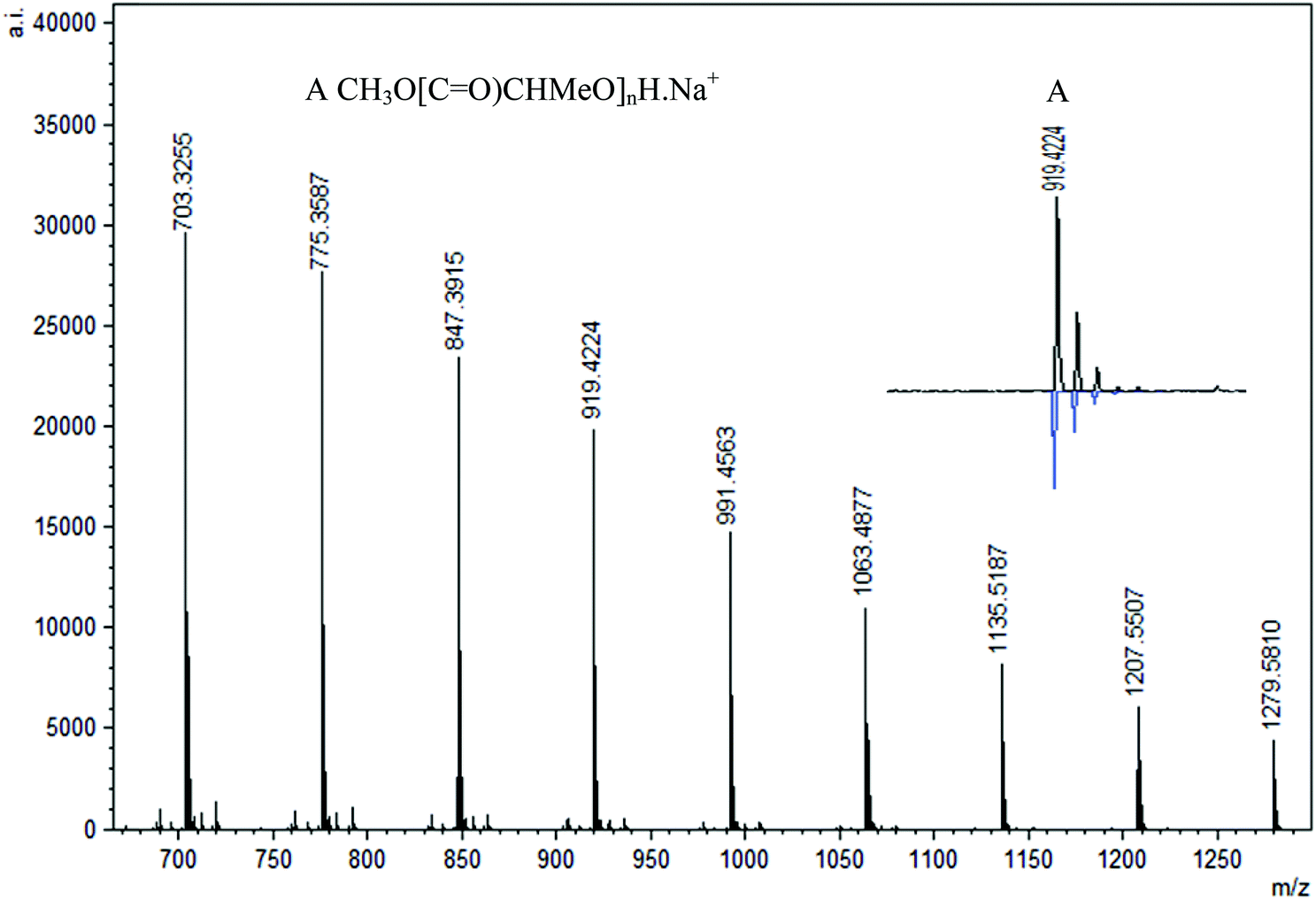
MALDI-TOF analysis of PLA was conducted with 2,5-dihydroxybenzoic acid (DHBA) as the matrix with a ratio of 5:1 (matrix:PLA). Using reflectron mode, the peaks are separated by 72 mass units and three major repeating masses were observed in the case of polymers obtained in the absence of BnOH and 1 equiv. BnOH. As shown in Fig. 14, two intense peaks (B and C, n = 12, m/z = 919 and 935) were assigned as CH3O[C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CHMeO]nH·Na+ and CH3O[CO)CHMeO]nH·K+. In addition, a less intense series of peaks for cyclic polymer (A, n = 12, m/z = 903) clustered with a Na+ ion were seen, pointing to the presence of intrachain transesterification side reactions. The termination with methoxy groups probably stems from initiation of polymerization by nucleophilic attack of the phenolate oxygen on the carbonyl followed by quenching with methanol. Similar end-groups have been observed by others recently.79 The polymers formed with 2 and 4 equiv. BnOH show only one major repeating series (A, n = 12, m/z = 919) that corresponds to polymers capped with methoxy groups CH3O[CO)CHMeO]nH·Na+ without any evidence for cyclic formation or the benzyl ester groups (Fig. 15). This contrasts with the NMR data obtained, which showed the presence of the expected benzyl ester groups.
O)CHMeO]nH·Na+ and CH3O[CO)CHMeO]nH·K+. In addition, a less intense series of peaks for cyclic polymer (A, n = 12, m/z = 903) clustered with a Na+ ion were seen, pointing to the presence of intrachain transesterification side reactions. The termination with methoxy groups probably stems from initiation of polymerization by nucleophilic attack of the phenolate oxygen on the carbonyl followed by quenching with methanol. Similar end-groups have been observed by others recently.79 The polymers formed with 2 and 4 equiv. BnOH show only one major repeating series (A, n = 12, m/z = 919) that corresponds to polymers capped with methoxy groups CH3O[CO)CHMeO]nH·Na+ without any evidence for cyclic formation or the benzyl ester groups (Fig. 15). This contrasts with the NMR data obtained, which showed the presence of the expected benzyl ester groups.
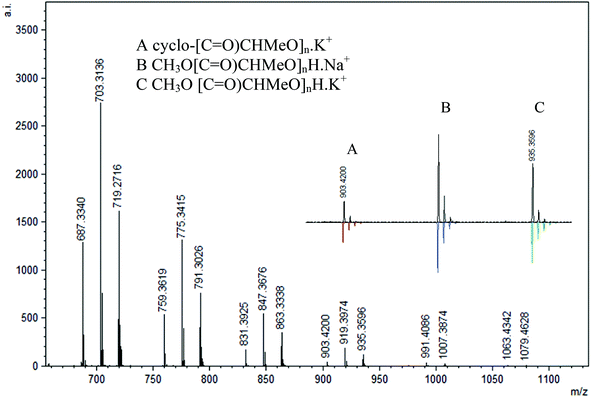 |
| | Fig. 14 Representative region of the MALDI-TOF mass spectrum (reflectron mode) of PLA formed using 3 under the following conditions: CH2Cl2 (5 mL), 2.95 mmol LA, 250LA:1Li:0BnOH, 25 °C (similar spectra obtained for PLA from other reactions using no BnOH). | |
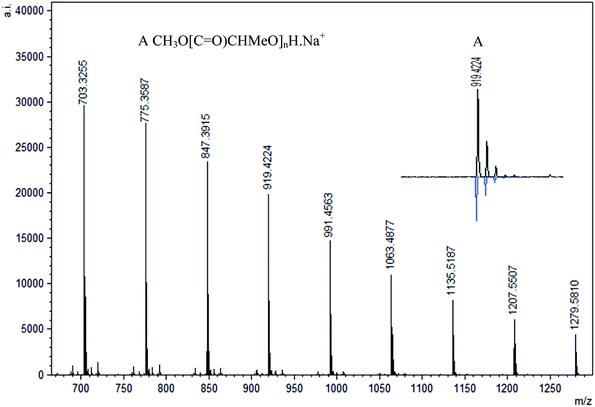 |
| | Fig. 15 Representative region of the MALDI-TOF mass spectrum (reflectron mode) of PLA formed using 1 under the following conditions: CH2Cl2 (5 mL), 2.95 mmol LA, 250LA:1Li:2BnOH, 25 °C. | |
Mechanistic proposal
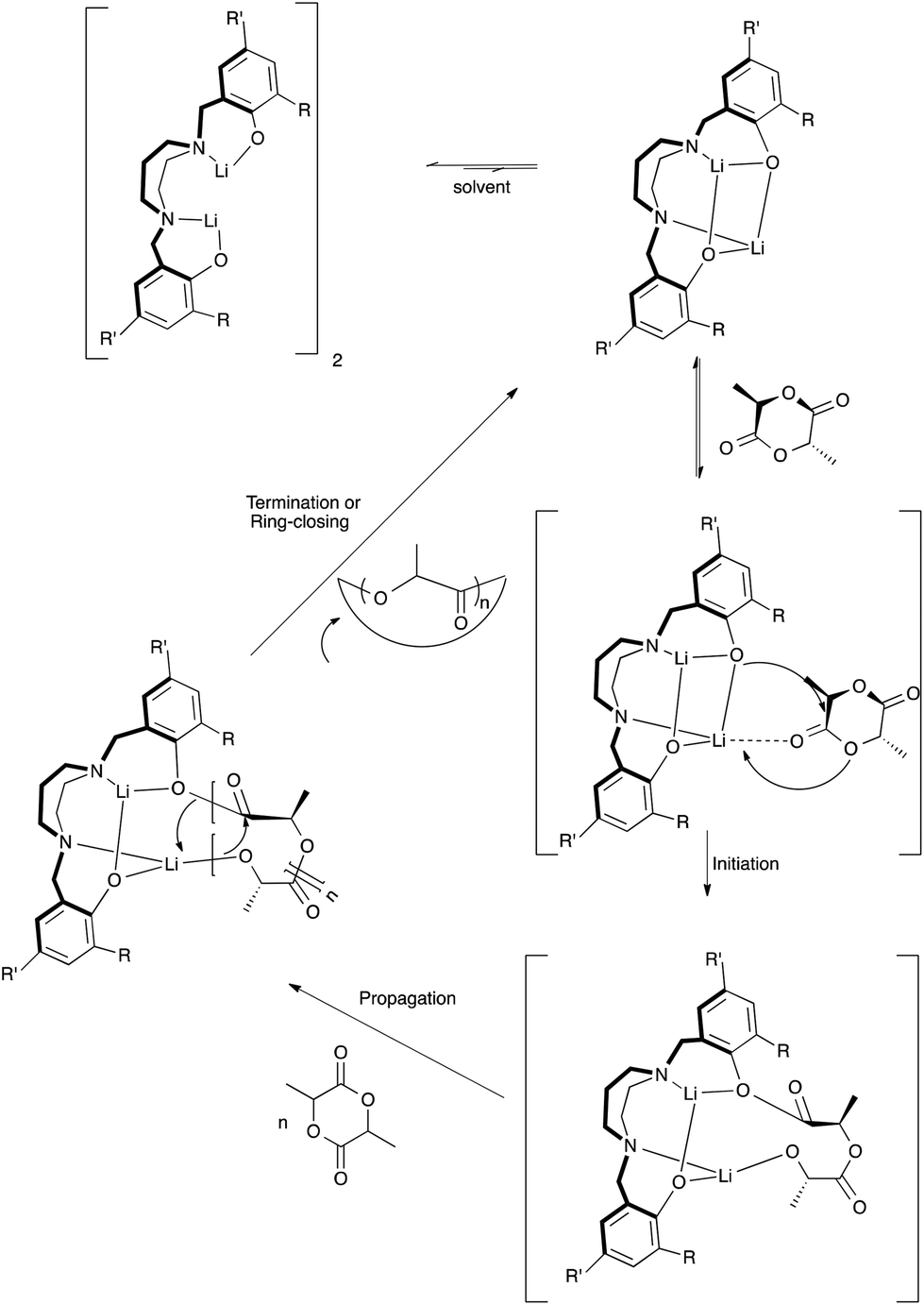
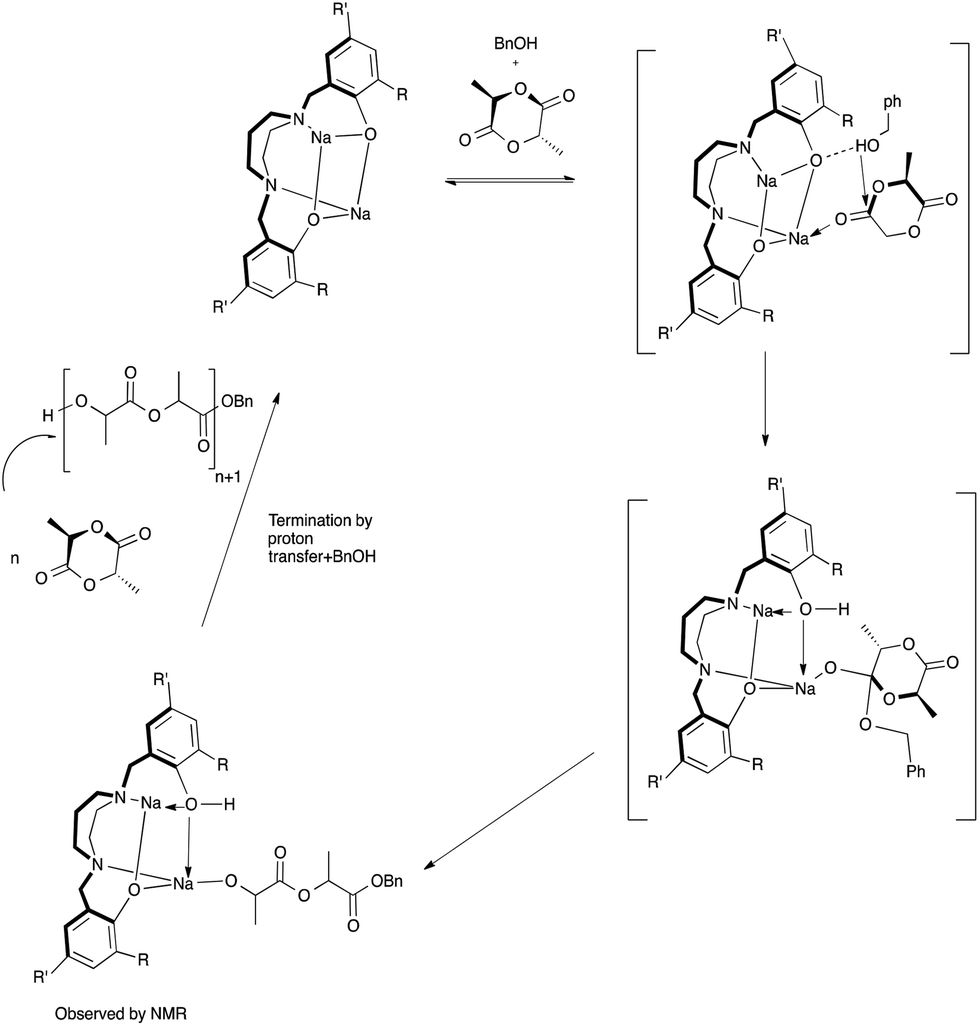
In the absence of BnOH, the initiation step in the ROP of rac-lactide could be proceeding via coordination insertion of the monomer into the metal–phenoxide bond as shown in Fig. 16 and described in the literature for related metal–phenolate systems.38,40,49,80–84 Comparison of 7Li NMR spectra recorded in the absence and presence of rac-lactide suggests the formation of an additional Li species upon addition of a small amount of rac-lactide with the growth of a new peak at 1.30 ppm (Fig. S17†). This supports the monomer coordination step of the mechanisms described herein but it should also be noted that these 7Li NMR spectra were obtained without BnOH. To investigate the ROP reaction in the presence of BnOH, stoichiometric reactions with 1 in CD2Cl2 at room temperature were monitored by 1H NMR spectroscopy. The spectra of a 1:1 (per 2 metal centers) reaction of 1 and BnOH confirms the formation of both {N2O2BuBuPip}H (11.05 ppm) and BnO–Li, the latter appearing as overlapping peaks around 7.34 ppm (Fig. S18, 19 and 20†). Similar observations have also been reported for related amino-phenolate systems by Chen and co-workers.43 Addition of 1 equiv. of rac-lactide should yield benzyl-2-((2-hydroxypropanoyl)oxy)propanoate, the product of lactide ring-opening. Unfortunately, we were unable to isolate this or identify resonances belonging to this species. In spite of this, we propose that ring-opening polymerization is occurring through a coordination–insertion mechanism, where benzyl alcohol is sufficiently activated by the lithium centers and protonates the phenolate group to yield a new lithium complex, followed by attack of the benzyl alkoxide group at the carbonyl group of rac-lactide (Fig. 17).38,40,49,80–84 We also note that the reactivity of BnOLi formed in situ affords similar polymerization data (Table 2, entry 23). From DOSY NMR experiments above, the predominant Li species present in CD2Cl2 (in the absence of lactide and BnOH) is the tetralithium complex. Therefore, it is likely that the alcohol also plays a role in assisting the dissociation process in solution, as we assume this will occur under reaction conditions to allow space for the growing polymer chain. Further data would be required in order to confirm this assumption.
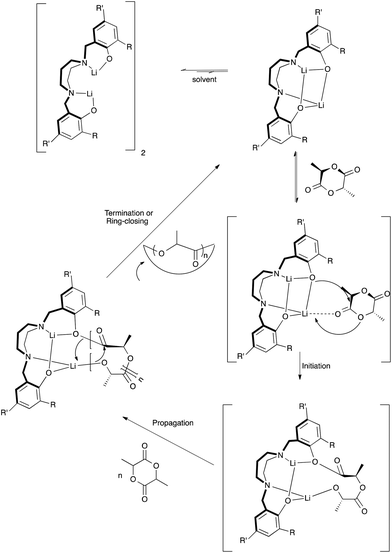 |
| | Fig. 16 Proposed mechanism of the ROP of rac-LA in the absence of BnOH initiated by lithium and sodium diamino-bis(phenolate) complexes (THF omitted for clarity, coordination sphere of the metals will be completed by either LA or THF). | |
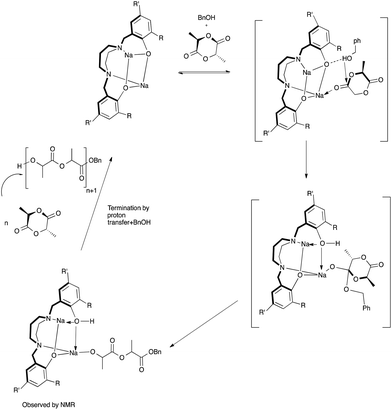 |
| | Fig. 17 Proposed mechanism of the ROP of rac-LA in the presence of BnOH initiated by sodium diamino-bis(phenolate) complexes (THF omitted for clarity, coordination sphere of the metals will be completed by either LA or THF). | |
Experimental
General experimental conditions
All operations were carried out under an atmosphere of dry, oxygen-free nitrogen using standard Schlenk techniques or an MBraun Labmaster glove box. Anhydrous THF, benzene were distilled from sodium benzophenone ketyl under nitrogen. Toluene and pentane were purified by an MBraun Manual Solvent Purification System. Most reagents were purchased either from Aldrich or Alfa Aesar and used without further purification. rac-Lactide was purchased from Alfa Aesar and dried over Na2SO4 in THF, recrystallized and stored under nitrogen in ampules. Benzyl alcohol was purchased from Alfa Aesar and dried over activated 4 Å molecular sieves, distilled under reduced pressure and stored under nitrogen in an ampule prior to use. Deuterated solvents were purchased from Cambridge Isotope Laboratories and purified and degassed through freeze–vacuum–thaw cycles and stored under nitrogen in ampules fitted with Teflon valves. NaH was washed twice with hexane, as it was purchased as a suspension in mineral oil. Aqueous formaldehyde (37 wt%) was purchased from Fisher Scientific and also used without further purification.
Instrumentation
1H, 13C and 1H{1H} NMR spectra were recorded on a Bruker Avance 500 or 300 MHz spectrometer at 25 °C (unless otherwise stated) and were referenced internally using the residual proton and 13C resonances of the solvent. 7Li NMR was recorded on a Bruker 300 MHz spectrometer and referenced externally to LiCl in D2O. MALDI-TOF MS spectra were obtained using an Applied Biosystems 4800 MALDI TOF/TOF Analyzer equipped with a reflectron, delayed ion extraction and high performance nitrogen laser (200 Hz operating at 355 nm). Samples were prepared in the glove box and sealed under nitrogen in a Ziploc© bag for transport to the instrument at a concentration of 10.0 mg mL−1 in toluene. For ligands and complexes, anthracene was used as the matrix, which was mixed at a concentration of 10.0 mg mL−1. For the polymers, mass spectra were recorded in reflectron mode and DHBA was used as the matrix and purified tetrahydrofuran was used as the solvent for depositing analytes onto the instrument's plate. The matrix was dissolved in THF at a concentration of 10 mg mL−1. Polymer was dissolved in THF at approximately 1 mg mL−1. The matrix and polymer solutions were mixed together at a ratio of 5 to 1; 1 μL of this was spotted on the MALDI plate and left to dry. Images of mass spectra were prepared using mMass™ software (http://www.mmass.org). GPC analysis was performed in THF at 25 °C on a Wyatt Triple Detection (triple angle light scattering, viscometry and refractive index) system with a Agilent 2600 series LC for sample and solvent handling, and two Phenogel 103 Å 300 × 4.60 mm columns. Samples were dissolved in THF at a concentration of 1 mg mL−1, left to equilibrate for ∼2 h and passed through syringe filters before analysis. An eluent flow rate of 0.30 mL min−1 and 100 μL injection volume were used. Molecular weights (g mol−1) were determined by triple detection using a dn/dc value of 0.049 mL g−1. Conversions were determined by integration of the methyl signals due to the residual rac-lactide and produced poly-(rac-lactide). Elemental analyses were performed by Canadian Microanalytical Service Ltd, Delta, BC Canada or at the Ocean Sciences Centre, St. John's, NL Canada.
Diffusion NMR measurements were performed on a Bruker Avance 500 NMR spectrometer equipped with a 5 mm TXI probe and a z-gradient coil with a maximum strength of 5.35 G cm−1 at 298 K. The sample was run in CD2Cl2 at a concentration of 10.0 mM. The 90° pulse length and the relaxation time T1 of the sample were determined before running the DOSY experiment. A standard 2D sequence with double stimulated echo and spoil gradient (DSTEBPGP3s) was used. The relaxation delay was set at 10 s. The gradient strength was calibrated by using the self-diffusion coefficient of residual HOD in D2O (1.9 × 10−9 m2 s−1). For each experiment, the gradient strength was increased from 2–95% in 32 equally spaced steps with 16 scans per increment. Values of δ (gradient pulse length) and Δ (diffusion time) were optimized on the sample to give an intensity of between 5 and 10% of the initial intensity at 95% gradient strength and were set to 1 ms and 100 ms respectively. The solvent peak was used as an internal standard to measure the viscosity of each sample, D0(CD2Cl2) = 3.28 × 10−9 m2 s−1 and η0 = 0.413 cp at 298 K.85 The data were plotted using DynamicCenter (Bruker) and the diffusion coefficient (D) was extracted by fitting a mono exponential function (ln(I/I0) = −γ2δ2G2(Δ − δ/3)Dt) with the data analysis component of the software. The hydrodynamic radius of the complex (rH,PGSE) was calculated using the procedure outlined by Carpentier, Sarazin and co-workers.39 An average of the values of Dt (9.86 × 10−10 m2 s−1 ± 0.65) found for 3 separate peaks in the 1H PGSE NMR spectrum was used in the calculations. rH(CD2Cl2) = 2.49 Å was used.86
Synthesis and characterization
Synthesis of H2[N2O2BuBuPip].
A mixture of 2,4-di-tert-butylphenol (24.4 g, 0.123 mol), 37% w/w formaldehyde (10.0 mL, 123 mmol) and homopiperazine (6.22 g, 0.0615 mol) in water (100 ml) was stirred and heated to reflux for 24 h. Upon cooling to room temperature, solvents were decanted from the resulting white solid, which was recrystallized from methanol and chloroform to afford a pure white powder (32.4 g, 98%). Anal. calc'd for C35H56N2O2: C, 78.31; H, 10.51; N, 5.22. Found: C, 78.19; H, 10.33; N, 5.11. 1H NMR (300 MHz, 298 K, CDCl3) δ 1.25 (18H, s, ArC–C(CH3)3), 1.40 (18H, s, ArC–C(CH3)3), 1.89 (2H, quintet, 3JHH = 6.04 Hz, N–CH2{CH2}CH2–N), 2.75 (4H, s, N–CH2{CH2}CH2–N), 2.80 (4H, t, 3JHH = 6 Hz, N–CH2CH2–N), 3.75 (4H, s, ArC–CH2–N), 6.79 (2H, d, 2JHH = 2.3 Hz, ArH), 7.19 (2H, d, 2JHH = 2.3, ArH), 11.03 (2H, s, OH). 13C {1H} NMR (300 MHz, 298 K, C6D6) δ 27.2 (N–CH2{CH2}CH2–N), 30.4 (ArC–C–(CH3)3), 32.4 (ArC–C–(CH3)3), 34.7 (ArC–C–(CH3)3), 35.7 (ArC–C–(CH3)3), 53.2 (N–CH2CH2–N), 54.9 (N–CH2{CH2}CH2–N), 63.1 (ArC–CH2–N), 122.1 (ArC–CH2–N), 123.6 (ArCH), 124.0 (ArCH), 136.5 (ArC–C(CH3)3), 141.1 (ArC–C(CH3)3), 155.6 (ArC–O). MS (MALDI-TOF) m/z (%, ion): 536.32 (100, H2[N2O2BuBuPip]+˙).
Synthesis of H2[N2O2BuMePip].
This was prepared in a similar manner to H2[N2O2BuBuPip] to yield a pure white powder (26.1 g, 93.6%). Anal. calc'd for C29H44N2O2: C, 76.95; H, 9.80; N, 6.19. Found: C, 77.11; H, 9.87; N, 6.12. 1H NMR (300 MHz, 298 K, CDCl3) δ 1.43 (18H, s, ArC–C(CH3)3), 1.89 (2H, quintet, 3JHH = 6.01 Hz, N–CH2{CH2}CH2–N), 2.25 (6H, s, ArC–CH3), 2.77 (4H, s, N–CH2{CH2}CH2–N), 2.82 (4H, t, 3JHH = 6.1 Hz, N–CH2CH2–N), 3.75 (4H, s, ArC–CH2–N, 6.66 (2H, s, ArH), 7.01 (2H, s, ArH), 11.0 (2H, s, OH). 13C {1H} NMR (300 MHz, 298 K, CDCl3) δ 21.0 (ArC–CH3), 27.0 (N–CH2{CH2}CH2–N), 29.7 (ArC–C–(CH3)3), 34.8 (ArC–C–(CH3)3), 53.2 (N–CH2CH2–N), 54.6 (N–CH2{CH2}CH2–N), 62.2 (ArC–CH2–N), 122.1 (ArC–CH2–N), 127.0 (ArCH), 127.4 (ArCH), 127.5 (ArC–CH3), 136.6 (ArC–C–(CH3)3), 154.6 (ArC–O). MS (MALDI-TOF) m/z (%, ion): 452.31(100, H2[N2O2BuMePip]+˙).
Synthesis of 1.
H2[O2N2]BuBuPip (2.01 g, 3.7 mmol) was dissolved in THF (50 mL) and cooled to −78 °C. n-Butyllithium (1.6 M, 5.0 ml, 8.14 mmol) was slowly added via cannula to afford a yellow solution, which was warmed to room temperature and stirred for 72 h. Solvent was removed under vacuum; the product was washed with cold pentane (10 mL). The product was then filtered and dried under vacuum to yield (2.36 g, 97%) of a pale yellow product. Anal. calc'd for C35H54Li2N2O2(0.8Li2O·0.35C5H12): C, 73.83; H, 9.81; N, 4.69. Found: C, 73.74; H, 10.03; N, 4.91. 1H NMR (300 MHz, 298 K, C6D6) δ 1.19 (8H, s, CH2, THF), 1.47 (18H, s, ArC–C(CH3)3), 1.68 (18H, s, ArC–C(CH3)3), 1.89 (6H, br, N–CH2{CH2}CH2–N), 2.71 (4H, s, N–CH2CH2–N), 3.38 (8H, s, CH2, THF), 3.58 (4H, s, ArC–CH2–N), 7.05 (2H, d, 2JHH = 2.2 Hz, ArH), 7.6 (2H, d, 2JHH = 2.2 Hz, ArH). 1H NMR (500 MHz, 298 K, C5D5N) δ 1.42 (18H, s, ArC–C(CH3)3), 1.53 (18H, s, ArC–C(CH3)3), 1.63 (4H, m, CH2, 3JHH = 6.5 Hz, THF), 1.68 (6H, s, CH2{CH2}CH2–N), 2.77 (4H, br, N–CH2CH2–N), 3.7 (4H, m, CH2, 3JHH = 6.5 Hz, THF), 3.86 (4H, s, ArC–CH2–N), 7.18 (2H, s, ArH), 7.53 (2H, s, ArH). 13C {1H} NMR (300 MHz, 298 K, CDCl3) δ 27.0 (CH2, THF), 29.8 (ArC–C–(CH3)3), 31.9 (ArC–C–(CH3)3), 34.4 (ArC–C–(CH3)3), 35.1 (ArC–C–(CH3)3), 53.3 (N–CH2CH2–N), 54.7 (N–CH2{CH2}CH2–N), 62.7 (ArC–CH2–N), 100.2 (ArC–CH2–N), 121.4 (ArCH), 123.6 (ArCH), 135.9 (ArC–C(CH3)3), 140.8 (ArC–C(CH3)3). 13C {1H} NMR (300 MHz, 298 K, C6D6) δ 25.6 (CH2, THF), 27.4 (N–CH2{CH2}CH2–N), 31.0 (ArC–C–(CH3)3), 32.8 (ArC–C–(CH3)3), 34.5 (ArC–C–(CH3)3), 35.7 (ArC–C–(CH3)3), 51.0 (N–CH2CH2–N), 53.4 (N–CH2{CH2}CH2–N), 62.8 (ArC–CH2–N), 68.5 (CH2, THF), 123.9 (ArC–CH2–N), 126.1 (ArCH), 127.2 (ArCH), 134.0 (ArC–C(CH3)3), 136.9 (ArC–C(CH3)3), 164.2 (ArC–O). 13C {1H} NMR (300 MHz, 298 K, C5D5N) δ 26.3 (CH2, THF), 27.4 (N–CH2{CH2}CH2–N), 31.1 (ArC–C–(CH3)3), 32.8 (ArC–C–(CH3)3), 34.5 (ArC–C–(CH3)3), 35.9 (ArC–C–(CH3)3), 51.8 (N–CH2CH2–N), 53.9 (N–CH2{CH2}CH2–N), 62.8 (ArC–CH2–N), 68.3 (CH2, THF), 126.4 (ArCH), 127.5 (ArCH), 132.8 (ArC–C(CH3)3), 137.5 (ArC–C(CH3)3), 166.0 (ArC–O). 7Li {1H} NMR (C6D6, 298 K, δ) 1.58. MS (MALDI-TOF) m/z (%, ion): 548.4 (100, Li2[N2O2BuBuPip]+˙).
Synthesis of 2.
H2[O2N2]BuBuPip (2.01 g, 3.7 mmol) was dissolved in THF (50 mL) and cooled to −78 °C. Sodium hydride (0.357 g, 14.8 mmol) was dissolved in THF (20 mL) and slowly added via cannula to afford a white suspension solution, which was warmed and stirred for 72 h. Solvent was removed under vacuum; the product was washed with cold pentane (10 mL). The product was then filtered and dried under vacuum to yield (2.36 g, 98%) of a deep yellow product. Anal. calc'd for C70H108Na4N4O24 (3C4H8O): C, 71.48; H, 9.66; N, 4.07. Found: C, 71.78; H, 9.38; N, 4.14 1H NMR (300 MHz, 298 K, C6D6) δ 1.28 (8H, m, 3JHH = 6.6 Hz, CH2, THF), 1.29 (18H, s, ArC–C(CH3)3), 1.70 (18H, s, ArC–C(CH3)3), 1.90 (2H, br, N–CH2{CH2}CH2–N), 2.47 (4H, br, N–CH2{CH2}CH2–N), 2.81 (2H, br, N–CH2CH2–N), 3.03 (2H, br, N–CH2CH2–N), 3.25 (8H, m, 3JHH = 6.6 Hz, CH2, THF), 4.02 (4H, br, ArC–CH2–N), 6.99 (2H, d, 2JHH = 2.3 Hz, ArH), 7.51 (2H, d, 2JHH = 2.3 Hz, ArH). 13C {1H} NMR (300 MHz, 298 K, C5D5N) δ 26.3 (CH2, THF), 27.9 (N–CH2{CH2}CH2–N), 31.2 (ArC–C–(CH3)3), 33.1 (ArC–C–(CH3)3), 34.5 (ArC–C–(CH3)3), 36.1 (ArC–C–(CH3)3), 54.2 (N–CH2CH2–N), 56.7 (N–CH2{CH2}CH2–N), 64.7 (ArC–CH2–N), 68.3 (CH2, THF), 123.3 (ArC–CH2–N), 126.7 (ArCH), 127.1 (ArCH), 130.4 (ArC–C(CH3)3), 137.7 (ArC–C(CH3)3), 168.8 (ArC–O). 13C {1H} NMR (300 MHz, 298 K, CDCl3) δ (ppm) 25.8 (CH2, THF), 27.0 (N–CH2{CH2}CH2–N), 29.8 (ArC–C–(CH3)3), 31.9 (ArC–C–(CH3)3), 34.4 (ArC–C–(CH3)3), 35.1 (ArC–C–(CH3)3), 53.3 (N–CH2CH2–N), 54.7 (N–CH2{CH2}CH2–N), 62.7 (ArC–CH2–N), 68.2 (CH2, THF), 100.2 (ArC–CH2–N), 121.5 (ArCH), 123.7 (ArCH), 135.8 (ArC–C(CH3)3), 140.9 (ArC–C(CH3)3), 154.3 (ArC–O). MS (MALDI-TOF) m/z (%, ion): 580.3 (100, Na2[N2O2BuBuPip]+˙).
Synthesis of 3.
This was prepared in a similar manner to 1 to yield (2.06 g, 97%) of a pale yellow product. Anal. calc'd for C29H42Li2N2O2 (1.95Li2O·1.2C5H12): C, 68.98; H, 9.33; N, 4.60. Found: C, 68.64; H, 9.73; N, 5.00. 1H NMR (300 MHz, 298 K, C6D6) δ 1.30 (8H, m, 3JHH = 6.6 Hz, CH2, THF), 1.66 (18H, s, ArC–C(CH3)3), 1.96 (6H, m, N–CH2{CH2}CH2–N), 2.41 (6H, s, ArC–CH3), 2.75 (4H, m, N–CH2CH2–N), 3.51 (8H, m, 3JHH = 6.6 Hz, CH2, THF), 3.61 (4H, s, ArC–CH2–N), 6.85 (2H, d, 2JHH = 2.3 Hz, ArH), 7.35 (2H, d, 2JHH = 2.3 Hz, ArH). 13C {1H} NMR (300 MHz, 298 K, C6D6) δ 21.6 (ArC–CH3), 25.8 (CH2, THF), 27.7 (N–CH2{CH2}CH2–N), 30.8 (ArC–C–(CH3)3), 35.4 (ArC–C–(CH3)3), 51.1 (N–CH2CH2–N), 53.5 (N–CH2{CH2}CH2–N), 62.2 (ArC–CH2–N), 68.5 (CH2, THF), 120.4 (ArC–CH2–N), 126.5 (ArCH), 131.3 (ArC–CH3), 137.6 (ArC–C–(CH3)3), 164.5 (ArC–O). 7Li {1H} NMR (C6D6, 298 K, δ) 1.60. MS (MALDI-TOF) m/z (%, ion): 464.3 (100, Li2[N2O2BuMePip]+˙).
Synthesis of 4.
This was prepared in a similar manner to 2 to yield (2.02 g, 96%) of a yellow product. Anal. calc'd for C58H84Na4N4O4 (3C4H8O): C, 69.51; H, 9.00; N, 4.63. Found: C, 69.23; H, 9.03; N, 4.94. 1H NMR (300 MHz, 298 K, C6D6) δ 1.25 (8H, m, 3JHH = 6.7 Hz, CH2, THF), 1.73 (18H, s, ArC–C(CH3)3), 2.34 (6H, s, ArC–CH3), 2.99 (4H, br, N–CH2{CH2}CH2–N), 3.25 (8H, m, 3JHH = 6.7 Hz, CH2, THF), 4.09 (4H, br, ring NCH2Ar), 6.83 (2H, s, ArH), 7.23 (2H, s, ArH). 1H NMR (500 MHz, 298 K, C5D5N) δ 1.16 (6H, s, CH2, THF), 1.59 (18H, s, ArC–C(CH3)3), 1.95 (2H, s, N–CH2{CH2}CH2–N), 2.24 (4H, m, N–CH2{CH2}CH2–N), 2.45 (6H, s, ArC–CH3), 2.94 (4H, m, N–CH2CH2–N), 3.28 (2H, s, NCH2Ar), 3.58 (2H, s, CH2, THF), 4.40 (2H, s, NCH2Ar), 6.97 (2H, s, ArH), 7.30 (2H, s, ArH). 13C {1H} NMR (300 MHz, 298 K, CDCl3) δ 20.96 (ArC–CH3), 25.78 (CH2, THF), 26.98 (N–CH2{CH2}CH2–N), 29.72 (ArC–C–(CH3)3), 34.74 (ArC–C–(CH3)3), 53.20 (N–CH2CH2–N), 54.66 (N–CH2{CH2}CH2–N), 62.12 (ArC–CH2–N), 68.13 (CH2, THF), 122.12 (ArC–CH2–N), 126.90 (ArCH), 127.47 (ArCH), 136.55 (ArC–C–(CH3)3), 154.41 (ArC–O). MS (MALDI-TOF) m/z (%, ion): 497.2 (10, Na2[N2O2BuMePip]+˙), 475.2 (25, Na[N2O2BuMePip]+˙).
X-ray crystallography
Crystals of 1 and 4 were mounted on low temperature diffraction loops. All measurements were made on a Rigaku Saturn70 CCD diffractometer using graphite monochromated Mo-Kα radiation, equipped with a SHINE optic. A summary of the collection details and refinement results can be found in Table S1.† For both structures, H-atoms were introduced in calculated positions and refined on a riding model while all non-hydrogen atoms were refined anisotropically. While refinement of 4 proceeded normally, in the structure of 1, two disordered t-butyl groups were present ([C60–C62] and [C63–65] with respective occupancies 0.647(14):0.353(14), and [C67–69] and [C70–72] with respective occupancies 0.761(10):0.239(10)). Similar anisotropic restraints were applied to these groups, as well as to one THF molecule (O5, C77–C80). Further disorder was treated by the Platon87 Squeeze procedure which was applied to recover 119 electrons per unit cell in two voids (total volume 813 Å3); that is 59.5 electrons per formula unit. Disordered lattice solvent toluene molecules (50 electrons per C7H8; one molecule per formula unit) were present prior to the application of Squeeze, however, a satisfactory point atom model could not be achieved. Note that there are two well-ordered toluene molecules associated with each formula unit that were not removed from the model using Squeeze.
Crystal data for 1.
C103H156Li4N4O7 (M = 1590.15 g mol−1), triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 16.592(2) Å, b = 18.655(2) Å, c = 19.067(2) Å, α = 103.219(7)°, β = 98.581(7)°, γ = 109.095(8)°, V = 5265.8(10) Å3, Z = 2, T = 163(2) K, μ(MoKα) = 0.061 mm−1, Dcalc = 1.003 g cm−3, 43063 reflections measured (6° ≤ 2θ ≤ 53°), 21429 unique (Rint = 0.0454) which were used in all calculations. The final R1 was 0.1130 (>2σ(I)) and wR2 was 0.3597 (all data). CCDC no. 1410026.
(no. 2), a = 16.592(2) Å, b = 18.655(2) Å, c = 19.067(2) Å, α = 103.219(7)°, β = 98.581(7)°, γ = 109.095(8)°, V = 5265.8(10) Å3, Z = 2, T = 163(2) K, μ(MoKα) = 0.061 mm−1, Dcalc = 1.003 g cm−3, 43063 reflections measured (6° ≤ 2θ ≤ 53°), 21429 unique (Rint = 0.0454) which were used in all calculations. The final R1 was 0.1130 (>2σ(I)) and wR2 was 0.3597 (all data). CCDC no. 1410026.
Crystal data for 4.
C66H100N4Na4O6 (M = 1137.45 g mol−1), monoclinic, space group P21/n (no. 14), a = 12.898(4) Å, b = 13.741(4) Å, c = 18.695(6) Å, β = 101.518(4)°, V = 3246.7(17) Å3, Z = 2, T = 163 K, μ(MoKα) = 0.096 mm−1, Dcalc = 1.164 g cm−3, 24304 reflections measured (4.61° ≤ 2θ ≤ 54.206°), 7155 unique (Rint = 0.0374) which were used in all calculations. The final R1 was 0.0543 (I > 2σ(I)) and wR2 was 0.1653 (all data). CCDC no. 1410027.
Polymerization procedures
Typical bulk polymerization procedure.
A monomer:initiator ratio employed was 100:1 and the reactions were conducted at 130 °C. A Schlenk tube equipped with magnetic stir bar was charged in a glovebox with the required amount of LA (0.50 g, 3.5 mmol) and initiator (0.02–0.023 g, 0.035 mmol). The reaction vessel was sealed, brought out of the glovebox and immersed in an oil bath that was preheated to 130 °C. At the desired time an aliquot was withdrawn from the flask for 1H NMR analysis to determine the monomer conversion. The vial was then placed in an ice bath to halt the reaction and solidify the polymer. The resulting solid was dissolved in dichloromethane and the polymer, precipitated with acidified methanol. Centrifugation was applied where needed for better separation of the solids. Solvents were decanted and the white solids were dried in vacuo followed by drying in a vacuum oven at 40 °C overnight.
Typical solution polymerization procedure.
The reaction mixtures were prepared in a glovebox and subsequent operations were performed with standard Schlenk techniques. A Schlenk tube containing a stir bar and the monomer (0.85 g, 5.9 mmol) in solvent (CH2Cl2, toluene or THF) was prepared. A second Schlenk tube was prepared containing a stir bar and the catalyst (0.007–0.0085 g, 0.0118 mmol) and a solution of BnOH (127 μL, 0.0118 mmol), if appropriate in 10 mL of solvent (CH2Cl2, toluene). Then the lactide solution was transferred by cannula to the complex mixture. Timing of the reaction began when all the lactide was transferred. An aliquot of the reaction solution was taken for NMR spectroscopic analysis, and the reaction was quenched immediately by the addition of methanol. The resulting solid was dissolved in dichloromethane and the polymer precipitated with excess cold methanol. Solvents were decanted and the white solids were dried in vacuo followed by drying in a vacuum oven at 40 °C overnight.
Conclusions
In conclusion, we have prepared and characterized tetrametallic lithium and sodium diamino-bis(phenolate) complexes. At room temperature, these complexes demonstrate good activity for ring-opening polymerization of rac-lactide both in the absence and presence of benzyl alcohol to yield polymers with narrow dispersities. The effect of solvents on the ROP reactions was studied. The sodium complexes outperform their lithium analogues especially in the presence of BnOH. We note that the effect of BnOH on the lithium initiators is negligible. The molecular weight of PLAs can be tuned according to the monomer:alcohol ratio, where increasing the amount of BnOH results in proportionally lower molecular weight polymers. GPC data of the produced polymers point to well-controlled polymerizations but evidence for transesterfication is seen in mass spectra of the polymers. On the basis of stoichiometric model reactions the polymerization occurs via a coordination–insertion mechanism.
Acknowledgements
NSERC of Canada, Memorial University, RDC-NL and CFI are thanked for operating and instrument grants. Special thanks to the Saudi Arabian Cultural Bureau in Canada and Taif University (Saudi Arabia) for financial support. We thank Yann Sarazin (Université de Rennes 1) for valuable suggestions and Céline Scheider (Memorial University) for assistance with NMR experiments.
References
- M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. K. Mohanty, Prog. Polym. Sci., 2013, 38, 1653–1689 CrossRef CAS.
- M. Singhvi and D. Gokhale, RSC Adv., 2013, 3, 13558–13568 RSC.
- A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC.
- B. Gupta, N. Revagade and J. Hilborn, Prog. Polym. Sci., 2007, 32, 455–482 CrossRef CAS.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- S. Slomkowski, S. Penczek and A. Duda, Polym. Adv. Technol., 2014, 25, 436–447 CrossRef CAS.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- J.-W. Rhim, H.-M. Park and C.-S. Ha, Prog. Polym. Sci., 2013, 38, 1629–1652 CrossRef CAS.
- I. Armentano, N. Bitinis, E. Fortunati, S. Mattioli, N. Rescignano, R. Verdejo, M. A. Lopez-Manchado and J. M. Kenny, Prog. Polym. Sci., 2013, 38, 1720–1747 CrossRef CAS.
- R. A. Auras, B. Harte, S. Selke and R. Hernandez, J. Plast. Film Sheeting, 2003, 19, 123–135 CrossRef CAS.
- Y. Ohya, A. Takahashi and K. Nagahama, Adv. Polym. Sci., 2012, 247, 65–114 CrossRef CAS.
- K. Hamad, M. Kaseem, H. W. Yang, F. Deri and Y. G. Ko, Polym. Lett., 2015, 9, 435–455 CrossRef CAS.
- O. Wichmann, R. Sillanpää and A. Lehtonen, Coord. Chem. Rev., 2012, 256, 371–392 CrossRef CAS.
- C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832–4846 RSC.
- W. Yi and H. Ma, Dalton Trans., 2014, 43, 5200–5210 RSC.
- J. P. Davin, J.-C. Buffet, T. P. Spaniol and J. Okuda, Dalton Trans., 2012, 41, 12612–12618 RSC.
- E. L. Marshall, V. C. Gibson and H. S. Rzepa, J. Am. Chem. Soc., 2005, 127, 6048–6051 CrossRef CAS PubMed.
- L. Wang and H. Ma, Macromolecules, 2010, 43, 6535–6537 CrossRef CAS.
- L. E. Breyfogle, C. K. Williams, J. V. G. Young, M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2006, 928–936 RSC.
- W.-C. Hung and C.-C. Lin, Inorg. Chem., 2009, 48, 728–734 CrossRef CAS PubMed.
- Y. Sarazin, V. Poirier, T. Roisnel and J.-F. Carpentier, Eur. J. Inorg. Chem., 2010, 3423–3428 CrossRef CAS.
- A. D. Schofield, M. L. Barros, M. G. Cushion, A. D. Schwarz and P. Mountford, Dalton Trans., 2009, 85–96 RSC.
- X. Zhang, T. J. Emge and K. C. Hultzsch, Organometallics, 2010, 29, 5871–5877 CrossRef CAS.
- H.-Y. Chen, L. Mialon, K. A. Abboud and S. A. Miller, Organometallics, 2012, 31, 5252–5261 CrossRef CAS.
- K. Devaine-Pressing, J. H. Lehr, M. E. Pratt, L. N. Dawe, A. A. Sarjeant and C. M. Kozak, Dalton Trans., 2015, 44, 12365–12375 RSC.
- D. J. Darensbourg, W. Choi, O. Karroonnirun and N. Bhuvanesh, Macromolecules, 2008, 41, 3493–3502 CrossRef CAS.
- B. Liu, T. Roisnel, J.-P. Guégan, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 18, 6289–6301 CrossRef CAS PubMed.
- B. Gao, R. Duan, X. Pang, X. Li, Z. Qu, Z. Tang, X. Zhuang and X. Chen, Organometallics, 2013, 32, 5435–5444 CrossRef CAS.
- Z. Qu, R. Duan, X. Pang, B. Gao, X. Li, Z. Tang, X. Wang and X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1344–1352 CrossRef CAS.
- X. Pang, R. Duan, X. Li, Z. Sun, H. Zhang, X. Wang and X. Chen, Polym. Chem., 2014, 5, 6857–6864 RSC.
- L. Postigo, M. a. d. C. Maestre, M. E. G. Mosquera, T. Cuenca and G. Jiménez, Organometallics, 2013, 32, 2618–2624 CrossRef CAS.
- N. Ikpo, S. M. Barbon, M. W. Drover, L. N. Dawe and F. M. Kerton, Organometallics, 2012, 31, 8145–8158 CrossRef CAS.
- M. O. Miranda, Y. DePorre, H. Vazquez-Lima, M. A. Johnson, D. J. Marell, C. J. Cramer and W. B. Tolman, Inorg. Chem., 2013, 52, 13692–13701 CrossRef CAS PubMed.
- I. Yu, A. Acosta-Ramírez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758–12773 CrossRef CAS PubMed.
- D. C. Aluthge, B. O. Patrick and P. Mehrkhodavandi, Chem. Commun., 2013, 49, 4295–4297 RSC.
- R. K. Dean, A. M. Reckling, H. Chen, L. N. Dawe, C. M. Schneider and C. M. Kozak, Dalton Trans., 2013, 42, 3504–3520 RSC.
- S.-C. Rosca, D.-A. Rosca, V. Dorcet, C. M. Kozak, F. M. Kerton, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2013, 42, 9361–9375 RSC.
- N. Ikpo, C. Hoffmann, L. N. Dawe and F. M. Kerton, Dalton Trans., 2012, 41, 6651–6660 RSC.
- Y. Huang, Y.-H. Tsai, W.-C. Hung, C.-S. Lin, W. Wang, J.-H. Huang, S. Dutta and C.-C. Lin, Inorg. Chem., 2010, 49, 9416–9425 CrossRef CAS PubMed.
- L. Wang, X. Pan, L. Yao, N. Tang and J. Wu, Eur. J. Inorg. Chem., 2011, 632–636 CrossRef.
- C.-A. Huang, C.-L. Ho and C.-T. Chen, Dalton Trans., 2008, 3502–3510 RSC.
- C.-A. Huang and C.-T. Chen, Dalton Trans., 2007, 5561–5566 RSC.
- F. M. Kerton, C. M. Kozak, K. Lüttgen, C. E. Willans, R. J. Webster and A. C. Whitwood, Inorg. Chim. Acta, 2006, 359, 2819–2825 CrossRef CAS.
- B.-T. Ko and C.-C. Lin, J. Am. Chem. Soc., 2001, 123, 7973–7977 CrossRef CAS PubMed.
- W. Clegg, M. G. Davidson, D. V. Graham, G. Griffen, M. D. Jones, A. R. Kennedy, C. T. O'Hara, L. Russo and C. M. Thomson, Dalton Trans., 2008, 1295–1301 RSC.
- Z. Janas, T. Nerkowski, E. Kober, L. B. Jerzykiewicz and T. Lis, Dalton Trans., 2012, 41, 442–447 RSC.
- E. Kober, R. Petrus, P. Kocięcka, Z. Janas and P. Sobota, Polyhedron, 2015, 85, 814–823 CrossRef CAS.
- F. M. García-Valle, R. Estivill, C. Gallegos, T. Cuenca, M. E. G. Mosquera, V. Tabernero and J. Cano, Organometallics, 2015, 34, 477–487 CrossRef.
- W.-Y. Lu, M.-W. Hsiao, S. C. N. Hsu, W.-T. Peng, Y.-J. Chang, Y.-C. Tsou, T.-Y. Wu, Y.-C. Lai, Y. Chen and H.-Y. Chen, Dalton Trans., 2012, 41, 3659–3667 RSC.
- X. Xu, X. Pan, S. Tang, X. Lv, L. Li, J. Wu and X. Zhao, Inorg. Chem. Commun., 2013, 29, 89–93 CrossRef CAS.
- B. Calvo, M. G. Davidson and D. García-Vivó, Inorg. Chem., 2011, 50, 3589–3595 CrossRef CAS PubMed.
- X. Xu, Y. Yao, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 3743–3751 CrossRef CAS PubMed.
-
N. Ikpo, PhD Thesis, Memorial University of Newfoundland, 2012.
- S. L. Hancock, R. Gati, M. F. Mahon, E. Y. Tshuva and M. D. Jones, Dalton Trans., 2014, 43, 1380–1385 RSC.
- S. L. Hancock, M. F. Mahon and M. D. Jones, Chem. Cent. J., 2013, 7, 135–135 CrossRef PubMed.
- S. L. Hancock, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 2033–2037 RSC.
- J. Baldamus and E. Hecht, Z. Anorg. Allg. Chem., 2003, 629, 188–191 CrossRef CAS.
- M. Sankaralingam and M. Palaniandavar, Dalton Trans., 2014, 43, 538–550 RSC.
- S. L. Hancock, M. D. Jones, C. J. Langridge and M. F. Mahon, New J. Chem., 2012, 36, 1891–1896 RSC.
- S. L. Hancock, M. F. Mahon, G. Kociok-Kőhn and M. D. Jones, Eur. J. Inorg. Chem., 2011, 4596–4602 CrossRef CAS.
- R. Mayilmurugan, B. N. Harum, M. Volpe, A. F. Sax, M. Palaniandavar and N. C. Mösch-Zanetti, Chem. – Eur. J., 2011, 17, 704–713 CrossRef CAS PubMed.
- C. Fernandes, R. W. A. Franco, L. M. Lube, S.-H. Wei, L. L. Mendes, R. W. A. Franco, L. M. Lube, J. H. Reibenspies, D. J. Darensbourg and A. Horn Jr., J. Braz. Chem. Soc., 2014, 25, 1050–1061 Search PubMed.
- R. Mayilmurugan, M. Sankaralingam, E. Suresh and M. Palaniandavar, Dalton Trans., 2010, 39, 9611–9625 RSC.
- R. Mayilmurugan, K. Visvaganesan, E. Suresh and M. Palaniandavar, Inorg. Chem., 2009, 48, 8771–8783 CrossRef CAS PubMed.
- T. N. Rao, J. Reedijk, J. Vanrijn, G. C. Verschoor, A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 Search PubMed.
- R. Wegner, B. Krebs, S. Uhlenbrock, R. Wegner and B. Krebs, J. Chem. Soc., Dalton Trans., 1996, 3731–3736 Search PubMed.
- G. Murphy, C. O'Sullivan, B. Murphy and B. Hathaway, Inorg. Chem., 1998, 37, 240–248 CrossRef CAS.
- A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479–489 RSC.
- M. W. Drover, J. N. Murphy, J. C. Flogeras, C. M. Schneider, L. N. Dawe and F. M. Kerton, Polyhedron, 2015, 102, 60–68 CrossRef CAS.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- C. Paetz and R. Hagen, Chem. Ing. Tech., 2014, 86, 519–523 CrossRef CAS.
- S. Bian, S. Abbina, Z. Lu, E. Kolodka and G. Du, Organometallics, 2014, 33, 2489–2495 CrossRef CAS PubMed.
- N. Ikpo, L. N. Saunders, J. L. Walsh, J. M. B. Smith, L. N. Dawe and F. M. Kerton, Eur. J. Inorg. Chem., 2011, 5347–5359 CrossRef CAS.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- K. A. M. Thakur, R. T. Kean, E. S. Hall, J. J. Kolstad, T. A. Lindgren, M. A. Doscotch, J. I. Siepmann and E. J. Munson, Macromolecules, 1997, 30, 2422–2428 CrossRef CAS.
- M.-H. Thibault and F.-G. Fontaine, Dalton Trans., 2010, 39, 5688–5697 RSC.
- N. Maudoux, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Organometallics, 2014, 33, 5740–5748 CrossRef CAS.
- G. K. Fukin, A. V. Cherkasov, N. Ajellal, T. Roisnel, M. A. Sinenkov, G. K. Fukin, A. V. Cherkasov, F. M. Kerton, J.-F. Carpentier and A. A. Trifonov, New J. Chem., 2011, 35, 204–212 RSC.
- C. E. Willans, M. A. Sinenkov, G. K. Fukin, K. Sheridan, J. M. Lynam, A. A. Trifonov and F. M. Kerton, Dalton Trans., 2008, 3592–3598 RSC.
- A. K. Sutar, T. Maharana, S. Dutta, C.-T. Chen and C.-C. Lin, Chem. Soc. Rev., 2010, 39, 1724–1746 RSC.
- H. E. Dyer, S. Huijser, A. D. Schwarz, C. Wang, R. Duchateau and P. Mountford, Dalton Trans., 2008, 32–35 RSC.
- T.-L. Yu, B.-H. Huang, W.-C. Hung, C.-C. Lin, T.-C. Wang and R.-M. Ho, Polymer, 2007, 48, 4401–4411 CrossRef CAS.
- H. Kato, T. Saito, M. Nabeshima, K. Shimada and S. Kinugasa, J. Magn. Reson., 2006, 180, 266–273 CrossRef CAS PubMed.
- D. Zuccaccia and A. Macchioni, Organometallics, 2005, 24, 3476–3486 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1410026 and 1410027. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03119b |
| ‡ Current Address: Department of Chemistry, Wilfrid Laurier University, Waterloo, Ontario, Canada. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a
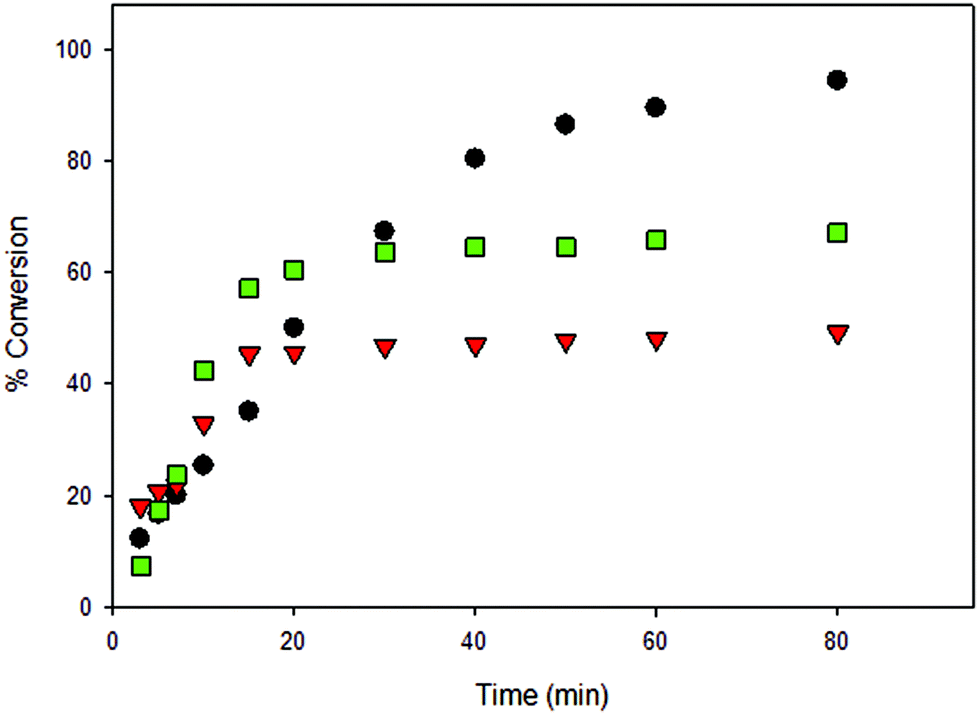
 toluene (30 mL),
toluene (30 mL),  THF (20 mL).
THF (20 mL).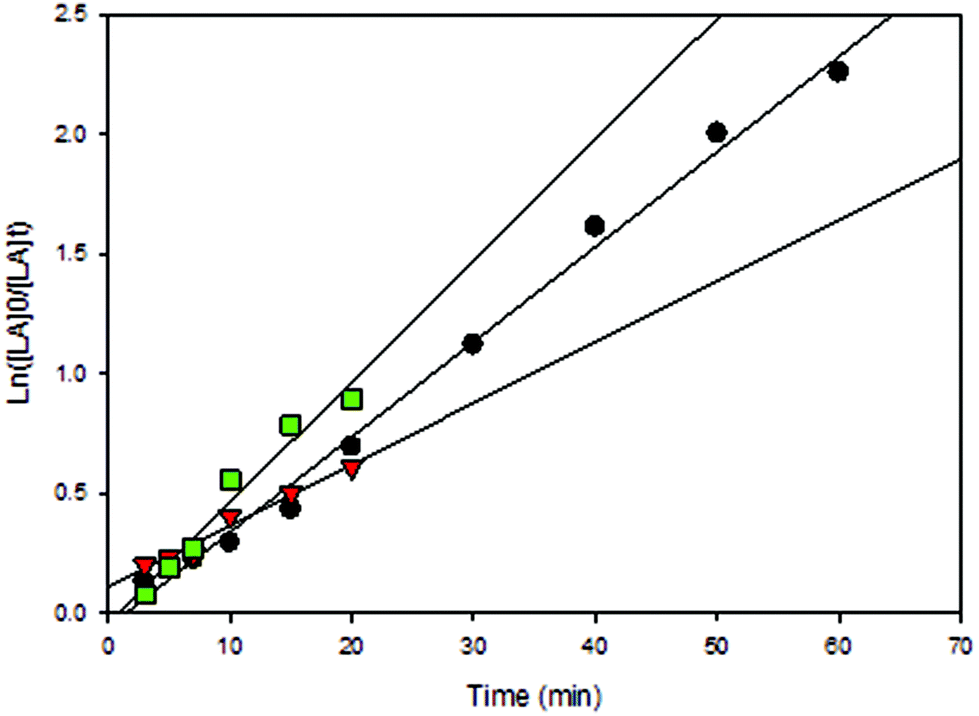
 toluene (30 mL, 0.0506x − 0.0448, R2 = 0.96),
toluene (30 mL, 0.0506x − 0.0448, R2 = 0.96),  THF (20 mL
THF (20 mL  toluene (y = 0.0255x + 0.1097, R2 = 0.9742).
toluene (y = 0.0255x + 0.1097, R2 = 0.9742).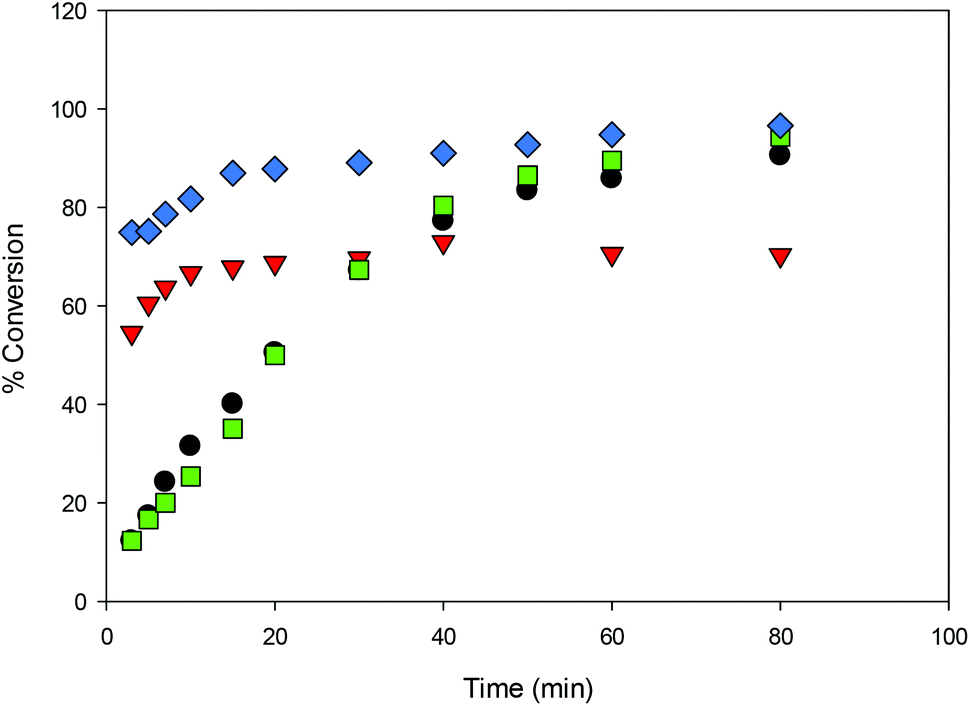
 2,
2,  3,
3,  4.
4.
 3 (y = 0.0395x − 0.0471, R2 = 0.9951).
3 (y = 0.0395x − 0.0471, R2 = 0.9951).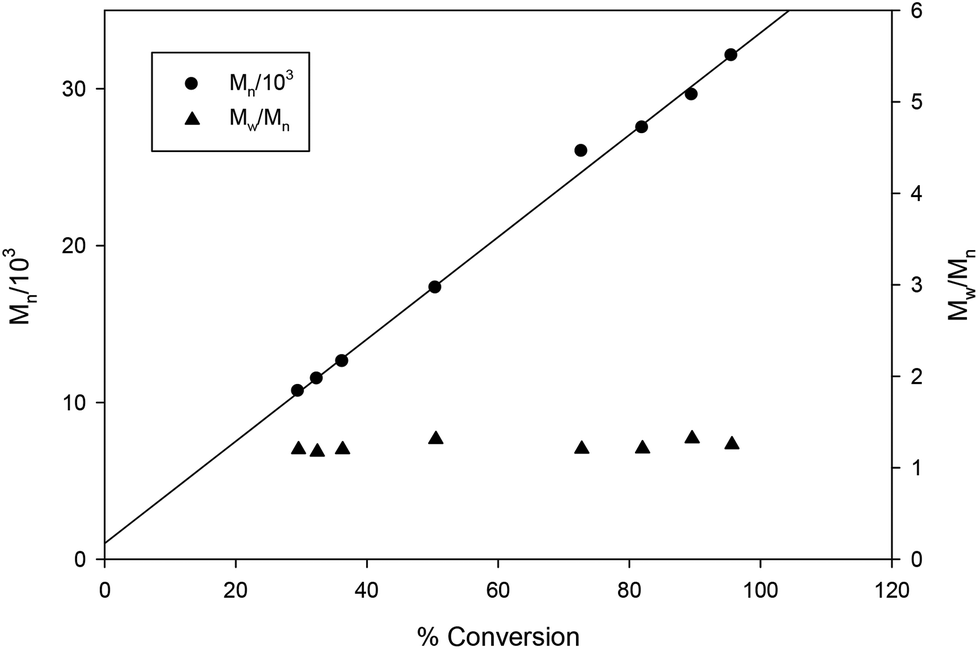
 2 eq. BnOH (y = 0.0259x + 0.2026, R2 = 0.9944),
2 eq. BnOH (y = 0.0259x + 0.2026, R2 = 0.9944),  4 eq. BnOH (y = 0.0812x + 0.2446, R2 = 0.9963).
4 eq. BnOH (y = 0.0812x + 0.2446, R2 = 0.9963).