
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new class of deep-blue emitting Cu(I) compounds – effects of counter ions on the emission behavior†
Timo
Gneu߇
a,
Markus J.
Leitl‡
b,
Lars H.
Finger
 a,
Hartmut
Yersin
*b and
Jörg
Sundermeyer
*a
a,
Hartmut
Yersin
*b and
Jörg
Sundermeyer
*a
aPhilipps-Universität Marburg, Materials Science Centre and Fachbereich Chemie, Hans-Meerwein-Straße 4, 35032 Marburg, Germany. E-mail: jsu@staff.uni-marburg.de; Fax: +49 (0)6421 28-25711; Tel: +49 (0)6421 28-25693
bUniversität Regensburg, Institut für Physikalische und Theoretische Chemie, Universitätsstr. 31, 93053, Regensburg, Germany. E-mail: hartmut.yersin@ur.de; Fax: +49 (0)941 943-4488; Tel: +49 (0)941 943-4464
First published on 9th October 2015
Abstract
Three deep blue emitting Cu(I) compounds, [Cu(PPh3)tpym]PF6, [Cu(PPh3)tpym]BF4, and [Cu(PPh3)tpym]BPh4 (tpym = tris(2-pyridyl)methane, PPh3 = triphenylphosphine) featuring the tripodally coordinating tpym and the monodentate PPh3 ligands were studied with regard to their structural and photophysical properties. The compounds only differ in their respective counter ions which have a strong impact on the emission properties of the powder samples. For example, the emission quantum yield can be significantly increased for the neat material from less than 10% to more than 40% by exchanging BPh4− with PF6−. These effects can be linked to different molecular packings which depend on the counter ion. In agreement with these results, it was found that the emission properties also strongly depend on the surrounding matrix environment which was elucidated by investigating photophysical properties of the compounds as powders, doped into a polymer matrix, and dissolved in a fluid solution, respectively. The observed differences in the emission behavior can be explained by different and pronounced distortions that occur in the excited state. These distortions are also displayed by density functional theory (DFT) calculations.
Introduction
In the last few decades, significant research attention has been devoted to the development of new emitter materials for organic light-emitting diodes (OLEDs) and light-emitting electrochemical cells (LEECs).1–14 A breakthrough in this field was achieved when the potential of 3rd row transition metal complexes was recognized for these applications. Such compounds can display strong spin–orbit coupling (SOC) which can result in high phosphorescence emission quantum yields and short emission decay times of only a few microseconds.9,15–18 Most importantly, the involvement of the triplet state in the emission process and the fast intersystem crossing (ISC) from the lowest excited singlet to the lowest triplet state allow the use of all injected excitons, singlets and triplets, for the generation of light in electroluminescent devices through the triplet harvesting effect.19–21 As a consequence, the efficiency increases by a factor of four compared to conventional purely organic, fluorescent emitters which can only utilize singlet excitons. However, efficient triplet emitters are based on expensive and rare noble metals such as iridium and platinum.1,4,5,7–9,11,13,16,21–29 Furthermore, developing efficient and long-term stable blue light emitters with this class of compounds remains challenging due to energetically relatively low-lying metal centered dd* states that provide a path for non-radiative decay to the ground state or even molecular decomposition.30In recent years, compounds based on low-cost and more abundant 1st row transition metal copper have come into the focus of research.9,10,14,31–38 At first sight, emitters based on Cu(I) seem rather unsuitable for application in electroluminescent devices due to the significantly smaller SOC constant of copper compared to platinum or iridium,39 for example. Thus, Cu(I) complexes frequently exhibit triplet decay times of several 100 microseconds or more.9,14,22,31–33,35,40–49 This would lead to pronounced saturation effects, if these materials are applied as emitters in electroluminescent devices.50 However, the long triplet decay time is not effective if the compounds exhibit only a small energy separation ΔE(S1–T1) between the first excited singlet S1 and the triplet state T1. Then, an efficient thermal population (up-ISC) of the S1 state from the energetically lower lying T1 state can occur at ambient temperature. As the spin-allowed S1 → S0 transition shows a significantly higher oscillator strength than the spin-forbidden T1 → S0 transition, the overall decay time decreases distinctly by this process and decay times of only a few μs can be reached.9,14,22,31–35,40–45,51,52 This emission mechanism corresponds to a thermally activated delayed fluorescence (TADF).53 In an electroluminescent device, this mechanism allows utilizing all injected excitons for the generation of light. Accordingly, the excitation is harvested and emitted essentially via the singlet S1 state. Thus, this mechanism is called the singlet harvesting effect.9,14,22,31–33,35,40,54–56 Additionally, in Cu(I) complexes the d-shell of the copper ion is fully occupied (d10 electron configuration) and therefore, low-lying dd* states that could quench the emission and would be a source of molecular decomposition do not occur. This renders Cu(I) complexes promising candidates for the realization of efficient and long-term stable blue light emitters for OLEDs and LEECs.
Compared to emitters based on Pt(II) and Ir(III), which preferably exhibit square-planar or octahedral coordination geometries, respectively, Cu(I) compounds show a richer structural diversity.14,31–36,38,40,43,45,54,56–80 However, so far photophysical investigations have been mainly focused on mononuclear complexes with two bidentate ligands,32,33,37,40,70–72,77–83 mononuclear complexes with one mono and one bidentate ligand,34,38,43,63–65 and on dinuclear complexes in which the two copper centers are bridged by halides.14,35,36,56,59–62 To our knowledge, only very few Cu(I) complexes with tripodal ligands have been studied with regard to their photophysical properties.54,57,58
Herein, we report the new cationic tripodally coordinated deep-blue emitting Cu(I) complex [Cu(PPh3)tpym]+ (PPh3 = triphenylphosphine, tpym = tris(2-pyridyl)methane). Interestingly, the emission properties distinctly depend on the counter ion. Therefore, we discuss properties of the three powder materials [Cu(PPh3)tpym]PF6 (1), [Cu(PPh3)tpym]BF4 (2), and [Cu(PPh3)tpym]BPh4 (3) and compare the results with those for the compounds doped into polymethylmethacrylate (PMMA). All compounds were characterized chemically by NMR spectroscopy, IR spectroscopy, mass spectrometry, elemental analysis, and X-ray analysis. In addition, density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations were performed for the cationic part [Cu(PPh3)tpym]+ to gain further insight into the electronic structure of this complex.
Results and discussion
Synthesis
The copper(I) compounds [Cu(PPh3)tpym]PF6 (1), [Cu(PPh3)tpym]BF4 (2), and [Cu(PPh3)tpym]BPh4 (3) were prepared in a two-step synthesis. In the first step, the tripodal ligand tris(2-pyridyl)methane (tpym) was prepared according to a literature method (Scheme 1).84 In the second step, compounds 1 and 2 were synthesized by reaction of the corresponding copper(I) salts [Cu(MeCN)4]PF6 and [Cu(MeCN)4]BF4, respectively, with PPh3 and tpym. The yields for both compounds are quantitative. Compound 3 was prepared with an overall yield of 35% in three in situ steps (Scheme 2). In the first step, the complex [Cu(Cl)tpym] was formed by reaction of CuCl with tpym. Then, by adding NaBPh4 an anion exchange between the chloride and the tetraphenylborate anion is accomplished. Finally, by adding PPh3 compound 3 was obtained. | ||
| Scheme 1 Synthesis of the methane derivative tris(2-pyridyl)methane (tpym) according to ref. 84. | ||
 | ||
| Scheme 2 Synthesis of the copper(I) compounds. | ||
X-ray crystal structures
Single crystals suitable for X-ray structure determination could be obtained for all three investigated compounds (1, 2, and 3) from a saturated chloroform solution by layering with n-pentane at ambient temperature. The crystallographic data and structure refinement details are summarized in Table 1; selected bond distances and angles are listed in Table 2. The molecular structures of 1–3 are shown in Fig. 1. The three compounds crystallize in different crystal systems, compound 1 in the monoclinic, compound 2 in the trigonal, and compound 3 in the orthorhombic crystal system. | ||
| Fig. 1 Molecular structures of 1–3 (thermal ellipsoids with 50% probability) resulting from X-ray analyses. Hydrogen atoms (except for H16) and solvent molecules are omitted for clarity. | ||
| 1 | 2 | 3 | |
|---|---|---|---|
| [Cu(PPh3)tpym]PF6·CHCl3 | [Cu(PPh3)tpym]BF4·0.5 CHCl3 | [Cu(PPh3)tpym]BPh4·2 CHCl3 | |
| Habitus | Plate | Needle | Plate |
| Color | Colorless | Colorless | Colorless |
| Formula | C35H29Cl3CuF6N3P2 | C34.50H28.50BCl1.50CuF4N3P | C60H50BCl6CuN3P |
| fw [g mol−1] | 837.44 | 719.60 | 1131.05 |
| Crystal size [mm3] | 0.220 × 0.180 × 0.090 | 0.291 × 0.043 × 0.041 | 0.319 × 0.267 × 0.064 |
| Crystal system | Monoclinic | Trigonal | Orthorhombic |
| Space group | P21 |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
Pna21 |
| a [Å] | 8.5970(3) | 12.8635(6) | 19.4837(7) |
| b [Å] | 18.9423(8) | 12.8635 | 11.1499(4) |
| c [Å] | 11.5950(5) | 67.361(3) | 24.6713(9) |
| α [°] | 90 | 90 | 90 |
| β [°] | 107.2000(10) | 90 | 90 |
| γ [°] | 90 | 120 | 90 |
| Cell volume [Å3] | 1803.77(13) | 9652.9(10) | 5359.6(3) |
| Z | 2 | 12 | 4 |
| D calc [Mg m−3] | 1.542 | 1.485 | 1.402 |
| Abs. coeff. [mm−1] | 0.978 | 0.907 | 0.780 |
| F(000) | 848 | 4404 | 2328 |
| T [K] | 100(2) | 100(2) | 100(2) |
| λ [Å] | 0.71069 | 0.71069 | 0.71069 |
| Reflns collected | 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 555 555 |
28280 |
54250 |
| Indep. reflns | 7884 | 2458 | 11572 |
| Obs. reflns [II > 2(I)] | 7265 | 1742 | 9836 |
| Reflns used for refin. | 7884 | 2458 | 11572 |
| Abs. correction | Multi-scan | Multi-scan | Multi-scan |
| GOF | 1.030 | 1.037 | 1.069 |
| wR2 | 0.0643 | 0.0958 | 0.0662 |
| R 1 [I > 2σ(I)] | 0.0290 | 0.0383 | 0.0294 |
| 1 | 2 | 3 | |
|---|---|---|---|
|
a Since compound 2 crystallizes in the space group Rc, the pyridine and phenyl groups are crystallographically imposed symmetry equivalent with respect to each other. Nevertheless, for better comparability we use the same atom labeling scheme for compound 2 as for 1 and 3.
|
|||
| Cu1–N1 | 2.047(3) | 2.073(2) | 2.056(3) |
| Cu1–N2 | 2.080(3) | 2.073(2) | 2.048(3) |
| Cu1–N3 | 2.075(3) | 2.073(2) | 2.098(3) |
| Cu1–P1 | 2.160(1) | 2.158(1) | 2.152(1) |
| C16–C1 | 1.527(4) | 1.522(3) | 1.520(4) |
| C16–C6 | 1.524(4) | 1.522(3) | 1.526(4) |
| C16–C11 | 1.520(4) | 1.522(3) | 1.518(4) |
| P1–Cu1–N1 | 124.9(1) | 125.3(1) | 132.1(1) |
| P1–Cu1–N2 | 123.4(1) | 125.3(1) | 120.3(1) |
| P1–Cu1–N3 | 127.5(1) | 125.3(1) | 121.3(1) |
| N1–Cu1–N2 | 89.6(1) | 90.0(1) | 90.4(1) |
| N1–Cu1–N3 | 91.1(1) | 90.0(1) | 89.2(1) |
| N2–Cu1–N3 | 89.1(1) | 90.0(1) | 92.3(1) |
| P1–Cu1–C16 | 177.6(0) | 180.0(0) | 172.6(1) |
The copper(I) centers are coordinated by the phosphine ligand PPh3 and the three N atoms of tpym in a distorted tetrahedral configuration. The bending of the PPh3 ligand from the C16–Cu1 axis (compare Fig. 1) is different for the three compounds. For compound 2, there is no bending of the PPh3 ligand (P1–Cu1–C16 = 180.0(0)°). For compound 1, with an angle of P1–Cu1–C16 = 177.6 (0)°, the PPh3 bending is small, but for compound 3, with an angle of P1–Cu1–C16 = 172.6 (1)°, the bending is clearly displayed. Thus it seems that the bending of the PPh3 ligand from the C16–Cu1 axis increases with the increasing size of the counter anion (BF4 < PF6 < BPh4). The crystal packing diagram of compound 3 with the most pronounced PPh3 bending reveals that this bending is a result of the interaction of the PPh3 ligand with the counter anion BPh4 (see Fig. S3, ESI†). One phenyl group of BPh4 is directly oriented towards the PPh3 ligand of the adjacent copper complex. The resulting steric repulsion is decreased by the bending of the PPh3 ligand. For compounds 1 and 2 with the smaller counter anions PF6 and BF4, the interaction of the PPh3 group of the Cu(I) complex with its neighboring molecules is rather balanced. This can be supported by the performed DFT calculations (see below) which indicate that in the absence of counter ions and neighboring molecules the angle P1–Cu1–C16 amounts to 180°.
It is remarked that we have performed similar investigations for neutral Cu(I) complexes with tripodal ligands (similar to tpym) previously but with a halide (Cl, Br, or I) instead of the PPh3 ligand at the fourth coordination site. Also for these compounds a bending of the monodentate (halide) ligand has been observed in the crystal structures.54
Computational studies
Quantum chemical calculations have been carried out for the cationic complex [Cu(PPh3)tpym]+ using density functional theory (DFT) and time-dependent density functional theory (TDDFT) with the hybrid functional B3LYP85–87 and the basis set def2-SVP.88,89 For [Cu(PPh3)tpym]+, the ground state (S0), the first excited triplet state (T1), and the first excited singlet state (S1) geometries were calculated. The results are displayed in Fig. 2. Frequency calculations confirm that these three optimized structures are minima on the potential energy surface.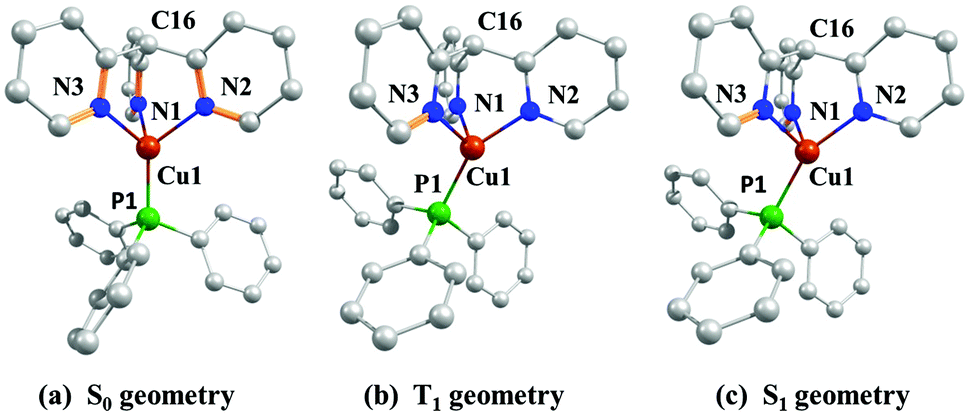 | ||
| Fig. 2 Optimized geometries of the ground state S0 (a), the first excited triplet state T1 (b), and the first excited singlet state S1 (c) of [Cu(PPh3)tpym]+. Calculations were performed on the B3LYP/def2-SVP level of theory. All hydrogen atoms are omitted for clarity. | ||
For the S0 geometry, it was found that the atom P1 of the PPh3 group lies on the axis that is defined by the atoms C16 and Cu1 (Fig. 2a) (angle P1–Cu1–C16 = 180°). The bond lengths of the three Cu–N bonds are almost equal, amounting to 2.137 Å (Cu1–N1), 2.137 Å (Cu1–N2), and 2.139 Å (Cu1–N3). However, in the T1 (Fig. 2b) and S1 state geometries (Fig. 2c), the P1 atom, and thus, the whole PPh3 group, is bent away from the C16–Cu1 axis. For the T1 geometry, the PPh3 group is angled by about 26° (P1–Cu1–C16 = 153.8°) and for the S1 geometry by about 30° (P1–Cu1–C16 = 149.7°). Also, the three Cu–N bonds are no longer equal in the excited state geometries. In the T1 geometry, two Cu–N bonds are significantly shorter than in the S0 geometry, with bond lengths amounting to Cu1–N1 = 2.000 Å and Cu1–N2 = 1.965 Å, whereas the length of the third copper–nitrogen bond increases to Cu1–N3 = 2.167 Å. On the other hand, for the S1 geometry two Cu–N bonds become longer, with two equal bond lengths of Cu1–N1 = 2.149 Å and Cu1–N3 = 2.149 Å, and one Cu–N bond becomes significantly shorter, with a bond length of Cu1–N2 = 1.986 Å. These data and other important bond lengths and angles are summarized in Table 3.
| S0 | T1 | S1 | |
|---|---|---|---|
| Cu1–N1 | 2.137 | 2.000 | 2.149 |
| Cu1–N2 | 2.137 | 1.965 | 1.986 |
| Cu1–N3 | 2.139 | 2.167 | 2.149 |
| Cu1–P1 | 2.242 | 2.356 | 2.339 |
| P1–Cu1–N1 | 125.9 | 108.4 | 106.4 |
| P1–Cu1–N2 | 125.9 | 150.0 | 154.8 |
| P1–Cu1–N3 | 126.4 | 110.3 | 107.8 |
| N1–Cu1–N2 | 88.9 | 89.2 | 90.0 |
| N1–Cu1–N3 | 88.9 | 97.4 | 93.2 |
| N2–Cu1–N3 | 88.9 | 90.5 | 89.8 |
| P1–Cu1–C16 | 179.7 | 153.8 | 149.7 |
Furthermore, it was found that the highest occupied molecular orbital (HOMO) is mainly located at the Cu(I) center whereas the lowest unoccupied molecular orbital (LUMO) is distributed over two of the three pyridine moieties of the tpym ligand (compare Fig. 3).
 | ||
| Fig. 3 Highest occupied (HOMO) and lowest unoccupied molecular orbitals (LUMO) for [Cu(PPh3)tpym]+ calculated for the ground state geometry. Calculations were performed on the B3LYP/def2-SVP level of theory. All hydrogen atoms are omitted for clarity. | ||
TDDFT calculations reveal that transitions between these two frontier orbitals largely determine the first excited singlet S1 and triplet T1 states which leads to the assignment of these states as metal-to-ligand charge-transfer (MLCT) states. This allows us to give an explanation for the occurrence of geometry distortions in the S1 and T1 states: On excitation, a significant amount of charge is transferred from the Cu(I) metal center to the ligand. As a consequence, the copper center is formally partially oxidized from Cu(I) to Cu(II). As Cu(II) prefers a planar coordination environment in contrast to Cu(I) (which favors tetrahedral coordination), such an oxidation is connected with pronounced structural reorganizations. For example, for Cu(I) complexes with two bidentate ligands, this is displayed by a flattening distortion from a tetrahedral to a more planar coordination geometry.71,77,90–92 In the case of the cationic Cu(I) complex presented in this study, the distortion is represented by the bending of the PPh3 group as described above.
The pronounced charge transfer character of the S1 and T1 states has another important consequence. Due to the distinct spatial separation of HOMO and LUMO, the spatial overlap between these two frontier orbitals is small. As a consequence, the exchange integral is also small. Accordingly, the energy separation ΔE(S1–T1) between the first excited singlet and triplet state is small. From TDDFT calculations it was found that this energy separation amounts only to ΔE(S1–T1) = 810 cm−1. For such a small energy splitting a thermally activated delayed fluorescence (TADF) is expected to occur at ambient temperature, which is also indicated by the studies presented below.
Photophysical studies
In Fig. 4, electronic absorption spectra of the compounds [Cu(PPh3)tpym]PF6 (1), [Cu(PPh3)tpym]BF4 (2), and [Cu(PPh3)tpym]BPh4 (3) are displayed. In addition, the absorption spectra of the tpym and the PPh3 ligands are also shown. All spectra were recorded under ambient conditions for the compounds dissolved in dichloromethane, except for PPh3 for which the absorption was measured in acetonitrile.93 | ||
| Fig. 4 Absorption spectra of the three compounds 1–3 and the tpym ligand recorded in dichloromethane solution under ambient conditions. PPh3 absorption has been measured under the same conditions but in acetonitrile solution.93 | ||
The absorption spectra of compounds 1–3 show similar spectral shapes. All spectra exhibit intense high energy absorption bands in the wavelength region below 280 nm with peaks at 229 nm, 248 nm, and 260 nm, respectively. The corresponding electronic transitions are identified to originate from ligand centered (LC) π–π* transitions of the tpym and the PPh3 ligands, which show intense absorptions in this spectral range. At longer wavelengths, distinctly weaker absorption bands are observed which are not present in the spectrum of the tpym or PPh3 ligands. Therefore, these bands can be attributed to transitions that are of metal-to-ligand charge transfer (MLCT) character. This assignment is also in agreement with DFT and TDDFT calculations discussed in the previous section which predict low lying MLCT states and agrees with literature reports of other Cu(I) complexes.9,14,22,33,40–45,51,52
It is not surprising that the absorption spectra of all three compounds exhibit similar spectral shapes as the investigated substances differ only in their respective counter anions. In a dilute solution, the counter anions do not interact with the Cu(I) complexes and therefore, do not influence the absorption behavior. Furthermore, the BF4− and PF6− ions are not expected to show absorption in the investigated wavelength range. In contrast, the phenyl groups of BPh4− show distinct absorption in the wavelength range between 230 nm and 260 nm which explains the differences in the spectra of substance 3 when compared to those of 1 and 2.
In fluid dichloromethane (DCM) solution, the Cu(I) complexes are not emissive at ambient temperature, even if oxygen is carefully removed from the solution by repeatedly applying a freeze–pump–thaw process. In contrast, emission is observed for complexes doped into a polymethylmethacrylate (PMMA) matrix. The corresponding emission spectra are displayed in Fig. 5 and are found to be essentially identical for all the compounds. They are broad and featureless, being in agreement with the MLCT character of the emissive state,31–35,40,63–65 with a peak at λmax = 470 nm. Also, the emission quantum yield of ΦPL = 7% is equal for all the compounds (compare Table 4). These results show that the counter anions do not have an influence on the emission behavior of the Cu(I) complexes doped into the PMMA matrix which presumably is due to the spatial separation of the counter anions and the Cu(I) complex in the PMMA matrix at low doping concentrations. The relatively small value of ΦPL is related to the non-rigid environment in the PMMA polymer. This allows significant quenching of the excited states as explained below. Interestingly, for the powder samples with different counter anions one finds distinct differences in the emission properties. The spectra of the three compounds are clearly shifted relative to each other with emission maxima lying at λmax(1) = 466 nm, λmax(2) = 449 nm, and λmax(3) = 452 nm (Fig. 5). Also, the emission quantum yields vary distinctly amounting to ΦPL(1) = 43%, ΦPL(2) = 19%, and ΦPL(3) = 7%, at ambient temperature.
 | ||
| Fig. 5 Normalized emission spectra of the investigated compounds 1–3 doped into a PMMA matrix and as powders. All spectra were recorded under ambient conditions. The samples were excited at λexc = 350 nm. | ||
| 1 | 2 | 3 | ||
|---|---|---|---|---|
| Powder | λ max (300 K) [nm] | 466 | 449 | 452 |
| τ (300 K) [μs] | 14 | 7.5* | 5.4* | |
| Φ PL (300 K) [%] | 43 | 19 | 7 | |
| k r (300 K) [s−1] | 3.1 × 104 | 2.5 × 104 | 1.3 × 104 | |
| k nr (300 K) [s−1] | 4.1 × 104 | 1.1 × 105 | 1.7 × 105 | |
| λ max (77 K) [nm] | 478 | 462 | 462 | |
| τ (77 K) [μs] | 26 | 19* | 25* | |
| PMMA | λ max (300 K) [nm] | 470 | 470 | 470 |
| Φ PL (300 K) [%] | 7 | 7 | 7 |
Presumably, these differences are a consequence of the interaction of the counter anions with the Cu(I) complex due to their proximity in the solid phase. In the crystals of the compounds, these interactions are displayed in different packings and different geometries of the [Cu(PPh3)tpym]+ complex, depending on the counter anion (compare crystal structures discussed above and ESI Fig. S1–S3†). For example, the orientation of the PPh3 group compared to the rest of the molecules is strongly dependent on the counter anion and thus, influences the emission properties. Similar effects have been investigated previously.94–97 It is reasonable to assume that in the powder phase, the counter anions have similar effects on the compounds’ geometries.
A comparison of the emission properties in different matrices reveals an interesting trend. In the powder phase, compound 1 exhibits an emission quantum yield of 43%, but when doped into a PMMA matrix the quantum yield amounts only to 7% and in solution, the quantum yield is ≪1%. This is related to increasing distortions that Cu(I) complexes undergo on excitation with decreasing matrix rigidity.9,31,40,55,60 As already discussed in the previous section, the compounds show structural distortion on excitation, especially, a bending of the PPh3 group away from the C16–Cu1 axis. As a consequence, the potential energy curves for the excited and the ground states are shifted with respect to each other. Accordingly, the non-radiative rate increases due to an increase of the Franck–Condon factors that govern these deactivation processes to the ground state.9,31,98,99 In matrices with low rigidity, such as fluid solutions, this effect is particularly pronounced. In more rigid environments, geometry distortions are much less distinct resulting in higher quantum yields. In powder, geometry distortions upon excitation are partly suppressed resulting in higher quantum yields of the Cu(I) compounds. Interestingly, here it is shown that also the counter anion (and the resulting molecular packing) has an influence on the emission quantum yield of the powder samples. Thus, the counter anions prevent large geometry distortions if they limit the available space for distortions in the molecular packing.
It is remarked that the emission spectra of the compounds as powders show a clear blue shift of the order of 10 nm on heating from T = 77 K to ambient temperature (1: 540 cm−1, 2: 620 cm−1, 3: 480 cm−1). This is an indication that a thermally activated delayed fluorescence (TADF) occurs. According to this emission mechanism, the singlet state S1 is populated at ambient temperature and contributes to the emission (Fig. 6). As this state lies energetically higher, a blue shift of the emission with increasing temperature is expected. The energy separation of ΔE(S1–T1) = 810 cm−1, as found from TDDFT calculations, is in fairly good agreement with the experimental values resulting from the spectral shifts and therefore supports this assignment. Furthermore, the thermal population of the singlet state should result in an increase of the radiative rate on heating as the spin-allowed S1 → S0 transition carries significantly more allowedness than the spin-forbidden T1 → S0 transition. Indeed, on heating, a slight increase of the radiative rate from kr (77 K) = 2.7 × 104 s−1 to kr (300 K) = 3.1 × 104 s−1 is observed for compound 1. However, this increase is smaller than that observed for other TADF materials. Presumably, this is mainly due to the fact that the triplet state itself exhibits comparably fast deactivation to the ground state with a decay time of only 26 μs for compound 1. This value is significantly shorter than that found for other TADF systems for which triplet decay times can be several hundred μs or even longer.31–33,41
 | ||
| Fig. 6 Emission decay paths for compound 1 at different temperatures. At low temperatures (T = 77 K), only the lowest excited triplet state T1 emits, while at ambient temperature (T = 300 K), an additional radiative decay path via thermal population of the S1 state is opened. | ||
Conclusion and outlook
In this contribution, we have presented a new class of emitter materials based on a Cu(I) complex with a tripodal ligand. As powders, the three compounds [Cu(PPh3)tpym]X (X = PF6 (1), BF4 (2), and BPh4 (3)) display bright emission in the deep-blue range of the spectrum. For example, at ambient temperature, the emission of [Cu(PPh3)tpym]PF6 (1) peaks at 466 nm and the emission quantum yield amounts to 43%. However, if this compound is doped into a polymer PMMA matrix, the emission is slightly red-shifted to 470 nm, but the quantum yield is drastically reduced to 7%. In fluid dichloromethane solution, the compound is not emissive. This indicates that molecular reorganizations on excitation which can easily occur in non-rigid environments are a major source of non-radiative relaxation to the ground state. For other Cu(I) compounds, it has been shown that limiting such distortions can lead to drastic increases of the emission quantum yields even in solution. For example, for the cationic complexes [Cu(POP)(dmbpy)]+ and [Cu(POP)(tmbpy)]+ (POP = bis[2-(diphenylphosphino)-phenyl]ether, dmbpy = 4,4′-dimethyl-2,2′-bipyridyl, tmbyp = 4,4′,6,6′-tetramethyl-2,2′-bipyridyl)40 it has been demonstrated that the introduction of two sterically demanding methyl groups can significantly reduce geometry distortions on excitation and therefore, cause a drastic increase of the emission quantum yield by almost a factor of ten. This strategy could also be applied in future investigations to increase the quantum yield of the compounds presented in this study, for example, by introducing sterically demanding groups that prohibit the bending distortion of the triphenylphosphine group.Furthermore, we have demonstrated that different counter ions and molecular packings can have a strong impact on the emission behavior of solid samples. For example, the powder of the compound [Cu(PPh3)tpym]PF6 exhibits an emission quantum yield of 43% and an emission maximum of 466 nm at ambient temperature, whereas for [Cu(PPh3)tpym]BPh4 the emission is blue shifted to 452 nm and the emission quantum yield is decreased to 7%. These results should be taken into consideration, if solid state samples of other Cu(I) complexes are being investigated.
Experimental
General remarks
Syntheses and handling of air- and moisture-sensitive substances were carried out using standard Schlenk- and glovebox-techniques. Solvents were dried using standard procedures100 and stored over Al2O3/molecular sieves 3 Å/R3-11G catalyst (BASF). The starting materials were obtained from commercial sources and used as received. The following materials were prepared according to literature procedures: Copper(I) chloride,101 tetrakis(acetonitrile)copper(I) hexafluorophosphate,102 tetrakis(acetonitrile)copper(I) tetrafluoroborate,103 and tris(2-pyridyl)methane (tpym).84NMR spectra were recorded at 300 K on a Bruker DPX 250, Bruker ARX 300, Bruker DRX 400, Bruker ARX 500, or Bruker DRX 500 using CDCl3 or CD3CN as the solvent. Chemical shifts are given with respect to tetramethylsilane (1H, 13C) and 85% phosphoric acid (31P). Calibration of 1H and 13C NMR spectra was accomplished with the deuterated solvent residual signals. 31P NMR spectra were calibrated externally (phosphoric acid). The numbering of the hydrogen and carbon atoms of the three compounds is shown in Fig. 7.
 | ||
| Fig. 7 Numbering of the compounds. | ||
Electrospray ionization (ESI) mass spectra were recorded on a Thermo Fisher Scientific LTQ FT Ultra using acetonitrile as the solvent. IR spectra were recorded on a Bruker Alpha FT-IR spectrometer using powder samples at ambient temperature. Intensities of the bands were characterized as follows: vs = very strong (0–50% transmission), s = strong (50–70% transmission), m = medium (70–90% transmission), w = weak (90–100% transmission). Elemental analysis was performed on an Elementar vario MICRO cube.
UV-Vis absorption measurements were carried out using a Varian Cary 300 double beam spectrometer. Emission spectra were recorded with a Fluorolog 3-22 (Horiba Jobin Yvon) spectrophotometer which was equipped with a cooled photo-multiplier (RCA C7164R). For the decay time measurements, the same photomultiplier was used in combination with a FAST ComTec multichannel scaler PCI card with a time resolution of 250 ps. As the excitation source for the decay time measurements, a pulsed diode laser (Picobrite PB-375L) with an excitation wavelength of λexc = 378 nm and a pulse width <100 ps was used. For absolute measurements of photoluminescence quantum yields at ambient temperature, a Hamamatsu Photonics (C9920-02) system was applied. Doping of polymethylmethacrylate (PMMA) films was performed by dissolving the respective complex (<1 wt%) and the polymer in dichloromethane. After this, the solution was spin-coated onto a quartz-glass plate.
All calculations were carried out with Gaussian09.104 As the functional B3LYP85–87 and as the basis set def2-SVP88,89 were used. As the starting geometry for optimization, a structure obtained from X-ray measurements was used. The optimization of the S0 and T1 structures was made by DFT methods and the optimization of the S1 structure was made by TDDFT methods. No symmetry constraints were applied. Vibrational frequency calculations confirm that all three optimized structures are minima on the potential energy surface.
The data collection for the single crystal structure determinations was performed on a Bruker D8 QUEST diffractometer by the X-ray service department of the Faculty of Chemistry, University of Marburg. The D8-QUEST is equipped with a Mo-Kα X-ray microsource (Incotec), a fixed chi goniometer and a PHOTON 100 CMOS detector. Bruker software (Bruker Instrument Service, APEX2, SAINT) was used for data collection, cell refinement and data reduction.105 The structures were solved with SIR-97,106 refined with SHELXL-2014107 and finally validated using PLATON108 software, all within the WinGX109 software bundle. Absorption corrections were applied within the APEX2 software (multi-scan).105 Graphic representations were created using Diamond 3.110 C-bound H-atoms were constrained to the parent site. In all graphics the displacement ellipsoids are shown for the 50% probability level, hydrogen atoms are shown with an arbitrary radius. CCDC 1415676–1415678 contain the supplementary crystallographic data for the structures reported in this paper.
Syntheses
Acknowledgements
The authors thank the German Ministry of Education and Research (BMBF), the European Research Council (ERC), the German Association of Chemical Industry (Verband der chemischen Industrie, VCI), and the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG), GRK 1782 “Functionalization of Semiconductors” for financial support.Notes and references
- Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Physics of Organic Semiconductors, ed. W. Brütting and C. Adachi, Wiley-VCH, Berlin, 2nd edn, 2012 Search PubMed.
- S. Reineke, M. Thomschke, B. Lüssem and K. Leo, Rev. Mod. Phys., 2013, 85, 1245–1293 CrossRef CAS.
- C.-L. Ho and W.-Y. Wong, New J. Chem., 2013, 37, 1665–1683 RSC.
- G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC.
- P.-T. Chou and Y. Chi, Chem. – Eur. J., 2007, 13, 380–395 CrossRef CAS PubMed.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS.
- G. J. A. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- D. Volz, M. Wallesch, C. Flechon, M. Danz, A. Verma, J. M. Navarro, D. M. Zink, S. Bräse and T. Baumann, Green Chem., 2015, 17, 1988–2011 RSC.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 124, 8300–8334 CrossRef.
- R. D. Costa, E. Ortí and H. J. Bolink, Pure Appl. Chem., 2011, 83, 2115–2128 CrossRef CAS.
- T. Hu, L. He, L. Duan and Y. Qiu, J. Mater. Chem., 2012, 22, 4206–4215 RSC.
- M. Wallesch, D. Volz, D. M. Zink, U. Schepers, M. Nieger, T. Baumann and S. Bräse, Chem. – Eur. J., 2014, 20, 6578–6590 CrossRef CAS PubMed.
- T. A. Niehaus, T. Hofbeck and H. Yersin, RSC Adv., 2015, 5, 63318–63329 RSC.
- A. Bossi, A. F. Rausch, M. J. Leitl, R. Czerwieniec, M. T. Whited, P. I. Djurovich, H. Yersin and M. E. Thompson, Inorg. Chem., 2013, 52, 12403–12415 CrossRef CAS PubMed.
- J. W. Facendola, M. Seifrid, J. Siegel, P. I. Djurovich and M. E. Thompson, Dalton Trans., 2015, 44, 8456–8466 RSC.
- W. Mroz, R. Ragni, F. Galeotti, E. Mesto, C. Botta, L. D. Cola, G. M. Farinola and U. Giovanella, J. Mater. Chem. C, 2015, 3, 7506–7512 RSC.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch and R. Czerwieniec, in Physics of Organic Semiconductors, ed. W. Brütting and C. Adachi, Wiley-VCH, 2012, pp. 371–424 Search PubMed.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- C.-L. Ho, H. Li and W.-Y. Wong, J. Organomet. Chem., 2014, 751, 261–285 CrossRef CAS.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC.
- G. Tan, S. Chen, N. Sun, Y. Li, D. Fortin, W.-Y. Wong, H.-S. Kwok, D. Ma, H. Wu, L. Wang and P. D. Harvey, J. Mater. Chem. C, 2013, 1, 808–821 RSC.
- T. Fischer, R. Czerwieniec, T. Hofbeck, M. M. Osminina and H. Yersin, Chem. Phys. Lett., 2010, 486, 53–59 CrossRef CAS.
- T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed.
- R. Czerwieniec, J. Yu and H. Yersin, Inorg. Chem., 2011, 50, 8293–8301 CrossRef CAS PubMed.
- R. Czerwieniec, K. Kowalski and H. Yersin, Dalton Trans., 2013, 42, 9826–9830 RSC.
- R. Czerwieniec and H. Yersin, Inorg. Chem., 2015, 54, 4322–4327 CrossRef CAS PubMed.
- T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS PubMed.
- M. J. Leitl, F. R. Küchle, H. A. Mayer, L. Wesemann and H. Yersin, J. Phys. Chem. A, 2013, 117, 11823–11836 CrossRef CAS PubMed.
- D. Volz, Y. Chen, M. Wallesch, R. Liu, C. Fléchon, D. M. Zink, J. Friedrichs, H. Flügge, R. Steininger, J. Göttlicher, C. Heske, L. Weinhardt, S. Bräse, F. So and T. Baumann, Adv. Mater., 2015, 27, 2538–2543 CrossRef CAS PubMed.
- A. Barbieri, G. Accorsi and N. Armaroli, Chem. Commun., 2008, 2185–2193 RSC.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed.
- S. L. Murov, G. L. Hug and I. Carmichael, Handbook of Photochemistry, Marcel Dekker, New York, 2nd edn, 1993 Search PubMed.
- C. L. Linfoot, M. J. Leitl, P. Richardson, A. F. Rausch, O. Chepelin, F. J. White, H. Yersin and N. Robertson, Inorg. Chem., 2014, 53, 10854–10861 CrossRef CAS PubMed.
- J. C. Deaton, S. C. Switalski, D. Y. Kondakov, R. H. Young, T. D. Pawlik, D. J. Giesen, S. B. Harkins, A. J. M. Miller, S. F. Mickenberg and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 9499–9508 CrossRef CAS PubMed.
- M. Osawa, I. Kawata, R. Ishii, S. Igawa, M. Hashimoto and M. Hoshino, J. Mater. Chem. C, 2013, 1, 4375–4383 RSC.
- M. Osawa, M. Hoshino, M. Hashimoto, I. Kawata, S. Igawa and M. Yashima, Dalton Trans., 2015, 44, 8369–8378 RSC.
- G. Blasse and D. R. McMillin, Chem. Phys. Lett., 1980, 70, 1–3 CrossRef CAS.
- G. Cheng, G. K.-M. So, W.-P. To, Y. Chen, C.-C. Kwok, C. Ma, X. Guan, X. Chang, W.-M. Kwok and C.-M. Che, Chem. Sci., 2015, 6, 4623–4635 RSC.
- X.-L. Chen, R. Yu, Q.-K. Zhang, L.-J. Zhou, X.-Y. Wu, Q. Zhang and C.-Z. Lu, Chem. Mater., 2013, 25, 3910–3920 CrossRef CAS.
- A. Tsuboyama, K. Kuge, M. Furugori, S. Okada, M. Hoshino and K. Ueno, Inorg. Chem., 2007, 46, 1992–2001 CrossRef CAS PubMed.
- Q. Zhang, T. Komino, S. Huang, S. Matsunami, K. Goushi and C. Adachi, Adv. Funct. Mater., 2012, 22, 2327–2336 CrossRef CAS.
- S. Igawa, M. Hashimoto, I. Kawata, M. Yashima, M. Hoshino and M. Osawa, J. Mater. Chem. C, 2013, 1, 542–551 RSC.
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801–6827 CrossRef CAS PubMed.
- Q. Zhang, J. Chen, X. Y. Wu, X. L. Chen, R. Yu and C. Z. Lu, Dalton Trans., 2015, 44, 6706–6710 RSC.
- H. Ohara, A. Kobayashi and M. Kato, Dalton Trans., 2014, 43, 17317–17323 RSC.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- T. Gneuß, M. J. Leitl, L. H. Finger, N. Rau, H. Yersin and J. Sundermeyer, Dalton Trans., 2015, 44, 8506–8520 RSC.
- D. M. Zink, D. Volz, T. Baumann, M. Mydlak, H. Flügge, J. Friedrichs, M. Nieger and S. Bräse, Chem. Mater., 2013, 25, 4471–4486 CrossRef CAS.
- D. Volz, M. Wallesch, S. L. Grage, J. Göttlicher, R. Steininger, D. Batchelor, T. Vitova, A. S. Ulrich, C. Heske, L. Weinhardt, T. Baumann and S. Bräse, Inorg. Chem., 2014, 53, 7837–7847 CrossRef CAS PubMed.
- A. Acosta, J. I. Zink and J. Cheon, Inorg. Chem., 2000, 39, 427–432 CrossRef CAS PubMed.
- V. Pawlowski, G. Knör, C. Lennartz and A. Vogler, Eur. J. Inorg. Chem., 2005, 2005, 3167–3171 CrossRef.
- K. Tsuge, Chem. Lett., 2013, 42, 204–208 CrossRef CAS.
- D. M. Zink, M. Bächle, T. Baumann, M. Nieger, M. Kühn, C. Wang, W. Klopper, U. Monkowius, T. Hofbeck, H. Yersin and S. Bräse, Inorg. Chem., 2013, 52, 2292–2305 CrossRef CAS PubMed.
- H. Araki, K. Tsuge, Y. Sasaki, S. Ishizaka and N. Kitamura, Inorg. Chem., 2005, 44, 9667–9675 CrossRef CAS PubMed.
- P. Aslanidis, P. J. Cox, S. Divanidis and A. C. Tsipis, Inorg. Chem., 2002, 41, 6875–6886 CrossRef CAS PubMed.
- V. A. Krylova, P. I. Djurovich, M. T. Whited and M. E. Thompson, Chem. Commun., 2010, 46, 6696–6698 RSC.
- V. A. Krylova, P. I. Djurovich, J. W. Aronson, R. Haiges, M. T. Whited and M. E. Thompson, Organometallics, 2012, 31, 7983–7993 CrossRef CAS.
- V. A. Krylova, P. I. Djurovich, B. L. Conley, R. Haiges, M. T. Whited, T. J. Williams and M. E. Thompson, Chem. Commun., 2014, 50, 7176–7179 RSC.
- P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99, 3625–3648 CrossRef CAS PubMed.
- P. M. Graham, R. D. Pike, M. Sabat, R. D. Bailey and W. T. Pennington, Inorg. Chem., 2000, 39, 5121–5132 CrossRef CAS PubMed.
- O. Horváth, Coord. Chem. Rev., 1994, 135–136, 303–324 CrossRef.
- C. Kutal, Coord. Chem. Rev., 1990, 99, 213–252 CrossRef CAS.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, Top. Curr. Chem., 2007, 280, 69–115 CrossRef CAS.
- D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243–266 CrossRef CAS.
- A. Vogler and H. Kunkely, Top. Curr. Chem., 2001, 213, 143–182 CrossRef CAS.
- V. W.-W. Yam, K. K.-W. Lo, W. K.-M. Fung and C.-R. Wang, Coord. Chem. Rev., 1998, 171, 17–41 CrossRef CAS.
- V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- A. Lavie-Cambot, M. Cantuel, Y. Leydet, G. Jonusauskas, D. M. Bassani and N. D. McClenaghan, Coord. Chem. Rev., 2008, 252, 2572–2584 CrossRef CAS.
- A. J. M. Miller, J. L. Dempsey and J. C. Peters, Inorg. Chem., 2007, 46, 7244–7246 CrossRef CAS PubMed.
- O. Moudam, A. Kaeser, B. Delavaux-Nicot, C. Duhayon, M. Holler, G. Accorsi, N. Armaroli, I. Seguy, J. Navarro, P. Destruel and J.-F. Nierengarten, Chem. Commun., 2007, 3077–3079 RSC.
- R. Venkateswaran, M. S. Balakrishna, S. M. Mobin and H. M. Tuononen, Inorg. Chem., 2007, 46, 6535–6541 CrossRef CAS PubMed.
- A. Robertazzi, A. Magistrato, P. de Hoog, P. Carloni and J. Reedijk, Inorg. Chem., 2007, 46, 5873–5881 CrossRef CAS PubMed.
- L.-Y. Zou, M.-S. Ma, Z.-L. Zhang, H. Li, Y.-X. Cheng and A.-M. Ren, Org. Electron., 2012, 13, 2627–2638 CrossRef CAS.
- G. Accorsi, N. Armaroli, B. Delavaux-Nicot, A. Kaeser, M. Holler, J.-F. Nierengarten and A. Degli Esposti, J. Mol. Struct. (THEOCHEM), 2010, 962, 7–14 CrossRef CAS.
- A. Maleckis, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 6618–6625 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. R. McMillin and K. M. McNett, Chem. Rev., 1998, 98, 1201–1220 CrossRef PubMed.
- I. I. Vorontsov, T. Graber, A. Y. Kovalevsky, I. V. Novozhilova, M. Gembicky, Y.-S. Chen and P. Coppens, J. Am. Chem. Soc., 2009, 131, 6566–6573 CrossRef CAS PubMed.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS PubMed.
- UV-VIS spectra of neutral bases and their protonated conjugate cationic acids in acetonitrile, http://tera.chem.ut.ee/~manna/spe/base.htm, (accessed July 27, 2015).
- H. Yersin, H. Otto and G. Gliemann, Theor. Chim. Acta, 1974, 33, 63–78 CrossRef CAS.
- D. Volz, M. Nieger, J. Friedrichs, T. Baumann and S. Bräse, Langmuir, 2013, 29, 3034–3044 CrossRef CAS PubMed.
- L. Zhang, B. Li and Z. Su, Langmuir, 2009, 25, 2068–2074 CrossRef CAS PubMed.
- Q. Benito, X. F. Le Goff, S. Maron, A. Fargues, A. Garcia, C. Martineau, F. Taulelle, S. Kahlal, T. Gacoin, J. P. Boilot and S. Perruchas, J. Am. Chem. Soc., 2014, 136, 11311–11320 CrossRef CAS PubMed.
- G. W. Robinson and R. P. Frosch, J. Chem. Phys., 1963, 38, 1187–1203 CrossRef CAS.
- W. Siebrand, J. Chem. Phys., 1967, 46, 440–447 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Elsevier, 4th edn, 1996 Search PubMed.
- R. N. Keller and H. D. Wycoff, in Inorganic Syntheses, McGraw-Hill, 1946, vol. 2, pp. 1–4 Search PubMed.
- G. J. Kubas, in Inorganic Syntheses, John Wiley & Sons, 1979, vol. 19, pp. 90–92 Search PubMed.
- E. J. Parish, H. Qin, B. H. Lipshutz and T. B. Petersen, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, 2009.
- Bruker AXS, Madison, Wisconsin, USA, 2012 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- K. Brandenburg and H. Putz, Crystal Impact GbR, Bonn, Germany, 2012 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Parts of the crystal structures and NMR spectra of 1–3 are given in Fig. S1–S11. CCDC 1415676–1415678. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03065j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |