
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron(II) spin crossover complexes with diaminonaphthalene-based Schiff base-like ligands: mononuclear complexes†
Charles
Lochenie
a,
Julia
Heinz
a,
Wolfgang
Milius
b and
Birgit
Weber
*a
aInorganic Chemistry II, Universität Bayreuth, Universitätsstraße 30, NW I, 95440 Bayreuth, Germany. E-mail: weber@uni-bayreuth.de
bInorganic Chemistry I, Universität Bayreuth, Universitätsstraße 30, NW I, 95440 Bayreuth, Germany
First published on 22nd September 2015
Abstract
The synthesis of new Schiff base-like ligands with extended π-system and their iron complexes is described. Some of the iron(II) complexes with N-heterocycles as axial ligands show spin crossover behaviour. The influence of the extended aromatic system on cooperative interactions is investigated by single crystal X-ray structure analysis, X-ray powder diffraction, and magnetic measurements. A combination of C–H⋯π and C–H⋯O interactions is made responsible for up to 10 K wide thermal hysteresis loops.
Introduction
Molecular switches continue to attract the interest of synthetic chemists due to potential applications in the field of sensing or memory devices.1 Iron(II) spin crossover (SCO) complexes belong to this class of molecular switches as they can be switched between the diamagnetic low-spin (LS) and the paramagnetic high-spin (HS) state.2 This switching process can be triggered by many different means. Next to physical stimuli like temperature, pressure, or light irradiation, the interaction with guest molecules3 (especially for porous materials) or phase transitions4 (e.g. for SCO materials with liquid crystalline properties) can initiate the spin transition. Of the different types of spin transition (gradual, abrupt, step-wise, with hysteresis) a special focus is set on spin transition with hysteresis, as this gives rise to bistability (memory effect) over a certain temperature region. Cooperative spin transitions with hysteresis are usually only observed in the solid state (bulk material or nanostructured materials), however it was recently shown that such phenomenon is also observable in solution.5 For the display of hysteretic behaviour, intermolecular interactions are needed in order to transfer the structural changes associated with the spin transition from one molecule to another. Different strategies can be used to realise those intermolecular interactions that were also applied to the Schiff base-like ligands used in our group. Building short contacts between the complex molecules is one strategy to design SCO complexes with hysteresis.6 If wider hysteresis loops are desired, a combination of short range and long range interactions as it is often obtained for 1D coordination polymers or ladder-like compounds is promising.7 In the last years we investigated in detail the impact of hydrogen bonds on the hysteresis width of SCO complexes.8,9 One interaction we have not used in our iron(II) complexes so far is the π–π-interaction. However, there are examples in literature, where π-interactions between extended aromatic systems increase the width of the hysteresis loop. One series with the general composition [Fe(L)2NCS2] (with L being a bidentate ligand 2,2′-bipyridine (bipy), phenantroline (phen), or dipyrido[3,2-a:2′3′-c]phenazine (dpp)) is discussed by Real and co-workers.10 The systematic increase of the aromatic part of the ligand leads to improved π-stacking and by this to wider hysteresis loops. Indeed, for the dpp ligand a 40 K wide thermal hysteresis loop is observed.11 Another example of a mononuclear complex with a 37 K wide hysteresis loop due to π-stacking was reported by Létard and co-workers.12 Consequently we decided to modify our Schiff base-like ligands through the introduction of a naphthalene based ligand backbone. In Scheme 1, the general structure of the new ligands, their iron complexes, and the used abbreviations are given. | ||
| Scheme 1 Pathway of synthesis of the SCO complexes described in this work and used abbreviations. | ||
Results
Syntheses
The spin crossover (SCO) complexes were produced in a three-step synthesis, whose synthetic pathway is given in Scheme 1. Firstly the new naphthalene-based Schiff base-like ligands H2Lx (1–4) were synthesised, then the precursor methanol complexes [FeLx(MeOH)n] (5–8) were formed by reaction with iron(II) acetate, which were finally converted to the target SCO complexes [FeLx(Lax)2] (9–18). Oxidation of the intermediate complexes [FeLx(MeOH)n] (5–8) in the air led to μ-oxido-bridged binuclear iron(III) complexes [μ-O-(FeLx)2] (19–20).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H of the chelate cycles. The rest of the proton signals can be found in their respective expected region. IR spectra show two strong characteristic signals for the CO vibrations, as well as a characteristic N–H band.
O bands shift compared to the free ligand, in agreement with the coordination of an iron metal centre. Also the colour of the compounds drastically changes towards dark brown/black in the solid state. This is due to strong charge transfer between the iron centre and the ligand, as observed for similar compounds in literature.14–16 It is usual for this type of complexes that two methanol molecules coordinate the iron(II) centre on axial positions, as shown for the phenyl-,17,18 dialkoxyphenyl-19,20 and dihydroxyphenyl-21 derivatives of the same compound class. However, it was found by elemental analysis that for the naphthalene derivatives discussed in this manuscript the amount of coordinated methanol is depending on the substituents of the equatorial ligand. When the substituents are ester functions, like with L1 (R = –OEt) and L4 (R = –OMe), the complex will have two methanol coordinated to the iron centre. When the substituents are ketone functions, like L2 (R = –Me) and L3 (R = –Ph), no coordinating methanol was found. Those results were confirmed by the determination of the crystal structure of [FeL2].
C–H of the chelate cycles. The rest of the proton signals can be found in their respective expected region. IR spectra show two strong characteristic signals for the CO vibrations, as well as a characteristic N–H band.
O bands shift compared to the free ligand, in agreement with the coordination of an iron metal centre. Also the colour of the compounds drastically changes towards dark brown/black in the solid state. This is due to strong charge transfer between the iron centre and the ligand, as observed for similar compounds in literature.14–16 It is usual for this type of complexes that two methanol molecules coordinate the iron(II) centre on axial positions, as shown for the phenyl-,17,18 dialkoxyphenyl-19,20 and dihydroxyphenyl-21 derivatives of the same compound class. However, it was found by elemental analysis that for the naphthalene derivatives discussed in this manuscript the amount of coordinated methanol is depending on the substituents of the equatorial ligand. When the substituents are ester functions, like with L1 (R = –OEt) and L4 (R = –OMe), the complex will have two methanol coordinated to the iron centre. When the substituents are ketone functions, like L2 (R = –Me) and L3 (R = –Ph), no coordinating methanol was found. Those results were confirmed by the determination of the crystal structure of [FeL2].
| Compound | SCO behaviour | χ M T (300 K) | χ M T (50 K) | γ HS (50 K) | T 1/2 | |
|---|---|---|---|---|---|---|
| 9 | [FeL2(py)2]·2.5H2O | HS | 3.87 | — | — | — |
| 10 | [FeL3(py)2]·py | Abrupt | 3.19 | 0.01 | 0 | 175 |
| 11 | [FeL4(py)2]·py | Two-step, gradual | 2.90 | 0.84 | 0.29 | 150, 80 |
| 12 | [FeL1(phpy)2] | Hysteresis, 10 K | 3.65 | 0.08 | 0 | ↓238, ↑248 |
| 13 | [FeL2(phpy)2]·2MeOH | HS | 3.66 | — | — | — |
| 14 | [FeL4(phpy)2] | Hysteresis, 10 K | 3.21 | 0.03 | 0 | ↓250, ↑260 |
| 15 | [FeL1(dmap)2] | Two-step, gradual, abrupt | 3.12 | 0.10 | 0.03 | 224, 149 |
| 16 | [FeL2(dmap)2] | HS | 3.39 | — | — | — |
| 17 | [FeL3(dmap)2] | HS | 3.32 | — | — | — |
| 18 | [FeL4(dmap)2] | HS | 3.31 | — | — | — |
Magnetism
The magnetic properties of the ten synthesised potential SCO complexes were investigated with a SQUID magnetometer, and are summarised in Table 1. Out of those complexes, five (samples 9, 13, 16, 17, 18) are HS compounds with a χMT value around 3.3 cm3 mol K−1 in the whole temperature range and no indication for SCO. The different values can be attributed to differences in the spin–orbit coupling. However, especially for the samples containing non-coordinating solvent molecules, small errors in the sample weight due to a solvent loss during the sample preparation are also possible. In the ESI, Fig. S1† the plot of χMT vs. T for those complexes is given. Compound 10 presents at 300 K a χMT value of 3.19 cm3 K mol−1, typical for iron(II) complexes in the HS state.14–21,41 Upon cooling, this value remains constant down to 175 K where compound 10 undergoes a complete abrupt spin transition (ST) towards the LS state (top of Fig. 1). Below 160 K the χMT value stays constant with a value of 0.01 cm3 K mol−1 until 10 K. Upon warming, the compound goes back to the HS state with the same T1/2 of 175 K. The ST properties of 10 are stable upon several measurement cycles.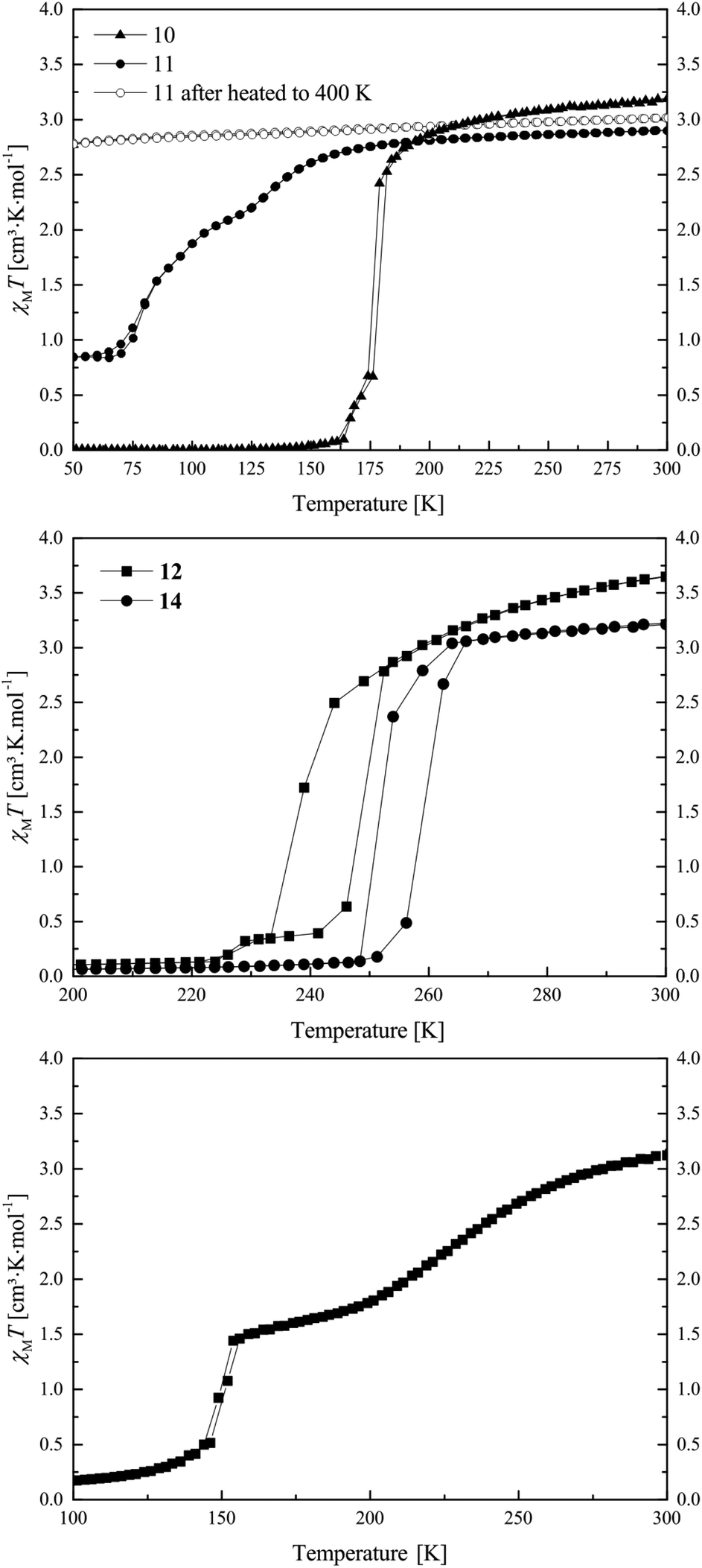 | ||
| Fig. 1 Magnetic susceptibility temperature product vs. temperature measurement for compounds 10 and 11 (top), 12 and 14 (middle), and 15 (bottom). | ||
Compound 11 exhibits at 300 K a χMT value of 2.90 cm3 K mol−1 which is in agreement with an iron(II) complex almost in the HS state (top of Fig. 1). Upon cooling, the χMT product stays constant until 170 K where compound 11 starts a gradual two-step SCO until 60 K, with T1 and T2, respectively, equal to 150 and 80 K. The SCO is incomplete with a residual HS fraction at 60 K of 0.29 (χMT = 0.84 cm3 K mol−1) which does not change down to 10 K. Upon warming, the compound 11 presents the same two-step transition towards the HS state at the same temperatures as upon cooling. When the compound is warmed above 380 K, the compound no longer exhibits SCO. Due to the conditions in the SQUID magnetometer (vacuum), uncoordinated as well as coordinated pyridine molecules could leave the sample, as already observed for other phenylene-based Fe(II) pyridine complexes.19,22 The loss of pyridine was confirmed with a TGA measurement, which is shown in the ESI: Fig. S2.† As a result, the compound does not undergo SCO anymore upon cooling. This phenomenon could explain why a rather high HS fraction was observed for the original sample: a deficiency in pyridine in the powder, resulting from drying the compound under vacuum after the synthesis, could be responsible for the HS fraction at low temperature.
Compounds 12 and 14 show very similar SCO properties: at 300 K both present, with 3.65 and 3.21 cm3 K mol−1, typical magnetic susceptibility with temperature product values for iron(II) HS complexes (middle of Fig. 1). Upon cooling, both show a rather abrupt ST with T1/2↓ = 238 K and T1/2↓ = 250 K, respectively, for compound 12 and 14.
Their χMT values at 50 K are with 0.08 and 0.03 cm3 K mol−1 in agreement with an iron(II) LS centre, and stay constant until 10 K. Upon heating, both samples show an abrupt ST back to the HS state at, respectively, T1/2↑ = 248 K and T1/2↑ = 260 K, revealing for both samples a 10 K hysteresis. As compounds 12 and 14 only differ by one substituent on the equatorial ligand (–OEt for 12 and –OMe for 14), and show the same SCO behaviour displaced by 12 K, one could see the direct influence of the substituents of the equatorial ligand on the crystal field strength of the complexes, but only if 12 and 14 are isostructural. The magnetic properties of 12 and 14 are stable upon heating until 400 K and can be cycled several times. This is a surprisingly high thermal stability for mononuclear complexes of this ligand type and can be explained with the results from X-ray structure analysis.
Compound 15 exhibits at 300 K a χMT value of 3.12 cm3 K mol−1, corresponding to an iron(II) HS compound. Upon cooling, the sample undergoes a two-step SCO with a gradual part starting at 273 K and ending at 189 K with a T1 of 224 K and an intermediate χMT value of 1.69 cm3 K mol−1 (γHS = 0.54, γHS was calculated as γHS = (χMT(50 K))/(χMT(300 K))) (bottom of Fig. 1). Then the sample undergoes a second abrupt SCO step with a T2 of 149 K. The magnetic susceptibility product remains constant with a value of 0.10 cm3 K mol−1 until 10 K, which is in agreement with an iron(II) centre in the LS state. Upon several temperature cycles, the magnetic properties of 15 are stable.
X-ray structure analysis
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and the asymmetric unit contains two H2L2 molecules, a water molecule and half a dioxane solvent molecule. ORTEP drawing of a H2L2 molecule is displayed in Fig. 2. Refinement of the ligand molecules and dioxane solvent molecule went smoothly, however the hydrogen atoms of the water molecule could not be refined. The ligand molecule can exist in two different tautomers: a keto–enamine form or an imino–enol form.
, and the asymmetric unit contains two H2L2 molecules, a water molecule and half a dioxane solvent molecule. ORTEP drawing of a H2L2 molecule is displayed in Fig. 2. Refinement of the ligand molecules and dioxane solvent molecule went smoothly, however the hydrogen atoms of the water molecule could not be refined. The ligand molecule can exist in two different tautomers: a keto–enamine form or an imino–enol form.
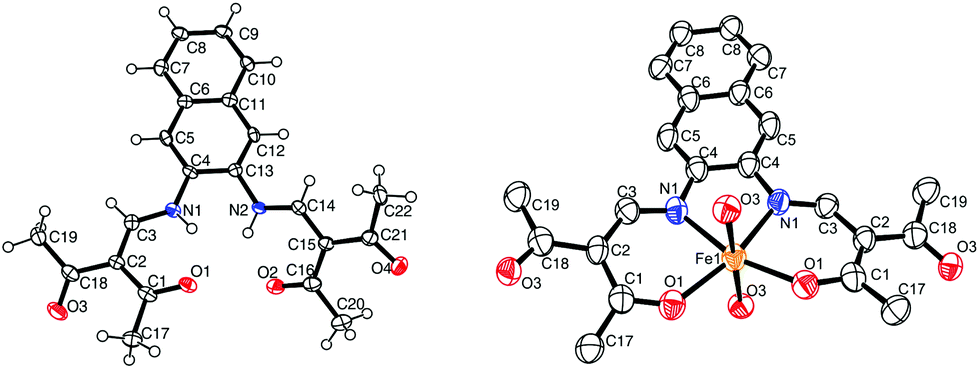 | ||
| Fig. 2 ORTEP drawing of 2 (left) and 6 (right). The thermal ellipsoids are shown at the 50% level. Hydrogen atoms and solvent molecules were omitted for clarity reasons. | ||
| Compound | 2·0.5 H2O·0.25 dioxane | 6 | 10HS | 10LS | 11 | 12 | 19 | 20 |
|---|---|---|---|---|---|---|---|---|
| CCDC | 1040379 | 1040378 | 1040374 | 1040375 | 1040380 | 1040376 | 1040375 | 1040377 |
| Sum formula | C46H48N4O10 | C22H20FeN2O4 | C47H39FeN5O4 | C47H39FeN5O4 | C37H35FeN5O6 | C46H42FeN4O6 | C48H48Fe2N4O13 | C44H40Fe2N4O13 |
| M r/g mol−1 | 816.88 | 432.5 | 793.68 | 793.68 | 701.55 | 802.69 | 1000.60 | 944.50 |
| Crystal system | Triclinic | Orthorhombic | Monoclinic | Monoclinic | Monoclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P |
Pbcn | P21/c | P21/c | P21/c |
P |
P |
P |
| a/Å | 7.8382(10) | 11.697(2) | 12.179(2) | 11.920(2) | 28.083(5) | 11.5899(7) | 13.273(3) | 9.027(1) |
| b/Å | 12.3303(11) | 15.864(3) | 24.655(4) | 24.712(4) | 12.058(6) | 11.9233(7) | 14.292(3) | 12.475(1) |
| c/Å | 21.170(3) | 10.566(2) | 16.589(2) | 16.280(2) | 22.071(5) | 14.3604(9) | 14.510(3) | 17.885(2) |
| α/° | 88.110(5) | 90 | 90 | 90 | 90 | 77.012(5) | 63.065(15) | 89.132(10) |
| β/° | 87.730(5) | 90 | 126.153(19) | 125.183(19) | 112.568(18) | 88.090(5) | 69.534(15) | 85.598(10) |
| γ/° | 86.920(5) | 90 | 90 | 90 | 90 | 84.122(5) | 78.915(16) | 85.422(10) |
| V/Å3 | 2040.4(16) | 1960.6(14) | 4022(2) | 3919(2) | 6902(3) | 1923.4(2) | 2297.1(9) | 2001.6(4) |
| Z | 2 | 4 | 4 | 4 | 8 | 2 | 2 | 2 |
| ρ/g cm−3 | 1.330 | 1.464 | 1.311 | 1.345 | 1.350 | 1.386 | 1.447 | 1.567 |
| μ/mm−1 | 0.094 | 0.801 | 0.426 | 0.437 | 0.490 | 0.449 | 0.702 | 0.801 |
| Crystal size | 0.02 × 0.09 × 0.14 | 0.103 × 0.114 × 0.124 | 0.100 × 0.163 × 0.262 | 0.100 × 0.163 × 0.262 | 0.03 × 0.100 × 0.12 | 0.07 × 0.100 × 0.19 | 0.23 × 0.24 × 0.24 | 0.292 × 0.309 × 0.380 |
| T/K | 100(2) | 133(2) | 180(2) | 133(2) | 175(2) | 133(2) | 133(2) | 133(2) |
| λ (MoKα)/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| θ-Range/° | 2.6–22.0 | 1.28–23.74 | 1.65–24.59 | 1.60–24.60 | 2.252–17.31 | 1.455–24.584 | 1.6–24.6 | 1.638–28.064 |
| Reflns. Collected | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 238 238 |
24192 |
47223 |
45921 |
26239 |
22912 |
27408 |
25770 |
| Indep. reflns. (Rint) | 4998 (0.074) | 1858 (0.4395) | 6730 (0.156) | 6540 (0.165) | 4159 (0.154) | 6444 (0.182) | 7688 (0.134) | 8940 (0.104) |
| Parameters | 541 | 134 | 514 | 514 | 403 | 514 | 604 | 568 |
| R 1 (F) (all data) | 0.0526 (0.1087) | 0.0851 (0.1601) | 0.0596 (0.1278) | 0.0571 (0.1158) | 0.0788 (0.1359) | 0.0748 (0.1224) | 0.0515 (0.1097) | 0.0861 (0.1222) |
| wR2 | 0.1299 | 0.1989 | 0.1205 | 0.1112 | 0.2073 | 0.1612 | 0.1049 | 0.2175 |
| GooF | 1.045 | 0.983 | 0.928 | 0.916 | 1.081 | 0.941 | 0.851 | 0.864 |
The crystal structure shows the molecule in its keto-enamine form as the C1–C2 and C15–C16 bonds can be attributed to single bonds (respectively 1.459(6) and 1.452(6)), and the C2–C3 and C14–C15 bonds can be attributed to double bonds (respectively 1.380(6) and 1.378(5)). This observation is in agreement with similar phenylene Schiff base-like ligand published in literature.18,23 Two intramolecular hydrogen bonds are present, both between the nitrogen of the enamine and the oxygen of the ketone (Table 3). π–π interactions are present between the stacked ligand molecules, with a distance of 3.55 Å between the centroids of the first ring of the naphthalene (C4–C5–C6–C11–C12–C13). This shows that the complexes produced with this ligand have potential to also form π–π interactions.
| Compound | D | H | A | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|---|---|---|
| a = −1 + x, 5/2 − y, −1/2 + z; b = x, 3/2 − y, −1/2 + z; c = x, 1/2 − y, −1/2 + z; d = 1 − x, 1/2 + y, 1/2 − z; e = −x, 1/2 + y, 1/2 − z; f = 1 − x, −1/2 + y, 1/2 − z; g = 1 + x, y, z; h = 2 − x, 1 − y, 1 − z; i = x, −1 + y, z; j = −1 + x, 1 + y, z.a Short contacts measured with mercury as the distances or angles exceed the threshold of PLATON. | |||||||
| 2·0.5 H2O·0.25 dioxane | N1 | H1 | O1 | 0.88 | 1.87 | 2.563(5) | 135 |
| N2 | H2 | O2 | 0.88 | 1.92 | 2.712(5) | 129 | |
| N31 | H31 | O31 | 0.88 | 1.93 | 2.566(5) | 128 | |
| N32 | H32 | O32 | 0.88 | 1.89 | 2.575(5) | 133 | |
| 10HS | C12 | H12 | O3a | 0.95 | 2.50 | 3.443(6) | 169 |
| 10LS | C12 | H12 | O3a | 0.95 | 2.41 | 3.361(6) | 174 |
| 11 | C105 | H105 | O105b | 0.95 | 2.33 | 3.20(2) | 152 |
| C212 | H212 | O206c | 0.95 | 2.36 | 3.22(2) | 150 | |
| C129 | H129 | O102d | 0.95 | 2.44 | 3.32(2) | 154 | |
| C229 | H229 | O201 | 0.95 | 2.67 | 3.56a | 156 | |
| C229 | H229 | O202 | 0.95 | 2.67 | 3.27a | 122 | |
| C224 | H224 | N302b | 0.95 | 2.58 | 3.52(2) | 175 | |
| C126 | H126 | N502 | 0.95 | 2.71 | 3.64a | 166 | |
| C304 | H304 | O203e | 0.95 | 2.56 | 3.33(3) | 139 | |
| C501 | H501 | O103f | 0.95 | 2.53 | 3.36(3) | 145 | |
| 12 | C45 | H45 | O5g | 0.95 | 2.70 | 3.24a | 117 |
| C46 | H46 | O5g | 0.95 | 2.46 | 3.135(7) | 128 | |
| 19 | C20 | H20A | O5h | 0.98 | 2.43 | 3.307(7) | 148 |
| C23 | H23A | O3i | 0.99 | 2.44 | 3.169(7) | 130 | |
| 20 | C22 | H22B | O33j | 0.98 | 2.55 | 3.359(9) | 140 |
| Fe–Neq | Fe–Oeq | Fe–Lax | Oeq–Fe–Oeq | Lax–Fe–Lax | |
|---|---|---|---|---|---|
| a Fe–Lax–Fe angle. | |||||
| 6 | 2.095(6) | 2.030(5) | 2.246(5) | 113.0(2) | 176.06(19) |
| 10HS | 2.063(4) | 1.989(4) | 2.138(5) | 106.98(14) | 174.22(17) |
| 2.040(4) | 2.018(4) | 2.226(5) | |||
| 10LS | 1.908(3) | 1.949(3) | 1.989(4) | 90.49(12) | 175.36(15) |
| 1.902(3) | 1.953(3) | 1.986(3) | |||
| 11 | 2.061(12) | 2.014(10) | 2.205(13) | 111.5(4) | 169.3(5) |
| 2.088(11) | 2.019(10) | 2.238(13) | 112.6(4) | 170.2(4) | |
| 2.096(11) | 2.004(10) | 2.218(12) | |||
| 2.089(11) | 2.012(10) | 2.276(12) | |||
| 12 | 1.903(4) | 1.943(3) | 1.996(4) | 87.79(14) | 173.85(16) |
| 1.906(4) | 1.933(3) | 2.004(4) | |||
| 19 | 2.066(4) | 1.951(3) | 1.771(3) | 92.46(13) | 148.18(19)a |
| 2.055(4) | 1.965(3) | 1.776(3) | 92.50(13) | ||
| 2.051(4) | 1.958(3) | ||||
| 2.069(3) | 1.961(3) | ||||
| 20 | 2.037(5) | 1.938(4) | 1.950(4) | 91.59(18) | 140.9(2)a |
| 2.055(5) | 1.964(4) | 1.954(4) | 90.18(17) | ||
| 2.067(4) | 1.954(4) | ||||
| 2.064(5) | 1.950(4) | ||||
Each 2D plane is succeeding each other along the axis [010] in a staggered fashion. Crystal packing pictures of the planes are shown in Fig. 3. Solvent accessible voids of 198.9 Å3 per unit cell (10%) were found by PLATON, however no solvent was found in the structure. Voids pictures are shown in Fig. 4. Although the structure shows voids, they seem to be inaccessible to solvent molecules, as the diameter of the “channels” is only 2 Å. Perhaps small gas molecules could be inserted in the network but this study falls out of the scope of this paper. Powder diffraction proved the bulk material to be isostructural to the crystals (ESI Fig. S3†).
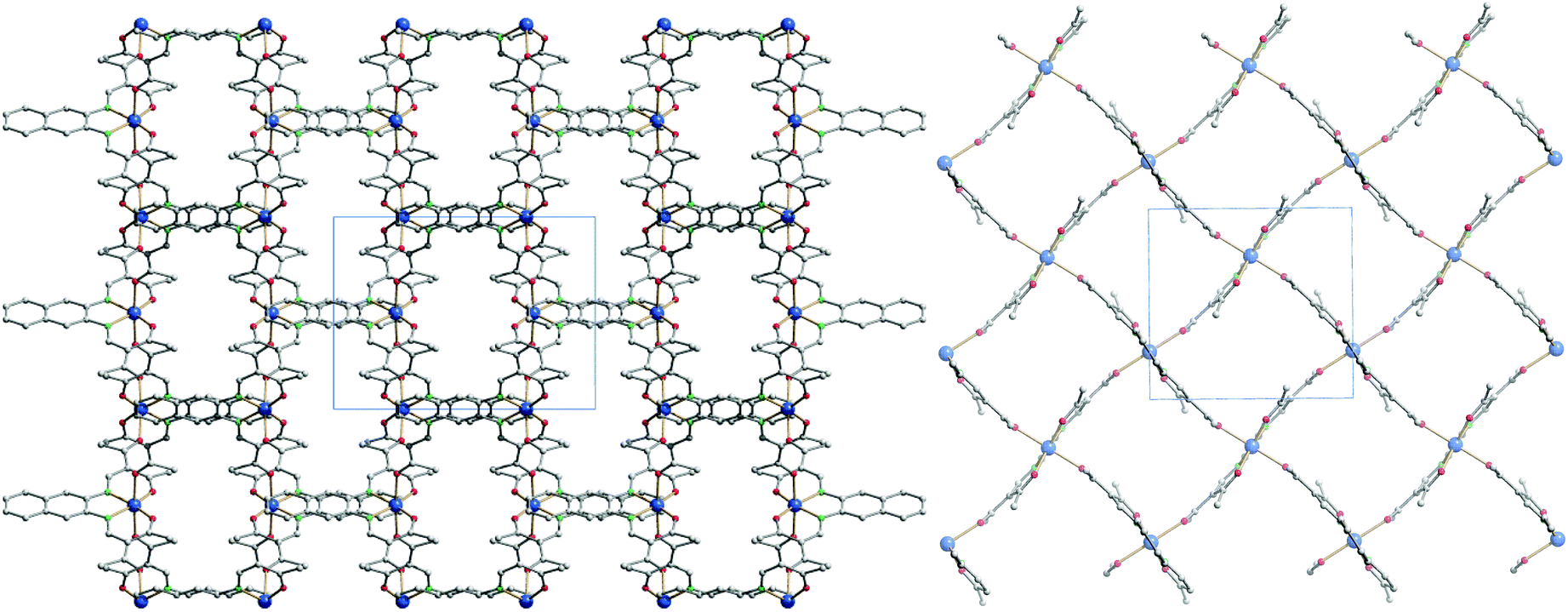 | ||
| Fig. 3 Crystal packing pictures of 6 along [001] (left) and picture of a single plane along [010] (right). Hydrogen atoms were omitted for clarity. | ||
 | ||
| Fig. 4 Solvent accessible voids pictures of 6 along [001] (left) and [100] (right). Hydrogen atoms were omitted for clarity. | ||
. The asymmetric unit of 10 contains one complex molecule and one non-coordinating pyridine solvent molecule, two different iron(II) complexes and two non-coordinating solvent pyridine molecules in the case of 11, and one complex molecule for 12. ORTEP drawings of the asymmetric units are displayed in Fig. 5. In all structures, the iron centres lie in a N4O2 coordination sphere, connected to the equatorial ligand and two (phenyl)pyridine molecules. The spin state of the iron centres can be determined by the value of the Oeq–Fe–Oeq angle which goes from ≈110° in the HS state to ≈90° in the LS state.24,25 Measured Oeq–Fe–Oeq angles for the HS structures (106.98(14)° for 10HS, 111.5(4)°, and 112.6(4)° for 11) and for the LS structures (90.49(12)° for 10LS and 87.79(14)° for 12) are in agreement with similar compounds in literature.8,26 Selected bond lengths and angles are listed in Table 4.
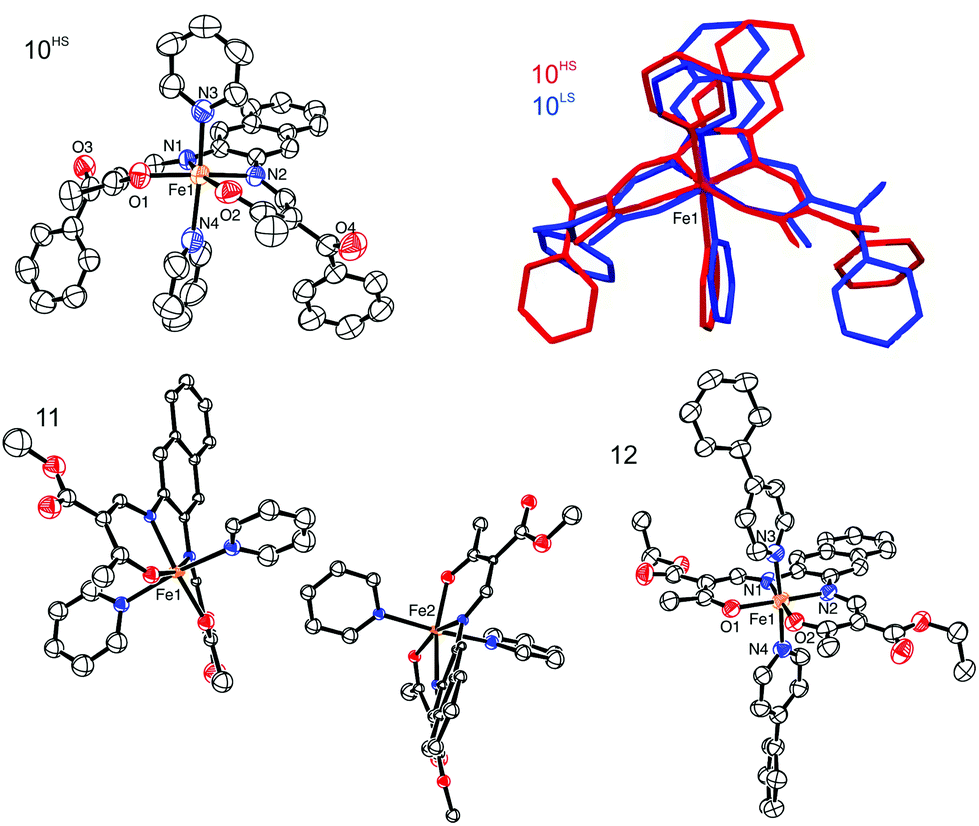 | ||
| Fig. 5 ORTEP drawing 10HS (top left), 11 (bottom left), and 12 (bottom right). The thermal ellipsoids are shown at the 50% level. Hydrogen atoms and non-coordinating solvent molecules were omitted for clarity reasons. The overlay of 10HS and 10LS is represented in the top right corner. | ||
Upon SCO, the volume of the cell of 10 is reduced by 2.5%, in agreement with the volume reduction of the coordination sphere of the iron centre as it switches from HS to LS state.27 The complex shows a strong saddle shape with an angle of 11.54° in the HS and 10.53° in the LS state between the FeN2O2 plane (Fe1–N1–N2–O1–O2) and the plane containing the naphthalene cycles. The change in the saddle angle comes from changes in lengths of intermolecular contacts between the complexes. Indeed, C–H⋯π interactions are observed between the aromatic rings of the naphthalene and aromatic hydrogen atoms of the axial pyridine ligands, leading to the formation of 1D chains propagating along the vector [001]. Additionally, the 1D chains are linked through C–H⋯O bridges linking the naphthalene ring (C12–H12) to the ketone group (O3) of a neighbouring molecule, along the vector [100]. Such C–H⋯O bridges are known to be structure defining in many cases.28 Contribution from both types of interactions leads to the formation of a 2D network in the a,c plane of the packing. Crystal packing and a scheme of the C–H⋯π interactions are shown in Fig. 6 and summarised in Tables 3 and 5. These intermolecular interactions have an influence on the slight change of the saddle angle of the molecule during SCO as the distance between the C–H and the aromatic rings partners in the interaction is increasing with the volume decrease of the coordination sphere.
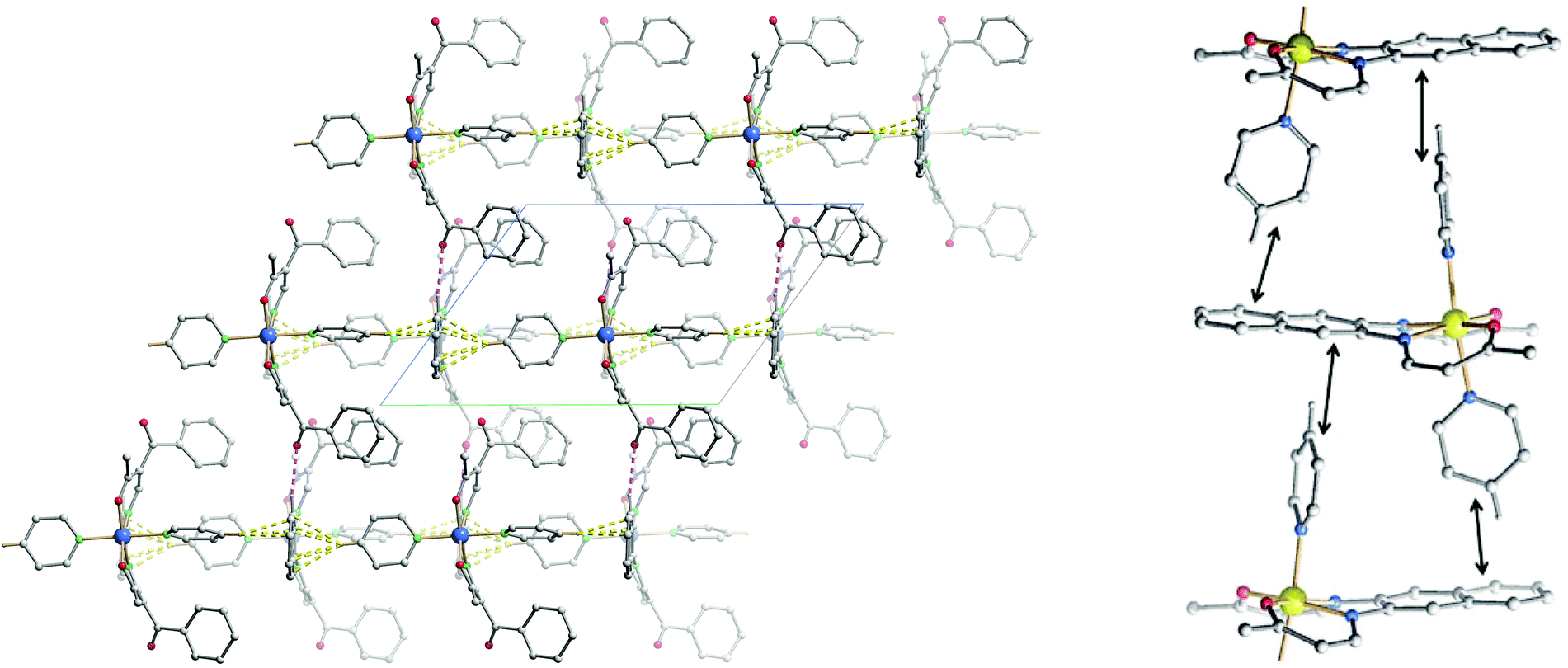 | ||
| Fig. 6 Left: crystal packing of 10HS along [0–10], hydrogen atoms not involved in short contacts were omitted for clarity reasons. C–H⋯π interactions are represented in dashed yellow lines, C–H⋯O in dashed pink lines. Right: excerpt of the crystal packing along [100], hydrogen atoms, non-coordinating solvent molecules, and substituents were omitted for clarity. Black double arrows indicate the C–H⋯π interactions positions. | ||
| Compound | X | Y | Cg | Y⋯Cg | X⋯Cg | X–Y⋯Cg |
|---|---|---|---|---|---|---|
| 10HS | C35 | H35 | C4–C6–C11–C13 | 2.41 | 3.358(7) | 174 |
| C40 | H40 | C6–C11 | 2.58 | 3.497(8) | 164 | |
| 10LS | C35 | H35 | C4–C6–C11–C13 | 2.42 | 3.364(6) | 170 |
| C40 | H40 | C6–C11 | 2.63 | 3.554(5) | 165 | |
| 11 | C131 | H131 | C104–C106–C111–C113 | 2.48 | 3.37(2) | 156 |
| C231 | H231 | C204–C206–C211–C213 | 2.65 | 3.48(2) | 147 | |
| 12 | C22 | O5 | C6–C11 | 3.807(4) | 3.560(6) | 69.1(3) |
The crystal packing of 11 presents a bi-layered structure: the different iron(II) complexes form planes perpendicular to vector [010], in a [Fe1Fe1–Fe2Fe2] fashion. The planes themselves are built by an intricate combination of C–H⋯O bridges between naphthalene and methyl ester substituents of neighbouring molecules along [001]; and, along [010], of C–H⋯O bridges and C–H⋯π interactions between axial pyridine and two neighbouring molecules. The latter C–H⋯O bridges connect to the oxygen atoms of the coordination sphere, which has been shown to have dramatic influence on SCO properties.9 The two different planes (Fe1 and Fe2) actually build the same network, although similar short contacts do not have the exact same distance, especially regarding C–H⋯O bridges connecting to coordinating oxygen (O101, O102, O201, O202). The non-coordinating pyridine solvent molecules occupy inter-planar position, binding to the planes through C–H⋯O and C–H⋯N bridges. Distances and angles of discussed short contacts are listed in Tables 3 and 5. The crystal packing of the 2D network is depicted in Fig. 7. The crystal packing of 12 is dominated by two strong interactions. Firstly, C–H⋯O bridges link one of the ethyl ester oxygens (O5) to a neighbouring phenylpyridine aromatic ring (C45–H45, C46–H46) and form a 1D chain of complexes along [100]. Secondly, an unusual overlap of the methyl ester group with the nearby naphthalene ring (–O6–C22O5⋯π) forming pairs of complexes.28c–f Additional weaker stacking between adjacent phenylpyridine aromatic rings, and between naphthalene rings creates a complex network between the Fe(II) spin centres. Crystal packing pictures as well as an ORTEP representation of the –O–CO⋯π and C–H⋯O bridges are shown in Fig. 8, distances and angles of discussed short contacts are listed in Tables 3 and 5.
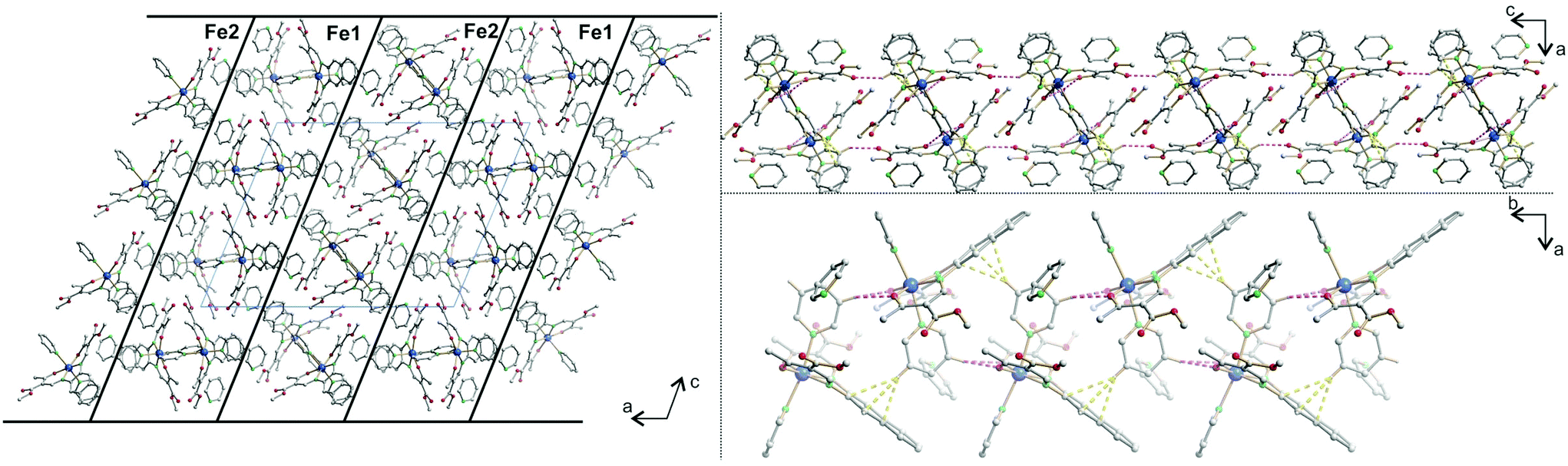 | ||
| Fig. 7 Crystal packing of 11 along [010] (left), illustrating the 2D network along [010] (top right) and [001] (bottom right); hydrogen atoms not involved in short contacts were omitted for clarity reasons. C–H⋯π interactions are represented in dashed yellow lines, C–H⋯O in dashed pink lines. | ||
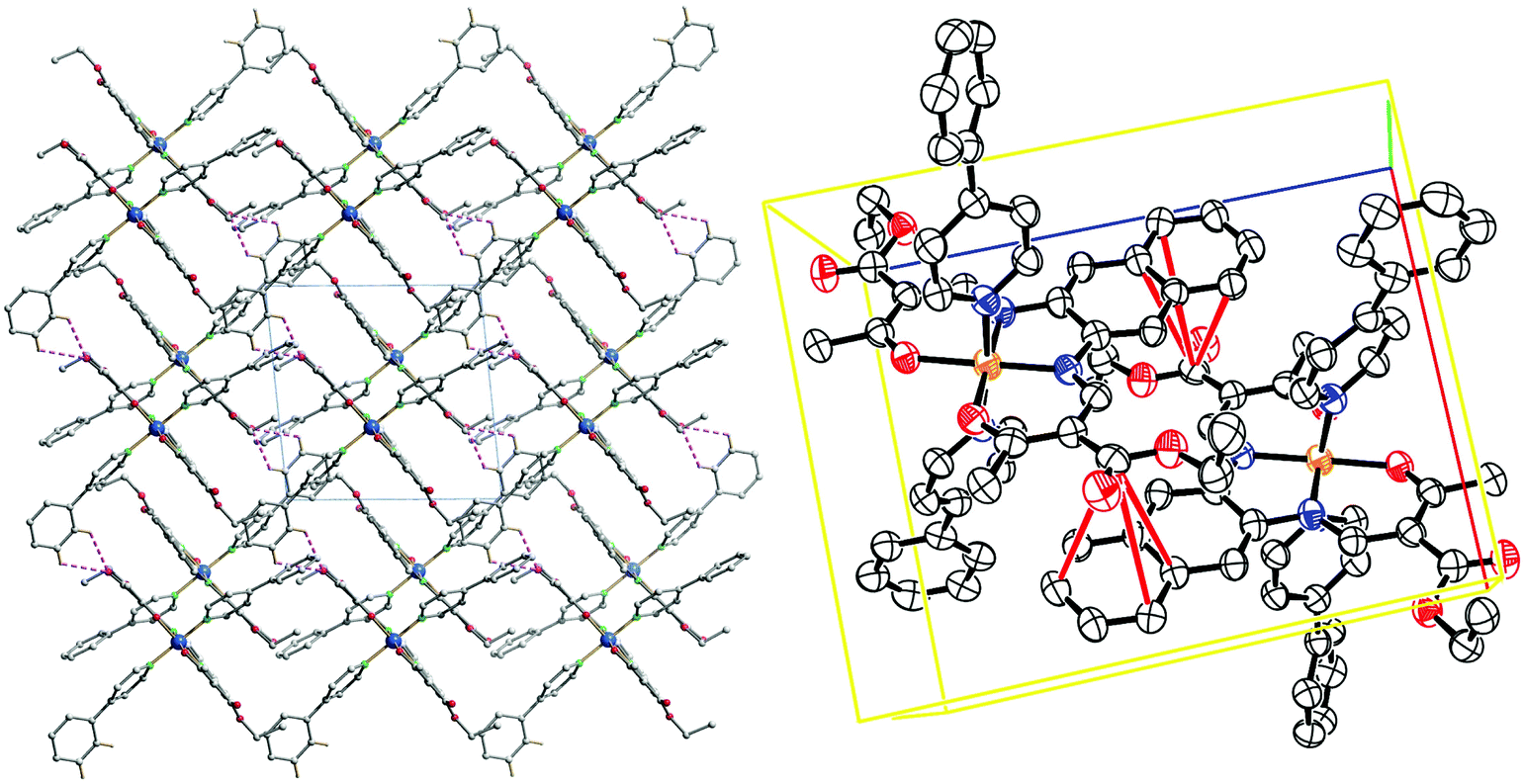 | ||
| Fig. 8 Crystal packing of 12 along [001] (left); C–H⋯O interactions represented in dashed pink lines; ORTEP representation of the –O–CO⋯π interactions, the thermal ellipsoids are shown at the 50% level. Hydrogen atoms were omitted for clarity reasons. | ||
, with an asymmetric unit containing one μ-oxido complex with the two iron(III) centres lying in a N2O3 coordination sphere. All square based pyramid iron(III) are connected through a μ-oxido bridge. The ORTEP drawing of the asymmetric units are shown in ESI, Fig. S6.† Bond lengths and angles within the coordination sphere are listed in Table 4, and are in agreement with similar published compounds.20,24 As for the SCO complexes, C–H⋯O bridges between ester side substituents can be found in the crystal packings, and the complexes are stacking in column fashion. Although the conformation of the two bridged iron complexes 19 and 20 are different, the crystal packing is actually similar. Pictures of the crystal packing are shown in ESI, Fig. S7.†
Discussion
Intermolecular interactions in the crystal packing result in cooperative interactions during the spin transition. These can result in abrupt spin transitions, thermal hysteresis loops, or step-wise spin transitions. Based on the structural data a deeper understanding of the different spin crossover properties is possible.Compound 10 shows an abrupt SCO behaviour (Fig. 1), which can be due to either to a phase transition during SCO, or due to other cooperative effects between the complexes in the solid state. The determination of the crystal structure in both spin states proved that no phase transition is involved during SCO. Cooperative interactions can arise from intermolecular contacts, and the packing of the crystal structure of 10 revealed, in both spin states, the formation of 1D chains through C–H⋯π interactions and C–H⋯O bridges between the complexes. Thus the Schiff base-like ligand was modified successfully and π-interactions are observed and lead to cooperative interactions between the complex molecules.
Compounds 12 and 14 show both an abrupt SCO with a hysteresis of 10 K. Powder diffraction of both samples in both spin states showed that no phase transition is involved through the ST, therefore attributing the hysteresis phenomena observed to cooperative effects. The powder diffraction patterns are shown in ESI (Fig. S4–S5†). In the crystal packing of 12, strong interactions between complexes are present, especially C–H⋯O bridges and –O–CO⋯π interactions connect all spin centres in a 3D network. This is responsible for the high cooperativity observed for these mononuclear complexes. It would have been of course interesting to have the structure of the HS state in order to pinpoint which interaction plays a determining role in the SCO. Unfortunately the low quality and low stability of the crystal at high temperature did not allow the determination of the structure in the HS state. Powder diffractograms of 12 and 14 were compared in order to determine if the compounds are isostructural, as their SCO behaviour is very similar and just shifted in temperature. Because of the complexity of the diffractograms, arising from the low symmetry of the crystal structures, no clear conclusions can be drawn about the isostructurality of samples 12 and 14.
Compounds 11 and 15 show both a two-step SCO. Such spin crossover are often related to different iron sites within the structure of the samples.29 The crystal structure of 11 confirmed this hypothesis as it shows a bi-layered packing with two different iron(II) centres arranged in layers separated by layers of non-coordinating pyridine solvent molecules. As no crystals suitable for X-ray analysis could be obtained for compound 15, a room temperature Mössbauer spectrum was measured. It shows an asymmetric doublet signal with the following hyperfine parameters: δ = 0.908(6), ΔEQ = 2.066(12) and Γ/2 = 0.174(13), which are in the region for an HS Fe(II) complex.6,16,29 The asymmetry of the peaks is characteristic for two slightly different doublet, as the right peak is larger than the left peak. It was however not possible to correctly fit the doublet signal with two sites. The Mössbauer spectrum is presented in the ESI, Fig. S8.†
For the crystal structures of the SCO complexes discussed in this work, C–H⋯π interactions and C–H⋯O bridges strongly influence the crystal packing. For the related phenylene based complexes, only C–H⋯O bridges were observed.15,16,25,29,30 The additional C–H⋯π interactions lead to a more frequent observation of interesting SCO properties. Out of the 10 naphthalene based complexes presented here, two showed a ST with a 10 K wide hysteresis and in two cases steps in the transition curve were observed. For comparison, for the 18 phenylene based mononuclear complexes reported so far, only one step-wise spin transition and two spin transitions with hysteresis (9 K and 70 K) were observed.25,31,32 The 70 K hysteresis does not fit into this comparison due to the use of imidazole as axial ligand that allowed the formation of an extended N–H⋯O hydrogen bond network between the complexes.31 First attempts to combine the naphthalene based Schiff base-like complexes with axial ligands suitable for the formation of hydrogen bond networks (like imidazole) are underway. The results obtained so far for the new ligands indicate that the use of an extended π-system was successful for the design of complexes with interesting SCO properties.
Experimental section
Synthesis
Methanol (MeOH) and ethanol (EtOH) were purified by distillation over magnesium under argon.33 Toluene and pyridine were of analytical grade and degassed under argon, pyridine was kept on a 4 Å molecular sieve. Ethoxymethyleneethylacetoacetate,34 ethoxymethyleneacetylacetone, ethoxymethylenebenzoylacetone,34 methoxymethylenemethylacetoacetate,18 and iron(II) acetate35 were synthesised as described in literature. Dioxane (Sigma, 99+%), 2,3-diaminonaphthalene (Alfa Aesar, 97%), 4-(dimethylamino)pyridine (dmap; Merck, 99%) and 4-phenylpyridine (phpy; Acros, 99%) were used without further purification. All syntheses with iron(II) were carried out under argon using Schlenk tube techniques. CHN analyses were measured with a Vario El III from Elementar Analysen-Systeme. Mass spectra were recorded with a Finnigan MAT 8500 with a data system MASPEC II.![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3156(b) (NH), 1654(s) (CO), 1648(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 438 (50) (H2L1+); elemental analysis calculated (found) for C24H26N2O6·C2H6O (438.47 g mol−1): C 64.45 (60.41), H 5.88 (5.95), N 6.17 (5.93). 1H-NMR (CDCl3, 300 MHz, ppm) δ = 13.03 (d, J = 12 Hz, –NH, 2H), 8.51 (d, J = 12 Hz, CH, 2H), 7.81 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 2H), 7.68 (s, Ar–H, 2H), 7.50 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 2H), 4.29 (qua, J = 7 Hz, –CH2–, 4H), 2.60 (s, –CH3, 6H), 1.36 (t, J = 7 Hz, –CH3, 6H).
= 3223(b) (NH), 1689(s) (CO), 1675(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 378 (11) (H2L2+); elemental analysis calculated (found) for C22H22N2O4·0.5C4H8O2 (422.47 g mol−1): C 68.23 (67.91), H 6.20 (5.88), N 6.63 (6.89). 1H-NMR (CDCl3, 300 MHz, ppm) δ = 13.06 (m, –NH, 2H), 8.24 (d, J = 11 Hz, CH, 2H), 7.83 (m, Ar–H, 2H), 7.66 (s, Ar–H, 2H), 7.53 (m, Ar–H, 2H), 2.59 (s, –CH3, 6H), 2.42 (s, –CH3, 6H).
= 3204(b) (NH), 1663(s) (CO), 1598(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 502 (78) (H2L3+); elemental analysis calculated (found) for C32H26N2O4·0.5CH4O (518.59 g mol−1): C 75.27 (74.90), H 5.442 (4.8), N 5.402 (5.377). 1H-NMR (CDCl3, 300 MHz, ppm) δ = 12.96 (d, J = 12 Hz, –NH, 2H), 7.92 (d, J = 12 Hz, CH, 2H), 7.76 (m, Ar–H, 4H), 7.68 (s, Ar–H, 2H), 7.48 (m, Ar–H, 10H), 2.56 (s, –CH3, 6H).
= 3265(b) (NH), 1652(s) (CO), 1622(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 410 (100) (H2L4+); elemental analysis calculated (found) for C22H22N2O6·0.5CH4O (426.4 g mol−1): C 63.37 (62.97), H 5.672 (4.765), N 6.569 (6.592). 1H-NMR (CDCl3, 300 MHz, ppm) δ = 13.67 (d, J = 12 Hz, –NH, 2H), 8.57 (d, J = 12 Hz, CH, 2H), 7.81 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 4H), 7.68 (s, Ar–H, 2H), 7.48 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 10H), 3.82 (s, –CH3, 6H), 2.60 (s, –CH3, 6H).
= 1614(s) (CO), 1597(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 492 (100) ([FeL1]+); elemental analysis calculated (found) for C26H32FeN2O8 (556.39 g mol−1): C 56.13 (55.42), H 5.80 (4.20), N 5.03 (5.20).
= 1685(s) (CO), 1584(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 432 (100) ([FeL2]+); elemental analysis calculated (found) for C22H20FeN2O4 (432.08 g mol−1): C 61.13 (60.43), H 4.66 (4.29), N 6.48 (6.36).
= 1629(s) (CO), 1575(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 556 (100) ([FeL3]+); elemental analysis calculated (found) for C32H24FeN2O4 (556.39 g mol−1): C 69.08 (68.37), H 4.35 (3.44), N 5.03 (4.997). There was a miscalculation on the theoretical values.
= 1620(s) (CO), 1598(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 528 (100) ([FeL4]+); elemental analysis calculated (found) for C24H28FeN2O8 (528.33 g mol−1): C 54.56 (54.17), H 5.34 (5.12), N 5.30 (5.26).
= 1612(s) (CO), 1596(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 432 (100) ([FeL2]+); elemental analysis calculated (found) for C32H35FeN4O6.5 (635.49 g mol−1): C 60.48 (60.46), H 5.55 (3.912), N 8.82 (7.865).
= 1623(s) (CO), 1584(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 556 (100) ([FeL3]+).
= 1601(s) (CO), 1582(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 464 (100) ([FeL4]+); elemental analysis calculated (found) for C35H34FeN5O6 (701.55 g mol−1): C 63.34 (62.06), H 5.03 (4.59), N 9.98 (9.624).
= 1614(s) (CO), 1587(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 432 (100) ([FeL2]+); elemental analysis calculated (found) for C36H46FeN4O6 (742.64 g mol−1): C 66.97 (68.48), H 5.14 (5.74), N 7.103 (6.90).
= 1612(s) (CO), 1596(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 464 (100) ([FeL4]+); elemental analysis calculated (found) for C44H38FeN4O6 (774.64 g mol−1): C 68.22 (68.68), H 4.94 (5.20), N 7.23 (7.18).
= 1603(s) (CO), 1574(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 492 (75) ([FeL1]+), 121 (100) (dmap+); elemental analysis calculated (found) for C38H44FeN6O6 (736.64 g mol−1): C 61.96 (62.35), H 6.02 (5.65), N 11.41 (11.65).
= 1606(s) (CO), 1569(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 431 (23) ([FeL2]+), 121 (100) (dmap+); elemental analysis calculated (found) for C36H40FeN6O4 (676.59 g mol−1): C 63.91 (63.74), H 5.96 (5.69), N 12.42 (12.34).
= 1617(s) (CO), 1583(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 556 (58) ([FeL3]+), 121 (100) (dmap+); elemental analysis calculated (found) for C46H44FeN6O4 (800.72 g mol−1): C 69.00 (69.73), H 5.54 (5.46), N 10.50 (10.11).
= 1614(s) (CO), 1596(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 464 (75) ([FeL4]+), 121 (100) (dmap+); elemental analysis calculated (found) for C36H40FeN6O6 (708.58 g mol−1): C 61.02 (59.20), H 5.69 (5.06), N 11.86 (11.48).
= 3156(b) (NH), 1654(s) (CO), 1648(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 438 (50) (H2L1+); elemental analysis calculated (found) for C24H26N2O6·C2H6O (438.47 g mol−1): C 64.45 (60.41), H 5.88 (5.95), N 6.17 (5.93). 1H-NMR (CDCl3, 300 MHz, ppm) δ = 13.03 (d, J = 12 Hz, –NH, 2H), 8.51 (d, J = 12 Hz, CH, 2H), 7.81 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 2H), 7.68 (s, Ar–H, 2H), 7.50 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 2H), 4.29 (qua, J = 7 Hz, –CH2–, 4H), 2.60 (s, –CH3, 6H), 1.36 (t, J = 7 Hz, –CH3, 6H).
= 3223(b) (NH), 1689(s) (CO), 1675(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 378 (11) (H2L2+); elemental analysis calculated (found) for C22H22N2O4·0.5C4H8O2 (422.47 g mol−1): C 68.23 (67.91), H 6.20 (5.88), N 6.63 (6.89). 1H-NMR (CDCl3, 300 MHz, ppm) δ = 13.06 (m, –NH, 2H), 8.24 (d, J = 11 Hz, CH, 2H), 7.83 (m, Ar–H, 2H), 7.66 (s, Ar–H, 2H), 7.53 (m, Ar–H, 2H), 2.59 (s, –CH3, 6H), 2.42 (s, –CH3, 6H).
= 3204(b) (NH), 1663(s) (CO), 1598(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 502 (78) (H2L3+); elemental analysis calculated (found) for C32H26N2O4·0.5CH4O (518.59 g mol−1): C 75.27 (74.90), H 5.442 (4.8), N 5.402 (5.377). 1H-NMR (CDCl3, 300 MHz, ppm) δ = 12.96 (d, J = 12 Hz, –NH, 2H), 7.92 (d, J = 12 Hz, CH, 2H), 7.76 (m, Ar–H, 4H), 7.68 (s, Ar–H, 2H), 7.48 (m, Ar–H, 10H), 2.56 (s, –CH3, 6H).
= 3265(b) (NH), 1652(s) (CO), 1622(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 410 (100) (H2L4+); elemental analysis calculated (found) for C22H22N2O6·0.5CH4O (426.4 g mol−1): C 63.37 (62.97), H 5.672 (4.765), N 6.569 (6.592). 1H-NMR (CDCl3, 300 MHz, ppm) δ = 13.67 (d, J = 12 Hz, –NH, 2H), 8.57 (d, J = 12 Hz, CH, 2H), 7.81 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 4H), 7.68 (s, Ar–H, 2H), 7.48 (dd, J3 = 6 Hz, J4 = 3 Hz, Ar–H, 10H), 3.82 (s, –CH3, 6H), 2.60 (s, –CH3, 6H).
= 1614(s) (CO), 1597(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 492 (100) ([FeL1]+); elemental analysis calculated (found) for C26H32FeN2O8 (556.39 g mol−1): C 56.13 (55.42), H 5.80 (4.20), N 5.03 (5.20).
= 1685(s) (CO), 1584(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 432 (100) ([FeL2]+); elemental analysis calculated (found) for C22H20FeN2O4 (432.08 g mol−1): C 61.13 (60.43), H 4.66 (4.29), N 6.48 (6.36).
= 1629(s) (CO), 1575(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 556 (100) ([FeL3]+); elemental analysis calculated (found) for C32H24FeN2O4 (556.39 g mol−1): C 69.08 (68.37), H 4.35 (3.44), N 5.03 (4.997). There was a miscalculation on the theoretical values.
= 1620(s) (CO), 1598(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 528 (100) ([FeL4]+); elemental analysis calculated (found) for C24H28FeN2O8 (528.33 g mol−1): C 54.56 (54.17), H 5.34 (5.12), N 5.30 (5.26).
= 1612(s) (CO), 1596(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 432 (100) ([FeL2]+); elemental analysis calculated (found) for C32H35FeN4O6.5 (635.49 g mol−1): C 60.48 (60.46), H 5.55 (3.912), N 8.82 (7.865).
= 1623(s) (CO), 1584(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 556 (100) ([FeL3]+).
= 1601(s) (CO), 1582(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 464 (100) ([FeL4]+); elemental analysis calculated (found) for C35H34FeN5O6 (701.55 g mol−1): C 63.34 (62.06), H 5.03 (4.59), N 9.98 (9.624).
= 1614(s) (CO), 1587(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 432 (100) ([FeL2]+); elemental analysis calculated (found) for C36H46FeN4O6 (742.64 g mol−1): C 66.97 (68.48), H 5.14 (5.74), N 7.103 (6.90).
= 1612(s) (CO), 1596(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 464 (100) ([FeL4]+); elemental analysis calculated (found) for C44H38FeN4O6 (774.64 g mol−1): C 68.22 (68.68), H 4.94 (5.20), N 7.23 (7.18).
= 1603(s) (CO), 1574(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 492 (75) ([FeL1]+), 121 (100) (dmap+); elemental analysis calculated (found) for C38H44FeN6O6 (736.64 g mol−1): C 61.96 (62.35), H 6.02 (5.65), N 11.41 (11.65).
= 1606(s) (CO), 1569(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 431 (23) ([FeL2]+), 121 (100) (dmap+); elemental analysis calculated (found) for C36H40FeN6O4 (676.59 g mol−1): C 63.91 (63.74), H 5.96 (5.69), N 12.42 (12.34).
= 1617(s) (CO), 1583(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 556 (58) ([FeL3]+), 121 (100) (dmap+); elemental analysis calculated (found) for C46H44FeN6O4 (800.72 g mol−1): C 69.00 (69.73), H 5.54 (5.46), N 10.50 (10.11).
= 1614(s) (CO), 1596(s) (CO) cm−1; MS (DEI-(+), 70 eV) m/z (%): 464 (75) ([FeL4]+), 121 (100) (dmap+); elemental analysis calculated (found) for C36H40FeN6O6 (708.58 g mol−1): C 61.02 (59.20), H 5.69 (5.06), N 11.86 (11.48).
X-Ray structure analysis
The intensity data of 2·0.5C4H8O·0.5H2O were collected with a Bruker D8 Venture diffractometer, the intensity data of 5, 10HS, 10LS, 11, 12, 19 and 20 were collected with a Stoe IPDS II diffractometer, both diffractometer using graphite-monochromated MoKα radiation. The data were corrected for Lorentz and polarization effects. The structures were dissolved by direct methods (SIR-97)36 and refined by full-matrix least-square techniques against Fo2 − Fc2 (SHELXL-97).37 All hydrogen atoms were calculated in idealised positions with fixed displacement parameters. ORTEP-III38 was used for the structure representation, SCHAKAL-9939 to illustrate molecule packing and CrystalExplorer40 for the representation of the voids. CCDC 1040373–1040380 contain the supplementary crystallographic data for this paper.Magnetic measurements
Magnetic susceptibility data were collected using a MPMSXL-5 SQUID magnetometer under an applied field of 0.5 T over the temperature range 2 to 400 K in the settle mode. The samples were placed in gelatin capsules held within a plastic straw. The data were corrected for the diamagnetic contributions of the ligands by using tabulated Pascal's constants and of the sample holder.41Conclusions
In this work we presented the synthesis of a new ligand system for iron(II) spin crossover complexes with an increased potential for the observation of π-interactions due to the extended aromatic naphthalene unit compared to the previously used benzene unit. Ten new complexes with a high potential for the observation of spin crossover were synthesised and 50% of those complexes showed the desired properties. Of those, three showed a cooperative spin transition with a maximum hysteresis width of 10 K. The X-ray structure analysis of 10 and 12 revealed C–H⋯π interactions, C–H⋯O bridges, and –O–CO⋯π interactions as reason for the abrupt spin transition, with hysteresis in the case of 12. The next step will be to extend the aromatic system further, e.g. by going to phenazine-derivatives42 in order to further increase the potential for π-stacking. Another possibility is to link the naphtalene-based Schiff base-like complexes to 1D chains and by this combine rigid linkers with π-stacking. This will be part of a following paper.
Acknowledgements
Financial supports from the German Science foundation (SFB840; project A10) and the University of Bayreuth are acknowledged. We thank S. Albrecht and P. Mayer (University of Munich) for the collection of single crystal XRD date of 1a.Notes and references
-
(a) O. Kahn, Science, 1998, 279, 44–48 CrossRef CAS
; (b) J.-F. Létard, P. Guionneau and L. Goux-Capes, in Spin Crossover in Transition Metal Compounds I-III, ed. P. Gütlich and H. Goodwin, Springer, Berlin/Heidelberg, 2004, pp. 221–249 Search PubMed
-
(a)
Spin-Crossover Materials, ed. M. A. Halcrow, John Wiley & Sons Ltd, Chichester, 2013 Search PubMed
-
(a) P. D. Southon, L. Liu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 CrossRef CAS PubMed
-
(a) S. Schlamp, B. Weber, A. D. Naik and Y. Garcia, Chem. Commun., 2011, 47, 7152–7154 RSC
- C. Gandolfi, G. G. Morgan and M. Albrecht, Dalton Trans., 2012, 41, 3726–3730 RSC
- B. Weber, E. S. Kaps, J. Obel, K. Achterhold and F. G. Parak, Inorg. Chem., 2008, 47, 10779–10787 CrossRef CAS PubMed
-
(a) W. Bauer, S. Schlamp and B. Weber, Chem. Commun., 2012, 48, 10222 RSC
- C. Lochenie, W. Bauer, A. P. Railliet, S. Schlamp, Y. Garcia and B. Weber, Inorg. Chem., 2014, 53, 11563–11572 CrossRef CAS PubMed
- B. Weber, W. Bauer, T. Pfaffeneder, M. M. Dîrtu, A. D. Naik, A. Rotaru and Y. Garcia, Eur. J. Inorg. Chem., 2011, 3193–3206 CrossRef CAS
- J. A. Real, A. B. Gaspar, V. Niel and M. C. Muñoz, Coord. Chem. Rev., 2003, 236, 121–141 CrossRef CAS
- Z. J. Zhong, J.-Q. Tao, Z. Yu, C.-Y. Dun, Y.-J. Liu and X.-Z. You, J. Chem. Soc., Dalton Trans., 1998, 327–328 RSC
- J.-F. Létard, P. Guionneau, E. Codjovi, O. Lavastre, G. Bravic, D. Chasseau and O. Kahn, J. Am. Chem. Soc., 1997, 119, 10861–10862 CrossRef
- L. Wolf and E.-G. Jäger, Z. Anorg. Allg. Chem., 1966, 346, 76–91 CrossRef CAS
-
(a) B. Weber, E. S. Kaps, C. Desplanches and J. Létard, Eur. J. Inorg. Chem., 2008, 2963–2966 CrossRef CAS
- B. Weber, E. S. Kaps, C. Desplanches, J.-F. Létard, K. Achterhold and F. G. Parak, Eur. J. Inorg. Chem., 2008, 4891–4898 CrossRef CAS
- B. Weber, E. Kaps, J. Weigand, C. Carbonera, J.-F. Létard, K. Achterhold and F. G. Parak, Inorg. Chem., 2008, 47, 487–496 CrossRef CAS PubMed
-
(a) B. R. Müller, G. Leibeling and E.-G. Jäger, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1999, 334, 389–394 CrossRef
- W. Bauer, T. Ossiander and B. Weber, Z. Naturforsch., B: Chem. Sci., 2010, 323–328 CAS
- S. Schlamp, P. Thoma and B. Weber, Eur. J. Inorg. Chem., 2012, 2759–2768 CrossRef CAS
- S. Schlamp, P. Thoma and B. Weber, Chem. – Eur. J., 2014, 20, 6462–6473 CrossRef CAS PubMed
-
(a) W. Bauer, C. Lochenie and B. Weber, Dalton Trans., 2014, 43, 1990–1999 RSC
- B. Weber, J. Obel, D. Henner-Vasquez and W. Bauer, Eur. J. Inorg. Chem., 2009, 5527–5534 CrossRef CAS
- C. Lochenie, S. Schlamp, A. P. Railliet, K. Robeyns, B. Weber and Y. Garcia, CrystEngComm, 2014, 16, 6213 RSC
- B. Weber and E.-G. Jäger, Eur. J. Inorg. Chem., 2009, 465–477 CrossRef CAS
- B. Weber, Coord. Chem. Rev., 2009, 253, 2432–2449 CrossRef CAS
- S. Schlamp, K. Dankhoff and B. Weber, New J. Chem., 2014, 38, 1965–1972 RSC
-
P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, in Spin Crossover in Transition Metal Compounds I–III, ed. P. Gütlich and H. Goodwin, Springer, Berlin/Heidelberg, 2004, vol. II, p. 785 Search PubMed
-
(a) T. Steiner and G. R. Desiraju, Chem. Commun., 1998, 891–892 RSC
- B. Weber, C. Carbonera, C. Desplances and J.-F. Létard, Eur. J. Inorg. Chem., 2008, 1589–1598 CrossRef CAS
- B. Weber, E. Kaps, J. Obel and W. Bauer, Z. Anorg. Allg. Chem., 2008, 634, 1421–1426 CrossRef CAS
- B. Weber, W. Bauer and J. Obel, Angew. Chem., Int. Ed., 2008, 47, 10098–10101 CrossRef CAS PubMed
- C. Lochenie, W. Bauer, S. Schlamp, P. Thoma and B. Weber, Z. Anorg. Allg. Chem., 2012, 638, 98–102 CrossRef CAS
-
H. G. O. Becker, Organikum, Organisch-chemisches Grundpraktikum, Johann Ambrosius Barth, Berlin, 19th edn, 1993 Search PubMed
- L. Claisen, Justus Liebigs Ann. Chem., 1897, 297, 1–98 CrossRef CAS
- B. Weber, R. Betz, W. Bauer and S. Schlamp, Z. Anorg. Allg. Chem., 2011, 637, 102–107 CrossRef CAS
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
-
(a)
C. K. Johnson and M. N. Burnett, ORTEP-III, Oak-Ridge National Laboratory, Oak-Ridge, TN, 1996 Search PubMed
-
E. Keller, Schakal-99, University of Freiburg, Freiburg, Germany, 1999 Search PubMed
-
(a)
S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackmann, CrystalExplorer, University of Wesertn Australia, 2012 Search PubMed
-
O. Kahn, Molecular Magnetism, VCH, New York, N.Y, 1993 Search PubMed
- C. Lochenie, K. G. Wagner, M. Karg and B. Weber, J. Mater. Chem. C, 2015, 3, 7925–7935 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Magnetic susceptibility measurements of compounds 9, 13, 16, 17 and 18; powder diffraction patterns of samples 6, 12 and 14, TGA measurement of 11 and ORTEP drawings of 19 and 20. CCDC 1040373–1040380. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03048j |
| This journal is © The Royal Society of Chemistry 2015 |