
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The carboboration of Me3Si-substituted alkynes and allenes with boranes and borocations†
James R.
Lawson
,
Valerio
Fasano
,
Jessica
Cid
,
Inigo
Vitorica-Yrezabal
and
Michael J.
Ingleson
*
School of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: Michael.ingleson@manchester.ac.uk
First published on 8th September 2015
Abstract
The 1,1-carboboration of 1-Me3Si-1-alkynes is the dominant reaction observed using [PhBCl(2-DMAP)][AlCl4], 1, and PhBCl2 electrophiles, with highly substituted vinyl pinacol boronate esters isolated post esterification. Other aryl and heteroaryl congeners of both 1 and PhBCl2 have a limited scope in the 1,1-carboboration of 1-Me3Si-1-alkynes, with desilylboration more prevalent. PhBCl2 converts Me3Si-substituted allenes to allylboranes via a formal 1,3-carboboration with Me3Si-migration. [Cl2B(2-DMAP)][AlCl4] reacts with a number of 1-Me3Si-1-alkynes by desilylboration, whilst with Me3Si-ethyne a 1,1-boroamination reaction proceeds, which with excess boron electrophile is followed by an intermolecular desilylboration to form a tricationic-borate. The use of excess 1-Me3Si-1-propyne relative to 1 (and a thienyl congener of 1) formed 2-boradienes in low yields from the reaction with two equivalents of alkyne. Vinyl borocations ligated by 2,6-lutidine of the general formula, [(vinyl)BCl(2,6-lutidine)][AlCl4] formed 1-boradienes with 1-Me3Si-1-alkynes.
Introduction
The 1,1-carboboration of alkynes has received increased interest in recent years following the discovery of the 1,1-carboboration of terminal and internal alkynes with the strong electrophile RB(C6F5)2 (R = C6F5, alkyl).1 Addition of RB(C6F5)2 to a terminal alkyne induces a 1,2-shift of H (or a hydrocarbyl for internal alkynes) along the alkynyl backbone with subsequent migration of the R group from boron to carbon resulting in 1,1-carboboration.2 This reaction has been developed into a useful route to arylboranes by benzannulation,3 and to highly substituted vinyl boranes that are complementary to those obtained from hydroboration.4 1,1-carboboration was pioneered by Wrackmeyer albeit with weaker boron electrophiles, such as trialkylboranes, for the 1,1-carboboration of activated alkynes, e.g., alkynes substituted with heavier group 14 substituents (Scheme 1, top).5 Wrackmeyer and co-workers reported extensively on this topic and determined the relative reactivity of a range of substituted alkynes toward BEt3 to be R3Pb > R3Sn > R3Ge > R3Si (TMS), with R3C-substituted alkynes not amenable.5 These studies predominantly used trialkylboranes; in contrast, the utilisation of vinyl and aryl boranes (excluding B(C6F5)3) for the 1,1-vinyl-boration or 1,1-aryl-boration of alkynes is rare with only limited examples reported using BPh3 or PhB(C6F5)2.6,7 One report particularly relevant to this work showed that Ph2BX (X = Cl or Br) effected the 1,1-carboboration of 1-TMS-1-hexyne (Scheme 1, top).8 It should be noted that with (chloro)x(aryl)3−xB compounds TMS substitution appears essential for 1,1-carboboration, with PhBCl2 and terminal alkynes reacting via 1,2-halo- or 1,2-carboboration instead.9 To the best of our knowledge the outcome from combining TMS-alkynes and (hydrocarbyl)BCl2 has not been reported to date.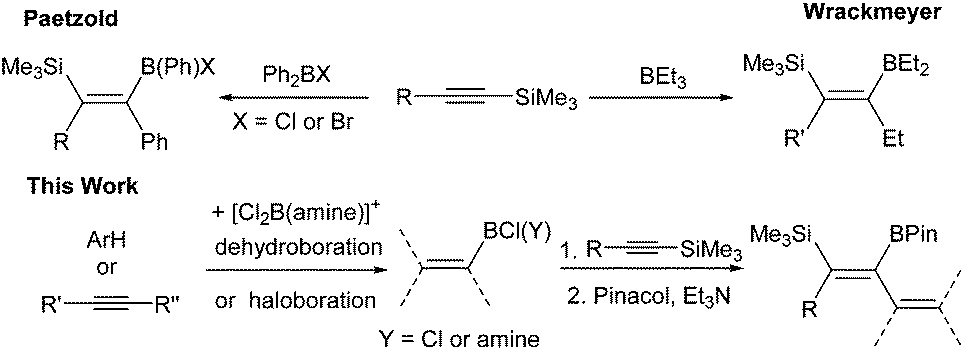 | ||
| Scheme 1 Relevant early 1,1-carboborations and the approach in this work. | ||
We envisaged combining arene borylation,10 or alkyne haloboration,11 using BCl3 derived borocations (to produce arylBCl2 and [vinylBCl(amine)]+, respectively) with 1,1-carboboration to generate synthetically useful highly substituted vinyl (or dienyl) boronate esters after esterification (Scheme 1, bottom).12 This process may proceed directly from the organoBCl2 or require enhancement of electrophilicity at boron by formation of a borocation. We have previously demonstrated that borocations are effective for the 1,2-haloboration and 1,2-carboboration of alkynes, with no 1,1-elementoboration observed.11,13 For example, terminal alkynes and the boronium salt [(Ph)ClB(2-DMAP)][AlCl4] (2-DMAP = 2-N,N-dimethylamino-pyridine) react only by 1,2-chloroboration.11 Based on the previous 1,1-carboboration studies with neutral boranes it was hypothesised that TMS-substituted alkynes would preferentially undergo 1,1-elementoboration over 1,2-elementoboration when combined with organoBCl2 or borocation compounds. Support for this comes from the work of Curran and co-workers on the 1,1-hydroboration of 1-TMS-1-alkynes using borenium equivalents such as (NHC)BH2(NTf)2.14 Herein is reported our studies using arylBCl2 and aryl and vinyl containing borocations synthesised by electrophilic borylation to effect the carboboration of TMS-substituted alkynes.
Results and discussion
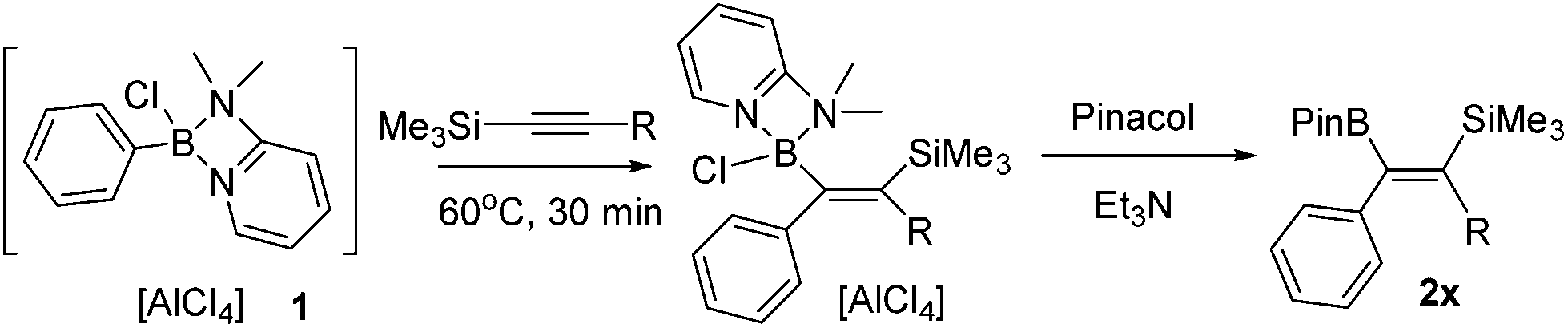
Vinylboronate ester formation
Studies commenced with the boronium cation [Ph(Cl)B(2-DMAP)][AlCl4], 1.11 The combination of 1 and equimolar 1-TMS-1-propyne at 20 °C resulted in a slow reaction generating a single new silicon containing compound (δ29Si −6.05) with minimal desilylboration observed (only a low intensity TMSCl resonance present in the 1H NMR spectra). Heating the reaction to 60 °C in CH2Cl2 in a sealed tube for extended periods (>1 h) led to complex mixtures, however heating for 30 minutes at 60 °C led to one major new product possessing identical resonances to that observed in the 20 °C reaction, with unreacted 1 also remaining. Esterification of the reaction mixture after 30 minutes with pinacol/Et3N led to two major boron containing products, PhBPin (derived from unreacted 1) and a new product isolable by column chromatography. NMR and mass spectroscopy confirmed this to be the product from the 1,1-carboboration of 1-TMS-1-propyne, formed as the E-isomer exclusively, 2a (Table 1). This reactivity was extended to a number of other 1-TMS-1-alkynes to yield 2b to 2e. Using these conditions PhBPin was observed as a minor product from esterification of unreacted 1 in all cases and required separation by column chromatography. The reaction of 1 with TMS-ethyne and trimethyl(3-methylbut-1-yn-1-yl)silane under analogous conditions led to complex intractable mixtures containing significant TMSCl (by 1H NMR spectroscopy).|
|
|||||
|---|---|---|---|---|---|
| Entry | Electrophile | T (°C) | Compound no. | R | Isolated yield (%) |
| a Performed in CH2Cl2 in sealed tubes fitted with J. Young valves. | |||||
| 1 | 1 | 60 | 2a | Me | 63 |
| 2 | 1 | 60 | 2b | Bu | 61 |
| 3 | 1 | 60 | 2c | C(Me)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
55 |
| 4 | 1 | 60 | 2d | Ph | 65 |
| 5 | 1 | 60 | 2e | p-Br-C6H4 | 56 |
With 1,1-carboboration observed from the combination of 1 and 1-TMS-1-alkynes the reaction of 1-TMS-1-propyne and [Cl2B(2-DMAP)][AlCl4], 3, was explored to determine if any 1,1-chloroboration occurred. Instead, this led to formation of TMSCl (by 1H and 29Si NMR spectroscopy) and a major new 11B resonance at +6.5 ppm. Esterification enabled identification of the alkynyl pinacol boronate ester confirming desilylboration (eqn (1)). Desilylboration was observed also on combination of 3 with 1-phenyl-2-TMS-acetylene and with 1-TMS-1-hexyne. Whilst in situ NMR spectra pre-esterification indicated desilylboration and alkynylborane formation is the dominant reaction outcome, the isolated yields post pinacol esterification were consistently low due to the susceptibility of alkynyl-B species to protodeboronation.11
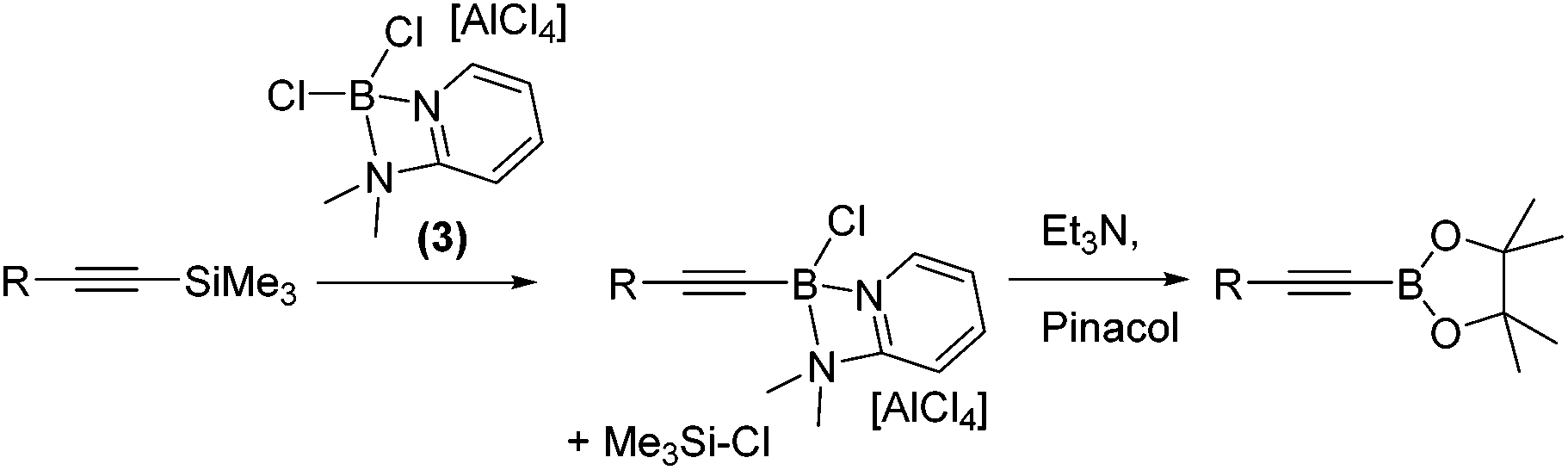 | (1) |
In contrast, the combination of 3 and TMS-ethyne only formed the alkynyl-borocation as a minor species (by NMR spectroscopy). The major soluble product contained a vinylic singlet at 6.65 ppm in the 1H NMR spectrum consistent with alkyne elemento-boration. Crystalline solid spontaneously deposited from CH2Cl2 solutions as the reaction proceeded with a concomitant increase in the quantity of TMSCl and a decrease in the vinylic singlet (by 1H NMR spectroscopy). The amount of precipitate was increased by using an excess of 3 (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio of 3:TMS-ethyne). X-ray diffraction studies on multiple crystals consistently produced poor quality data due to low crystal quality but an unambiguous connectivity map was obtained (Fig. 1, bottom left). This revealed the compound to be the tricationic borate, 4 formed from 1,1-boroamination of TMS-ethyne and desilylboration. Due to the low data quality detailed discussion of structural metrics of 4 is not warranted.
:4 ratio of 3:TMS-ethyne). X-ray diffraction studies on multiple crystals consistently produced poor quality data due to low crystal quality but an unambiguous connectivity map was obtained (Fig. 1, bottom left). This revealed the compound to be the tricationic borate, 4 formed from 1,1-boroamination of TMS-ethyne and desilylboration. Due to the low data quality detailed discussion of structural metrics of 4 is not warranted.
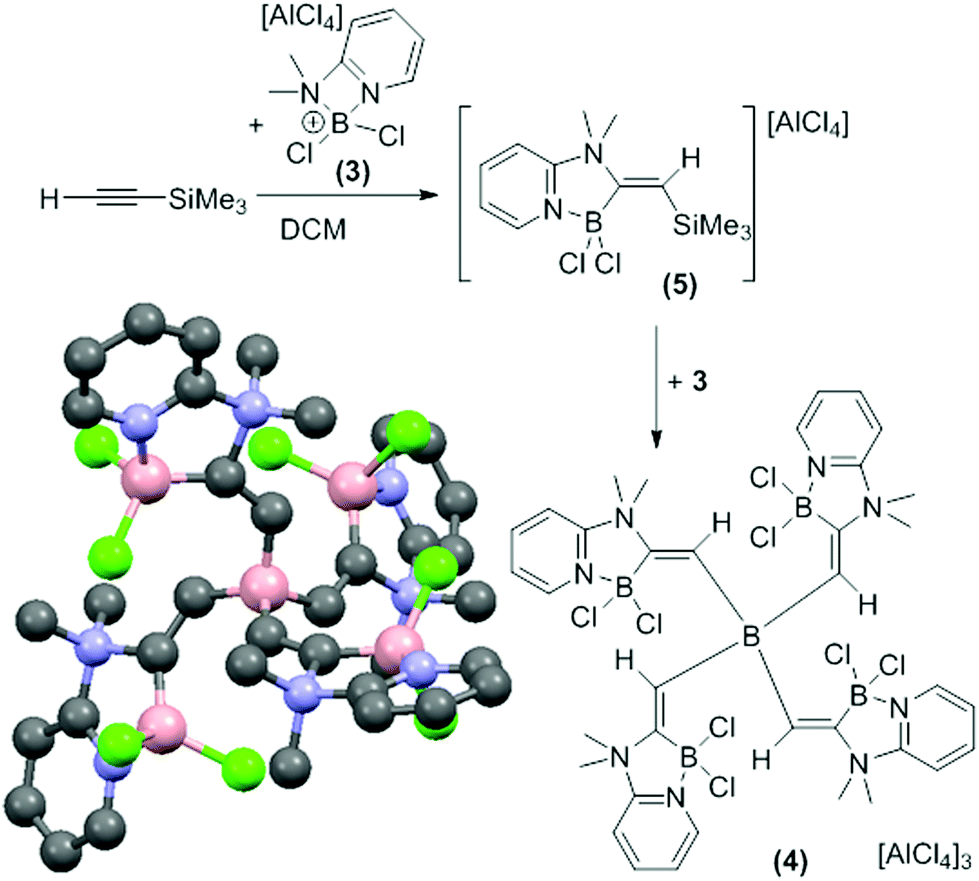 | ||
| Fig. 1 Formation of the tricationic borate 4 by 1,1-boroamination. Bottom left, solid state structure of 4 (anions and hydrogens omitted). | ||
Compound 4 was confirmed as the major component of the CH2Cl2 insoluble material by elemental microanalysis. Furthermore, on dissolution of the crystalline solid in CD3CN two major resonances at +3.9 ppm and −15.4 ppm in the 11B NMR spectrum were observed consistent with four coordinate cationic and anionic boron centres, respectively. The formation of 4 suggested that the CH2Cl2 soluble product formed is 5, the product from 1,1-boroamination of TMS-ethyne prior to intermolecular desilylboration (Fig. 1). Whilst 5 could not be isolated analytically pure (due to contamination with 4, the alkynyl-borocation and [H(2-DMAP)][AlCl4]) multinuclear NMR spectroscopy is fully consistent with this formulation with NOE spectroscopy confirming the regio- and stereo-chemistry and indicating that the desilylboration of 5 to form 4 occurs with retention. The reactivity disparity between TMS-ethyne and other 1-TMS-1-alkynes studied is attributed to a less stabilised vinyl cation formed on interaction of 3 with TMS-ethyne which presumably favours rapid TMS-migration leading to 5 as the initial product and not the alkynyl-borocation from desilylboration. It is noteworthy that the reaction of [Cl2B(2,6-lutidine)][AlCl4], 6, a borocation where the amine does not contain a pendant nucleophile, with 1-TMS-alkynes, including TMS-ethyne, led predominantly to desilylboration in all cases.
Whilst the reactivity of 1-TMS-1-alkynes with BCl3 proceeds by desilylboration15 the analogous reactivity with PhBCl2 has not been explored to the best of our knowledge. To determine if PhBCl2 and 1 react comparably with 1-TMS-1-alkynes equimolar 1-TMS-1-propyne and PhBCl2 were combined in CH2Cl2 at 20 °C. This resulted in a rapid reaction producing a single new product identified by multinuclear NMR spectroscopy as the product from 1,1-carboboration. Post esterification 2a was isolated in a higher yield than when using 1. A substrate scope exploration (Table 2) confirmed that the 1,1-carboboration of 1-TMS-1-alkynes with PhBCl2 consistently proceeds in higher yield than when using 1 and does not require purification by chromatography post esterification. Furthermore, the structure of 2e was also confirmed by a single crystal X-ray diffraction study (Table 2, right). It is noteworthy that trimethyl(4-phenylbut-1-yn-1-yl)silane (entry 7) resulted in only 1,1-carboboration with no 6-endo-dig cyclisation as recently reported for related alkynes and BCl3.16 The facile formation of 2a–2h by 1,1-carboboration represents an alternative to transition metal catalysed borosilyation of alkynes for accessing these versatile intermediates.17 Attempts to extend this reaction to 1-triisopropylsilyl-1-propyne resulted in no reaction, whilst combining PhBCl2 and 1-(PhMe2Si)-1-propyne resulted predominantly in desilylboration products (by observation of PhMe2SiCl by 1H and 29Si NMR spectroscopy) and multiple other currently unidentified products.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Electrophile | t (h) | T (°C) | Compound no. | R | Isolated yield (%) |
| a Isolated yield not obtained due to intractable minor contaminants of (heteroaryl)BPin. | ||||||
| 1 | PhBCl2 | 1 | 20 | 2a | Me | 88 |
| 2 | PhBCl2 | 6 | 20 | 2b | Bu | 79 |
| 3 | PhBCl2 | 24 | 20 | 2c | C(Me)CH2 |
77 |
| 4 | PhBCl2 | 20 | 20 | 2d | Ph | 85 |
| 5 | PhBCl2 | 20 | 20 | 2e | p-Br-C6H4 | 68 |
| 6 | PhBCl2 | 2 | 20 | 2f | iPr | 76 |
| 7 | PhBCl2 | 1 | 20 | 2g | CH2CH2Ph | 37 |
| 8 | PhBCl2 | 1 | 60 | 2h | H | 30 |
| 9 | 7a | 1 | 20 | 2i | Me | —a |
| 10 | 7b | 0.5 | 20 | 2j | Me | —a |
| 11 | 7c | 1 | 20 | 2k | Me | 68 |
| 12 | 7d | 1 | 20 | 2l | Me | 61 |
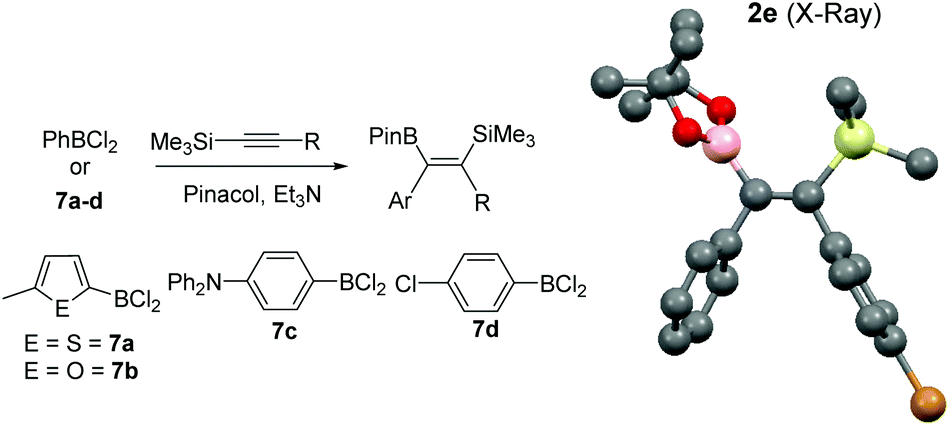
To increase the scope, variation of the aryl substituent on the borane was explored. ArylBCl2 and heteroarylBCl2 species are readily accessible by electrophilic arene borylation.10 Using established methodologies 2-methylthiophene, 2-methylfuran, chlorobenzene and triphenylamine were all borylated to produce the respective (hetero)arylBCl2 compounds 7a–7d (Table 2) in good conversion as determined by multi-nuclear NMR spectroscopy.10a,b Removal of reaction solvent (CH2Cl2 or 1,2-Cl2C6H4) and extraction of 7a–d into hexanes was sufficient to enable subsequent reaction with 1-TMS-1-propyne without any additional purification steps. This led to the formation of the desired 1,1-carboboration products which were esterified to form a single regio- and stereo-isomer of the respective vinyl pinacol boronate esters (entries 9–12). The products derived from carboboration using 7a and 7b were repeatedly contaminated with minor quantities of heteroarylBPin (from esterfication of unreacted 7a and 7b) which in our hands proved challenging to separate from 2i and 2j. The 1,1-carboboration reaction using longer times (3 h) for 7a and 1-TMS-1-propyne led to considerably more complex NMR spectra and intractable products post esterification.
Attempts to extend 1,1-carboboration using 7a–7d to other 1-TMS-1-alkynes, specifically 1-TMS-2-phenylacetylene and trimethyl(3-methylbut-1-yn-1-yl)silane, instead led to desilylboration being the dominant reaction pathway (by 1H, 11B and 29Si NMR spectroscopy). To preclude the disparity between PhBCl2 and the four (hetero)arylBCl2 compounds 7a–d being due to any impurities in commercially sourced PhBCl2 or impurities in (hetero)arylBCl2 synthesised by electrophilic borylation, benzene was borylated in 1,2-Cl2C6H4 using 4-N,N-trimethylaniline, BCl3 and two equivalents of AlCl3 to form PhBCl2.10c This reaction mixture was dried and PhBCl2 extracted into hexane and found to form 2f on addition of trimethyl(3-methylbut-1-yn-1-yl)silane, a substrate that 7a–d react with predominantly by desilylboration. Therefore the greater prevalence for desilylboration using 7a–d is attributed to the modified electrophilicity of the borane and the different migratory propensity of the (hetero)aryl group (relative to phenyl), indicating that the 1,1-carboboration of 1-TMS-1-alkynes using (hetero)arylBCl2 compounds is somewhat limited in scope.
PhBCl2 was effective for the carboboration of silylated allenes with 8a and 8b (Scheme 2) undergoing carboboration with TMS migration producing only a single allylBCl2 product (by multinuclear NMR spectroscopy). With no intermediates observed we attribute the reaction outcome to a 1,1-carboboration followed by a rapid intramolecular sigmatropic 1,3-boron shift to form the more thermodynamically stable less hindered allylBCl2 species.18 This can be subsequently pinacol protected and the resultant boronate esters 9a and 9b isolated. This enables access to complementary boronate ester isomers to that produced by the hydroboration of closely related TMS-allenes where TMS migration does not occur.19
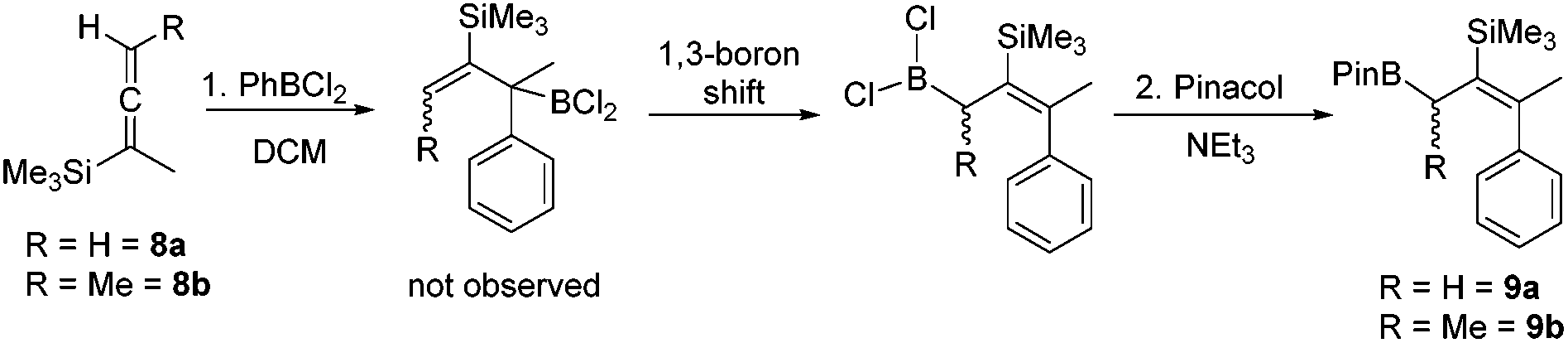 | ||
| Scheme 2 The carboboration of TMS-allenes with PhBCl2. | ||
Boradiene formation using borocations
To explore potential scope expansion further [5-methyl-2-(BCl(2-DMAP))-thiophene][AlCl4], 10, was synthesised by addition of 2-DMAP and AlCl3 to 7a. The addition of 1-TMS-1-propyne to 10 resulted in a slow reaction at 20 °C that after 18 hours produced four new TMS resonances one of which was attributable to the 1,1-carboboration product (by 1H, 11B and 29Si NMR spectroscopy). One of the other new compounds derived from 10/7a could be formed as a greater component of the reaction mixture when an excess of 1-TMS-1-propyne (5 equivalents) was used with heating to 60 °C for 18 h. Post esterification, isolation by column chromatography and analysis by NMR and mass spectroscopy enabled it to be identified as the 2-boradiene 11a, from reaction of 10 with two equivalents of 1-TMS-1-propyne (Scheme 3). Under identical conditions 1 also reacted with excess 1-TMS-1-propyne to produce the 2-boradiene 11b as a minor product. In contrast, heating PhBCl2 with excess (5 equiv.) 1-TMS-1-propyne led to no observable 2-boradiene after esterification with pinacol, instead complex mixtures were produced with significant TMSCl observed in situ indicating desilylboration. Under a range of conditions 11a and 11b were formed only as minor products from 10 and 1 (with a maximum 13 and 15% isolated yield, respectively) with the 1,1-carboboration products, 2i and 2a, being the major species isolated post esterification. 2-Boradienes related to 11x have been previously synthesised by Wrackmeyer and co-workers from the reaction of BEt3 with two equivalents of 1-R3Sn-1-alkynes.20 Precise isomer assignment for 11x was based on 1D and 2D NMR spectroscopy, most notably NOESY indicated a trans-disposition of TMS and PinB moieties in the 2-boradiene. This is in contrast to that in 2x suggesting that 1,1-carboboration is not the first step in 2-boradiene formation, a hypothesis supported by the fact that resonances for the 1,1- carboboration products increase in intensity as the reaction progresses, suggesting it is not an intermediate in diene formation. Instead we propose that 1-TMS-1-propyne is activated to an intermolecular attack by a second equivalent of alkyne by interaction with the borocation and this ultimately leads to the observed 2-boradiene structures. Related borane activation of alkynes towards external π nucleophiles have been reported previously.2b,21 As repeated attempts to crystallise 11a and 11b failed in our hands to support the proposed diene structure, particularly correlating the structure with the multiple NOE interactions observed, the structure of 11b was optimised at the M06-2X/6-311G(d,p)(PCM:DCM) level. This revealed that steric bulk forces a significant dihedral angle in the diene (CC–CC = 60.24°) and thus short distances (<4 Å) were observed in the calculated structure for all the observed NOE interactions, supporting this isomer assignment. Attempts to form other 2-boradienes using different (hetero)arylboronium cations or different TMS-alkynes all led to complex mixtures and lower conversions than that observed for 11a and 11b.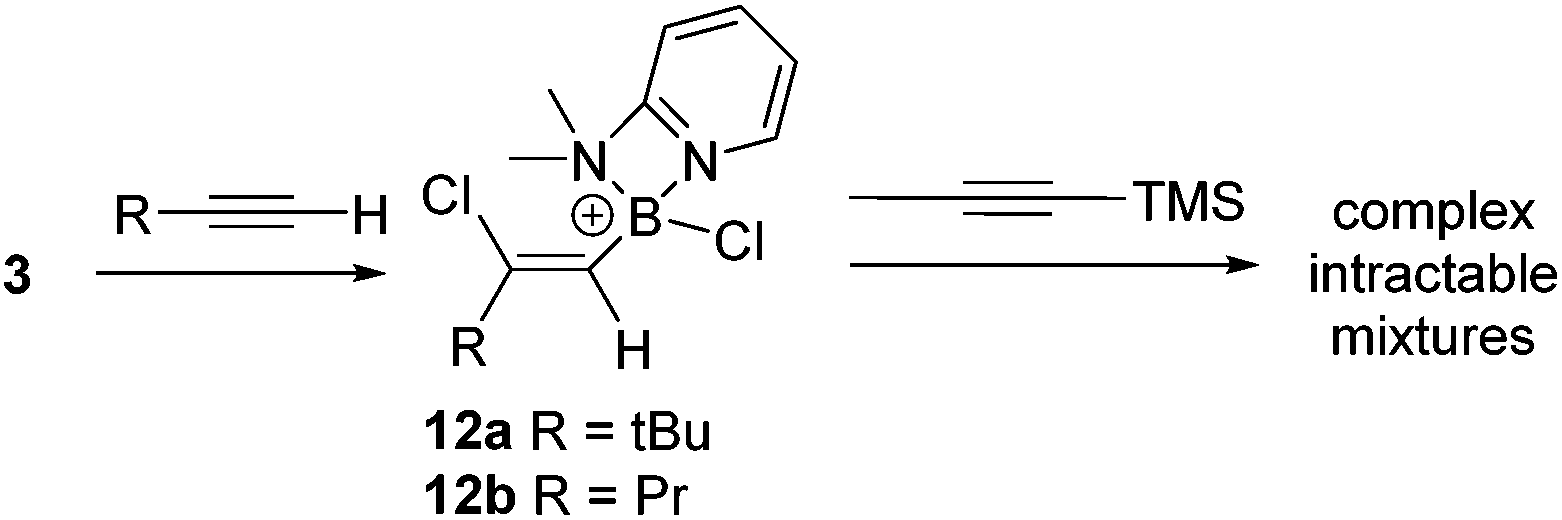 | (2) |
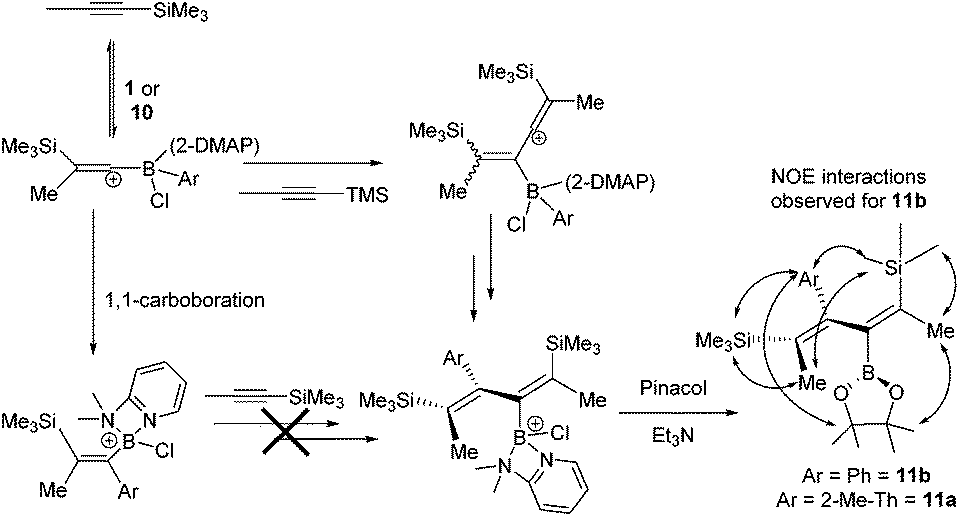 | ||
| Scheme 3 2-Boradiene formation from 1 and 10 and 1-TMS-1-propyne (shown as vinyl cation intermediates, π complexes of {Me3Si}+ are also feasible). | ||
Subsequently, the one pot, two step reaction of 3 with terminal alkynes (proceeding by 1,2-haloboration as previously reported to form 12x, eqn (2))11 followed by addition of 1-TMS-1-propyne was explored as an alternative route to 2-boradienes. The addition of 1-TMS-1-propyne to 12a or 12b gave no reaction (by NMR spectroscopy) at room temperature after 18 hours. When the reaction mixture was heated to 60 °C for 1 h multiple new species were observed in the 1H NMR spectrum, including [(2-DMAP)H]+, as well as four new 29Si resonances (one corresponding to TMSCl) and new 11B resonances at +55 and +66 ppm. Esterification and attempts to purify the resultant complex mixture failed to deliver pure products in our hands. A more electrophilic vinyl-borocation was targeted to enable room temperature reactivity with 1-TMS-1-alkynes and potentially avoid the complex mixtures observed with 12x at 60 °C. Thus the reactivity of [(vinyl)BCl(2,6-lutidine)]+ cations, made via haloboration of alkynes with 6, with 1-TMS-1-alkynes was explored.
In a one pot two step reaction 6 was used to separately haloborate tBu-acetylene and phenylacetylene followed by addition of one equivalent 1-TMS-1-propyne, which did not lead to any significant TMSCl formation at short reactions times (<1 h by NMR spectroscopy) in each case. The initial haloboration step is rapid (complete in <5 minutes with both terminal alkynes) whilst the subsequent reaction with 1-TMS-1-propyne is slower it did proceed to form carboboration products (Scheme 4). Running the reaction for longer times at 20 °C (≥2 h) resulted in significant TMSCl formation, whilst attempts to use greater equivalents of 1-TMS-1-propyne also led to more TMSCl formation; thus optimized conditions of 1.2 equivalents of 1-TMS-1-propyne and a 1 h reaction duration were found to minimise the amount of unreacted haloboration compounds (12a–b) remaining and TMSCl formation. Post esterification the boradiene products 13a–b could be separated from the vinyl-pinacol boronate esters (formed from esterification of unreacted 12a–b) with NMR spectroscopy consistent with a 1-boradiene formulation formed from a 1,2-carboboration reaction (Scheme 4). Notably a 4JHH coupling of 1 Hz is observed between the methyl and the vinyl-H in both 13a and 13b confirming the connectivity as this coupling would not be observed in the 1,1-carboboration products. Whilst 1,2-carboboration is less documented that 1,1-carboboration several recent examples have been reported,22 including using borocations.13,23 The diene structure (Scheme 4) expected from 1,2-carboboration was confirmed by NOESY and in the absence of crystalline material (which was unobtainable in our hands) supported by optimising the structure of 13b at the M06-2X/6-311G(d,p)(PCM:DCM) level. This indicated that 13b is a non-planar diene (CC–CC = 39.93°) and that all observed NOE interactions correspond to calculated H⋯H distances of <4 Å. We attribute the reactivity disparity between 2-DMAP (2-boradienes) and 2,6-lutidine (1-boradienes) borocations to the greater steric demand of 2,6-lutidine which disfavours formation of a more sterically hindered 2-boradiene. This is consistent with calculations on a model complex (at the M06-2X/6-311G(d,p)(PCM:DCM) level) which show that the 2-boradiene B is 5 kcal mol−1 higher in energy than the 1-boradiene isomer A (Scheme 4). Furthermore, in contrast to 1,1-carboboration reactions with BEt3,5 the 1,2-vinylboration to form A is unlikely to be reversible at 20 °C. This is indicated by the conversion of the model compound 12Me (where R = Me) to A being found to be exergonic by 7.7 kcal mol−1, thus the barrier to the reverse process (retro-vinylboration) will be significantly higher than that for the forward reaction (which requires at least 1 h for significant conversion).
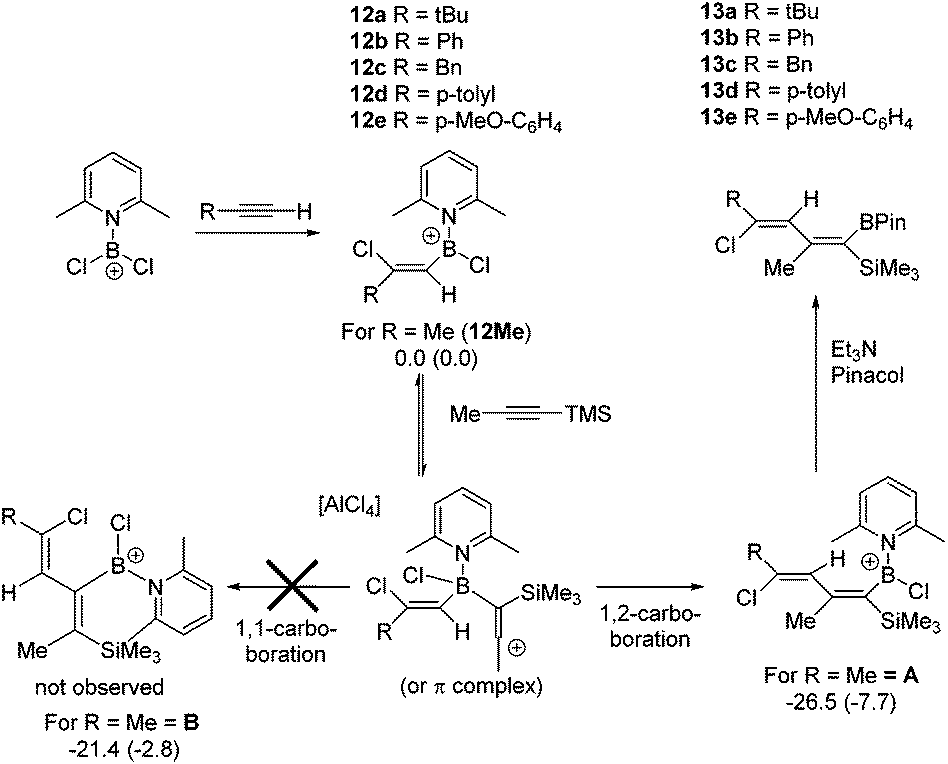 | ||
| Scheme 4 1-Boradiene formation by haloboration and 1,2-carboboration. Electronic energies (Gibbs free energies at 293 K) shown in kcalmol−1 for the model system where R = Me (all energies relative to 12Me and 1-TMS-1-propyne at infinite distance). | ||
As bora-dienes are useful species for a range of subsequent synthetic transformations,24 the broader applicability of this reaction was explored initially looking at other terminal alkynes. 12c–12e were all readily produced by haloboration with 6 and underwent 1,2-carboboration to form 13c–13e, however the isolated yields of 13x are poor to moderate (23–59%), whilst 13d and 13e could not be separated from reaction by-products. The propensity of other 1-TMS-1-alkynes to undergo 1,2-carboboration, specifically 1-TMS-2-phenylacetylene and 1-TMS-1-hexyne, were investigated using 12a, however the reaction was slower (by in situ NMR spectroscopy) and resulted in lower conversions to the desired boradiene and more unidentified by-products. Finally, the use of an internal alkyne, 3-hexyne, was investigated, which as previously reported underwent facile haloboration with 6,11 but subsequent reaction with 1-TMS-1-propyne resulted in a low conversion to the 1-boradiene product which was isolated as the pinacol boronate ester, 13f in only 10% yield. The low conversions with more substituted systems is presumably due to the increased steric crowding resulting in the slower formation of the 1,2-carboboration products and thus increased formation of by-products derived from desilylboration.
Conclusions
The carboboration of 1-TMS-1-alkynes with aryldichloroboranes and aryl-substituted and vinyl-substituted borocations has been demonstrated to yield highly substituted vinyl and dienyl boronate esters post esterification. However, due to carboboration occurring in competition with desilylboration and diene formation, coupled with further reactions proceeding subsequent to the initial carboboration (e.g., desilylboration), complex mixtures are often produced that limit the overall utility of this reaction. Variation in borane and borocation structure is therefore essential to preclude desilylboration to generate more general and higher yielding 1-TMS-1-alkyne carboboration protocols.Experimental
General considerations
All manipulations of air and moisture sensitive species were performed under an atmosphere of argon or nitrogen using standard Schlenk and glovebox techniques. Glassware was dried in a hot oven overnight and heated under vacuum before use. Hexane, ortho-dichlorobenzene, d1-chloroform, d2-dichloromethane, 2,6-lutidine, Et3N and were dried over calcium hydride and distilled under vacuum. Pentane and dichloromethane were dried by passing through an alumina drying column incorporated into an MBraun SPS800 solvent purification system. All solvents were degassed and stored over molecular sieves (3 Å) under an inert atmosphere. Compounds 1 and 3 were synthesised according to the published procedures.11 All other materials were purchased from commercial vendors and used as received. NMR spectra were recorded with a Bruker AV-400 spectrometer (400 MHz 1H; 100 MHz 13C; 128 MHz 11B; 376.50 MHz 19F; 104.3 MHz 27Al; 79.5 MHz 29Si). 1H NMR chemical shifts are reported in ppm relative to protio impurities in the deuterated solvents and 13C NMR using the solvent resonances unless otherwise stated. 11B NMR spectra were referenced to external BF3:Et2O, 19F to Cl3CF, 27Al to Al(NO3)3 in D2O (Al(D2O)63+), 29Si to Si(CH3)4. Resonances for the carbon directly bonded to boron are not observed in the 13C{1H} NMR spectra due to quadrupolar effects. GC-MS analysis was performed on an Agilent Technologies 7890A GC system equipped with an Agilent Technologies 5975C inert XL EI/CI MSD with triple axis detector. The column employed was an Agilent J&W HP-5 ms ((5%-phenyl)-methylpolysiloxane) of dimensions: length, 30 m; internal diameter, 0.250 mm; film, 0.25 μm. Mass spectra were recorded on a Waters QTOF mass spectrometer. X-ray data for compounds 2e and 4 was collected at a temperature of 150 K using Mo-Kα radiation on an Agilent Supernova, equipped with an Oxford Cryosystems Cobra nitrogen flow gas system. Elemental analysis of air sensitive compounds was performed by London Metropolitan University service. Repeated attempts to obtain satisfactory elemental analyses for a range of the pinacol boronate esters repeatedly gave results with low carbon content, even when using V2O5 as an additional oxidant.
General procedures for 1,1-carboboration reactions
:hexane, 1:1) was used to separate the desired product from the by-products.
(E)-(1-Phenyl-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)prop-1-en-2-yl) trimethylsilane (2a). The product was isolated as a yellow oil (Route 1: 24 mg, 63%). (Route 2: 424 mg, 88%).
1H NMR (400 MHz, CDCl3) δ 7.30 (t, 2H, 3J(H,H) = 7.2 Hz), 7.18 (t, 1H, 3J(H,H) = 7.2 Hz), 7.06 (d, 2H, 3J(H,H) = 7.2 Hz), 1.68 (s, 3H), 1.23 (s, 12H), 0.23 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 150.92, 143.54, 128.42, 127.86, 125.55, 83.50, 25.05, 20.58, 0.00; 11B NMR (128.4 MHz, CDCl3): δ 30.2; 29Si NMR (79.5 MHz, CDCl3) δ −4.22 ppm. MS: (M + Na+m/z) calculated for C18H29O2SiBNa = 339.1928 Found = 339.1923.
(E)-(1-Phenyl-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)hex-1-en-2-yl) trimethylsilane (2b). The product was isolated as a yellow oil (Route 1: 27 mg, 61%). (Route 2: 431 mg, 79%).
1H NMR (400 MHz, CDCl3) δ 7.29 (t, 2H), 7.18 (t, 1H), 7.07 (d, 2H), 2.07–2.03 (m, 2H), 1.21 (s, 12H), 1.19–1.10 (m, 4H), 0.72 (t, 2H), 0.26 (9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 155.70, 143.59, 128.16, 127.67, 125.35, 83.42, 33.49, 32.49, 24.90, 22.81, 13.77, 0.81; 11B NMR (128.4 MHz, CDCl3): δ 30.4; 29Si NMR (79.5 MHz, CDCl3) δ −4.60 ppm. MS: (GC, M + H+, m/z) 359.3.
(E)-(3-Methyl-1-phenyl-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)buta-1,3-dien-2-yl)trimethyl silane (2c). The product was isolated as a yellow oil ((Route 1: 23 mg, 55%). (Route 2: 403 mg, 77%).
1H NMR (400 MHz, CDCl3) δ 7.24–7.15 (m, 5H), 4.69 (m, 1H), 4.38 (m, 1H), 1.42 (s, 1H), 1.23 (s, 12H), 0.24 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 158.70, 147.58, 142.92, 134.70, 128.37, 127.19, 125.58, 83.72, 25.00, 24.17, 0.27; 11B NMR (128.4 MHz, CDCl3): δ 31.1; 29Si NMR (79.5 MHz, CDCl3) δ −6.59 ppm. MS: (GC, M + Na+m/z) 365.3.
(E)-(1,2-Diphenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)vinyl)trimethylsilane (2d). The product was isolated as a yellow oil (Route 1: 29 mg, 65%). (Route 2: 489 mg, 85%).
1H NMR (400 MHz, CDCl3) δ 7.06–7.00 (m, 4H), 6.96–6.93 (m, 4H), 6.75–6.73 (m, 2H), 1.29 (s, 12H), 0.17 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 156.92, 144.48, 142.51, 128.84, 128.10, 127.19, 127.11, 125.22, 124.57, 83.91, 25.11, 0.36; 11B NMR (128.4 MHz, CDCl3): δ 30.6; 29Si NMR (79.5 MHz, CDCl3) δ −5.00 ppm. MS: (GC m/z) 378.2 MS: (M + Na+m/z) 401.4.
(E)-(1-(4-Bromophenyl)-2-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)vinyl)trimethylsilane (2e). The product was isolated as a yellow oil (Route 1: 31 mg, 56%). (Route 2: 102 mg, 68%, reaction performed on a 1/4 scale of the general procedure).
1H NMR (400 MHz, CDCl3) δ 7.17 (d, 2H, 3J(H,H) = 8.2 Hz), 7.07–6.91 (m, 5H), 6.63 (d, 2H, 3J(H,H) = 8.2 Hz), 1.28 (s, 12H), 0.17 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 155.55, 143.50, 142.16, 130.36, 129.80, 128.68, 128.08, 127.33, 125.47, 83.98, 25.08, 0.34; 11B NMR (128.4 MHz, CDCl3): δ 30.0; 29Si NMR (79.5 MHz, CDCl3) δ −4.92 ppm. MS: (GC, M + H+m/z) 458.0.
(E)-(3-Methyl-1-phenyl-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)but-1-en-2-yl)trimethylsilane (2f). The product was isolated as orange crystals (100 mg, 76%, reaction performed on a 1/4 scale of the general procedure.
1H NMR (400 MHz, CDCl3) δ 7.13 (t, 2H, 3J(H,H) = 7.0 Hz), 7.03 (t, 1H, 3J(H,H) = 7.0 Hz), 6.94 (d, 2H, 3J(H,H) = 7.0 Hz), 2.57 (septet, 1H, 3J(H,H) = 7.0 Hz), 1.02 (s, 12H), 0.81 (d, 6H, 3J(H,H) = 7.0 Hz), 0.16 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 157.97, 143.70, 128.05, 127.89, 125.57, 83.56, 33.79, 24.92, 22.28, 2.67; 11B NMR (128.4 MHz, CDCl3): δ 30.5; 29Si NMR (79.5 MHz, CDCl3) δ −5.83 ppm. MS: (GC, M + Na+m/z) 367.4.
(E)-(1,4-Diphenyl-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)but-1-en-2-yl)trimethylsilane (2g). Trimethyl(4-phenylbut-1-yn-1-yl)silane (45 μL, 0.20 mmol, 1.0 eq.) was dissolved in DCM (0.6 ml) in a J. Youngs tube, PhBCl2 (29 μL, 0.22 mmol, 1.1 eq.) was then added and the reaction sealed and rotated. After 1 h, a cooled solution of pinacol (26 mg, 0.22 mmol, 1.1 eq.) and excess of triethylamine were added to the reaction mixture. The reaction mixture was then concentrated under reduced pressure, the resultant crude was dissolved in pentane, filtered and concentrated. The residue was then purified by flash chromatography (petroleum ether
:DCM 70:30), affording 2g (30 mg, 37%) as a white solid.
1H NMR (400 MHz, CDCl3) δ 7.31 (t, J = 7.6 Hz, 2H), 7.22 (d, J = 7.34 Hz, 1H), 7.13–7.18 (m, 2H), 7.07–7.12 (m, 3H), 6.85 (d, J = 7.0 Hz, 2H), 2.40–2.47 (m, 2H), 2.28–2.36 (m, 2H), 1.22 (s, 12H), 0.31 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3): δ 154.4, 143.2, 142.4, 128.1, 128.0, 127.9, 127.8, 125.6, 125.5, 83.5, 36.6, 36.2, 24.9, 0.7; 11B NMR (128.4 MHz, CDCl3): δ 30.9 (s) ppm; 29Si NMR (79.5 MHz, CDCl3): δ −4.4 (s) ppm; MS (GC, [M − CH3]+, m/z) 391.3; accurate mass: ([M + H]+ 407.2583).
(Z)-Trimethyl(2-phenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)vinyl)silane (2h). Trimethylsilylacetylene (48 μL, 0.33 mmol, 1.0 eq.) was dissolved in DCM in a J. Youngs tube, PhBCl2 (49 μL, 0.37 mmol, 1.1 eq.) was then added and the tube sealed. After 1 h at 60 °C a cooled solution of pinacol (44 mg, 0.37 mmol, 1.1 eq.) and excess of triethylamine were added to the reaction mixture. The reaction mixture was then concentrated under reduced pressure and the crude was dissolved in pentane, filtered and concentrated. The residue was then purified by flash chromatography (petroleum ether
:DCM 70:30), affording 2h (30 mg, 30%) as a colourless oil.
1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 8.1 Hz, 2H), 7.30 (t, J = 7.8 Hz, 2H), 7.21–7.26 (m, 1H), 6.74 (s, 1H), 1.34 (s, 12H), 0.23 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3): δ 150.6, 145.5, 128.0, 126.9, 126.7, 83.8, 25.1, 0.3; 11B NMR (128 MHz, CDCl3): δ 30.2 (s) ppm; 29Si NMR (79 MHz, CDCl3): δ −9.2 (s) ppm; MS (GC, [M − CH3]+, m/z) 287.1; accurate mass: ([M − CH3]+ 287.1635).
(E)-(1-(5-Methylthiophen-2-yl)-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)prop-1-en-2-yl)trimethylsilane (2i). [BCl2(2-DMAP)][AlCl4] (100 mg, 2.7 mmol, 1 eq.) was combined with 2-methylthiophene (26 μl, 2.7 mmol, 1 eq.) in DCM (0.5 ml) in a J. Youngs NMR tube which was then sealed and heated at 60 °C for 1 hour. NMR spectroscopy confirmed formation of 2-methylthiophene-BCl2 which was extracted into hexane (10 ml). To this, 1-TMS-1-propyne (80 μl, 5.4 mmol, 2 eq.) was added with the reaction mixture turning deep orange. The reaction mixture was esterified after 1 hour by addition of excess Et3N (0.1 ml) and pinacol (96 mg, 3 eq.). The crude product was extracted into pentane (20 ml) and filtered through a 1 inch plug of silica affording the desired product contaminated with 2-methyl-5-BPin-thiophene. The data below are for 2i with resonances for the minor by-product omitted.
1H NMR (400 MHz, CDCl3) δ 6.63 (m, 1H), 6.57 (d, 1H, 3J(H,H) = 7.1 Hz), 2.46 (s, 3H), 1.93 (s, 3H), 1.29 (s, 12H), 0.22 (s, 9H); 13C NMR (100.06 MHz, CDCl3) δ 148.27, 139.11, 135.82, 126.00, 122.91, 83.22, 25.01, 20.37, 15.86, 0.02 ppm; 11B NMR (128.4 MHz, CDCl3): δ 30.0; 29Si NMR (79.5 MHz, CDCl3) δ −3.92 ppm. MS (GC, M+, m/z): 336.4.
(E)-(1-(5-Methylfuran-2-yl)-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)prop-1-en-2-yl)trimethylsilane (2j). [BCl2(2-DMAP)][AlCl4] (100 mg, 2.7 mmol, 1 eq.) was combined with 2-methylfuran (24 μl, 2.7 mmol, 1 eq.) in DCM (0.5 ml) in a J. Youngs NMR tube. NMR spectroscopy confirmed formation of 2-methylfuran-BCl2 in <5 min which was then extracted into hexane (10 ml). To this 1-TMS-1-propyne (40 μl, 2.7 mmol, 1 eq.) was added with the reaction turning deep orange. The reaction mixture was esterified after 30 minutes with excess Et3N (0.1 ml) and pinacol (96 mg, 3 eq.). The crude product was extracted into pentane and filtered through a 1 inch plug of silica affording the desired product contaminated with 2-methyl-5-BPin-furan. The data below are for 2j with resonances for the minor by-product omitted.
1H NMR (400 MHz, CDCl3) δ 6.17 (d, 1H, 3J(H,H) = 3.2 Hz), 5.99 (m, 1H), 2.28 (s, 3H), 2.02 (s, 3H), 1.37 (s, 12H), 0.21 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 155.78, 153.22, 152.09, 109.07, 102.72, 82.98, 25.03, 21.25, 14.76, 0.98 ppm; 11B NMR (128.4 MHz, CDCl3): δ 30.2; 29Si NMR (79.5 MHz, CDCl3) δ −3.92 ppm. MS (GC, M+, m/z): 320.3.
(E)-N,N-Diphenyl-4-(1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-(trimethylsilyl)prop-1-en-1-yl)aniline (2k). [2,6-lutBCl2][AlCl4] (80 mg, 2.2 mmol, 1 eq.) was generated in situ (from lut-BCl3 and AlCl3) and combined with triphenylamine (55 mg, 2.2 mmol, 1 eq.) in DCM (0.5 ml) in a J. Youngs NMR tube which was then sealed and rotated at room temperature for 2 hours resulting in the solution turning brown. NMR spectroscopy confirmed formation of triphenylamine-BCl2, which was extracted into hexane (10 ml). To this 1-TMS-1-propyne (66 μl, 4.5 mmol, 2 eq.) was added. After 30 minutes the reaction mixture was esterified with excess Et3N (0.1 ml) and pinacol (78 mg, 3 eq.). The crude product was extracted into pentane (20 ml) and filtered through a 1 inch plug of silica afforded the product. (74 mg, 68%).
1H NMR (400 MHz, CDCl3) δ 7.07–6.77 (m, 14H), 1.58 (s, 3H), 1.07 (s, 12H), 0.05 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 150.41, 147.97, 145.25, 137.85, 129.38, 129.08, 123.97, 123.69, 122.35, 83.49, 25.04, 20.63, −0.03; 11B NMR (128.4 MHz, CDCl3): δ 30.3; 29Si NMR (MHz, CDCl3) δ −4.29 ppm. MS: (GC, M+, m/z) 483.3.
(E)-(1-(4-Chlorophenyl)-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)prop-1-en-2-yl)trimethylsilane (2l). DMT-BCl3 (100 mg, 1 eq.) and AlCl3 (111 mg, 2.1 eq.) were combined in dichlorobenzene (0.5 ml) in a J. Youngs NMR tube which was then sealed and heated to 100 °C for 18 h. NMR spectroscopy confirmed formation of 4-chloro-phenyl-BCl2, which was extracted into hexane (10 ml). To this hexane solution 1-TMS-1-propyne (117 μl, 2 eq.) was added turning the solution brown. After 40 minutes the reaction mixture was then esterified with excess Et3N (0.1 ml) and pinacol (140 mg, 3 eq.). The crude product was extracted into pentane (20 ml) and filtered through a 1 inch plug of silica affording the product. (85 mg, 61%).
1H NMR (400 MHz, CDCl3) δ 7.03 (d, 2H, 3J(H,H) = 8.2 Hz), 6.75 (d, 2H, 3J(H,H) = 8.2 Hz), 1.44 (s, 3H), 1.00 (s, 12H), 0.00 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 152.57, 136.11, 131.30, 129.82, 128.03, 83.59, 24.99, 20.77, 0.00; 11B NMR (128.4 MHz, CDCl3): δ 30.0; 29Si NMR (79.5 MHz, CDCl3) δ −4.04 ppm. MS: (GC, M+, m/z) 350.1.
Compound 4. A 5
:4 ratio of [BCl2(2-DMAP)][AlCl4] (100 mg, 0.27 mmol, 5 eq.), to trimethylsilylacetylene (31 μl, 0.22 mmol, 4 eq.) was dissolved in CH2Cl2 in a J. Young's NMR tube. On standing a crystalline solid precipitated out of solution. Removal of the solvent and washing with pentane allowed isolation of the crystals 21 mg, 27% (based on boron content).
1H NMR (400 MHz, CD3CN, −40 °C): δ 8.91 (d, 1H), 8.71 (t, 1H), 8.28 (d, 1H), 8.16 (t, 1H), 7.89 (s, 1H), 3.78 (s, 3H), 3.45 (s, 3H); 11B NMR (128.4 MHz, CD3CN): δ 3.90, −15.44; 13C{1H} NMR (100.6 MHz, CD3CN): δ 154.09, 150.15, 143.80, 130.47, 119.85 ppm. Expected (%) for C36H44Al3B5N8Cl20 C = 30.18 H = 3.10 N = 7.82, Found (%) C = 29.94, H = 2.97, N = 7.65.
Compound 5. To a 1
:1 suspension of [BCl2(2-DMAP)][AlCl4] (100 mg, 0.27 mmol, 1 eq.) in CH2Cl2 in a J. Young's NMR tube was added trimethylsilylacetylene (38 μl, 0.27 mmol, 1 eq.) and tube was sealed and rotated at room temperature for 18 hours. The solvent was removed under reduced pressure, and the product redissolved in DCM.
1H NMR (400 MHz, CD2Cl2): δ 8.99 (d, 1H), 8.88 (t, 1H), 8.56 (d, 1H), 8.25 (t, 1H), 6.65 (s, 1H), 3.80 (s, 6H), 0.38 (s, 9H); δ11B NMR (128.4 MHz, CD2Cl2): δ 2.5 (s); 29Si NMR (79.5 MHz, CH2Cl2) δ −3.6 ppm. Due to difficulties in purifying this compound, the 13C NMR spectra were complicated by numerous minor species. Accurate elemental analysis could not be obtained due to the impure nature of the material from each attempt to isolate this product.
(E)-(3-phenyl-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)but-2-en-2-yl)trimethylsilane (9a). 3-(Trimethylsilyl)-1,2-butadiene (25 μL, 0.15 mmol, 1.0 eq.) was dissolved in DCM in a J. Youngs tube and PhBCl2 (22 μL, 0.16 mmol, 1.1 eq.) was added. After 1 h a cooled solution of pinacol (19 mg, 0.16 mmol, 1.1 eq.) and excess triethylamine were added to the reaction mixture. The reaction mixture was then dried under reduced pressure and the crude was dissolved in pentane, filtered and concentrated. The residue was purified by flash chromatography (petroleum ether
:DCM 60:40), affording 9a (21 mg, 43%) as a white solid.
1H NMR (400 MHz, CDCl3) δ 7.29 (t, J = 7.2 Hz, 2H), 7.19 (t, J = 7.4 Hz, 1H), 7.14 (d, J = 7.0 Hz, 2H), 2.10 (s, 3H), 1.57 (s, 2H), 1.23 (s, 12H), 0.23 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3): δ 146.0, 145.1, 131.3, 128.0, 127.6, 125.9, 82.9, 25.3, 24.8, 0.4; 11B NMR (128 MHz, CDCl3): δ 33.7 (s) ppm; 29Si NMR (79.5 MHz, CDCl3): δ −5.8 (s) ppm. MS: (GC, M+, m/z) 330.2, accurate mass: [M]+ 330.2181.
(E)-(2-Phenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pent-2-en-3-yl)trimethylsilane (9b). 2-(Trimethylsilyl)-2,3-pentadiene (25 μL, 0.13 mmol, 1.0 eq.) was dissolved in DCM in a J. Youngs tube and PhBCl2 (20 μL, 0.14 mmol, 1.1 eq.) was added. The reaction mixture was heated at 60 °C and after 24 h a cooled solution of pinacol (17 mg, 0.14 mmol, 1.1 eq.) and excess triethylamine were added to the reaction mixture. The reaction mixture was then dried under reduced pressure and the crude was dissolved in pentane, filtered and concentrated. The residue was purified by flash chromatography (petroleum ether
:DCM 60:40), affording 9b (10 mg, 22%) as a colourless oil.
1H NMR (400 MHz, CDCl3) δ 7.29 (t, J = 7.2 Hz, 2H), 7.20 (t, J = 7.3 Hz, 1H), 7.14 (d, J = 6.9 Hz, 2H), 2.08 (s, 3H), 2.04 (q, J = 7.5 Hz, 1H), 1.22 (s, 12H), 0.92 (d, J = 7.5 Hz, 3H), 0.26 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3): δ 146.4, 145.8, 138.4, 128.0, 127.8, 125.8, 82.9, 25.8, 25.3, 24.7, 16.3, 1.6; 11B NMR (128 MHz, CDCl3): δ 34.5 (s) ppm; 29Si NMR (79 MHz, CDCl3): δ −6.6 (s) ppm. MS: (GC, [M − CH3]+, m/z) 329.2, accurate mass: [M]+ 344.2337.
((2Z,4Z)-3-(5-Methylthiophen-2-yl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)hexa-2,4-diene-2,5-diyl)bis(trimethylsilane) (11a). To a suspension of [Cl2B(2-DMAP)][AlCl4] (50 mg, 0.14 mmol, 1 eq.) in DCM (0.5 ml) in a J. Youngs NMR tube was added 2-methylthiophene (14 μl 0.15 mmol, 1.1 eq.). The reaction was heated at 60 °C for 1 h when borylation was confirmed by NMR spectroscopy. Sequential addition of equimolar 2-DMAP and AlCl3 then generated [ClB2-methylthiophene(2-DMAP)][AlCl4]. To this 1-TMS-1-propyne (90 μl, 0.6 mmol, 5 eq.) was addedand the tube was sealed and heated to 60 °C. After 18 h an excess of triethylamine (0.1 ml) and pinacol (30 mg, 0.24 mmol, 2 eq.) were added and the solvent was removed under reduced pressure. Pentane (20 ml) was used to extract the product which was then passed through a 1 inch plug of silica. Column chromatography (DCM
:hexane, 1:1) was used to separate the desired diene from the 1,1-carboboration product. The product was isolated as a yellow oil (8 mg, 22%).
1H NMR (400 MHz, CDCl3) δ 6.59 (d, 1H, 3J(H,H) = 3.5 Hz), 6.51 (m, 1H), 2.42 (s, 3H), 1.96 (s, 3H), 1.81 (s, 3H), 1.20 (s, 6H), 1.17 (s, 6H), 0.08 (s, 9H), −0.03 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 147.42, 146.02, 144.67, 139.85, 135.77, 126.83, 123.83, 83.06, 24.65, 24.59, 22.23, 20.49, 15.44, −0.13, −0.54; 11B NMR (128.4 MHz, CDCl3): δ 30.0; 29Si NMR (79.5 MHz, CDCl3) δ −5.98 ppm. MS: (GC, M+, m/z) 448.6 g mol−1.
((2Z,4Z)-3-Phenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)hexa-2,4-diene-2,5-diyl)bis(trimethylsilane) (11b). To a suspension of [PhBCl(2-DMAP)][AlCl4] (50 mg, 0.12 mmol, 1 eq.) in DCM (0.5 ml) in a J. Youngs NMR tube was added 1-TMS-1-propyne (90 μl, 0.6 mmol, 5 eq.). The tube was sealed and then heated to 60 °C. After 18 h an excess of triethylamine (0.1 ml) and pinacol (30 mg, 0.24 mmol, 2 eq.) were added and the solvent was removed under reduced pressure leaving a yellow/orange oil. Pentane (20 ml) was used to extract the product which was then passed through a 1 inch plug of silica. Column chromatography (DCM
:hexane, 1:1) was used to separate the desired diene from the 1,1-carboboration product. The product was isolated as a yellow oil (8 mg, 15%).
1H NMR (400 MHz, CDCl3) δ 7.22–7.20 (m, 5H), 1.98 (s, 3H), 1.85 (s, 3H), 1.16 (s, 6H), 1.11 (s, 6H), 0.09 (s, 9H), −0.15 (s, 9H) ppm; 13C NMR (100.06 MHz, CDCl3) δ 144.00, 133.75, 130.67, 129.96, 127.37, 126.72, 125.59, 83.08, 24.72, 22.40, 20.86, 0.01 (TMS resonances coincident); 11B NMR (128.4 MHz, CDCl3): δ 29.6; 29Si NMR (MHz, CDCl3) δ −6.00 ppm. MS: (m/z): Calculated [M]+ = 429.2811. Measured = 429.2812.
General procedure for vinylboration with 6
LutBCl3 (50 mg, 0.22 mmol) was suspended in anhydrous CH2Cl2 (5 ml) in a J. Young's NMR tube and AlCl3 (30 mg, 0.22 mmol) added causing dissolution to form a yellow solution. To this the appropriate alkyne (0.22 mmol) was added. The reaction mixture was then sealed and rotated at room temperature. After 10 min 1-TMS-1-propyne (40 μl, 0.26 mmol) was added and the tube resealed and rotated for a further 45 minutes. Then the solution was esterified with excess triethylamine (0.5 ml) and pinacol (2 eq.). The solvent was removed under reduced pressure, leaving a yellow oil. Hexane (30 ml) was used to extract the product, which was filtered. This left a mixture of the desired diene and the haloboration derived vinyl by-product used to determine NMR conversion before purification by column chromatography.:1 hexane:DCM eluent and isolated as a yellow oil (18 mg, 23%).
1H NMR (400 MHz, CDCl3) δ 6.24 (q, 1H, 4J (H,H) = 1 Hz), 1.78 (d, 3H, 4J (H,H) = 1 Hz), 1.29 (s, 12H), 1.22 (s, 9H), 0.19 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 154.05, 144.78, 123.72, 83.26, 38.72, 28.94, 25.36, 20.73, 0.28; 11B NMR (128.4 MHz, CDCl3): δ 29.41; 29Si NMR (79.5 MHz, CDCl3) δ −4.64 ppm. MS: (GC, M+m/z) 356.3 g mol−1.
:1 hexane:DCM eluent and isolated as a yellow oil (49 mg, 59%).
1H NMR (400 MHz, CDCl3) δ 7.69 (m, 2H), 7.38–7.30 (m, 3H), 6.94 (q, 1H, 4J (H,H) = 1.2 Hz), 1.91 (d, 3H, 4J (H,H) = 1.2 Hz), 1.33 (s, 12H), 0.25 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 155.01, 147.37, 136.44, 129.00, 128.31, 127.32, 120.10, 84.07, 24.25, 22.87, 0.41; 11B NMR (128.4 MHz, CDCl3): δ 29.16 29Si NMR (79.5 MHz, CDCl3) δ −4.18 ppm. MS: (GC, M+, m/z) 376.2.
:1 hexane:DCM eluent and isolated as a yellow oil (27 mg, 31%).
1H NMR (400 MHz, CDCl3) δ 7.33–7.23 (m, 5H), 6.37 (q, 1H, 4J (H,H) = 1 Hz), 3.74 (s, 2H), 1.84 (d, 3H, 4J (H,H) = 1 Hz), 1.30 (s, 12H), 0.20 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 156.72, 138.00, 132.80, 129.37, 128.85, 128.28, 126.56, 83.41, 45.64, 25.32, 21.43, 0.36; 11B NMR (128.4 MHz, CDCl3): δ 29.5 ppm; 29Si NMR (79.5 MHz, CDCl3) δ −4.40 ppm. MS: (GC, M+, m/z) 390.2.
1H NMR (400 MHz, CDCl3) δ 7.42–7.38 (m, 2H), 7.01–6.99 (m, 2H), 6.72 (m, 1H), 1.72 (m, 3H), 1.21 (s, 3H), 1.15 (s, 12H), 0.07 (s, 9H); 11B NMR (128.4 MHz, CDCl3): δ 29.5; 29Si NMR (79.5 MHz, CDCl3) δ −4.24 ppm.
1H NMR (400 MHz, CDCl3) δ 7.61–7.59 (m, 2H), 6.89–6.87 (m, 2H), 6.81 (m, 1H), 3.83 (s, 3H), 1.88 (m, 3H), 1.32 (s, 12H), 0.23 (s, 9H); 11B NMR (128.4 MHz, CDCl3): δ 30.0; 29Si NMR (79.5 MHz, CDCl3) δ −4.28 ppm.
:1 hexane:DCM eluent and isolated as a yellow oil (8 mg, 10%).
1H NMR (400 MHz, CDCl3) δ 2.50–2.39 (m, 2H), 2.26–2.16 (m, 2H), 1.74 (s, 3H), 1.24 (s, 12H), 1.14 (t, 3H), 0.95 (t, 3H), 0.19 (s, 9H); 13C{1H} NMR (100.06 MHz, CDCl3) δ 153.05, 139.87, 128.54, 82.65, 27.98, 26.10, 24.62, 24.39, 20.72, 12.65, 12.30; 11B NMR (128.4 MHz, CDCl3): δ 29.30; 29Si NMR (79.5 MHz, CDCl3) δ −4.75 ppm.
Acknowledgements
The research leading to these results has received funding from the University of Manchester, the EPSRC (EP/J000973/1 and EP/K03099X/1) and the European Research Council (FP/2007–2013/ERC Grant Agreement 305868). MJI acknowledges the Royal Society (for the award of a University Research Fellowship) The authors would also like to acknowledge the use of the EPSRC UK National Service for Computational Chemistry Software (NSCCS) at Imperial College London in carrying out this work.Notes and references
- For initial reports on 1,1-carboboration using fluorinated boranes see: (a) C. Chen, F. Eweiner, B. Wibbeling, R. Fröhlich, S. Senda, Y. Ohki, K. Tatsumi, S. Grimme, G. Kehr and G. Erker, Chem. – Asian J., 2010, 4711 Search PubMed; (b) C. Jiang, O. Blacque and H. Berke, Organometallics, 2010, 29, 125 CrossRef CAS; (c) C. Fan, W. E. Piers, M. Parvez and R. McDonald, Organometallics, 2010, 29, 5132 CrossRef CAS; (d) C. Chen, G. Kehr, R. Fröhlich and G. Erker, J. Am. Chem. Soc., 2010, 132, 13594 CrossRef CAS PubMed.
- For recent reviews see: (a) G. Kehr and G. Erker, Chem. Commun., 2012, 48, 1839 RSC; (b) R. Melen, Chem. Commun., 2014, 50, 1161 RSC.
- For a recent synthetic use of 1,1-carboboration for benzannulation see: (a) R. Liedtke, F. Tenberge, C. G. Daniliuc, G. Kehr and G. Erker, J. Org. Chem., 2015, 80, 2240 CrossRef CAS PubMed.
- C. Chen, T. Voss, R. Fröhlich, G. Kehr and G. Erker, Org. Lett., 2011, 13, 62 CrossRef CAS PubMed.
- (a) B. Wrackmeyer, Coord. Chem. Rev., 1995, 145, 125 CAS; (b) B. Wrackmeyer, Heteroat. Chem., 2006, 17, 188 CrossRef CAS.
- For select reactions of BPh3 in 1,1-carboborations: With 1-TMS-1-alkynes see: (a) R. Köster, G. Seidel and B. Wrackmeyer, Chem. Ber., 1989, 122, 1825 CrossRef. With stannylated alkynes see: (b) B. Wrackmeyer, O. L. Tok and W. Milius, Z. Naturforsch., B: Chem. Sci., 2007, 62, 1509 CAS; (c) B. Wrackmeyer, P. Thoma, S. Marx, T. Bauer and R. Kempe, Eur. J. Inorg. Chem., 2014, 2103 CrossRef CAS. With Te-alkynes see: (d) F. A. Tsao, A. J. Lough and D. W. Stephan, Chem. Commun., 2015, 51, 4287 RSC; (e) F. A. Tsao and D. W. Stephan, Dalton Trans., 2015, 44, 71 RSC.
- For an example with PhB(C6F5)2: R. Liedtke, M. Harhausen, R. Fröhlich, G. Kehr and G. Erker, Org. Lett., 2012, 14, 1448 CrossRef CAS PubMed.
- R.-J. Binnewirtz, H. Klingenberger, R. Welte and P. Paetzold, Chem. Ber., 1983, 116, 1271 CrossRef CAS.
- M. Lappert and B. Prokai, J. Organomet. Chem., 1963, 1, 384 CrossRef.
- (a) A. Del Grosso, M. D. Helm, S. A. Solomon, D. Caras-Quintero and M. J. Ingleson, Chem. Commun., 2011, 47, 12459 RSC; (b) V. Bagutski, A. Del Grosso, J. Ayuso-Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474 CrossRef CAS PubMed; (c) A. Del Grosso, J. Ayuso Carrillo and M. J. Ingleson, Chem. Commun., 2015, 51, 2878 RSC.
- J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon and M. J. Ingleson, Angew. Chem., Int. Ed., 2013, 52, 7518 CrossRef CAS PubMed.
- For synthetic applications of boronic acid derivatives see: Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine, ed. D. G. Hall, Wiley-VCH, Weinheim, New York, 2011 Search PubMed.
- I. A. Cade and M. J. Ingleson, Chem. – Eur. J., 2014, 20, 12874 CrossRef CAS PubMed.
- A. Boussonniere, X. Pan, S. J. Geib and D. P. Curran, Organometallics, 2013, 32, 7445 CrossRef CAS.
- D. A. Singleton and S.-W. Leung, J. Organomet. Chem., 1997, 544, 157 CrossRef CAS.
- A. J. Warner, J. R. Lawson, V. Fasano and M. J. Ingleson, Angew. Chem., Int. Ed., 2015, 54, 11245 CrossRef CAS PubMed.
- For metal catalysed alkyne borosilylation see: (a) T. Ohmura, K. Oshima, H. Taniguchi and M. Suginiome, J. Am. Chem. Soc., 2010, 132, 12194 CrossRef CAS PubMed; (b) J. Jiao, K. Hyodo, H. Hu, K. Nakajima and Y. Nishihara, J. Org. Chem., 2014, 79, 285 CrossRef CAS PubMed and references therein.
- A related 1,1-carboboration followed by a 1,3-boron shift has been observed previously with B(C6F5)3: M. M. Hansmann, R. L. Melen, F. Rominger, A. S. K. Hashmi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 777 CrossRef CAS PubMed.
- K. K. Wang, Y. G. Gu and C. Liu, J. Am. Chem. Soc., 1990, 112, 4424 CrossRef CAS.
- B. Wrackmeyer and R. Zentgraf, J. Chem. Soc., Chem. Commun., 1978, 402 RSC.
- (a) M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6422 CrossRef CAS; (b) B.-H. Xu, G. Kehr, R. Fröhlich and G. Erker, Organometallics, 2011, 30, 5080 CrossRef CAS.
- S. Roscales and A. G. Csaky, Org. Lett., 2015, 17, 1605 CrossRef CAS PubMed.
- M. Devillard, R. Brousses, K. Miqueu, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5722 CrossRef CAS PubMed.
- L. Eberlin, F. Tripoteau, F. Carreaux, A. Whiting and B. Carboni, Beilstein J. Org. Chem., 2014, 10, 237 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra, coordinates for calculated structures and crystallographic data. CCDC 1416375 and 1416376. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03003j |
| This journal is © The Royal Society of Chemistry 2016 |