DOI:
10.1039/C5DT02994E
(Paper)
Dalton Trans., 2015,
44, 17189-17200
Syntheses and properties of phosphine-substituted ruthenium(II) polypyridine complexes with nitrogen oxides†
Received
4th August 2015
, Accepted 27th August 2015
First published on 28th August 2015
Abstract
Four novel phosphine-substituted ruthenium(II) polypyridine complexes with nitrogen oxides—trans(P,NO2)-[Ru(trpy)(Pqn)(NO2)]PF6 (trans-NO2), cis(P,NO2)-[Ru(trpy)(Pqn)(NO2)]PF6 (cis-NO2), [Ru(trpy)(dppbz)(NO2)]PF6 (PP-NO2), and cis(P,NO)-[Ru(trpy)(Pqn)(NO)](PF6)3 (cis-NO)—were synthesised (trpy = 2,2′:6′,2′′-terpyridine, Pqn = 8-(diphenylphosphanyl)quinoline, and dppbz = 1,2-bis(diphenylphosphanyl)benzene). The influence of the number and position of the phosphine group(s) on the electronic structure of these complexes was investigated using single-crystal X-ray structural analysis, UV-vis absorption spectroscopy, and electrochemical measurements. The substitution lability of the nitrogen oxide ligand of each complex is discussed in comparison with that of the corresponding acetonitrile complexes.
Introduction
Ruthenium(II) polypyridine complexes are widely studied materials because of their contributions to fundamental coordination chemistry, including electrochemistry, photochemistry, and photophysics,1 and their potential applications in energy conversion,2 luminescent sensors,3 electroluminescence displays,4 and biotechnology.5 Of particular interest are the Ru(II) complexes [Ru(TL)(BL)(L)]n+ (TL = tridentate polypyridine ligand, BL = bidentate polypyridine ligand, and L = monodentate labile ligand) given their catalytic activity for various reactions, such as oxidation,6,7 reduction,8–11 and photo-induced reactions.12
Phosphine-containing ruthenium(II) complexes are also attractive for potential applications in energy conversion systems13 and catalysis14–17 owing to the σ-donating and π-accepting abilities of the phosphine ligands. Thus, it should be possible to develop ruthenium complexes that have novel and tunable properties and reactivity by designing mixed polypyridyl/phosphine complexes. There have been several reports on the syntheses of ruthenium(II) polypyridine complexes containing monodentate phosphine ligands16,18,19 and their catalytic activities.19d,20 However, ruthenium(II) polypyridine complexes of the type [Ru(TL)(BL)(L)]n+ bearing bidentate ligands with P and N donors have not been investigated.
In our previous report,21 we synthesised and structurally characterised for the first time a series of phosphine-containing ruthenium(II) polypyridine complexes of the type [Ru(TL)(BL)(L)]n+, with L = acetonitrile, TL = 2,2′:6′,2′′-terpyridine (trpy), and BL = 8-(diphenylphosphanyl)quinoline (Pqn) or 1,2-bis(diphenylphosphanyl)benzene (dppbz) (Scheme 1). The influence of the number and position of phosphine donors on the structures and electronic properties was characterised, and unique isomerisation behaviours of these complexes were observed. The coordinating phosphorus ligands played a crucial role in these isomerisation reactions, and the results encouraged us to investigate in more detail the influence of the P atom on the physical properties of Ru-based metal complexes.
 |
| | Scheme 1 Structures of a tridentate ligand (trpy), bidentate ligands (Pqn and dppbz), and metal complexes (trans-NO2, cis-NO2, PP-NO2, and cis-NO) used in this study. | |
In this study, we investigated the reaction of phosphine-containing ruthenium complexes with the nitrogen oxides, NO and NO2−, because of their biological roles as signalling molecules22 and reservoirs,23 in addition to the fundamental interest in their coordination chemistry. Here, we show the syntheses, structural characterisation, and electrochemical and spectroscopic properties of a series of ruthenium(II) polypyridine complexes containing Pqn or dppbz with nitric oxides. Three novel nitrito-κN complexes—trans(P,NO2)- and cis(P,NO2)-[Ru(trpy)(Pqn)(NO2)]PF6 (trans-NO2 and cis-NO2), and [Ru(trpy)(dppbz)(NO2)]PF6 (PP-NO2)—were successfully synthesised. Described here are the single-crystal X-ray structural determinations, UV-vis absorption spectra, and electrochemical measurements for these complexes, and the preparation of the nitrosyl complex cis(P,NO)-[Ru(trpy)(Pqn)(NO)](PF6)3 (cis-NO) from cis-NO2 and its properties are examined. The present study allows us to probe systematically the chemical and structural properties of phosphine-containing ruthenium complexes with nitrogen oxides by the use of the geometric isomers of the Pqn complex. Additionally, other reactivity properties and comparisons with those of the corresponding acetonitrile complexes are presented.
Results and discussion
Syntheses and characterisation
The synthetic procedures to obtain trans-NO2, cis-NO2, and PP-NO2 are shown in Scheme 2. The precursors, trans-MeCN, cis-MeCN, and PP-MeCN, were synthesised according to the method that we previously reported.21 The reaction of the respective acetonitrile complexes with excess NaNO2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethanol:water at 100 °C gave the corresponding nitrito-κN complexes (trans-NO2, cis-NO2, and PP-NO2).24,26b The resulting products were characterised by 1H NMR and 31P{1H} NMR spectroscopy and elemental analysis.
:1 mixture of ethanol:water at 100 °C gave the corresponding nitrito-κN complexes (trans-NO2, cis-NO2, and PP-NO2).24,26b The resulting products were characterised by 1H NMR and 31P{1H} NMR spectroscopy and elemental analysis.
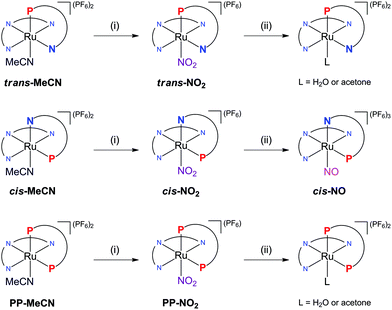 |
| | Scheme 2 Syntheses of trans-NO2, cis-NO2, PP-NO2, and cis-NO. (i) NaNO2 in a 1:1 mixture of ethanol:water at 100 °C. (ii) HPF6 in acetone at 0 °C. | |
The 31P{1H} NMR spectra of trans-NO2 and cis-NO2 in CD3CN displayed singlets at δ 53.10 and 54.06, respectively, showing upfield shifts (Δδ = 5.70 and 1.90) compared to the spectra of the corresponding acetonitrile complexes, trans-MeCN and cis-MeCN (δ 58.80 and 55.96). The 31P{1H} NMR spectrum of PP-NO2 in CD3CN afforded two doublets at δ 62.65 and 68.59 with coupling constants of 14.2 Hz, again showing upfield shifts (Δδ = 5.85 and 1.18, respectively) compared with the spectrum of PP-MeCN (68.57 and 69.77, 2JP–P = 20.2 Hz).
Conversions of the nitrito-κN complexes to the corresponding ruthenium nitrosyls were attempted by adding an excess of HPF6 to acetone solutions of the nitrito-κN species at 0 °C. The preparations of trans-NO and PP-NO from trans-NO2 and PP-NO2, respectively, were not successful because of the instability of the nitrosyl complex or of a reaction intermediate under acidic conditions (for details, see Fig. S1 and S2 in the ESI†). However, cis-NO was isolated in 85% yield and was characterised by 1H and 31P{1H} NMR spectroscopy and elemental analysis. The 31P{1H} NMR spectrum of cis-NO in acetone-d6 gave a singlet at δ 54.23. cis-NO immediately converted to a solvent-coordinated complex in acetonitrile (Fig. S3 in the ESI†) but was meta-stable in weaker-coordinating solvents, such as acetone, γ-butyrolactone and ethylene glycol (Fig. S4 in the ESI†). The difference in the stability of these nitrosyl complexes will be discussed in the “Substitution lability of nitrogen oxide” section.
Crystal structures
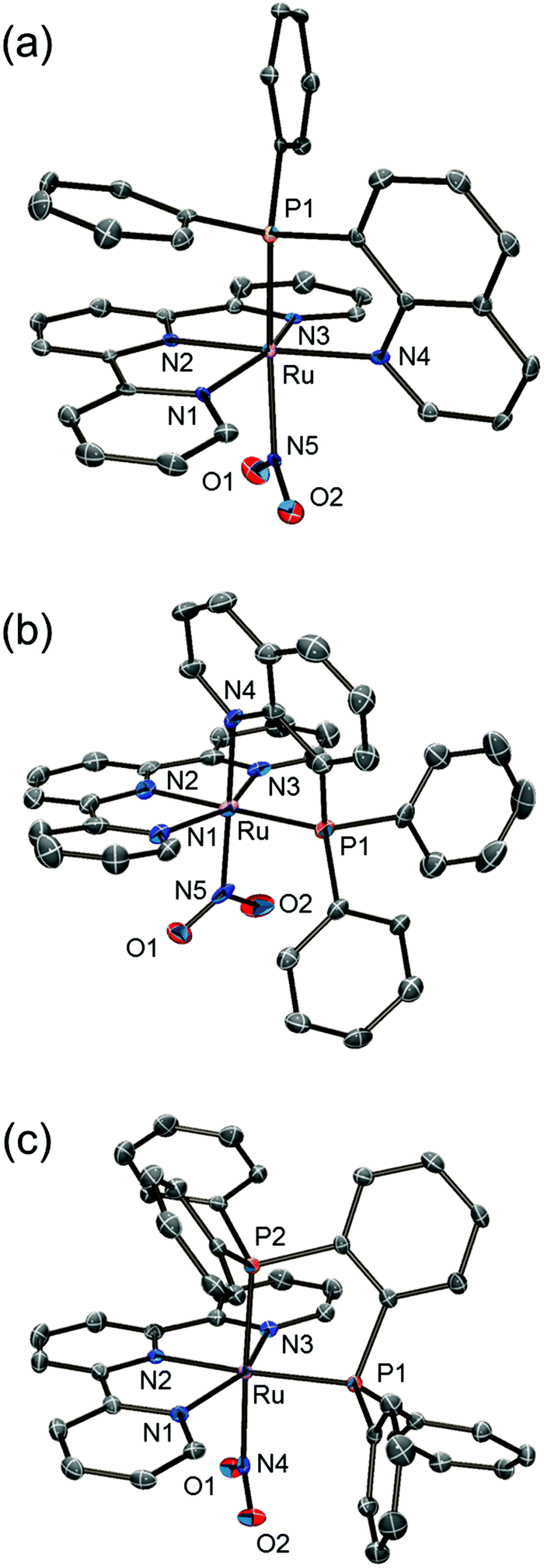
Single crystals of trans-NO2 and PP-NO2 suitable for structural determination were obtained by recrystallisation from diethyl ether/methanol/acetonitrile. Single crystals of the cis-nitrito-κN complex were obtained as the BPh4– salt, cis(P,NO2)-[Ru(trpy)(Pqn)(NO2)]BPh4 (cis-NO2′), by adding an excess of NaBPh4, instead of NH4PF6, after the reaction. The molecular structures of trans-NO2, cis-NO2′, and PP-NO2 determined by single-crystal X-ray crystallography and the summary of crystallographic data are shown in Fig. 1 and Table 1, respectively. The asymmetric unit of the monoclinic P21/n crystal of trans-NO2 contained one cationic ruthenium complex, one PF6 anion, and one acetonitrile molecule. The cis-NO2′ crystallised with two crystallographically independent ruthenium complexes, two BPh4− anions, and one acetonitrile molecule as the crystal solvent in the asymmetric unit of the triclinic P1 space group. The asymmetric unit of the monoclinic P21/c crystal of PP-NO2 contained one cationic ruthenium complex, one PF6 anion, and two methanol molecules. For each complex, the ratio of ruthenium to counter anion indicated that the oxidation state of the ruthenium centre was +2. The coordination geometry of each Ru atom was that of a distorted octahedron composed of a meridionally coordinated terpyridine ligand, a bidentate ligand, and a nitrito ligand.
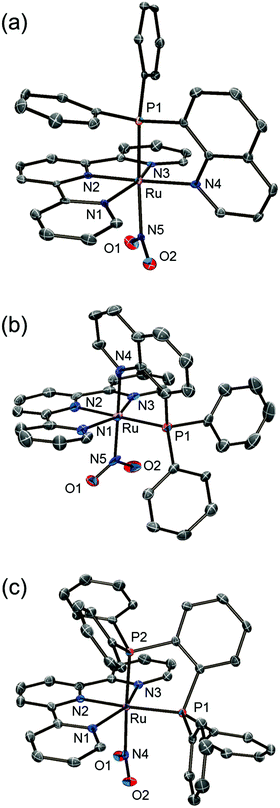 |
| | Fig. 1 ORTEP drawings (50% probability level) of cationic complexes in (a) trans-NO2, (b) cis-NO2′, and (c) PP-NO2. Hydrogen atoms are omitted for clarity. | |
Table 1 Crystallographic data for trans-NO2, cis-NO2′, and PP-NO2
| Complex |
trans-NO2·CH3CN |
cis-NO2′·0.5CH3CN |
PP-NO2·2CH3OH |
|
R
1 = −∑||Fo| − |Fc||/∑|Fo|.
wR2 = [∑(w(Fo2 − Fc2)2)/∑w(Fo2)2]1/2.
|
| Formula |
C38H30F6N6O2P2Ru |
C61H48.5BN5.5O2PRu |
C47H43F6N4O4P3Ru |
| Formula weight |
879.69 |
1033.40 |
1035.83 |
| Crystal colour, habit |
Red, needle |
Orange-red, platelet |
Orange, block |
| Crystal system |
Monoclinic |
Triclinic |
Monoclinic |
| Crystal size, mm3 |
0.30 × 0.05 × 0.05 |
0.15 × 0.15 × 0.10 |
0.15 × 0.10 × 0.05 |
| Space group |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
|
a, Å |
12.1909(17) |
10.2764(2) |
17.1049(6) |
|
b, Å |
11.9003(17) |
21.2343(4) |
13.0763(6) |
|
c, Å |
25.260(4) |
24.3802(4) |
20.1547(8) |
|
α, ° |
90 |
77.0060(10) |
90 |
|
β, ° |
99.579(3) |
78.9530(10) |
106.853(3) |
|
γ, ° |
90 |
74.9360(10) |
90 |
|
V, Å3 |
3613.5(9) |
4955.30(16) |
4314.4(3) |
|
Z
|
4 |
4 |
4 |
|
D
calc, g cm−3 |
1.617 |
1.385 |
1.595 |
|
μ, mm−1 |
0.599 |
0.400 |
0.552 |
|
F(000) |
1776 |
2132 |
2112 |
|
R
1a |
0.0596 |
0.0337 |
0.0397 |
| wR2b |
0.1678 |
0.1071 |
0.0941 |
| Goodness-of-fit S |
1.032 |
1.178 |
1.004 |
The bond distances between the ruthenium and nitrogen atoms of the nitrito ligand of trans-NO2 and PP-NO2 were 2.146(4) (Ru1–N5) and 2.124(2) Å (Ru1–N4), respectively (Fig. 2), and were longer than those found in [Ru(trpy)(bpm)(NO2)]PF6 (2.034(5) Å, bpm = 2,2′-bipyrimidine).25 By contrast, the Ru–N(NO2) distances in cis-NO2′ (2.0362(18) and 2.0290(18) Å for Ru1–N5 and Ru2–N10, respectively, Fig. 2) were similar to those of [Ru(trpy)(bpm)(NO2)]PF6 (2.034(5) Å). These results indicated a stronger trans influence of the phosphorus atom of Pqn or dppbz compared with that of the nitrogen atom of bpm or bpy. This tendency was also observed in trans-MeCN, cis-MeCN, and PP-MeCN in our previous study.21
 |
| | Fig. 2 Comparison of bond distances (Å) around the ruthenium centres of trans-NO2, cis-NO2′, and PP-NO2. | |
UV-vis absorption spectra
Fig. 3 shows the UV-Vis absorption spectra of the nitrito-κN complexes, trans-NO2, cis-NO2, and PP-NO2, in acetonitrile solution. The spectral data for these complexes and related compounds are listed in Table 2. All complexes displayed intense absorption bands in the UV region that were assigned to ligand-based π–π* transitions. Additionally, a moderately intense band was observed in the visible region for each complex. TD-DFT calculations that were performed at the B3LYP/LANL2DZ and B3LYP/SDD level of theory indicated that the visible region band could be assigned to metal-to-ligand charge transfer (MLCT) transitions from the dπ orbitals of ruthenium to the π* orbitals of trpy and Pqn or dppbz (for details, see Table S1 and Fig. S5–9 in the ESI†). The molar absorption coefficient of PP-NO2 was nearly half of those of trans-NO2 and cis-NO2. The absorption maximum (λmax) of the MLCT transition of trans-NO2, cis-NO2, and PP-NO2 was 443, 431, and 402 nm, respectively, and was blue-shifted compared with that of [Ru(trpy)(bpm)(NO2)]PF6,25 suggesting the stabilisation of the dπ orbitals of the ruthenium centre upon introduction of the phosphine donors. Note that the MLCT band of cis-NO2 was more blue-shifted than that of trans-NO2 despite their isomeric relationship. A similar pattern was noted for the acetonitrile complexes, trans-MeCN, cis-MeCN, and PP-MeCN.21
 |
| | Fig. 3 UV-Vis absorption spectra of trans-NO2, cis-NO2, and PP-NO2 in acetonitrile at room temperature. | |
Table 2 UV-Vis absorption data (λmax/nm (10−3ε/M−1 cm−1)) in acetonitrile and infrared data (v/cm−1) for trans-NO2, cis-NO2, PP-NO2, and related compounds at room temperature
| Complex |
λ
max
|
IR |
| |
|
v
as(NO2) |
v
s(NO2) |
|
Ref. 26.
Ref. 25.
Absorption shoulder.
Data not collected.
|
|
trans-NO2
|
443 (8.02), 327c, 303 (31.0), 281 (23.9), 271 (24.1) |
1349 |
1304 |
|
cis-NO2
|
431 (7.95), 331c, 315 (29.7), 282 (26.9), 276c |
1339 |
1286 |
|
PP-NO2
|
402 (3.82), 332 (12.6), 307 (17.7), 283 (16.5), 273 (17.5) |
1354 |
1311 |
| [Ru(trpy)(bpy)(NO2)]PF6a |
472 |
|
|
| [Ru(trpy)(bpm)(NO2)]PF6b |
470 (6.50), 362 (6.10), 330c, 308 (25.6), 264 (23.3) |
1342 |
1286 |
The UV-vis absorption spectra of the cis-isomers with different ligands L, cis(P,L)-[Ru(trpy)(Pqn)(L)]n+ (L = Cl−, NO2−, MeCN, and NO+), in ethylene glycol solution are shown in Fig. 4. These complexes each exhibited ligand-based π–π* transitions in the UV region and MLCT transitions in the visible region. The MLCT transition of cis-NO was observed at λmax = 383 nm and is comparable to similar Ru–NO complexes (Table 3). For the different Ls, the lowest energy MLCT bands of the cis-isomers are in the following order: L = Cl− (468 nm) < NO2− (423 nm) < MeCN (413 nm) < NO+ (383 nm). This result can be attributed to the competing σ-donor/π-acceptor properties of the respective ligands.
 |
| | Fig. 4 UV-Vis absorption spectra of cis(P,X)-[Ru(trpy)(Pqn)(L)]n (L = Cl−, NO2−, MeCN, NO+) in ethylene glycol at room temperature. | |
Table 3 UV-Vis absorption data (λmax/nm (10−3ε/M−1 cm−1)) in acetonitrile, infrared data (v/cm−1), and redox potentials (E1/2/V vs. Fc/Fc+) in acetonitrile for cis-NO and related compounds at room temperature
| Complex |
λ
max
|
v
N–O
|
E
1/2(NO+/NO˙) |
E
1/2(NO˙/NO−) |
|
UV-Vis absorption data and redox potentials in ethylene glycol and γ-butyrolactone, respectively.
Ref. 25.
Ref. 24.
Absorption shoulder.
E
pc value for the irreversible process.
Data not collected.
|
|
cis-NO
|
383 (8.38), 329 (17.9), 323d, 287 (23.7), 279d |
1929 |
0.05 |
−0.61e |
| [Ru(trpy)(bpm)(NO)](PF6)3b |
362 (5.12), 331d, 312d, 291 (8.93), 265 (10.8) |
1957 |
0.17 |
−0.47 |
|
trans(P,P)-[Ru(trpy)(PPh3)2(NO)](ClO4)3c |
393d, 330 (32) |
1900 |
0.12 |
|
Electrochemical properties
The cyclic voltammograms (CVs) of trans-NO2, cis-NO2, and PP-NO2 are shown in Fig. 5, and electrochemical data for these complexes and related compounds are listed in Table 4. The CVs were measured in 0.1 M tetraethylammonium perchlorate (TEAP)/acetonitrile. cis-NO2 displayed one reversible oxidation wave in the positive region at E1/2 = 0.79 V vs. ferrocene/ferrocenium (Fc/Fc+), which was assigned to a Ru(III)/Ru(II) redox couple. By contrast, trans-NO2 and PP-NO2 exhibited one irreversible (Epa = 0.79 for trans-NO2 and 0.95 V for PP-NO2) and one reversible (E1/2 = 0.97 for trans-NO2 and 1.27 V for PP-NO2) redox waves in the positive region. The former irreversible oxidation peak can be attributed to oxidation of the (Ru–NO2)+ centre, and the latter reversible wave was observed exactly at the same potential as the Ru(III)/Ru(II) redox couple of the corresponding acetonitrile complex (E1/2 = 0.97 for trans-MeCN and 1.27 V for PP-MeCN, Table 4 and Fig. S10b and S10d in the ESI†).21 These observations suggest that the one-electron oxidation of trans-NO2 and PP-NO2 results in the release of NO2, owing to the labilising effect of the trans-phosphine in each case. This leads to the formation of the respective ruthenium(II) acetonitrile complexes, which are oxidised reversibly on further sweep to more positive potential (Scheme 3). In the absence of a trans-labilising phosphine, the oxidation of the (Ru–NO2)+ centre of cis-NO2 occurs at a similar potential to the trans-isomer, but reversibly (Fig. 5 and Table 4).
 |
| | Fig. 5 Cyclic voltammograms of trans-NO2, cis-NO2, and PP-NO2 (0.5 mM) in 0.1 M TEAP/acetonitrile under an Ar atmosphere (WE: GC, CE: Pt wire, RE: Ag/Ag+; scan rate: 100 mV s−1). | |
 |
| | Scheme 3 Electrochemical behaviour of nitrito-κN complexes. | |
Table 4 Redox potentials (V vs. Fc/Fc+) in acetonitrile for trans-NO2, cis-NO2, PP-NO2, and related compounds at room temperature
| Complex |
Red. |
Ox. |
| |
E(1) |
E(2) |
E(3) |
E(1) |
E(2) |
|
Ref. 21.
These reductions induced the dissociation of NO2− and the formation of MeCN-coordinated species.
NO2 was released upon these oxidation processes, and subsequent coordination of MeCN resulted in the formation of corresponding acetonitrile complexes.
cis-MeCN underwent isomerization to trans-MeCN upon reduction.
E
pc values for the irreversible processes.
E
pa values for the irreversible processes.
Redox processes correspond to MeCN-coordinated complexes.
|
|
trans-NO2
|
−1.77b |
|
−2.13e,g |
0.79c,f |
0.97g |
|
cis-NO2
|
−1.70 |
−1.99 |
— |
0.79 |
— |
|
PP-NO2
|
−1.70b,e |
|
−2.49g |
0.95c,f |
1.27g |
|
trans-MeCN
|
−1.70 |
−1.77 |
−2.13 |
0.97 |
— |
|
cis-MeCN
|
|
|
|
1.05 |
— |
|
PP-MeCN
|
−1.50 |
−1.46 |
−2.49 |
1.27 |
— |
In the negative potential region, cis-NO2 displayed two reversible reduction waves at −1.70 and −1.99 V, which were assigned to the trpy/trpy− and Pqn/Pqn− redox couple, respectively. trans-NO2 exhibited two redox waves at E1/2 = −1.77 and −2.13 V, and these redox potentials were the same as that of trans-MeCN (Table 4 and Fig. S10a in the ESI†). PP-NO2 displayed two irreversible waves (Epc = −1.70 V and Epa = −1.47 V) and one reversible (E1/2 = −2.49 V) redox wave in the negative region. The reversible wave at E1/2 = −2.49 V and the irreversible anodic wave at Epa = −1.47 V were similar to those observed for PP-MeCN (Table 4 and Fig. S10c in the ESI†). These redox behaviours of trans-NO2 and PP-NO2 revealed that the reduction of these complexes induced the dissociation of NO2− and the formation of MeCN-coordinated species; this behaviour was similar to that observed in the positive potential region. The electrochemical behaviours of the nitrito-κN complexes are summarised in Scheme 3.
cis-NO displayed one reversible redox wave at E1/2 = 0.05 V and one irreversible reduction peak at Epc = −0.61 V (Fig. 6). Comparison with similar nitrosyl compounds24,25 revealed that the former redox wave was attributed to the NO+/NO˙ redox couple and the latter peak could be assigned to the reduction of NO˙ to NO−. Note that a Ru(III)/Ru(II) redox couple was not observed in the potential region between −1.6 and 1.5 V due to the low HOMO energy level originating from the poor donating ability of the NO+ ligand.
 |
| | Fig. 6 Cyclic voltammogram of cis-NO (0.5 mM) in 0.1 M TEAP/γ-butyrolactone under an Ar atmosphere (WE: GC, CE: Pt wire, RE: Ag/Ag+; scan rate: 100 mV s−1). | |
Cyclic voltammograms of cis-isomers with various ligands L are shown in Fig. 7. The redox potentials of a Ru(III)/Ru(II) redox couple for each complex were observed at 0.49, 0.79, and 1.05 V for cis(P,Cl)-[Ru(trpy)(Pqn)(Cl)]+ (cis-Cl), cis-NO2, and cis-MeCN, respectively. This result indicated the increase in the HOMO energy level by electron donation from monodentate ligands and was consistent with the UV-vis absorption spectroscopy.
 |
| | Fig. 7 Cyclic voltammograms of cis-Cl, cis-NO2, and cis-MeCN (0.5 mM) in 0.1 M TEAP/acetonitrile under an Ar atmosphere (WE: GC, CE: Pt wire, RE: Ag/Ag+; scan rate: 100 mV s−1). | |
Photostability of a nitrosyl complex
The photostability of cis-NO was investigated by UV-vis absorption spectroscopy and by using a Sievers nitric oxide analyser (NOA) to evaluate NO release. When a solution of cis-NO in ethylene glycol (0.05 mM) was irradiated at λirr = 355 nm, the slow spectral changes seen in Fig. 8 were observed. The quantum yields of NO release, ΦNO, were quite low, 0.0048 in air saturated solution and 0.003 under helium (ESI Fig. S11 and S12†). These ΦNO values are about two orders of magnitude smaller than that measured by Silva et al. for the photolysis of the analogous ruthenium nitrosyl complex, [Ru(tpy)(bpy)(NO)]3+ (bpy = 2,2′-bipyridine).27 Notably, exhaustive photolysis led to a nearly quantitative conversion to a spectrum analogous to that of trans-L (L = solvent, see Fig. S13 in the ESI†), which suggests that the photodissociation of NO and the isomerization of the complex from cis to trans form proceeds in a step-wise manner. The result is consistent with the multi-step spectral change shown in Fig. 8. It should further be noted that a Ru(II) complex, rather than the Ru(III) species is obtained upon NO photodissociation from cis-NO (ESI Fig. S13†). There are several possible explanations, one being that the Ru(III) intermediate is readily reduced by the solvent. Another is that the principal photo-reaction is release of NO+ rather than NO27 owing to phosphine stabilization of the low-valent Ru(II) state. However, this question was not explored in greater detail.
 |
| | Fig. 8 UV-Vis absorption spectra of cis-NO in ethylene glycol at room temperature during photolysis using a Nd/YAG laser operating at 355 nm. “Times” indicates the number of the laser pulses used to irradiate the sample. | |
Substitution lability of nitrogen oxide
The substitution lability of the monodentate ligand L in the [Ru(TL)(BL)(L)]n+-type complexes is an important factor in determining the reactivity of the complexes in various catalytic and photo-induced reactions. Several experimental results described above enabled us to discuss in detail the lability of the monodentate ligand in the complexes. First, the reaction of cis-NO2 with HPF6 afforded the desired cis-NO. However, similar reactions of trans-NO2 and PP-NO2 resulted in the formation of trans-MeCN and PP-MeCNvia the dissociation of a monodentate labile ligand, N(O)OH or NO+ (probably the former). Second, UV-vis absorption spectroscopy revealed that the nitrito ligands of trans-NO2, cis-NO2, and PP-NO2 did not dissociate, even in strongly coordinating solvents such as acetonitrile, whereas ligand exchange reactions of trans-MeCN and PP-MeCN easily occurred under analogous conditions (Fig. S14 in the ESI†). cis-NO was not stable in coordinating solvent and was easily converted to a solvent-coordinated form (Fig. S4 in the ESI†).
The difference in lability of the oxidised and reduced state can also be clarified by the results of the electrochemical measurements. In the one-electron oxidised states, trans-NO2 and PP-NO2 were labile and were converted to the solvent-coordinated forms, trans-MeCN and PP-MeCN, respectively. By contrast, cis-NO2 was inert during the oxidation process, and a reversible redox wave was observed in the electrochemical measurement. Similarly, in the reduced states, the nitrito ligands of trans-NO2, and PP-NO2 easily dissociated, although cis-NO2 was stable during the whole electrochemical process. However, this stability of cis-NO2 was quite different from that of cis-MeCN: the acetonitrile ligand of cis-MeCN became labile upon reduction, and the dissociation of the ligand resulted in the isomerisation of cis-MeCN to trans-MeCN.21 The stability of the monodentate labile site for each complex is shown in Scheme 4.
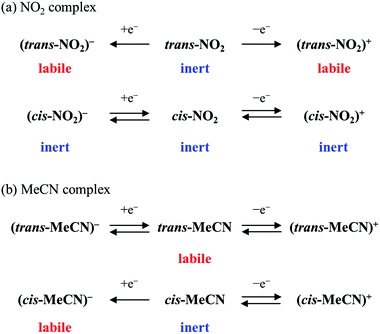 |
| | Scheme 4 Changes in lability upon oxidation or reduction. | |
The difference in the lability of these complexes can be explained by considering the following factors. First, the σ-donor character of the phosphine group significantly elongates the bond length between the ruthenium centre and the ligand trans to the phosphine group. This trans influence of the phosphine group was clearly observed in the X-ray structure for the series of nitrito-κN complexes; the bond distances between the ruthenium and nitrogen atom of the nitrite ligand are 2.141(3), 2.124(2), and ca. 2.03 Å for trans-NO2, PP-NO2, and cis-NO2, respectively. Therefore, the trans-isomers and PP complexes exhibited greater lability compared with the corresponding cis-isomers. Second, the electron donation from labile ligands can stabilise Ru–L bonds. The donating ability of labile ligands was confirmed by the comparison of the HOMO energy levels obtained from the electrochemical measurements of the cis-isomers and follows the order NO2− > MeCN > NO+ and was in accordance with the stability of the complexes. Third, oxidation decreases the electron density of the Ru centre, and the π back-donating ability of Ru centres should be weakened. DFT calculations revealed that π back-donation from Ru to the nitrito ligand occurs and stabilises the Ru–L bond (Fig. S6 in the ESI†). Finally, the reduction of the complexes stabilises the five-coordinated species, as rationalised for the Ru(II)–MeCN complexes in our previous report.21 The formation of five-coordinated species in MeCN results in ligand exchange in trans-NO2 and PP-NO2 or isomerisation of cis-MeCN to trans-MeCN, whereas no observable chemical process exists in the case of trans-MeCN and PP-MeCN. These results suggest that the (1) number and position of P atom(s), (2) coordinating ability of the monodentate ligand, and (3) oxidation state of the complexes are all factors in defining the lability of complexes.
Conclusions
This study describes the syntheses, crystal structures and spectroscopic and electrochemical properties of a series of phosphine-substituted ruthenium(II) polypyridine complexes with nitrogen oxides. Three nitrito-κN complexes, trans-NO2, cis-NO2, and PP-NO2, were synthesised by the reaction of the corresponding acetonitrile complex with NaNO2 in an ethanol/water mixed solution, and a nitrosyl complex, cis-NO, was obtained by the reaction of cis-NO2 with HPF6 in acetone. Crystallographic, spectroscopic, and electrochemical analyses for these complexes revealed that the σ-donating and π-accepting characters of the phosphine ligands clearly affected the dσ and dπ orbitals of the ruthenium centre, respectively. The investigation of the substitution lability of the monodentate ligand of each complex suggested that the (1) number and position of the phosphine groups, (2) coordinating ability of the monodentate ligand, and (3) oxidation state of the metal centre are all factors in determining the lability of the complex. As a further extension of our studies, investigations on various catalytic and photo-induced reactions of the phosphine-substituted ruthenium(II) polypyridine complexes are in progress in our laboratories.
Experimental
Materials
NaNO2 was purchased from Kanto Chemical Co., Inc. NH4PF6 and HPF6 were purchased from Wako Pure Chemical Industries, Ltd. All solvents and reagents were of the highest quality available and were used as received. cis(P,Cl)-[Ru(trpy)(Pqn)Cl]PF6 (cis-Cl), trans(P,MeCN)- and cis(P,MeCN)-[Ru(trpy)(Pqn)(MeCN)](PF6)2 (trans-MeCN and cis-MeCN), and [Ru(trpy)(dppbz)(MeCN)](PF6)2 (PP-MeCN) were prepared following methods found in the literature.21
Measurements
1H and 31P{1H} NMR spectra were recorded at room temperature on a JEOL JNM-LA500 spectrometer using tetramethylsilane as an internal reference for the 1H NMR spectra and phosphoric acid as an external reference for the 31P{1H} NMR spectra. UV-vis absorption spectra were obtained on a Shimadzu UV-2450SIM spectrophotometer at room temperature. Elemental analyses were carried out on a Yanagimoto MT-5 elemental analyser. Infrared data were obtained using a Perkin-Elmer Spectrum 100 FT-IR spectrometer. ESI-TOF mass spectra were recorded on a JEOL JMS-T100LC mass spectrometer. All the ESI-TOF mass spectrometric measurements were recorded in the positive ion mode at a cone voltage of 20 V. Typically, each sample solution was introduced in the spectrometer at a flow rate of 10 mL min−1 using a syringe pump. Cyclic voltammograms were measured at room temperature on a BAS ALS Model 650DKMP electrochemical analyser in acetonitrile ([complex] = 0.5 mM; 0.1 M tetraethylammonium perchlorate (TEAP)). A glassy carbon disk, platinum wire, and Ag/Ag+ electrode (Ag/0.01 M AgNO3) were used as the working, auxiliary, and reference electrodes, respectively. The redox potentials of the samples were calibrated against the redox signal for the ferrocene/ferrocenium (Fc/Fc+) couple. The photochemical experiments shown in Fig. 8 were made using a photolysis apparatus consisting of a LS-2134UTF Nd/YAG laser (Tokyo Instruments, INC.) with excitation provided by the third harmonic at λ = 355 nm. The pulse width was 5 ns, the beam diameter incident on the sample was 6 mm, and the repetition rate was 5 Hz.
Synthetic procedures
Synthesis of trans(P,NO2)-[Ru(trpy)(Pqn)(NO2)]PF6 (trans-NO2).
A mixture of trans-MeCN (26.8 mg, 0.0252 mmol) and NaNO2 (38.8 mg, 0.562 mmol) in ethanol (4 cm3)/water (4 cm3) was heated at 100 °C for 3 hours and then cooled to room temperature. Acetonitrile (2 cm3) and a NH4PF6 (188.4 mg, 1.16 mmol)/water (2 cm3) solution was added to the abovementioned solution. The resulting red solution was concentrated to ca. 5 cm3 under reduced pressure. The red product was collected by filtration and washed with water and diethyl ether. Yield 19.1 mg (0.0216 mmol, 86%). Single red crystals suitable for X-ray crystallography were grown by the slow diffusion of diethyl ether into a mixture of methanol and a few drops of acetonitrile solution of trans-NO2. ESI-TOF MS (positive ion, acetonitrile): m/z 694 ([Ru(trpy)(Pqn)(NO2)]+). 1H NMR (CD3CN): δ 6.56 (t, 2H, J = 9.0 Hz), 6.98 (t, 4H, J = 7.0 Hz), 7.14 (t, 2H, J = 7.0 Hz), 7.21 (t, 2H, J = 8.0 Hz), 7.65 (d, 2H, J = 5.0 Hz), 7.80 (t, 2H, J = 8.0 Hz), 7.91 (m, 3H), 8.03 (d, 2H, J = 8.0 Hz), 8.09 (t, 1H, J = 7.5 Hz), 8.23 (d, 2H, J = 8.0 Hz), 8.46 (d, 1H, J = 7.0 Hz), 8.75 (d, 1H, J = 8.0 Hz), 9.93 (d, 1H, J = 5.0 Hz). 31P{1H} NMR (CD3CN): δ 53.10 (s). FT-IR: νs(NO2) 1304, νas(NO2) 1349 cm−1. Anal. Found: C, 48.50; H, 3.59; N, 8.22. Calcd for C36H32F6N5O4.5P2Ru (trans-NO2·2.5H2O): C, 48.93; H, 3.65; N, 7.93.
Synthesis of cis(P,NO2)-[Ru(trpy)(Pqn)(NO2)]PF6 (cis-NO2).
This complex was prepared from cis-MeCN (26.0 mg, 0.0250 mmol) instead of trans-MeCN by a method similar to that for trans-NO2. Yield 19.5 mg (0.0221 mmol, 88%). ESI-TOF MS (positive ion, acetonitrile): m/z 694 ([Ru(trpy)(Pqn)(NO2)]+). 1H NMR (CD3CN): δ 6.81 (t, 2H, J = 6.5 Hz), 7.06 (m, 1H), 7.4 1 (d, 2H, J = 5.5 Hz), 7.52 (m, 4H), 7.63 (t, 2H, J = 7.5 Hz), 7.73 (m, 4H), 7.84 (t, 2H, J = 8.0 Hz), 7.92 (d, 1H, J = 5.5 Hz), 7.97 (t, 1H, J = 7.5 Hz), 8.18 (d, 1H, J = 8.0 Hz), 8.23 (d, 1H, J = 8.0 Hz), 8.36 (m, 3H), 8.55 (d, 2H, J = 8.0 Hz) 8.78 (t, 1H, J = 8.0 Hz). 31P{1H} NMR (CD3CN): δ 54.06 (s). FT-IR: νs(NO2) 1286, νas(NO2) 1339 cm−1. Anal. Found: C, 50.38; H, 3.44; N, 8.19. Calcd for C36H29F6N5O3P2Ru (cis-NO2·H2O): C, 50.47; H, 3.41; N, 8.18.
Synthesis of cis(P,NO2)-[Ru(trpy)(Pqn)(NO2)]BPh4 (cis-NO2′).
This complex was prepared by a method similar to that for cis-NO2 with an excess of NaBPh4 instead of NH4PF6. The product was recrystallised from dichloromethane and a small amount of acetonitrile/diethyl ether to afford orange-red crystals of cis-NO2′. Anal. Found: C, 70.54; H, 4.72; N, 6.92. Calcd for C60H48BN5O2.5PRu (cis-NO2′·0.5H2O): C, 70.52; H, 4.73; N, 6.85.
Synthesis of cis(P,NO)-[Ru(trpy)(Pqn)(NO)](PF6)3 (cis-NO).
cis-NO2 (22.4 mg, 0.0261 mmol) was dissolved in acetone (1 cm3). An excess of 60% HPF6 acid solution was added dropwise until the solution changed colour from red to yellow with the shielding of light in an ice-water bath. The resulting yellow solution was concentrated under reduced pressure, and 10 cm3 of diethyl ether was added to precipitate the product. Yield 27.4 mg (0.0223 mmol, 85%). 1H NMR (acetone-d6): δ 7.47 (t, 2H, J = 7.0 Hz), 7.66 (m, 1H), 7.80 (d, 2H, J = 6.0 Hz), 7.86 (m, 4H), 8.01 (t, 2H, J = 7.5 Hz), 8.12 (dd, 4H, J = 7.5, 13.0 Hz), 8.42 (t, 1H, J = 7.5 Hz), 8.49 (m, 2H), 8.55 (m, 1H), 8.80 (d, 1H, J = 8.0 Hz), 9.01 (m, 3H), 9.10 (m, 1H), 9.22 (m, 3H). 31P{1H} NMR (acetone-d6): δ 54.23 (s). FT-IR: νs(NO) 1929 cm−1. Anal. Found: C, 35.03; H, 2.95; N, 5.59. Calcd for C36H40F18N5O7.5P4Ru (cis-NO·6.5H2O): C, 35.16; H, 3.28; N, 5.70.
Synthesis of [Ru(trpy)(dppbz)(NO2)]PF6 (PP-NO2).
This complex was prepared from PP-MeCN (31.5 mg, 0.0263 mmol) instead of trans-MeCN by a method similar to that for trans-NO2. Yield 22.9 mg (0.0227 mmol, 86%). Single orange crystals suitable for X-ray crystallography were grown through the slow diffusion of diethyl ether into a mixture of methanol and a few drops of acetonitrile solution of PP-NO2. ESI-TOF MS (positive ion, acetonitrile): m/z 827 ([Ru(trpy)(dppbz)(NO2)]+). 1H NMR (CD3CN): δ 6.50 (m, 4H), 6.78 (m, 2H), 6.88 (m, 4H), 7.08 (d, 2H, J = 5.5 Hz), 7.17 (m, 2H), 7.45 (t, 4H, J = 7.5 Hz), 7.61 (m, 7H), 7.77 (m, 3H), 7.87 (t, 1H, J = 7.5 Hz), 8.03 (d, 2H, J = 8.0 Hz), 8.23 (m, 3H), 8.39 (t, 1H, J = 7.5 Hz). 31P{1H} NMR (CD3CN): δ 62.65 (d, 2JP–P = 14.2 Hz), 68.59 (d, 2JP–P = 14.2 Hz). FT-IR: νs(NO2) 1311, νas(NO2) 1354 cm−1. Anal. Found: C, 53.62; H, 4.05; N, 5.46. Calcd for C45H39F6N4O4P3Ru (PP-NO2·2H2O): C, 53.63; H, 3.90; N, 5.56.
X-ray crystallography
The X-ray data collection and processing was performed on a Kappa APEX II CCDC diffractometer by using graphite-monochromated Mo-Kα radiation (0.71075 Å) for trans-NO2 and PP-NO2. Single-crystal X-ray diffraction measurement of cis-NO2′ was performed with a RAXIS-RAPID Imaging Plate diffractometer equipped with confocal monochromated Mo-Kα (0.71075 Å) radiation, and the data were processed using RAPID-AUTO (Rigaku). The structure was solved by the direct methods using SIR-9228 and refined on F2 with the full-matrix least squares technique using SHELXL-2014.29 All non-hydrogen atoms were refined anisotropically. Molecular graphics were generated using ORTEP-3 for Windows30 and POV-RAY.31 A summary of the crystallographic data and structure refinement parameters is given in Table 1.
The crystallographic data have been deposited at the Cambridge Crystallographic Data Centre: deposition numbers CCDC 1040452, 1040453, and 1040454 for trans-NO2, cis-NO2′, and PP-NO2, respectively.
DFT calculations
Calculations were performed using the DFT method implemented in the Gaussian 09 package.32 The structures were fully optimised using the hybrid B3LYP method, which uses Becke's three-parameter exchange functional33 with the correlation energy functional of Lee, Yang, and Parr.34 All calculations were performed using the standard double-ζ type LANL2DZ basis set35a–c or SDD basis set35d implemented in Gaussian 09, without adding any extra polarisation or diffuse functions. The LANL2DZ basis set also uses a relativistic effective core potential (RECP) for the Ru atom to account for the scalar relativistic effects of the inner 28 core electrons ([Ar]3d10). All calculations were performed using the polarisable continuum model (PCM)36 to compute the structures in acetonitrile media. All stationary points were characterised as minima of the potential energy surface by their harmonic vibrational frequencies. The free energies at 298 K and 1 atm were obtained through thermochemical analysis of the frequency calculation using the thermal correction to Gibbs free energy as implemented in Gaussian 09. The excited states were calculated using the TDDFT37 method within the Tamm–Dancoff approximation as implemented in Gaussian 09. These calculations employ the hybrid B3LYP functional along with the basis sets described above. A minimum of 100 excited states was computed in each calculation. To obtain the simulated spectrum of each species, transition energies and oscillator strengths were interpolated by a Gaussian convolution with a common σ value of 0.2 eV.
Quantum yield measurements
Nitric oxide was detected and analysed using a GE Sievers model 280i nitric oxide analyser (NOA).38 Known volumes of the gases from the solution headspace were injected into the NOA purge vessel, and these gases were entrained to the detector using helium or medical grade air. The NO present in the sample was quantified using a calibration curve generated from the reaction of NaNO2 with acidic KI. Chemical actinometry was performed with ferric oxalate solutions.39 The photolysis source was the output from a 200 W high-pressure mercury lamp passed through an IR filter and collimated with lenses. An appropriate interference filter was used to select the desired λirr. A shutter shielded the sample from the arc lamp. A sample of a known volume in a 1 cm square cuvette with a magnetic stirring bar was irradiated for determined time periods.40 The NO quantum yields (ΦNO) were calculated based on the nitric oxide release measured using the NOA.
Acknowledgements
This work was supported by a Grant-in-Aid for Young Scientists (A) (No. 25708011) (to S.M.), a Grant-in-Aid for Challenging Exploratory Research (No. 26620160) (to S.M.), and a Grant-in-Aid for Young Scientists (A) (No. 15H05480) (to M.K) from the Japan Society for the Promotion of Science. This work was also supported by a Grant-in-Aid for Scientific Research on Innovative Areas “AnApple” (No. 25107526). Studies at UCSB were supported by a US National Foundation Grant (CHE-1058794 and CHE-1405062). We thank Dr Guang Wu of UCSB for the X-ray diffraction studies and Dr John Garcia of UCSB for confirming photochemical results.
Notes and references
-
(a) N. Sutin, J. Photochem., 1979, 10, 19–40 CrossRef CAS;
(b) K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS;
(c) E. S. Dodsworth and A. B. P. Lever, Chem. Phys. Lett., 1986, 124, 152–158 CrossRef CAS;
(d) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS;
(e) A. B. P. Lever, Inorg. Chem., 1990, 29, 1271–1285 CrossRef CAS;
(f) V. Balzani and A. Juris, Coord. Chem. Rev., 2001, 211, 97–115 CrossRef CAS;
(g) D. W. Thompson, J. F. Wishart, B. S. Brunschwig and N. Sutin, J. Phys. Chem. A, 2001, 105, 8117–8122 CrossRef CAS;
(h) S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS;
(i) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed;
(j) Q. Sun, S. Mosquera-Vazquez, Y. Suffren, J. Hankache, N. Amstutz, L. M. L. Daku, E. Vauthey and A. Hauser, Coord. Chem. Rev., 2015, 282–283, 87–99 CrossRef CAS PubMed.
-
(a) C. D. Clark and M. Z. Hoffman, Coord. Chem. Rev., 1997, 159, 359–373 CrossRef CAS;
(b) L. De Cola and P. Belser, Coord. Chem. Rev., 1998, 177, 301–346 CrossRef CAS;
(c) M. D. Ward and F. Barigelletti, Coord. Chem. Rev., 2001, 216–217, 127–154 CrossRef CAS;
(d) M. H. V. Huynh, D. M. Dattelbaum and T. J. Meyer, Coord. Chem. Rev., 2005, 249, 457–483 CrossRef CAS PubMed; H. Kon, K. Tsuge, T. Imamura, Y. Sasaki, S. Ishizaka, N. Kitamura and M. Kato, Dalton Trans., 2008, 1541–1543 Search PubMed;
(e) A. Lavie-Cambot, C. Lincheneau, M. Cantuel, Y. Leydet and N. D. McClenaghan, Chem. Soc. Rev., 2010, 39, 506–515 RSC;
(f) O. Filevich, B. García-Acosta and R. Etchenique, Photochem. Photobiol. Sci., 2012, 11, 843–847 RSC.
-
(a) F. G. Gao and A. J. Bard, J. Am. Chem. Soc., 2000, 122, 7426–7427 CrossRef CAS;
(b) J. N. Demas and B. A. DeGraff, Coord. Chem. Rev., 2001, 211, 317–351 CrossRef CAS;
(c) P. D. Beer and E. J. Hayes, Coord. Chem. Rev., 2003, 240, 167–189 CrossRef CAS;
(d) R. Martinez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419–4476 CrossRef PubMed;
(e) A. S. Polo, M. K. Itokazu and N. Y. M. Iha, Coord. Chem. Rev., 2004, 248, 1343–1361 CrossRef CAS PubMed;
(f) M. S. Vickers, K. S. Martindale and P. D. Beer, J. Mater. Chem., 2005, 15, 2784–2790 RSC;
(g) N. Haddour, J. Chauvin, C. Gondran and S. Cosnier, J. Am. Chem. Soc., 2006, 128, 9693–9698 CrossRef CAS PubMed;
(h) H. Wei and E. Wang, Trends Anal. Chem., 2008, 27, 447–459 CrossRef CAS PubMed;
(i) J. L. Delaney, C. F. Hogan, J. Tian and W. Shen, Anal. Chem., 2011, 83, 1300–1306 CrossRef CAS PubMed.
-
(a) G. J. Wilson, A. Launikonis, W. H. F. Sasse and A. W.-H. Mau, J. Phys. Chem. A, 1997, 101, 4860–4866 CrossRef CAS;
(b) J. A. Simon, S. L. Curry, R. H. Schmehl, T. R. Schatz, P. Piotrowiak, X. Jin and R. P. Thummel, J. Am. Chem. Soc., 1997, 119, 11012–11022 CrossRef CAS;
(c) A. D. Guerzo, S. Leroy, F. Fages and R. H. Schmehl, Inorg. Chem., 2002, 41, 359–366 CrossRef PubMed;
(d) D. S. Tyson, C. R. Luman, X. Zhou and F. N. Castellano, Inorg. Chem., 2001, 40, 4063–4071 CrossRef CAS PubMed;
(e) S. Bernhard, J. A. Barron, P. L. Houston, H. D. Abruña, J. L. Ruglovksy, X. Gao and G. G. Malliaras, J. Am. Chem. Soc., 2002, 128, 9693–9698 Search PubMed;
(f) S. Welter, K. Brunner, J. W. Hofstraat and L. D. Cola, Nature, 2003, 421, 54–57 CrossRef CAS PubMed;
(g) H. Shahroosvand, P. Abbasi, A. Faghih, E. Mohajerani, M. Janghouri and M. Mahmoudi, RSC Adv., 2014, 4, 1150–1154 RSC.
-
(a) J. K. Barton, Science, 1986, 233, 727–734 CrossRef CAS;
(b) C. Turro, S. H. Bossmann, Y. Jenkins, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1995, 117, 9026–9032 CrossRef CAS;
(c) H. B. Gray and J. R. Winkler, Annu. Rev. Biochem., 1996, 65, 537–561 CrossRef CAS PubMed;
(d) A. D. Guerzo and A. K.-D. Mesmaeker, Inorg. Chem., 2002, 41, 938–945 CrossRef PubMed;
(e) S. Le Gac, M. Foucart, P. Gerbaux, E. Defrancq, C. Moucheron and A. Kirsch-De Mesmaeker, Dalton Trans., 2010, 39, 9672–9683 RSC;
(f) H. Song, J. T. Kaiser and J. K. Barton, Nat. Chem., 2012, 4, 615–620 CrossRef CAS PubMed;
(g) H. Niyazi, J. P. Hall, K. O'Sullivan, G. Winter, T. Sorensen, J. M. Kelly and C. J. Cardin, Nat. Chem., 2012, 4, 621–628 CrossRef CAS PubMed;
(h) A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 RSC.
-
(a) T. J. Meyer and M. H. V. Huynh, Inorg. Chem., 2003, 42, 8140–8160 CrossRef CAS PubMed;
(b) E. Masllorens, M. Rodriguez, I. Romero, A. Roglans, T. Parella, J. Benet-Buchholz, M. Poyatos and A. Llobet, J. Am. Chem. Soc., 2006, 128, 5306–5307 CrossRef CAS PubMed;
(c) Y. Shiota, J. M. Herrera, G. Juhász, T. Abe, S. Ohzu, T. Ishizuka, T. Kojima and K. Yoshizawa, Inorg. Chem., 2011, 50, 6200–6209 CrossRef CAS PubMed;
(d) T. Kojima, K. Nakayama, K. Ikemura, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11692–11700 CrossRef CAS PubMed;
(e) Z. Hu, H. Du, W.-L. Man, C.-F. Leung, H. Liang and T.-C. Lau, Chem. Commun., 2012, 48, 1102–1104 RSC;
(f) Z. Hu, L. Ma, J. Xie, H. Du, W. W. Y. Lam and T.-C. Lau, New J. Chem., 2013, 37, 1707–1710 RSC.
-
(a) J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462–16463 CrossRef CAS;
(b) H.-W. Tseng, R. Zong, J. T. Muckerman and R. P. Thummel, Inorg. Chem., 2008, 47, 11763–11773 CrossRef CAS PubMed;
(c) S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 182–183 CrossRef CAS;
(d) M. Yoshida, S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 702–703 CrossRef CAS;
(e) S. Romain, L. Vigara and A. Llobet, Acc. Chem. Res., 2009, 42, 1944–1953 CrossRef CAS PubMed;
(f) J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. Murakami Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed;
(g) L. Duan, L. Tong, Y. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 3296–3313 RSC;
(h) D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218–227 RSC;
(i) M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
-
(a) H. Yamazaki, T. Hakamata, M. Komi and M. Yagi, J. Am. Chem. Soc., 2011, 133, 8846–8849 CrossRef CAS PubMed;
(b) J. L. Boyer, D. E. Polyansky, D. J. Szalda, R. Zong, R. P. Thummel and E. Fujita, Angew. Chem., Int. Ed., 2011, 50, 12600–12604 CrossRef CAS PubMed;
(c) S. K. Padhi, R. Fukuda, M. Ehara and K. Tanaka, Inorg. Chem., 2012, 51, 5386–5392 CrossRef CAS PubMed;
(d) M. Hirahara, M. Z. Ertem, M. Komi, H. Yamazaki, C. J. Cramer and M. Yagi, Inorg. Chem., 2013, 52, 6354–6364 CrossRef CAS PubMed.
-
(a) K. Tanaka and D. Ooyama, Coord. Chem. Rev., 2002, 226, 211–218 CrossRef CAS;
(b) J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed;
(c) Y. Tsukahara, T. Wada and K. Tanaka, Chem. Lett., 2010, 39, 1134–1135 CrossRef CAS;
(d) K. Kobayashi, T. Kikuchi, S. Kitagawa and K. Tanaka, Angew. Chem., Int. Ed., 2014, 52, 1–6 Search PubMed.
-
(a) Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Chem. Commun., 2011, 47, 12607–12609 RSC;
(b) Z. Chen, J. J. Concepcion, M. K. Brennaman, P. Kang, M. R. Norris, P. G. Hoertz and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15606–15611 CrossRef CAS PubMed;
(c) Z. Chen, P. Kang, M.-T. Zhang and T. J. Meyer, Chem. Commun., 2014, 50, 335–337 RSC;
(d) P. Kang, Z. Chen, A. Nayak, S. Zhang and T. J. Meyer, Energy Environ. Sci., 2014, 7, 4007–4012 RSC.
-
(a) A. Kobayashi, R. Takatori, I. Kikuchi, H. Konno, K. Sakamoto and O. Ishitani, Organometallics, 2001, 20, 3361–3363 CrossRef CAS;
(b) A. Kobayashi, H. Konno, K. Sakamoto, A. Sekine, Y. Ohashi, M. Iida and O. Ishitani, Chem. – Eur. J., 2005, 11, 4219–4226 CrossRef CAS PubMed;
(c) M. Kimura and K. Tanaka, Angew. Chem., Int. Ed., 2008, 47, 9768–9771 CrossRef CAS PubMed;
(d) Y. Matsubara, E. Fujita, M. D. Doherty, J. T. Muckerman and C. Creutz, J. Am. Chem. Soc., 2012, 134, 15743–15757 CrossRef CAS PubMed;
(e) Y. Matsubara, T. Kosaka, K. Koga, A. Nagasawa, A. Kobayashi, H. Konno, C. Creutz, K. Sakamoto and O. Ishitani, Organometallics, 2013, 32, 6162–6165 CrossRef CAS;
(f) J. Huang, J. Chen, H. Gao and L. Chen, Inorg. Chem., 2014, 53, 9570–9580 CrossRef CAS PubMed.
-
(a) R. W. Callahan and T. J. Meyer, Inorg. Chem., 1977, 574–581 CrossRef CAS;
(b) H. Hadadzadeh, M. C. DeRosa, G. P. A. Yap, A. R. Rezvani and R. J. Crutchley, Inorg. Chem., 2002, 41, 6521–6526 CrossRef CAS PubMed;
(c) M. G. Sauaia, R. G. de Lima, A. C. Tedesco and R. S. da Silva, J. Am. Chem. Soc., 2003, 125, 14718–14719 CrossRef CAS PubMed;
(d) Z. N. da Rocha, M. S. P. Marchesi, J. C. Molin, C. N. Lunardi, K. M. Miranda, L. M. Bendhack, P. C. Ford and R. S. da Silva, Dalton Trans., 2008, 4282–4287 RSC;
(e) A. C. Pereira, P. C. Ford, R. S. da Silva and L. M. Bendhack, Nitric Oxide, 2011, 24, 192–198 CrossRef CAS PubMed;
(f) T. A. Heinrich, A. C. Tedesco, J. M. Fukuto and R. S. da Silva, Dalton Trans., 2014, 43, 4021–4025 RSC;
(g) R. G. de Lima, B. R. Silva, R. S. da Silva and L. M. Bendhack, Molecules, 2014, 19, 9628–9654 CrossRef PubMed.
-
(a) T. Kinoshita, J. T. Dy, S. Uchida, T. Kubo and H. Segawa, Nat. Photonics, 2013, 7, 535–539 CrossRef CAS PubMed;
(b) R. Katoh and A. Furube, J. Photochem. Photobiol., C, 2014, 20, 1–16 CrossRef CAS PubMed.
-
(a) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS;
(b) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS;
(c) R. Noyori, Adv. Synth. Catal., 2003, 345, 15–32 CrossRef CAS PubMed;
(d) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS PubMed;
(e) A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418–514 CrossRef CAS PubMed;
(f) R. Noyori, Angew. Chem., Int. Ed., 2013, 52, 79–92 CrossRef CAS PubMed.
-
(a) P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1995, 34, 2039–2041 CrossRef CAS PubMed;
(b) H. Clavier and S. P. Nolan, Chem. – Eur. J., 2007, 13, 8029–8036 CrossRef CAS PubMed;
(c) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed;
(d) J. S. M. Samec, B. K. Keitz and R. H. Grubbs, J. Organomet. Chem., 2010, 695, 1831–1837 CrossRef CAS PubMed;
(e) S. P. Nolan and H. Clavier, Chem. Soc. Rev., 2010, 39, 3305–3316 RSC.
-
(a) C. M. Moore and N. K. Szymczak, Chem. Commun., 2013, 49, 400–402 RSC;
(b) K.-N. T. Tseng, J. W. Kampf and N. K. Szymczak, Organometallics, 2013, 32, 2046–2049 CrossRef CAS;
(c) K.-N. T. Tseng, A. M. Rizzi and N. K. Szymczak, J. Am. Chem. Soc., 2013, 135, 16352–16355 CrossRef CAS PubMed.
-
(a) D. K. Dutta and B. Deb, Coord. Chem. Rev., 2011, 255, 1686–1712 CrossRef CAS PubMed;
(b) C. S. Yi, J. Organomet. Chem., 2011, 696, 76–80 CrossRef CAS PubMed;
(c) I. Mellone, M. Peruzzini, L. Rosi, D. Mellmann, H. Junge, M. Beller and L. Gonsalvi, Dalton Trans., 2013, 42, 2495–2501 RSC.
-
(a) R. A. Leising, J. J. Grzybowski and K. J. Takeuchi, Inorg. Chem., 1988, 27, 1020–1025 CrossRef CAS;
(b) B. J. Coe, D. W. Thompson, C. T. Culbertson, J. R. Schoonover and T. J. Meyer, Inorg. Chem., 1995, 34, 3385–3395 CrossRef CAS;
(c) L. F. Szczepura, S. A. Kubow, R. A. Leising, W. J. Perez, M. H. V. Huynh, C. H. Lake, D. G. Churchill, M. R. Churchill and K. J. J. Takeuchi, Chem. Soc., Dalton Trans. Inorg. Chem., 1996, 7, 1463–1470 RSC;
(d) W. J. Perez, C. H. Lake, R. F. See, L. M. Toomey, M. R. Churchill, K. J. Takeuchi, C. P. Radano, W. J. Boyko and C. A. Bessel, J. Chem. Soc., Dalton Trans., 1999, 2281–2292 RSC;
(e) S. B. Billings, M. T. Mock, K. Wiacek, M. B. Turner, W. S. Kassel, K. J. Takeuchi, A. L. Rheingold, W. J. Boyko and C. A. Bessel, Inorg. Chim. Acta, 2003, 355, 103–115 CrossRef CAS;
(f) S. Sharma, S. K. Singh, M. Chandra and D. S. Pandey, J. Inorg. Biochem., 2005, 99, 458–466 CrossRef CAS PubMed.
-
(a) B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334–3341 CrossRef CAS;
(b) B. P. Sullivan, D. Conrad and T. J. Meyer, Inorg. Chem., 1985, 24, 3640–3645 CrossRef CAS;
(c) J. P. Otruba, G. A. Neyhart, W. J. Dressick, J. L. Marshall, B. P. Sullivan, P. A. Watkins and T. J. Meyer, J. Photochem., 1986, 35, 133–153 CrossRef CAS;
(d) R. A. Leising and K. J. Takeuchi, Inorg. Chem., 1987, 26, 4391–4393 CrossRef CAS;
(e) R. A. Leising, J. S. Ohman and K. J. Takeuchi, Inorg. Chem., 1988, 27, 3804–3809 CrossRef CAS;
(f) C. A. Bessel, J. A. Margarucci, J. H. Acquaye, R. S. Rubmo, J. Crandall, A. J. Jircitano and K. J. Takeuchi, Inorg. Chem., 1993, 32, 5779–5784 CrossRef CAS;
(g) M. R. Churchill, L. M. Krajkowski, M. H. V. Huynh and K. J. Takeuchi, J. Chem. Crystallogr., 1997, 27, 589–597 CrossRef CAS;
(h) M. Salierno, E. Marceca, D. S. Peterka, R. Yuste and R. Etchenique, J. Inorg. Biochem., 2010, 104, 418–422 CrossRef CAS PubMed;
(i) S. V. Litke, A. Y. Ershov and T. J. Meyer, J. Phys. Chem. A, 2011, 115, 14235–14242 CrossRef CAS PubMed;
(j) V. S. Miguel, M. Álvarez, O. Filevich, R. Etchenique and A. del Campo, Langmuir, 2012, 28, 1217–1221 CrossRef PubMed;
(k) R. Araya, V. Andino-Pavlovsky, R. Yuste and R. Etchenique, ACS Chem. Neurosci., 2013, 4, 1163–1167 CrossRef CAS PubMed.
-
(a) M. E. Marmion and K. J. Takeuchi, J. Chem. Soc., Dalton Trans., 1988, 2385–2391 RSC;
(b) C. A. Bessel, R. A. Leising and K. J. Takeuchi, J. Chem. Soc., Chem. Commun., 1991, 833–835 RSC;
(c) N. D. Schley, G. E. Dobereiner and R. H. Crabtree, Organometallics, 2011, 30, 4174–4179 CrossRef CAS.
- G. Nakamura, M. Okamura, M. Yoshida, T. Suzuki, H. D. Takagi, M. Kondo and S. Masaoka, Inorg. Chem., 2014, 53, 7214–7226 CrossRef CAS PubMed.
- J. Heinecke and P. C. Ford, Coord. Chem. Rev., 2010, 254, 235–247 CrossRef CAS PubMed.
- M. T. Gladwin, A. N. Schechter, D. B. Kim-Shapiro, R. P. Patel, N. Hogg, S. Shiva, R. O. Cannon III, M. Kelm, D. A. Wink, M. Graham Espey, E. H. Oldfield, R. M. Pluta, B. A. Freeman, J. R. Lancaster Jr., M. Feelisch and J. O. Lundberg, Nat. Chem. Biol., 2005, 1, 308–314 CrossRef CAS PubMed.
- R. A. Leising, S. A. Kubow and K. J. Takeuchi, Inorg. Chem., 1990, 29, 4569–4574 CrossRef CAS.
- P. Singh, J. Fiedler, S. Záliš, C. Duboc, M. Niemeyer, F. Lissner, T. Schleid and W. Kaim, Inorg. Chem., 2007, 46, 9254–9261 CrossRef CAS PubMed.
-
(a) D. W. Pipes and T. J. Meyer, Inorg. Chem., 1984, 23, 2466–2472 CrossRef CAS;
(b) W. R. Murphy, Jr., K. J. Takeuchi, M. H. Barley and T. J. Meyer, Inorg. Chem., 1984, 25, 1041–1053 CrossRef.
- R. G. de Lima, M. G. Sauaia, D. Bonaventure, A. C. Tedesco, L. M. Bendhack and R. S. da Silva, Inorg. Chim. Acta, 2006, 359, 2543–2549 CrossRef PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. J. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–566 CrossRef CAS.
- T. D. Fenn, D. Ringe and G. A. Petsko, J. Appl. Crystallogr., 2003, 36, 944–947 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision C.01), Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
-
(a)
T. H. Dunning, Jr. and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer, III, Plenum, New York, 1976 Search PubMed;
(b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed;
(c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS PubMed;
(d) D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS PubMed.
-
(a) M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS PubMed;
(b) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS PubMed;
(c) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- P. T. Burks, J. V. Garcia, R. GonzalezIrias, J. T. Tillman, M. Niu, A. A. Mikhailovsky, J. Zhang, F. Zhang and P. C. Ford, J. Am. Chem. Soc., 2013, 135, 18145–18152 CrossRef CAS PubMed.
-
(a)
J. G. Calvert and J. N. Pitts, Photochemistry, J. Wiley & Sons, New York, 1967, pp. 783–786 Search PubMed;
(b) G. Malouf and P. C. Ford, J. Am. Chem. Soc., 1977, 99, 7213–7221 CrossRef CAS.
- C. F. Works, C. J. Jocher, G. D. Bart, X. Bu and P. C. Ford, Inorg. Chem., 2002, 41, 3728–3739 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1040452–1040454. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02994e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a