
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stepwise assembly of mixed-metal coordination cages containing both kinetically inert and kinetically labile metal ions: introduction of metal-centred redox and photophysical activity at specific sites†
Ashley B.
Wragg
,
Alexander J.
Metherell
,
William
Cullen
and
Michael D.
Ward
*
Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: m.d.ward@sheffield.ac.uk
First published on 21st September 2015
Abstract
Stepwise preparation of the heterometallic octanuclear coordination cages [(Ma)4(Mb)4L12]16+ is reported, in which Ma = Ru or Os and Mb = Cd or Co (all in their +2 oxidation state). This requires initial preparation of the kinetically inert mononuclear complexes [(Ma)L3]2+ in which L is a ditopic ligand with two bidentate chelating pyrazolyl-pyridine units: in the complexes [(Ma)L3]2+ one terminus of each ligand is bound to the metal ion, such that the complex has three pendant bidentate sites at which cage assembly can propagate by coordination to additional labile ions Mb in a separate step. Thus, combination of four [(Ma)L3]2+ units and four [Mb]2+ ions results in assembly of the complete cages [(Ma)4(Mb)4L12]16+ in which a metal ion lies at each of the eight vertices, and a bridging ligand spans each of the twelve edges, of a cube. The different types of metal ion necessarily alternate around the periphery with each bridging ligand bound to one metal ion of each type. All four cages have been structurally characterised: in the Ru(II)/Cd(II) cage (reported in a recent communication) the Ru(II) and Cd(II) ions are crystallographically distinct; in the other three cages [Ru(II)/Co(II), Os(II)/Cd(II) and Os(II)/Co(II), reported here] the ions are disordered around the periphery such that every metal site refines as a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of the two metal atom types. The incorporation of Os(II) units into the cages results in both redox activity [a reversible Os(II)/Os(III) couple for all four metal ions simultaneously, at a modest potential] and luminescence [the Os(II) units have luminescent 3MLCT excited states which will be good photo-electron donors] being incorporated into the cage superstructure.
:50 mixture of the two metal atom types. The incorporation of Os(II) units into the cages results in both redox activity [a reversible Os(II)/Os(III) couple for all four metal ions simultaneously, at a modest potential] and luminescence [the Os(II) units have luminescent 3MLCT excited states which will be good photo-electron donors] being incorporated into the cage superstructure.
Introduction
The self-assembly and host–guest chemistry of coordination cages is an active and growing topic in modern supramolecular chemistry.1 These cage molecules are of interest due a combination of the syntheses of elaborate new structures from simple components by self-assembly methods, as well as the useful functional behaviour2 – ranging from drug delivery3 to catalysis4 – that can arise when guests bind in the central cavity. As the preparation of coordination cages is based on self-assembly, it follows that they require kinetically labile metal ions: without this, the necessary ability of the metal:ligand assembly to search out a thermodynamic minimum by forming, breaking and re-forming metal ligand bonds is not possible. The consequence is that, usually, just one type of metal ion is incorporated into the cage superstructure, and the vast majority of coordination cages – even those of the highest structural complexity – contain just one type of metal ion and one type of bridging ligand.1
This strongly limits the properties that a cage complex might display. If a cage is just required as a container whose only important attributes are structural (size, shape and so on) then the nature of the metal ion is of secondary importance. However if the properties of the metal ion are required as part of the functional behaviour of a cage (redox properties, magnetism, luminescence etc.)5 then the type of metal ion is fundamentally important, and it is significant that many transition metal ions that show fully reversible redox behaviour and desirable photophysical properties are second/third row metal ions whose kinetic inertness makes them difficult to use in traditional self-assembly processes (with a few notable exceptions).5
Accordingly we have set out to investigate the preparation of heterometallic cages in which different metal ions can be incorporated – with complete control – at different positions in the superstructure. Most commonly, heterometallic cages and related assemblies have been prepared by one of two strategies. The first involves use of unsymmetrical ligands which possess both hard and soft binding sites which will selectively bind to hard and soft metals, respectively.6,7 Thus, for example, Raymond and co-workers used a ligand containing both hard (catechol) and soft (phosphine) donors to assemble mixed metal (Ti4+/Pd2+) trigonal prismatic cages in which [Ti(catecholate)3]2− and trans-PdBr2(phosphine)2 units act as orthogonal assembly elements;6a Shionoya and co-workers exploited a pyridyl/catecholate ligand in an exactly similar way.6b The second method involves the use of metal ions with different coordination preferences.8–10 Early examples of this come from helicate complexes based on a mixture of octahedral and tetrahedral metal ions which occupy different positions along the helicate axis.8 A recent example of this principle in a coordination cage is the formation of a cubic cage in which octahedral tris-chelate Fe(III) ions form the corners, and square planar Cu(II) ions with four monodentate ligands occupy the centres of the faces.9 In this case the octahedral [Fe(NN)6]3+ units and the planar [CuL4]2+ units act as orthogonal assembly elements based on two different types of labile metal ion.
Both of these approaches allow the rational design and self-assembly of heterometallic structures with different metal ions at specific sites. However, in all members of our family of cage complexes,1c all of the metal coordination sites are octahedral tris-chelates with every metal ion being in a tris(pyrazolyl-pyridine) coordination environment. This precludes the two methods outlined above: the equivalence of all metal binding sites means that there is no basis for selecting which ion goes at which position in the cage on the basis of hard/soft considerations or different coordination geometries, so the necessary differentiation between sites does not exist. Accordingly we have investigated a different approach based on the use of pre-formed, kinetically stable, metal complex fragments with pendant binding sites; combination of these ‘complex ligands’ with additional labile ions in a second step results in assembly of the pre-formed fragments into a complete cage structure.11 We note that the use of a combination of ‘inert + labile’ components to control assembly of heteronuclear complexes with similar coordination sites is known in other contexts,12 but application of this method to assembly of large cages remains undeveloped.
The approach we have used is shown in Fig. 1 and exploits the structure of a [M8L12]X16 coordination cage which has an approximately cubic structure, with a metal ion at each vertex and a bridging ligand along each edge.13 This stepwise approach is based on the use of pre-formed and kinetically stable [RuL3]2+ and [OsL3]2+ units (in which the inert metal ion is labelled as Ma), whose pendant binding sites assemble in a separate step around Co(II) or Cd(II) ions (Mb) to give the set of four mixed-metal cages [(Ma)4(Mb)4L12]X16 cages. The presence of Ru(II) or Os(II) ions allows introduction of redox activity and, for Os(II), metal-centred luminescence into the cage assemblies. A preliminary communication outlining this strategy, and the crystal structure of the Ru4Cd4 cage, was published recently.11 In this follow-up paper we extend the strategy to use of Os(II) complex units as cage components, and report the synthesis and structures of several more mixed-metal cages prepared using this method. In addition we report the luminescence and electrochemical properties of the Os(II)-based cages which offer interesting possibilities for future functional behaviour of the cages2 in areas such as redox or luminescence reporting of guest binding or photocatalytic reactions on bound guests.
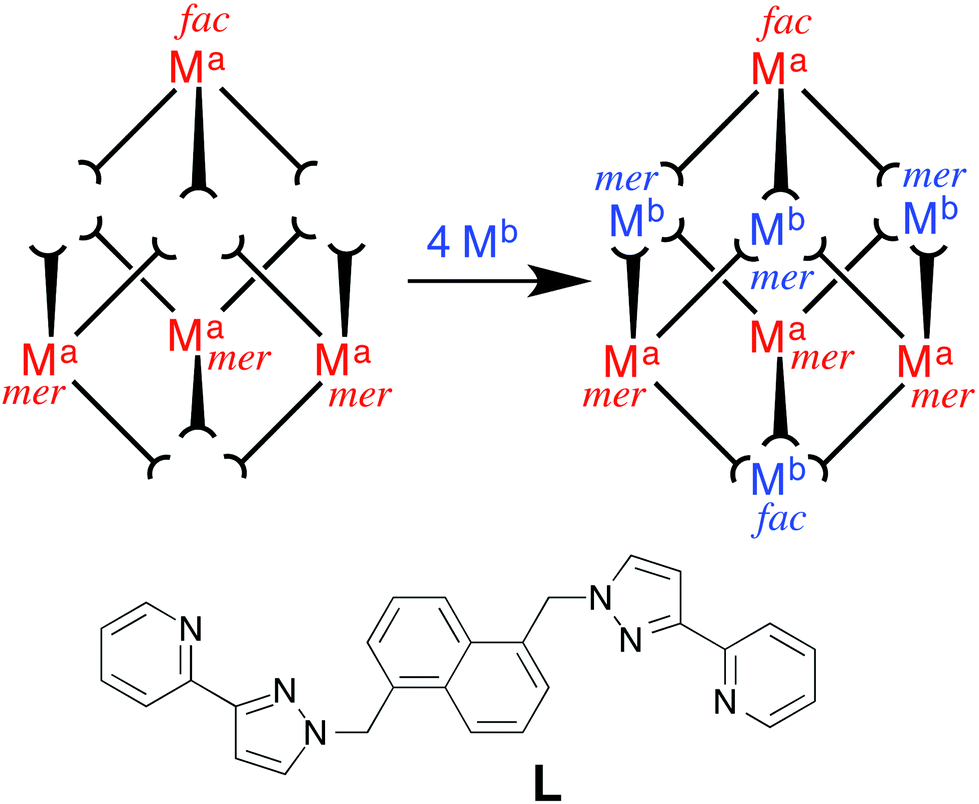 | ||
| Fig. 1 Sketch outlining the stepwise synthetic strategy used to prepare the heterometallic cubic cage complexes: viz combination of pre-formed, kinetically inert [(Ma)L3]2+ (Ma = Ru, Os) with additional labile ions (Mb)2+ (Mb = Co, Cd) in a 4:4 ratio to give octanuclear [(Ma)4(Mb)4L12]16+. | ||
Results and discussion
Mononuclear [ML3]2+ complexes as building blocks
We chose Ru(II) and Os(II) as the ‘inert’ metal complex vertices for a stepwise synthetic strategy given their very well developed coordination chemistry with a tris-diimine ligand set and the likelihood of useful redox- or luminescence-based behaviour. The first step was therefore to prepare the mononuclear ‘complex ligands’ [RuL3]X2 and [OsL3]X2 in which each ligand is coordinated through only one terminus and the complex has three pendant bidentate sites at which cage assembly can propagate. These complexes were prepared by reaction of >3 equivalents of the ligand L with one equivalent of Ru(dmso)4Cl2 or OsCl3 in ethylene glycol at reflux, followed by anion metathesis and chromatographic purification. As the preparation of [RuL3](PF6)2 was reported in the earlier communication,11 we focus the discussion here on [OsL3](PF6)2.Given the non-symmetrical nature of the pyrazolyl-pyridine chelates, [OsL3](PF6)2 necessarily forms as a mixture of fac and mer isomers. A 1:3 ratio is expected for statistical reasons unless there is some specific electronic or steric factor that favours one isomer, in which case a non-statistical distribution may result.14 The 1H NMR spectrum of [OsL3](PF6)2 (which is isolated following chromatography with no separation of the isomers) shows that we do in fact have the expected 1:3 fac:mer ratio of geometric isomers. In the threefold-symmetric fac isomer all three ligands are equivalent, but this product is only one third as abundant as the mer isomer in which all three ligands are inequivalent. The result is the presence of four ligand environments with equal abundance, which was clear from some regions of the 1H NMR spectrum (Fig. 2) although complete assignment was not possible due to the presence of 88 inequivalent proton environments with extensive regions of overlap.
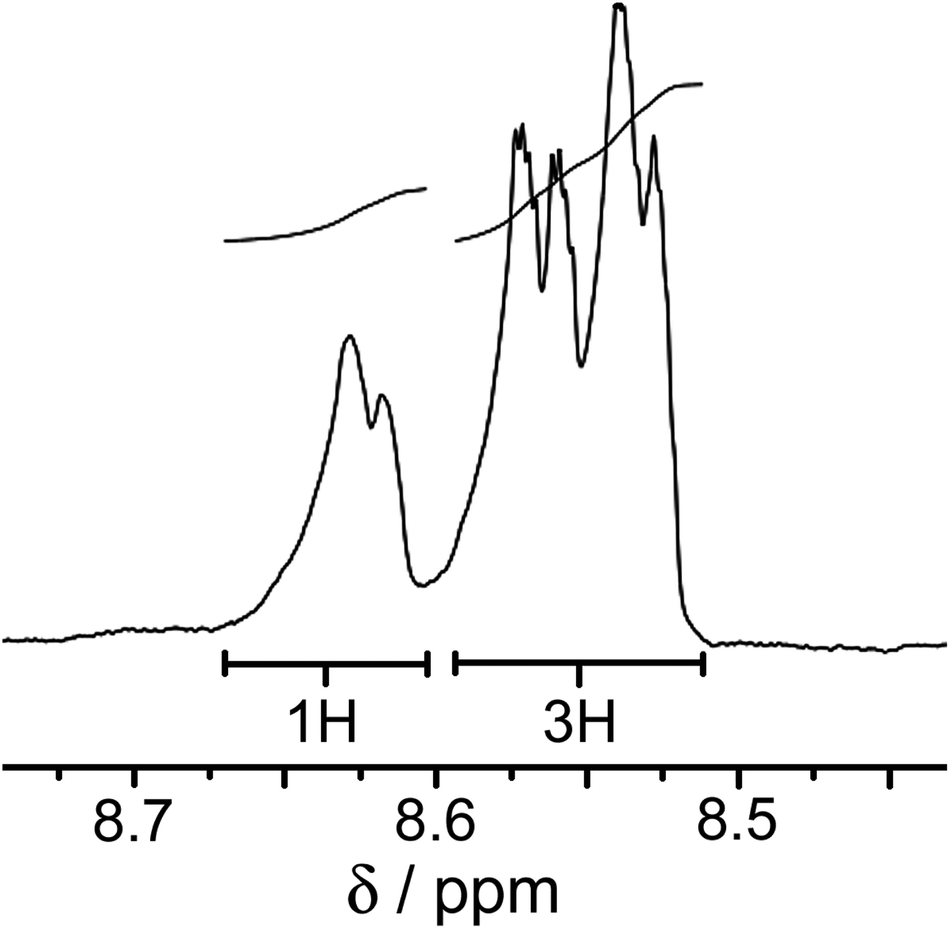 | ||
| Fig. 2 1H NMR signal for the set of pyridyl H6 protons of [OsL3](PF6)2 (in CD3CN) showing the presence of four independent ligand environments consistent with a statistical mixture of fac and mer isomers. | ||
These isomers do not require separating for our purposes, because this 1:3 fac:mer ratio of geometric isomers is precisely what is required for assembly of the complete cages. Other members of our cage family contain varying proportions of fac:mer metal complex units at the vertices,1c ranging from all fac15 to all mer,16 and various ratios in between, as required to facilitate any specific self-assembly. The cubic [M8L12]X16 cages that we use here happen to contain two fac vertices (at opposite ends of the long diagonal of the cube) and six mer vertices.13 Half of these sites will be occupied by the Os(II) or Ru(II) ions, and the positions of these must strictly alternate with the sites occupied by the labile partner ions, given that the synthetic strategy prevents two ions of the same type from being connected by a single bridging ligand. Thus the four kinetically inert Ru(II) or Os(II) subcomponents must contain a 1:3 fac:mer isomer ratio, which arises naturally from the synthesis and is exactly what is required for assembly of this cage type – which is one of the reasons why this cage type was chosen for this work. Stepwise assembly of other cages will require either preparation of pure fac or mer metal complex units as building blocks;17 or will require the isomers to be separated after synthesis of a mixture. This issue is however avoided here, which is helpful because a wide range of chromatographic conditions could not separate the fac and mer isomers of [OsL3](PF6)2.
[OsL3](PF6)2 (mix of isomers) shows a symmetric redox wave, assigned to a chemically reversible Os(II)/Os(III) couple, at +0.46 in MeCN (Fig. 3), which is identical to the Os(II)/Os(III) redox potential of [Os(bipy)3]2+.19 The fac and mer isomers are expected to have very similar redox potentials: these were not resolved in the voltammetric wave which is symmetric (equal cathodic and anodic peak currents) with ΔEp = 80 mV, and therefore behaves exactly like a normal one-electron reversible redox process. This redox potential is ca. 0.4 V less positive than the corresponding Ru(II)/Ru(III) couple,11 which is typical behaviour for isostructural Ru(II) and Os(II) complexes due to the lower ionisation energy of Os(II) compared to Ru(II) in a comparable environment.
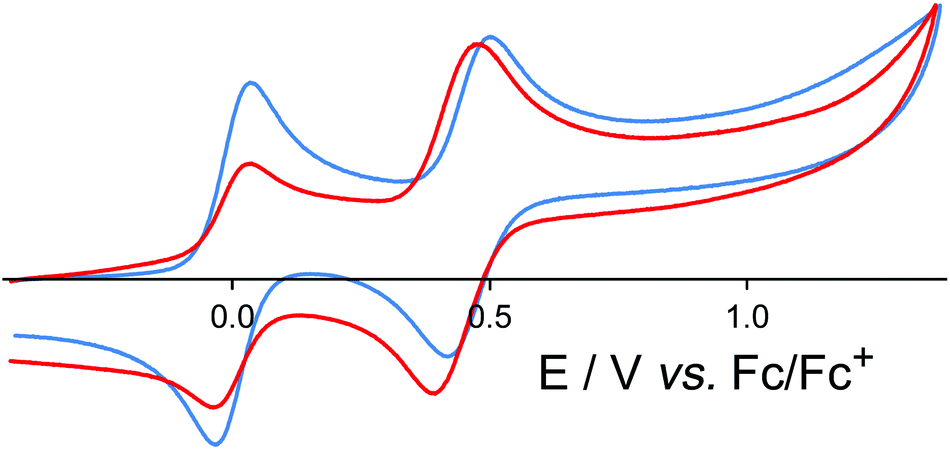 | ||
| Fig. 3 Cyclic voltammograms in MeCN of [OsL3](PF6)2 (blue trace) and [Os4Cd4L12](ClO4)16 (red trace) at a scan rate of 0.1 V s−1. In both cases the reversible wave at 0.0 V is internal ferrocene added as a reference. | ||
The UV/Vis absorption spectrum of [OsL3](PF6)2 (Fig. 4) shows the usual combination of spin-allowed 1MLCT absorptions around 400 nm, plus a less intense spin forbidden 3MLCT absorption manifold which provides a low-energy absorption tail in the 500–600 nm region, and high-energy ligand-centred transitions in the UV region. These 1MLCT and 3MLCT absorptions are at somewhat higher energy than in [Os(bipy)3]2+. Given that the Os-based d(π) orbitals are at similar energy in both cases (as shown by the identical Os(II)/Os(III) redox potentials of [Os(bipy)3]2+ and [OsL3]2+) it follows that the higher 1MLCT/3MLCT absorption energies in [OsL3](PF6)2 arise from a higher-lying ligand-centred LUMO of the pyrazolyl-pyridine unit compared to a bipy ligand, and this is reflected in the luminescence properties.
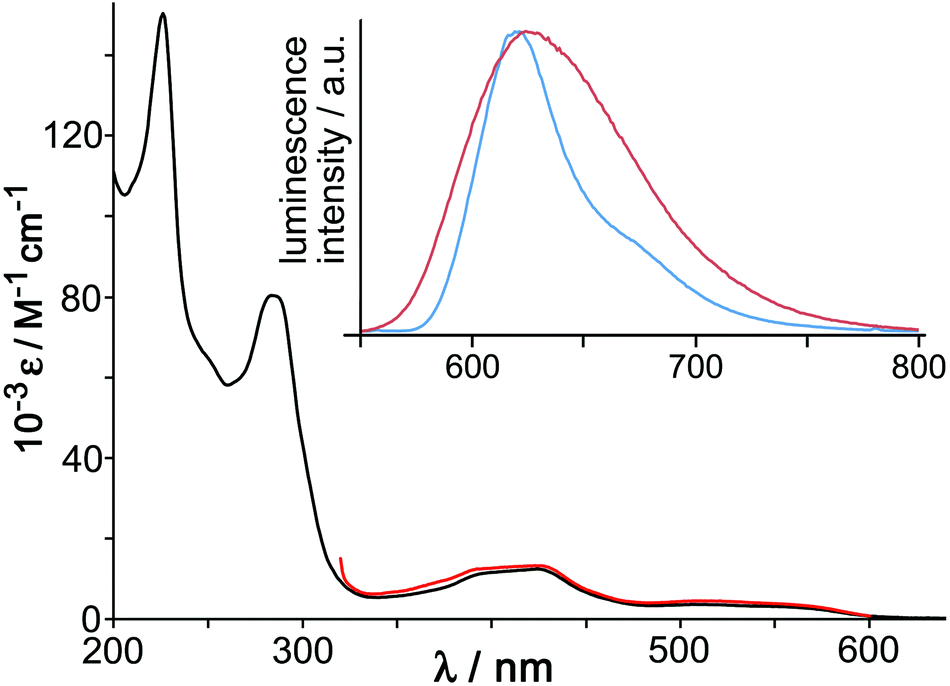 | ||
| Fig. 4 Main figure: UV/Vis absorption spectrum of [OsL3](PF6)2 in MeCN (black) with, overlaid in red, the excitation spectrum for the Os-based emission at 620 nm. Inset: luminescence spectra of [OsL3](PF6)2 in MeCN fluid solution at room temperature (red trace) and of [OsL3]Cl2 in EtOH/MeOH (4:1, v/v) glass at 77 K. | ||
[OsL3](PF6)2 shows a broad luminescence spectrum with a maximum at 625 nm in air-equilibrated MeCN (ϕ = 0.05) which we suggest arises from the 3MLCT state (Fig. 4, inset). This contrasts with [RuL3](PF6)2 which is non-luminescent in fluid solution at room temperature. This situation arises with Ru(II) tris-diimine complexes when the 3MLCT and d–d states are sufficiently similar in energy for the d–d state to provide a rapid deactivation pathway.18 However the greater ligand-field splitting associated with Os(II) compared to Ru(II) in the same coordination environment means that the d–d state is now too high in energy to provide a thermally accessible deactivation pathway, and the lowest-energy 3MLCT state now shows luminescence. This emission is typical for Os(II) tris-diimines but, consistent with what was observed in the absorption spectrum, is notably higher in energy than that of [Os(bipy)3]2+ in fluid solution (λem = 743 nm).19 Luminescence lifetime measurements in MeCN revealed the presence of two lifetime components [200 ns (minor component) and 72 ns (major component)] which is consistent with the presence of the two isomers.
The 77 K emission maximum of [OsL3]2+ (as its chloride salt to provide solubility in the MeOH/EtOH solvent mixture) shows that the luminescence maximum is sharpened and slightly blue-shifted with the highest energy feature at 620 nm (Fig. 4, inset). This is typical behaviour for 3MLCT excited states, arising because the lack of solvent repolarisation when the sample is frozen destabilises the excited state. From the highest energy emission feature at 77 K we can see that the 3MLCT energy is 16100 cm−1, compared to 14100 cm−1 for [Os(bipy)3]2+.19 As the excited-state energy content of photoexcited [OsL3](PF6)2 is 2000 cm−1 higher than that of [Os(bipy)3]2+, but the cost of oxidising it to Os(III) is the same, it follows that photo-excited [OsL3](PF6)2 should be a better electron donor than [Os(bipy)3]2+ by ca. 0.25 eV, which makes it a considerably better excited-state electron donor than the well-known [Ru(bipy)3]2+ unit.20
Preparation and structural characterisation of heterometallic cages
In our recent communication11 we reported the first example of our stepwise methodology for heterometallic cage assembly, demonstrating how [RuL3]2+ combined with four Cd(II) ions to complete assembly of the heterometallic cage [Ru4Cd4L12](ClO4)16 according to Fig. 1, with the Ru(II) and Cd(II) ions necessarily lying in strictly alternating positions around the periphery. This cage was structurally characterised, with the significantly different M–N bond distances at Ru(II) and Cd(II) clearly differentiating the different types of metal ion in the expected positions. We have now extended this further to a mixed second row/first row metal ion combination by preparation of [Ru4Co4L12](BF4)16. A 1:1 mixture of [RuL3](BF4)2 and Co(BF4)2 was crystallised from MeNO2/di(isopropyl)ether to afford X-ray quality orange crystals of the mixed-metal cage. ES mass spectrometry confirmed the formulation with a series of peaks corresponding to the species {Ru4Co4L12(BF4)16−x}x+, i.e. the intact octanuclear cation associated with varying numbers of anions. We note also that the UV/Vis spectrum (see Experimental section for details) contained an absorption at 398 nm assigned to the MLCT absorption of the Ru(II) units whose extinction coefficient is approximately four times that of mononuclear [RuL3](BF4)2.11
The molecular structure derived from crystallographic data is shown in Fig. 5. The basic structure of the cage is similar to that of other [M8L12]16+ cages with the same ligand, having a metal ion at each vertex and a bis-bidentate ligand spanning each of the twelve edges.13 Extensive inter-ligand π-stacking around the periphery involves alternating arrays of electron-rich (naphthyl) and electron-deficient (coordinated pyrazolyl-pyridine) groups. Metal–metal separations along the cube edges are 11.3–11.4 Å.
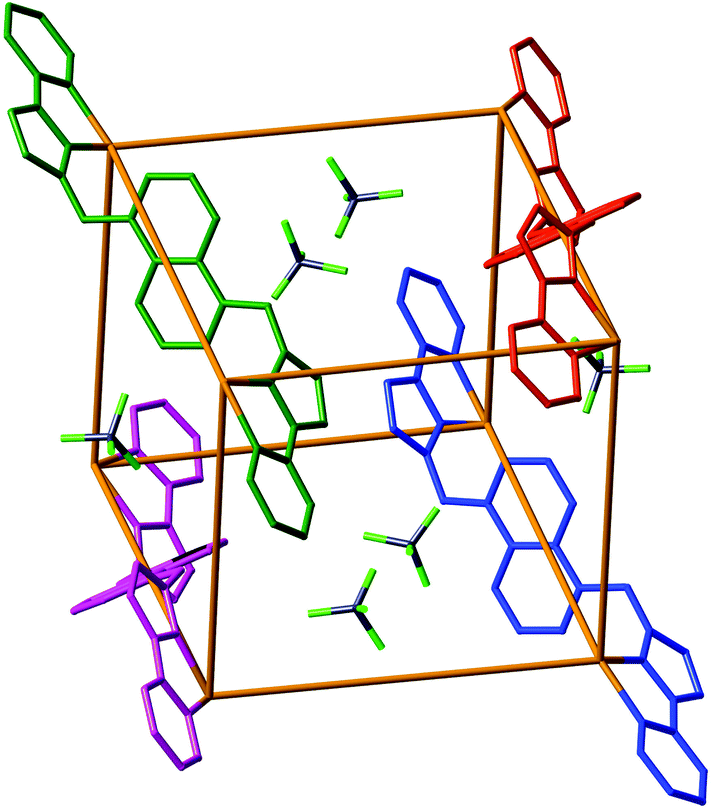 | ||
| Fig. 5 Partial structure of the complex cation of [Ru4Co4L12](BF4)16·3MeNO2. The metal ions lie at the corners of the cube with each vertex containing a 50:50 disordered mixture of Ru and Co atoms. Only four of the twelve bridging ligands – each one coloured separately for clarity – are included, as well as the [BF4]− anions which lie in the windows in the centre of the faces of the cubic cage assembly. Metal–N separations lie in the range 2.08–2.12 Å. | ||
In this case however, unlike with [Ru4Cd4L12](ClO4)16,11 the metal sites are indistinguishable crystallographically as the cage exhibits disorder over two orientations. If the two alternate sets of four positions in the cube superstructure (cf.Fig. 1) are labelled ‘A’ and ‘B’ then we have two possibilities: the cube can be oriented such that all four Ru(II) ions are in the A sites, and the four Co(II) ions are in the B sites: or it can be oriented such that all four Co(II) ions are in the A sites and the Ru(II) ions are in the B sites. This results in the metal ion occupancy at each site being 50% Ru and 50% Co. This affects only the metal ions and not the ligands; this disorder is presumably made easier by the fact that (high-spin) Co(II)–N and Ru(II)–N bond distances are similar, such that the positions of ligand atoms in the structure are fairly insensitive to which metal ion is at which terminus. The complex crystallises in space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) with only one sixth of the cage in the asymmetric unit, which therefore contains one complete mer tris-chelate metal ion and its coordination environment (50:50 mixture of Ru and Co), and one third of a fac tris-chelate metal ion which lies on a C3 axis that extends along the long diagonal of the cage, through both fac tris-chelate sites. Thus the cage has crystallographic S6 symmetry,13a with an inversion centre whose presence requires all metal sites to be equivalent. Of course for each individual molecule the inversion centre will be lost due to the inequivalence of the Co(II) and Ru(II) ions and the cage will have molecular C3 symmetry. Anions are located in the ‘windows’ in the centre of the faces, where they can participate in CH⋯F interactions with the ligands, but they do not occupy the central cavity. The cavity itself appears to be empty, but we note that the structure contained diffuse electron density which could not be refined and was removed from the refinement using the ‘SQUEEZE’ function in PLATON (see Experimental section and CIFs for details). Other structures of cages of this type, including the two described below, have included solvent molecules in the cavity close to the two hydrogen-bond donor sites associated with the fac tris-chelate vertices.13
with only one sixth of the cage in the asymmetric unit, which therefore contains one complete mer tris-chelate metal ion and its coordination environment (50:50 mixture of Ru and Co), and one third of a fac tris-chelate metal ion which lies on a C3 axis that extends along the long diagonal of the cage, through both fac tris-chelate sites. Thus the cage has crystallographic S6 symmetry,13a with an inversion centre whose presence requires all metal sites to be equivalent. Of course for each individual molecule the inversion centre will be lost due to the inequivalence of the Co(II) and Ru(II) ions and the cage will have molecular C3 symmetry. Anions are located in the ‘windows’ in the centre of the faces, where they can participate in CH⋯F interactions with the ligands, but they do not occupy the central cavity. The cavity itself appears to be empty, but we note that the structure contained diffuse electron density which could not be refined and was removed from the refinement using the ‘SQUEEZE’ function in PLATON (see Experimental section and CIFs for details). Other structures of cages of this type, including the two described below, have included solvent molecules in the cavity close to the two hydrogen-bond donor sites associated with the fac tris-chelate vertices.13
We next investigated the use of [OsL3]2+ as a building block for heterometallic cages in the same way; we note that Os(II)-containing coordination cages are almost unknown.21 Reaction of [OsL3](PF6)2 with Cd(ClO4)2 or Co(ClO4)2 in nitromethane afforded, after concentration and slow crystallisation of the solutions, red X-ray quality crystals of the mixed-metal cages [Os4Cd4L12](ClO4)16 and [Os4Co4L12](ClO4)16 respectively. The two structures are isostructural and isomorphous. Both show the same core [M8L12]16+ cage architecture as outlined above, and in both – as with the Ru4Co4 complex above – the metal ions exhibit twofold disorder such that all metal positions refine as a 1:1 mixture of Os/Cd or Os/Co respectively (Fig. 6). In [Os4Cd4L12](ClO4)16 a perchlorate ion (disordered over two closely-spaced positions) occupies the window in the centre of each face, and in this case we can see the O atoms of water molecule guests occupying the two fac tris-chelate H-bonding pockets on the interior surface of the cage, where several inwardly-directed CH protons converge.13c In [Os4Co4L12](ClO4)16, the [ClO4]− anions likewise occupy the windows in the centres of the cube faces, and again we can see two water molecules in the H-bonding pockets close to the fac tris-chelate metal vertices inside the cube (Fig. 7).
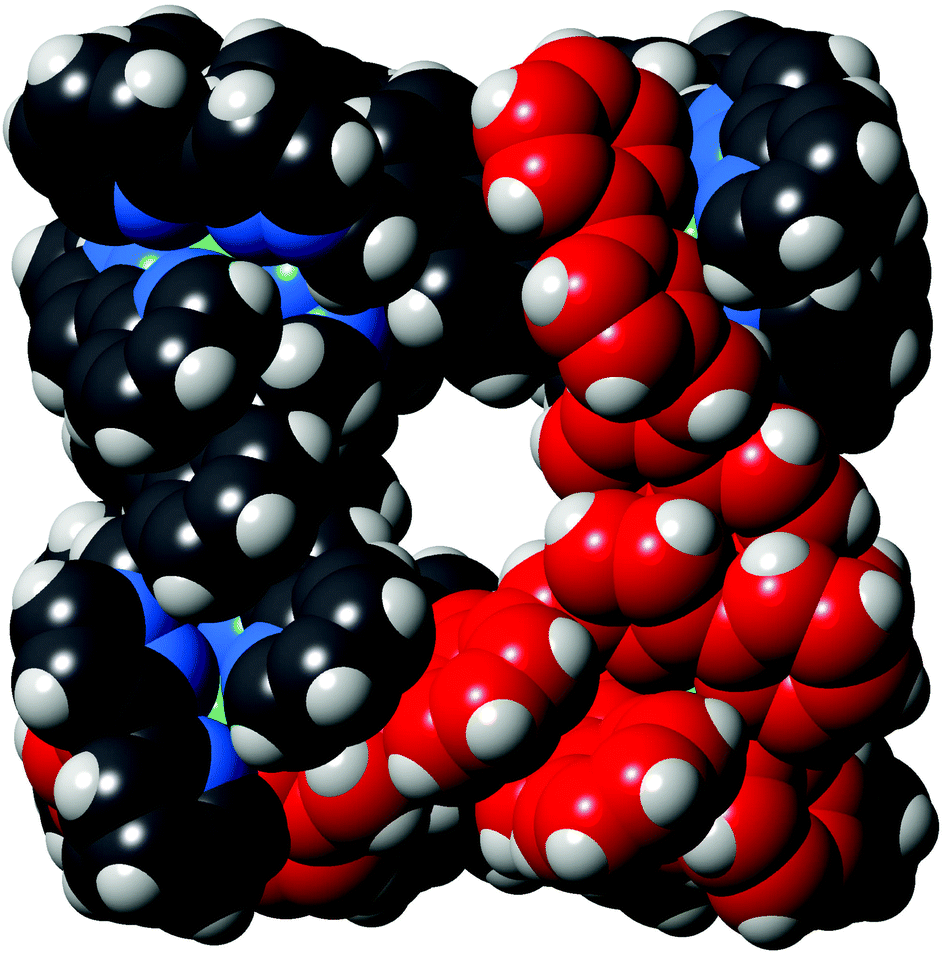 | ||
| Fig. 6 Space-filling view of the complex cation of [Os4Cd4L12](ClO4)16·H2O, with one of the pre-formed fac-[OsL3]2+ units highlighted in red. | ||
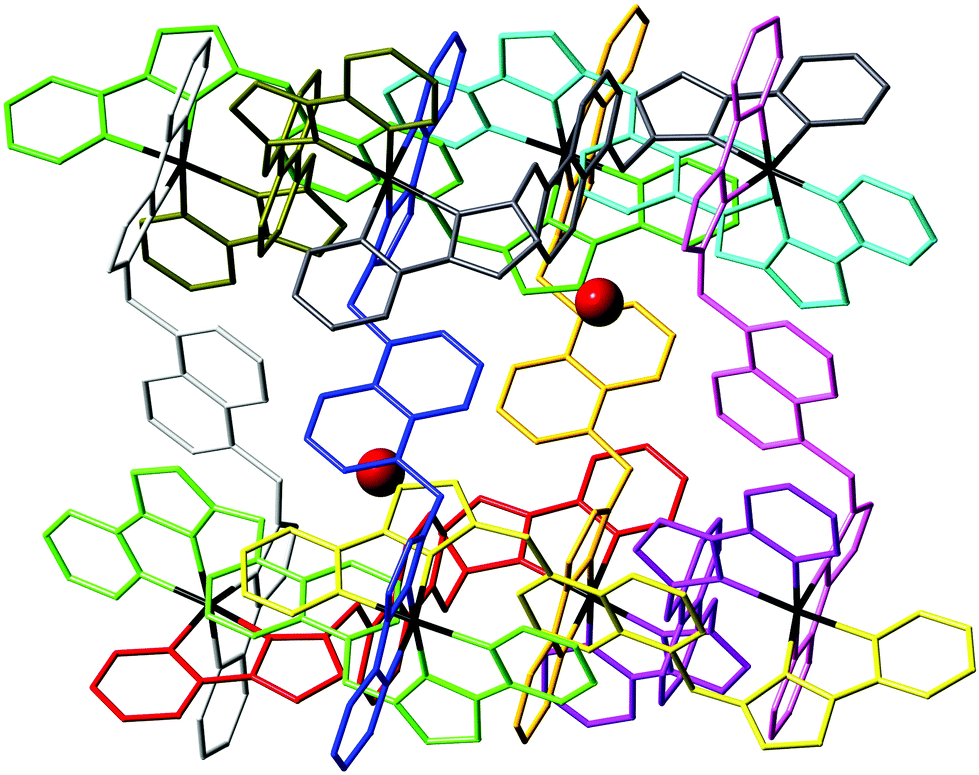 | ||
| Fig. 7 View of the complex cation of [Os4Co4L12](ClO4)16·H2O, with each ligand coloured separately for clarity, and the water molecule guests (50% site occupancy) that lie in the cavity included. | ||
In all three cases therefore we can confirm the basic structure crystallographically but disorder of the two types of metal ion prevents unambiguous identification of which metal ion is at which site. However, as mentioned above, the synthetic methods necessarily requires that the two types of metal ion strictly alternate around the periphery, and electrospray mass spectra confirm the formulations of the cage cations with masses and isotope patterns consistent with the expected mixture of four of each type of metal ion (Ru/Co, Os/Cd, Os/Co). Thus, for example, the ES mass spectrum of [Os4Cd4L12](ClO4)16 shows a series of peaks corresponding to the loss of different numbers of anions to give the sequence {Os4Cd4L12(ClO4)16−x}x+ and the other mixed-metal complexes behave similarly. Expansion of sample high-resolution ESMS signals for the Os4Cd4 and Os4Co4 cages are in Fig. 8.
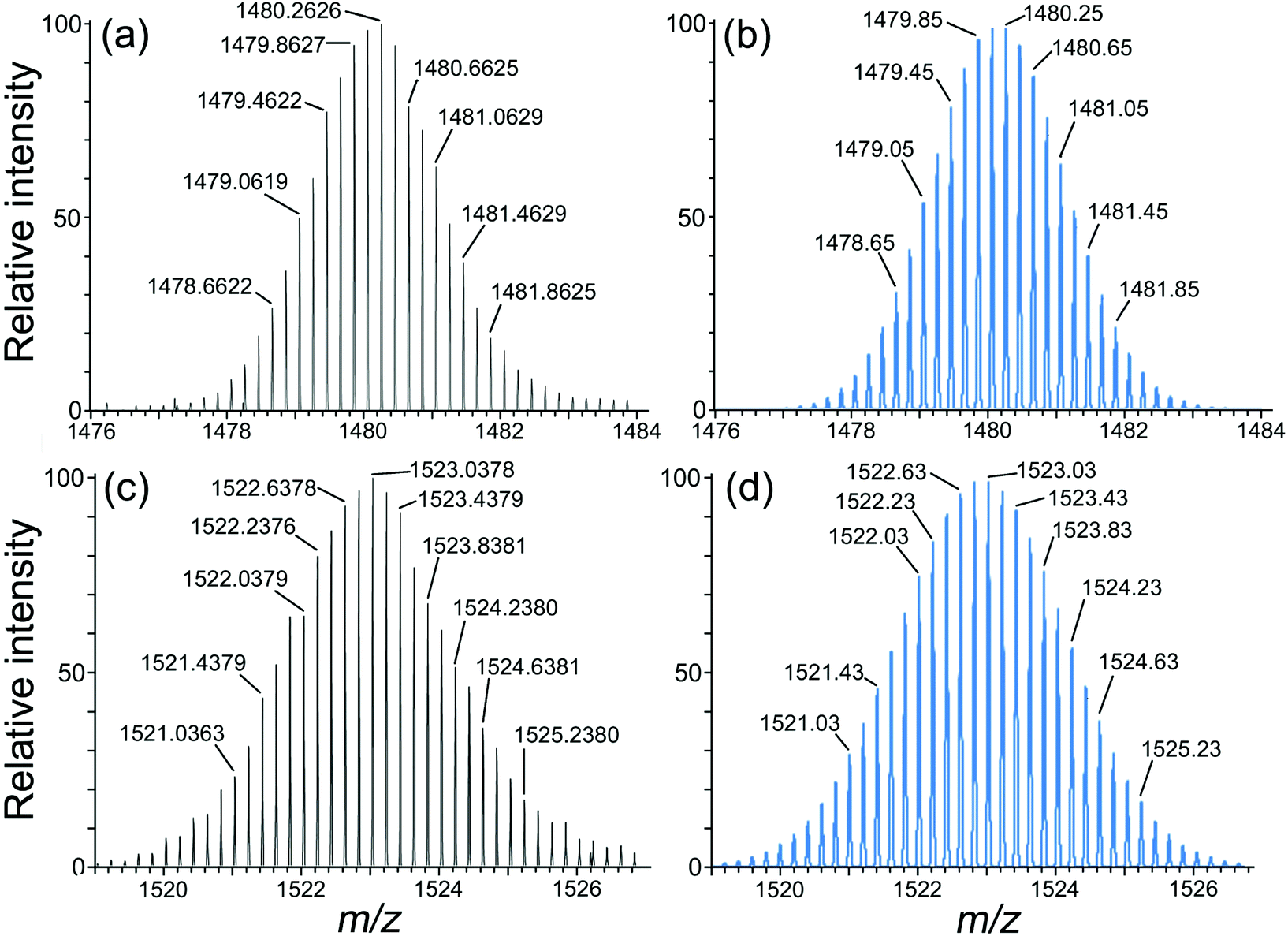 | ||
| Fig. 8 Expansions of high-resolution ESMS signals corresponding to the ions {[Os4M4L12](ClO4)11}5+ (M = Co, Cd). Top: M = Co [(a), observed; (b), simulated]. Bottom: M = Cd [(c) observed; (d), simulated]. Note the spacing of 0.2 mass units between adjacent peaks within each manifold. | ||
1H NMR spectroscopy was of limited value. We have used 1H NMR spectra successfully to characterise both diamagnetic (Cd8) and paramagnetic (Co8) cages of this family.13 The presence of two independent ligand environments, with no internal symmetry, in the homonuclear cages results in 44 1H signals. In the Cd8 cage enough of these could be resolved to show that the correct symmetry was present,13a and in the Co8 cage the paramagnetic high-spin Co(II) centres act as a shift reagent to spread the signals out over a range of ca. 200 ppm.13 However loss of the molecular inversion centre due to the presence of two types of metal ion means that there are now four magnetically inequivalent ligand environments, giving 88 independent 1H signals between 5 and 8.5 ppm for [Os4Cd4L12](ClO4)16. Whilst these are not individually resolved sufficiently to make any sensible attempts at assignments even at 500 MHz, a DOSY spectrum confirms that all of the signals have the same diffusion coefficient D and the molecule clearly retains its structural integrity in solution. Fig. 9 illustrates this by comparing the 1–D and DOSY spectra of mononuclear [OsL3](PF6)2 and the cage [Os4Cd4L12](ClO4)16 in MeCN, from which it is obvious that the larger complex has significantly slower diffusion in solution. For pseudo-spherical compounds D is proportional to 1/r: from Fig. 9 we can see that the difference in the logD values is ca. 0.5 implying that the cage complex has an effective radius ca. 3 times larger than the mononuclear starting material.
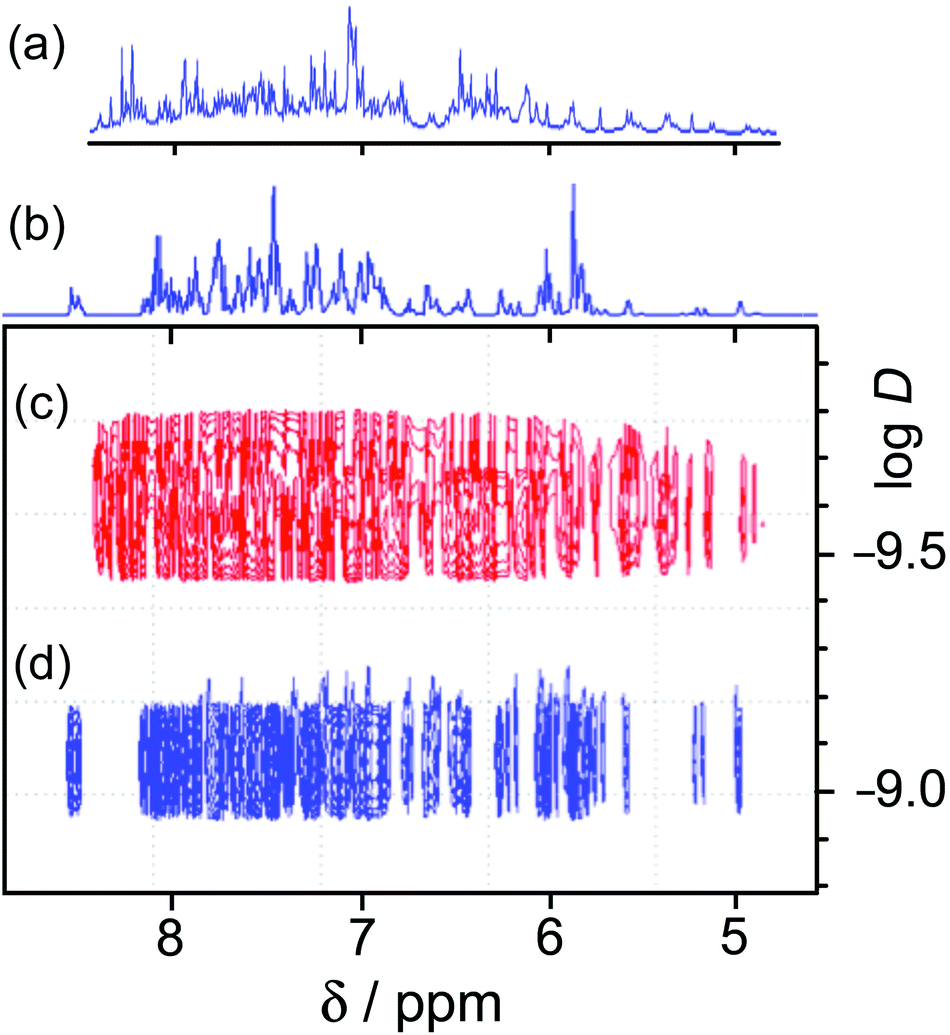 | ||
| Fig. 9 Comparison of 1–D and DOSY 1H NMR spectra for [OsL3](PF6)2 and [Os4Cd4L12](ClO4)16 in CD3CN: (a) 1–D spectrum of [Os4Cd4L12](ClO4)16; (b) 1–D spectrum of [OsL3](PF6)2; (c) DOSY spectrum of [Os4Cd4L12](ClO4)16; (d) DOSY spectrum of [OsL3](PF6)2. | ||
Redox and photophysical properties of [Os4Cd4L12](ClO4)16
In the earlier communication we showed how inclusion of Ru(II) ions in the [Ru4Cd4L12](ClO4)16 cage imparted reversible redox activity into the cage.11 In this section we focus on the new Os(II) containing cages which incorporate not just redox activity but also luminescence from the Os(II) centres. Given the potentially quenching effect of Co(II) ions due to their low-lying d–d states, we have used [Os4Cd4L12](ClO4)16 for these studies.Cyclic voltammetry of [Os4Cd4L12](ClO4)16 in MeCN gave very similar results as for mononuclear [OsL3](PF6)2, with a symmetric wave at +0.43 V vs. ferrocene/ferrocenium having a peak–peak separation of 80 mV (Fig. 3). Slightly surprisingly, given the 16+ positive charge on the cage, this redox potential is around 30 mV less positive than for mononuclear [OsL3](PF6)2. Clearly all four Os(II) centres oxidise and reduce essentially simultaneously with no electronic interaction between the redox centres, otherwise this wave would be either broader or split into multiple one-electron components. This modest redox potential allows the complex to oxidise slowly in air. The as-synthesised complex containing Os(II) centres is red, but it slowly turns green in solution when exposed to oxygen (Fig. 10); the collapse of the 1MLCT and 3MLCT bands in the visible region, and the growth of a weak long-wavelength band which extends into the red region beyond 900 nm [probably LMCT involving Os(III)], are both obvious. We note that the spectra in Fig. 10 could only be recorded from 380 nm at the high-energy end due to the limited solvent window of nitromethane which is the best solvent for this experiment as the oxidised complex precipitates from less polar solvents such as MeCN. Addition of ascorbic acid reversed the process and regenerates the spectrum of the fully reduced Os(II) form. The clear isosbestic point at ca. 600 nm confirms the chemical reversibility of the process. The stability of the cage in both [OsII4Cd4L12]16+ and [OsIII4Cd4L12]20+ forms is further confirmed by a DOSY spectrum: although the 1H NMR spectrum of the [OsIII4Cd4L12]20+ species after oxidation lacked resolution and could not be assigned, the DOSY spectrum shows no change in its diffusion coefficient compared to the starting complex (Fig. 11).
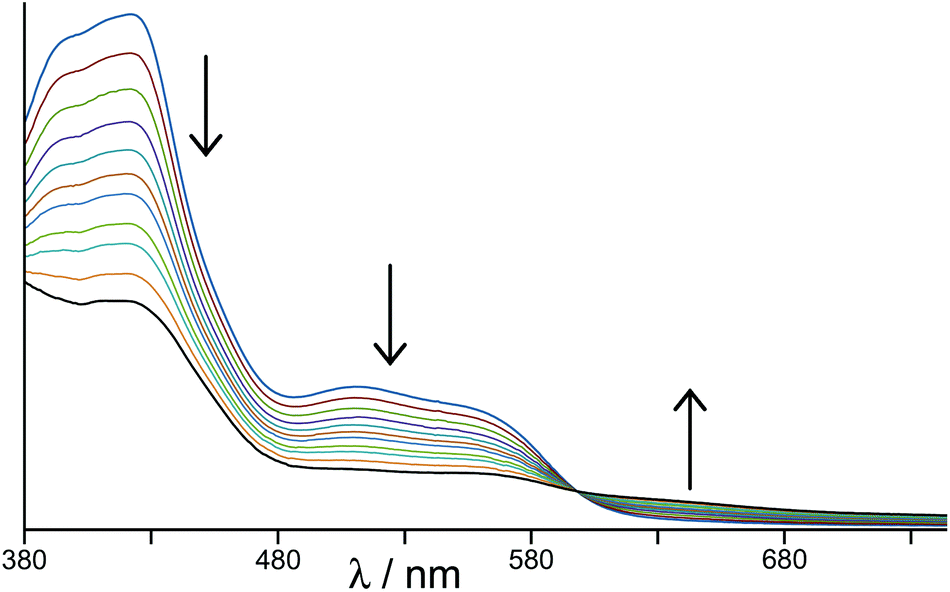 | ||
| Fig. 10 Change in electronic spectrum of [Os4Cd4L12](ClO4)16 as the four Os(II) centres oxidise in air to Os(III) (in MeNO2). | ||
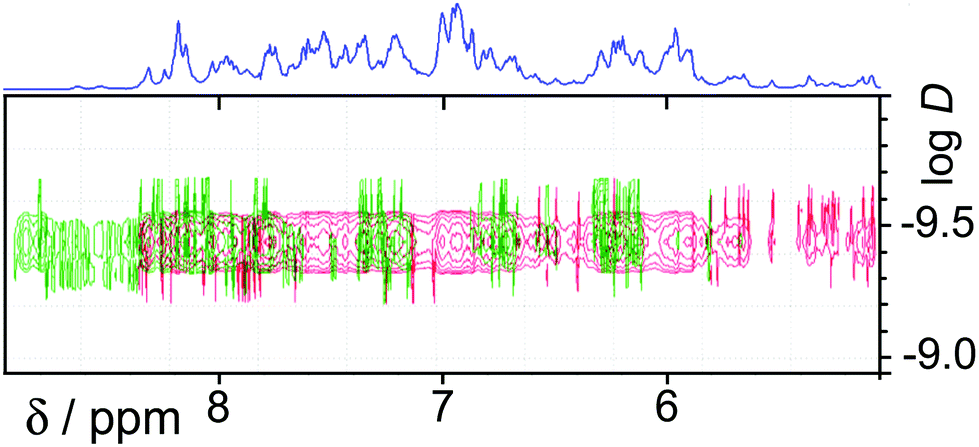 | ||
| Fig. 11 DOSY spectra of [Os4Cd4L12](ClO4)16 [red, Os(II) form] following aerial oxidation of the four Os(II) centres to Os(III) (green spectrum): there is no significant change in diffusion coefficient. | ||
[Os4Cd4L12](ClO4)16 also retains the photophysical properties of the component unit [OsL3](PF6)2. Its luminescence spectrum in solution is essentially identical to that of [OsL3](PF6)2 with a broad maximum at 625 nm in MeCN, with a quantum yield of 2.5% and, again, two lifetime components: 156 ns (minor) and 73 ns (major) which can be ascribed to the mixture of geometric isomers of the Os(II) units. Significantly, the strong naphthalene-based fluorescence characteristic of the free ligand, and which we also saw in the cage complex [Cd8L12](ClO4)16,13a is completely quenched; which implies the presence of (naphthyl) → Os(II) energy-transfer from the ligand array to the Os(II) ions at the vertices of the cage, presumably assisted by the aromatic π-stacking which brings naphthyl units into close association with Os(II) tris(pyrazolyl-pyridine) termini (see figures of crystal structures). The complex is not sufficiently soluble in solvents that give good low-temperature glasses to get a good 77 K luminescence spectrum, but based on the near-identical behaviour of the luminescence from mononuclear [OsL3](PF6)2 and the cage [Os4Cd4L12](ClO4)16 at room temperature, it is reasonable to assume that the 3MLCT energy of the chromophores in the cage is again 16100 cm−1 and that it will be a good excited-state photo-electron donor to electron-deficient guests that occupy the central cavity.
Conclusions
The stepwise synthetic method of cage assembly for which we reported the first example recently11 has been extended to complete the preparation of a set of four heterometallic self-assembled cubic cages, in which four kinetically inert ions [Ru(II) or Os(II)] and four kinetically labile ions [Cd(II) or Co(II)] alternate around the periphery of the cage superstructure. There are two particularly important features of the Os(II)-containing cages which will be exploited in future work. The first is the reversible redox activity at modest potential, which allows a four-electron redox swing to change the charge on the cage between 16+ and 20+; as guest binding in organic solvents is driven by polar interactions between the guest and the interior surface of the cage,13 this may provide a mechanism to modulate guest binding for controlled uptake/release. The second is the photophysical activity, with the four Os(II) units – which are good photo-electron donors in their excited states – surrounding the cavity where guests will bind, which opens the door for one-electron or even multi-electron photoinduced interactions between the cage and bound guests.Experimental section
General details
Metal salts and all organic reagents were purchased from Alfa or Sigma-Aldrich and used as received. NMR spectra were recorded on Bruker DRX 500 MHz, Bruker AV-III 400 MHz or AV-I 250 MHz instruments. UV/Vis absorption spectra were measured on a Varian Cary 50 spectrophotometer. Steady-state luminescence spectra were measured on a Jobin-Yvon Fluoromax 4 fluorimeter, using 1 cm cuvettes with samples sufficiently dilute to have an optical density of no more than 0.1 at the excitation wavelength. Os-based luminescence lifetimes were measured in air-equilibrated MeCN solutions by the time-correlated single-photon counting method, using an Edinburgh Instruments Mini-τ instrument, with fitting of the decay data to give lifetimes using the supplied software. Cyclic voltammetry was performed using an Autolab PGSTAT100 potentiostat controlled via the General Purpose Electrochemical System (GPES) version 4.9 software. A standard three-electrode configuration was used with Pt-bead working and counter electrodes and a Ag/AgCl reference; the base electrolyte was 0.1 M Bu4NPF6, and ferrocene was used as an internal calibrant. The ligand L was prepared according to the published method.13a Metal salts and other reagents were purchased from Alfa or Sigma Aldrich and used as received.Low-resolution electrospray mass spectra were recorded on a Micromass LCT instrument. High-resolution mass spectra (e.g.Fig. 8) were recorded using an electron transfer dissociation (ETD) enabled ThermoFisher-Scientific Orbitrap Elite, equipped with an HESI source (ThermoFisher Scientific). Samples were delivered by syringe pump for direct infusion at 5 μL min−1. Electrospray ionization was carried out at 4 kV with a source temperature of 50 °C, sheath gas set at 5, ion transfer capillary at 275 °C, and S-lens setting of 60%. MS1 spectra were acquired at a resolving power of 240000 with an automatic gain control (AGC) target value of 1 × 106 ions by the Orbitrap detector, and a trap fill time of 500 ms over a range of 750–2000 m/z.
Preparation of [OsL3](PF6)2
A mixture of OsCl3·3H2O (0.20 g, 0.57 mmol) and L (1.30 g, 2.85 mmol) in ethylene glycol was heated to reflux under N2 for 12 hours. After cooling to room temperature a saturated aqueous solution of KPF6 was added to precipitate the crude product, which was collected by filtration. The filtrate was washed copiously with water and then desiccated overnight. The crude solid was dissolved in acetonitrile and purified by column chromatography on silica by elution with MeCN/water/saturated aqueous KNO3 (100:4:2, v/v/v). The main red band was collected and solvent was removed to give a dark red/orange solid. The product was dissolved in water and aqueous KPF6 was added to precipitate pure [OsL3](PF6)2 (mixture of fac and mer isomers). The solid was washed with water before drying in vacuo. Yield: 0.25 g, 0.14 mmol, 24%. ESMS: m/z 1664 (M − PF6)+, 760 (M − 2PF6)2+. Accurate mass, calculated for the complex dication: (C84H66N18Os)/2 = 759.2667; measured 759.2641. Elemental analysis: C, 55.9; H, 3.7; N, 13.4% (expected for C84H66F12N18OsP2: C, 55.8; H, 3.7; N, 14.0%). UV/Vis in MeCN [λmax/nm (10−3ε/M−1 cm−1)]: 570 (sh), 515 (sh), 425 (12.5), 400 (sh), 283 (80.4), 226 (149).
Preparation of [Os4Cd4L12](ClO4)16
A solution of [OsL3](PF6)2 (0.05 g, 0.028 mmol) and excess Cd(ClO4)2·6H2O (0.12 g, 0.28 mmol) in nitromethane was stirred overnight. The mixture was filtered over a small amount of L-ascorbic acid to ensure that sample was fully reduced [i.e. that any Os(III) from aerial oxidation was reduced back to Os(II), see main text], then crystallised by slow diffusion of di-isopropyl ether into the MeNO2 solution. The crystalline product was collected by filtration and washed with di-isopropyl ether, diethyl ether, and cold methanol. The remaining red crystalline precipitate was the pure product. X-Ray quality crystals were grown by slow diffusion of di-isopropyl ether into an MeNO2 solution. Yield: 0.05 g, 0.005 mmol, 21%. ESMS: m/z 1523.03 (M − 5ClO4)5+, 1059.47 (M − 7ClO4)7+, 801.93 (M − 9ClO4)9+ (see Fig. 8 for high-resolution ESMS). Elemental analysis: C, 49.0; H, 3.5; N, 13.0% (expected for C348H290Cd4Cl16N78O68Os4: C, 49.6; H, 3.5; N, 13.3%). UV/Vis in MeCN [λmax/nm (10−3ε/M−1 cm−1)]: 540 (sh), 426 (47.8), 400 (sh), 287 (309), 227 (557).Preparation of [Os4Co4L12](ClO4)16
This was prepared in the identical way to [Os4Cd4L12](ClO4)16 (above) but starting from [OsL3](PF6)2 (0.05 g, 0.028 mmol) and excess Co(ClO4)2·6H2O (0.10 g, 0.19 mmol). X-Ray quality crystals were grown by slow diffusion of di-isopropyl ether into an MeNO2 solution of the product. Yield: 0.04 g, 0.005 mmol, 19%. ESMS: m/z 1480 (M − 5ClO4)5+, 1217 (M − 6ClO4)6+, 1029 (M − 7ClO4)7+ (see Fig. 8 for high-resolution ESMS). Elemental analysis of material after removal of organic solvents in vacuo was consistent with uptake of a large number of water molecules into the voids in the crystals and is therefore of limited use.Preparation of [Ru4Co4L12](BF4)16
To a stirred solution of [RuL3](PF6)2 (0.003 g, 0.002 mmol) in nitromethane (20 cm3) was added an excess of Co(BF4)2·6H2O (0.004 g, 0.013 mmol). After stirring overnight, the mixture was evaporated to dryness and then washed with dichloromethane and methanol. Slow diffusion of diisopropyl ether into a solution of the powder in nitromethane gave the product as small orange blocks. Yield: 0.002 g, 50%. ESMS: m/z 1748 (M − 4BF4)4+, 1381 (M − 5 BF4)5+, 1136 (M − 6 BF4)6+, 962 (M − 7 BF4)7+, 831 (M − 8 BF4)8+, 729 (M − 9 BF4)9+. UV/Vis in MeCN [λmax/nm (10−3ε/M−1 cm−1)]: 398 (57), 285 (330), 227 (630). Elemental analysis of material after removal of organic solvents in vacuo was consistent with uptake of a large and variable number of water molecules into the voids in the crystals and is therefore of limited use.X-ray crystallography
For [Os4Cd4L12](ClO4)16·H2O and [Os4Co4L12](ClO4)16·H2O, diffraction data were collected at the EPSRC National Crystallography Service at the University of Southampton, using a Rigaku FR-E+ diffractometer equipped with a Saturn 724+ CCD detector, using high-intensity Mo-Kα radiation from either a rotating anode or a microfocus sealed-tube source.22 For [Ru4Co4L12](BF4)16·3MeNO2, data were collected on a Bruker Apex-II diffractometer at the University of Sheffield. In each case a crystal was removed from the mother liquor, coated with oil, and transferred rapidly to a stream of cold N2 on the diffractometer to prevent any decomposition due to solvent loss. In all cases, after integration of the raw data, and before merging, an empirical absorption correction was applied (SADABS)23 based on comparison of multiple symmetry-equivalent measurements. The structures were solved by direct methods and refined by full-matrix least squares on weighted F2 values for all reflections using the SHELX suite of programs.24 Pertinent crystallographic data are collected in Table 1.| Complex | [Ru4Co4L12](BF4)16·3MeNO2 | [Os4Cd4L12](ClO4)16·H2O | [Os4Co4L12](ClO4)16·H2O |
|---|---|---|---|
| a The value of R1 is based on ‘observed’ data with I > 2σ(I); the value of wR2 is based on all data. | |||
| Formula | C339H273B16Co4F64N75O6Ru4 | C336H266Cd4Cl16N72O65Os4 | C336H266Cl16Co4N72O65Os4 |
| Molecular weight | 7522.27 | 8129.79 | 7915.91 |
| T, K | 100(2) | 100(2) | 100(2) |
| Crystal system | Trigonal | Trigonal | Trigonal |
| Space group |
R |
R |
R |
| a, Å | 34.091(2) | 28.987(8) | 28.9579(16) |
| b, Å | 34.091(2) | 28.987(8) | 28.9579(16) |
| c, Å | 35.5084(17) | 54.10(2) | 52.757(3) |
| V, Å3 | 35740(5) |
39357(28) |
38313(5) |
| Z | 3 | 3 | 3 |
| ρ, g cm−3 | 1.048 | 1.029 | 1.029 |
| Crystal size, mm3 | 0.2 × 0.1 × 0.1 | 0.2 × 0.15 × 0.06 | 0.05 × 0.05 × 0.05 |
| μ, mm−1 | 0.334 | 1.261 | 1.259 |
| Independent data, restraints, parameters | 8305/672/620 | 19846, 728, 618 | 14953, 576, 555 |
| Final R1, wR2a | 0.077, 0.246 | 0.153, 0.445 | 0.127, 0.448 |
In all cases crystals exhibited the usual problems of this type of structure, viz. weak scattering due to a combination of poor crystallinity, solvation, and disorder of anions/solvent molecules. All three structures contained large solvent-accessible voids whose volume was ca. 40% of the total unit cell volume. These showed in the refinement to contain diffuse electron density which could not meaningfully be modelled, ascribed to severely disordered solvent molecules as well as those anions that could not be located. This diffuse electron density was removed from the refinements using the SQUEEZE function in PLATON.25 As a typical example, in the structure of [Os4Cd4L12](ClO4)16·H2O the electron density removed using ‘SQUEEZE’ corresponded to ca. 900 electrons per complete cage, corresponding to the missing perchlorate anion plus 15– 26 solvent molecules [depending on the balance between di(isopropyl)ether (58 e−) and nitromethane (32 e−)]. The other two structures behave comparably and full details are in the individual CIFs. To assist in the refinements, the number of parameters was kept as low as possible by extensive use of geometric restraints on aromatic rings and anions (e.g. pyridyl and pyrazolyl rings were refined as idealised hexagons and pentagons, respectively; and perchlorate/tetrafluoroborate anions as idealised tetrahedra), as well as global restraints on aromatic displacement parameters. This helped to keep refinements stable.
As a consequence of this the refinements are of poor quality by normal small-molecule standards, but are quite typical for large cage structures of this type. We emphasise that in each case the basic structure and connectivity of the complex cation could be unambiguously determined with reasonable precision and we use the structures only for that purpose with no detailed analysis of structural minutiae. Full details are in the individual CIFs. CCDC numbers 1413546–1413548.
Acknowledgements
We thank the Leverhulme Trust (grant RPG-2013-308) and EPSRC (grant EP/K003224/1) for financial support. We also thank Dr R. Beniston for recording the high-resolution mass spectra. These mass spectrometry analyses were undertaken in biOMICS, the University of Sheffield Faculty of Science Mass Spectrometry Facility, supported by funding from Yorkshire Cancer Research (SHEND01), the European Structural Fund Programme, and the University of Sheffield Alumni Fund.References
- (a) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS PubMed; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed; (c) M. D. Ward, Chem. Commun., 2009, 4487 RSC; (d) J. J. Perry, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400 RSC; (e) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015 CrossRef CAS PubMed; (f) A. F. Williams, Coord. Chem. Rev., 2011, 255, 2104 CrossRef CAS PubMed; (g) Z. Laughrey and B. Gibb, Chem. Soc. Rev., 2011, 40, 363 RSC; (h) P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2012, 254, 1760 CrossRef PubMed; (i) R. J. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (j) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349 CrossRef CAS PubMed; (k) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed; (l) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC; (m) T. Nakamura, H. Ube and M. Shionoya, Chem. Lett., 2014, 42, 328 CrossRef; (n) H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293–294, 139 CrossRef CAS PubMed; (o) L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201 CrossRef CAS PubMed.
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619 RSC.
- (a) J. W. Yi, N. P. E. Barry, M. A. Furrer, O. Zava, P. J. Dyson, B. Therrien and B. H. Kim, Bioconjugate Chem., 2012, 23, 461 CrossRef CAS PubMed; (b) B. Therrien, Chem. – Eur. J., 2013, 19, 8378 CrossRef CAS PubMed; (c) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778 RSC; (d) W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 625 RSC.
- (a) C. J. Brown, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 17530 CrossRef CAS PubMed; (b) C. J. Hastings, M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 6938 CrossRef CAS PubMed; (c) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958 CrossRef CAS PubMed; (d) H. Vardhan and F. Verpoort, Adv. Synth. Catal., 2015, 357, 1351 CrossRef CAS PubMed; (e) X. Jing, C. He, Y. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 3967 CrossRef CAS PubMed; (f) C. García-Simón, R. Gramage-Doria, S. Raoufmoghaddam, T. Parella, M. Costas, X. Ribas and J. N. H. Reek, J. Am. Chem. Soc., 2015, 137, 2680 CrossRef PubMed; (g) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed.
- (a) O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334 CrossRef CAS PubMed; (b) K. Yamashita, M. Kawano and M. Fujita, Chem. Commun., 2007, 4102 RSC.
- (a) X. Sun, D. W. Johnson, D. L. Caulder, K. N. Raymond and E. H. Wong, J. Am. Chem. Soc., 2001, 123, 2752 CrossRef CAS PubMed; (b) S. Hiraoka, Y. Sakata and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 10058 CrossRef CAS PubMed.
- (a) W. J. Ramsay, T. K. Ronson, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 13439 CrossRef CAS PubMed; (b) F. E. Hahn, M. Offermann, C. SchulzeIsfort, T. Pape and R. Frohlich, Angew. Chem., Int. Ed., 2008, 47, 6794 CrossRef CAS PubMed; (c) H.-B. Wu and Q.-M. Wang, Angew. Chem., Int. Ed., 2009, 48, 7343 CrossRef CAS PubMed; (d) A. J. Metherell and M. D. Ward, RSC Adv., 2013, 3, 14281 RSC.
- E. C. Constable, A. J. Edwards, P. R. Raithby and J. V. Walker, Angew. Chem., Int. Ed. Engl., 1993, 32, 1465 CrossRef PubMed.
- F. Reichel, J. K. Clegg, K. Gloe, K. Gloe, J. J. Weigand, J. K. Reynolds, C.-G. Li, J. R. Aldrich-Wright, C. J. Kepert, L. F. Lindoy, H.-C. Yao and F. Li, Inorg. Chem., 2014, 53, 688 CrossRef CAS PubMed.
- (a) V. C. M. Smith and J.-M. Lehn, Chem. Commun., 1996, 2733 RSC; (b) M. M. J. Smulders, A. Jimenez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681 CrossRef CAS PubMed; (c) K. Li, L.-Y. Zhang, C. Yan, M. Pan, L. Zhang and C.-Y. Su, J. Am. Chem. Soc., 2014, 136, 4456 CrossRef CAS PubMed; (d) M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2013, 19, 10170 CrossRef CAS PubMed; (e) A. Galstyan, P. J. Sanz Miguel, K. Weise and B. Lippert, Dalton Trans., 2013, 42, 16151 RSC; (f) P. de Wolf, S. L. Heath and J. A. Thomas, Chem. Commun., 2002, 2540 RSC; (g) M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 17743 CrossRef CAS PubMed.
- A. J. Metherell and M. D. Ward, Chem. Commun., 2014, 50, 6330 RSC.
- (a) Y.-T. Chan, X. Li, J. Yu, G. A. Carri, C. N. Moorefield, G. R. Newkome and C. Wesdemiotis, J. Am. Chem. Soc., 2011, 133, 11967 CrossRef CAS PubMed; (b) H. Sato, A. Nakao and A. Yamagishi, New J. Chem., 2011, 35, 1823 RSC.
- (a) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed; (b) S. Turega, M. Whitehead, B. R. Hall, M. F. Haddow, C. A. Hunter and M. D. Ward, Chem. Commun., 2012, 48, 2752 RSC; (c) S. Turega, M. Whitehead, B. R. Hall, A. J. H. M. Meijer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2013, 52, 1122 CrossRef CAS PubMed.
- S. L. Dabb and N. C. Fletcher, Dalton Trans., 2015, 44, 4406 RSC.
- (a) I. S. Tidmarsh, B. F. Taylor, M. J. Hardie, L. Russo, W. Clegg and M. D. Ward, New J. Chem., 2009, 33, 366 RSC; (b) R. L. Paul, Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4883 CrossRef CAS PubMed.
- (a) S. P. Argent, H. Adams, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding, O. Mamula and M. D. Ward, Inorg. Chem., 2006, 45, 3905 CrossRef CAS PubMed; (b) Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 2002, 41, 2515 CrossRef CAS.
- (a) A. J. Metherell, W. Cullen, A. Stephenson, C. A. Hunter and M. D. Ward, Dalton Trans., 2014, 43, 71 RSC; (b) A. J. Metherell and M. D. Ward, Chem. Commun., 2014, 50, 10979 RSC; A. J. Metherell and M. D. Ward, Polyhedron, 2015, 89, 260 Search PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- L. De Cola, V. Balzani, F. Barigelletti, L. Flamigni, P. Belser, A. von Zelewsky, M. Frank and F. Vögtle, Inorg. Chem., 1993, 32, 5228 CrossRef CAS.
- C.-T. Lin and N. Sutin, J. Phys. Chem., 1976, 80, 97 CrossRef CAS.
- N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, J. Organomet. Chem., 2012, 705, 1 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- G. M. Sheldrick, SADABS: A program for absorption correction with the Siemens SMART system, University of Göttingen, Germany, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS; P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 194 CrossRef.
Footnote |
| † CCDC 1413546–1413548. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02957k |
| This journal is © The Royal Society of Chemistry 2015 |