
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRare-earth metal methylidene complexes with Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3 core structure†
Dorothea
Schädle
a,
Melanie
Meermann-Zimmermann
b,
Cäcilia
Maichle-Mössmer
a,
Christoph
Schädle
a,
Karl W.
Törnroos
c and
Reiner
Anwander
*a
aInstitut für Anorganische Chemie, Universität Tübingen, Auf der Morgenstelle 18, D-72076 Tübingen, Germany. E-mail: reiner.anwander@uni-tuebingen.de
bDezernat Forschung, Technische Universität Darmstadt, Karolinenplatz 5, D-64289 Darmstadt, Germany
cDepartment of Chemistry, University of Bergen, Allégaten 41, N-5007 Bergen, Norway
First published on 15th September 2015
Abstract
Trinuclear rare-earth metal methylidene complexes with a Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3 structural motif were synthesized by applying three protocols. Polymeric [LuMe3]n (1-Lu) reacts with the sterically demanding amine H[NSiMe3(Ar)] (Ar = C6H3iPr2-2,6) in tetrahydrofuran via methane elimination to afford isolable monomeric [NSiMe3(Ar)]LuMe2(thf)2 (4-Lu). The formation of trinuclear rare-earth metal tetramethyl methylidene complexes [NSiMe3(Ar)]3Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3(thf)3 (7-Ln; Ln = Y, Ho, Lu) via reaction of [LnMe3]n (1-Ln; Ln = Y, Ho, Lu) with H[NSiMe3(Ar)] is proposed to occur via an “intermediate” species of the type [NSiMe3(Ar)]LnMe2(thf)x and subsequent C−H bond activation. Applying Lappert's concept of Lewis base-induced methylaluminate cleavage, compounds [NSiMe3(Ar)]Ln(AlMe4)2 (5-Ln; Ln = Y, La, Nd, Ho) were converted into methylidene complexes 7-Ln (Ln = Y, Nd, Ho) in the presence of tetrahydrofuran. Similarly, tetramethylgallate complex [NSiMe3(Ar)]Y(GaMe4)2 (6-Y) could be employed as a synthesis precursor for 7-Y. The molecular composition of complexes 4-Ln, 5-Ln, 6-Y and 7-Ln was confirmed by elemental analyses, FTIR spectroscopy, 1H and 13C NMR spectroscopy (except for holmium derivatives) and single-crystal X-ray diffraction. The Tebbe-like reactivity of methylidene complex 7-Nd with 9-fluorenone was assessed affording oxo complex [NSiMe3(Ar)]3Nd3(μ3-O)(μ2-Me)4(thf)3 (8-Nd). The synthesis of 5-Ln yielded [NSiMe3(Ar)]2Ln(AlMe4) (9-Ln; Ln = La, Nd) as minor side-products, which could be obtained in moderate yields when homoleptic Ln(AlMe4)3 were treated with two equivalents of K[NSiMe3(Ar)].
Introduction
Multiple metal–carbon bonds, predominantly alkylidene units “M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR2”, continue to trigger immense research activities in organometallic synthesis and catalysis.1–8 Schrock's neopentylidene complex (tBuCH2)3Ta(CHtBu)9,10 marked the first example of a stable transition metal alkylidene complex, with the bulky tBu substituents at the α-carbon atom impeding intermolecular decomposition pathways. In contrast, methylidene “CH22−” species are sterically less protected against bimolecular reactions, and hence present a particular challenge and are found to be kinetically labile. Although authenticated by single-crystal X-ray diffraction, the first transition metal methylidene complex Cp2Ta(CH2)(CH3) decomposes bimolecularly forming ethylene complex Cp2Ta(C2H4)(CH3).11,12 Current progress in the field of early transition metal terminal methylidene complexes features Mindiola's group 4 and group 5 derivatives (PNP)MCH2(OAr) (M = Zr, Hf, PNP = N[2-P(iPr)2-4-CH3-C6H3]2, Ar = C6H3iPr2-2,6) and (ArO)2NbCH2(H2CPPh3) (Ar = C6H2(CHPh2)2-2,6-tBu-4).13,14 Alternatively, stabilization of the methylidene moiety is achieved by delocalizing the negative charge over multinuclear, homometallic or heterobimetallic Lewis-acid stabilized complexes.15–18 The most prominent example of a Lewis-acid stabilized heterobimetallic methylidene complex is the Tebbe reagent Cp2Ti[(μ2-CH2)(μ2-Cl)AlMe2],5,19 whose structural elucidation proved to be delicate.20 The Tebbe reagent also laid the ground work for a series of rare-earth metal variants, (L)Ln(III)(CH2)x(AlMe2R)y (L = monoanionic,21–25 neutral ligand;26–29 R = Me, ferrocenyl). Previous studies from our laboratory and others on rare-earth-metal(III) alkyl complexes (L)LnRR′ (L = C5Me5(= Cp*), NSiMe3(Ar), PhC(NC6H3iPr2-2,6)2; Ln = Y, La, Ho, Lu; R, R′ = Me, AlMe4, Cl) led to the isolation of “Lewis-acid-free” LnIII methylidene complexes with a striking Ln3(μ3-CH2) core structure (Scheme 1).30–32
CR2”, continue to trigger immense research activities in organometallic synthesis and catalysis.1–8 Schrock's neopentylidene complex (tBuCH2)3Ta(CHtBu)9,10 marked the first example of a stable transition metal alkylidene complex, with the bulky tBu substituents at the α-carbon atom impeding intermolecular decomposition pathways. In contrast, methylidene “CH22−” species are sterically less protected against bimolecular reactions, and hence present a particular challenge and are found to be kinetically labile. Although authenticated by single-crystal X-ray diffraction, the first transition metal methylidene complex Cp2Ta(CH2)(CH3) decomposes bimolecularly forming ethylene complex Cp2Ta(C2H4)(CH3).11,12 Current progress in the field of early transition metal terminal methylidene complexes features Mindiola's group 4 and group 5 derivatives (PNP)MCH2(OAr) (M = Zr, Hf, PNP = N[2-P(iPr)2-4-CH3-C6H3]2, Ar = C6H3iPr2-2,6) and (ArO)2NbCH2(H2CPPh3) (Ar = C6H2(CHPh2)2-2,6-tBu-4).13,14 Alternatively, stabilization of the methylidene moiety is achieved by delocalizing the negative charge over multinuclear, homometallic or heterobimetallic Lewis-acid stabilized complexes.15–18 The most prominent example of a Lewis-acid stabilized heterobimetallic methylidene complex is the Tebbe reagent Cp2Ti[(μ2-CH2)(μ2-Cl)AlMe2],5,19 whose structural elucidation proved to be delicate.20 The Tebbe reagent also laid the ground work for a series of rare-earth metal variants, (L)Ln(III)(CH2)x(AlMe2R)y (L = monoanionic,21–25 neutral ligand;26–29 R = Me, ferrocenyl). Previous studies from our laboratory and others on rare-earth-metal(III) alkyl complexes (L)LnRR′ (L = C5Me5(= Cp*), NSiMe3(Ar), PhC(NC6H3iPr2-2,6)2; Ln = Y, La, Ho, Lu; R, R′ = Me, AlMe4, Cl) led to the isolation of “Lewis-acid-free” LnIII methylidene complexes with a striking Ln3(μ3-CH2) core structure (Scheme 1).30–32
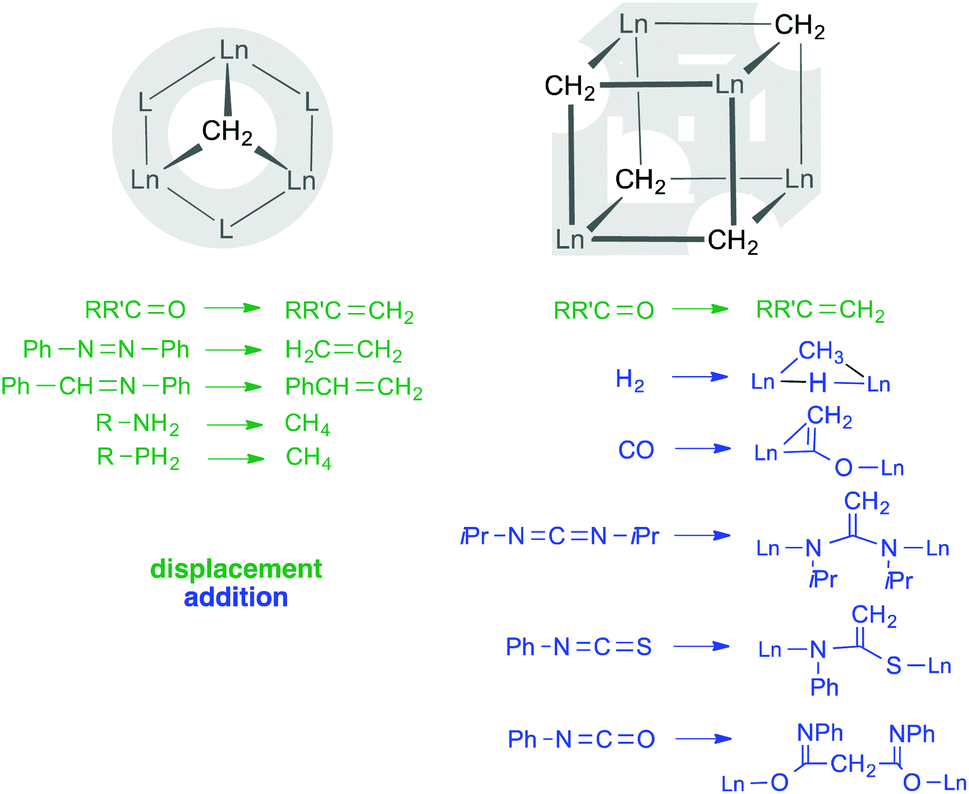 | ||
| Scheme 1 Stabilization and reactivity of CH22− methylidene moieties in trigonal pyramidal and cuboid arrangements with Ln(III) centres. | ||
Experimental and theoretical studies on homometallic trinuclear rare-earth metal methylidene complexes L3Ln3(μ3-CH2)(μ3-CH3)(μ2-CH3)3(thf)n (L = monoanionic ligand) revealed Tebbe-like reactivity in methylenation reactions (Scheme 1) along with methylation of the carbonylic functionalities.31–33 In sharp contrast, treatment of Cp′3Tm3(μ3-CH2)(μ3-CH3)(μ2-CH3)3 (Cp′ = C5Me4SiMe3) with a ketone would rather yield Cp′3Tm3(μ2-CH3)3[OC(CH3)(C6H4)2)] as a result of methylidene transfer and simultaneous ortho-metallation of benzophenone (not shown in Scheme 1).34 Moreover, Hou and coworkers accessed “Lewis-acid-free” cuboid clusters [Cp′Ln(μ3-CH2)]4via prolonged thermal treatment of the respective trinuclear complexes Cp′3Tm3(μ3-CH2)(μ3-CH3)(μ2-CH3)3 or Cp′3Lu3(μ2-CH3)6, respectively. While such Lewis-acid-free Ln(III)–CH22− clusters revealed interesting reactivity toward (electronically unsaturated) functional substrates (Scheme 1),35–37 seemingly less efforts were devoted to elucidate the formation of such rare-earth metal methylidene species. Herein, we present three synthesis approaches toward rare-earth metal tetramethyl methylidene complexes capable of methylenating ketones. The isolation of dimethyl complex [NSiMe3(Ar)]LuMe2(thf)2 hints to a mechanism with one crucial C–H bond activation step. Part of the work on methylidene complexes [NSiMe3(Ar)]3Ln3(μ3-CH2)(μ3-CH3)(μ2-CH3)3(thf)3 (7-Ln, Ln = Y, Ho, Lu) were recently communicated by our group.31
Results and discussion
We have shown previously that treatment of [LnMe3]n (1-Ln; Ln = Y, Ho, and Lu)31,38 suspended in n-hexane with one equivalent of H[NSiMe3(Ar)] and subsequent addition of excess thf at ambient temperature, selectively yields the trinuclear rare-earth metal complexes [NSiMe3(Ar)]3Ln3(μ3-CH2)(μ3-CH3)(μ2-CH3)3(thf)3 (7-Ln, Ln = Y, Ho, Lu; Scheme 2, route A).31 Due to the insolubility of 1-Ln in aliphatic solvents, immediate reaction with H[NSiMe3(Ar)] in n-hexane to form putative [NSiMe3(Ar)]LnMe2 did not occur. It is anticipated that the donor solvent (thf) is necessary to break up the polymeric network of 1-Ln, thus initiating the reaction. Re-investigation of the lutetium-reaction afforded single crystals of [NSiMe3(Ar)]LuMe2(thf)2 (4-Lu, 33%), which could be isolated together with precipitated [NSiMe3(Ar)]3Lu3(μ3-CH2)(μ3-Me)(μ2-Me)3(thf)3 (7-Lu, 19%). The 1H NMR spectrum of 4-Lu in thf-d8 at ambient temperature features one set of signals for the amido ligand and the coordinated thf donor molecules. The Lu–CH3 moieties appeared as one narrow singlet at −1.06 ppm (6H), indicating a highly fluxional nature of complex 4-Lu. Cooling a solution of 4-Lu in thf-d8 to −90 °C significantly shifted the resonances of the metal-bonded methyl groups, the silyl moiety and the methine groups (iPr) to lower field, while the signals for the aryl hydrogen atoms and the methyl groups appeared at higher fields (Fig. S9†). The high thermal sensitivity of compound 4-Lu allowed 13C{1H} NMR spectroscopy only at low temperatures (−35 °C) revealing the signal for the Lu–CH3 groups at 29.5 ppm.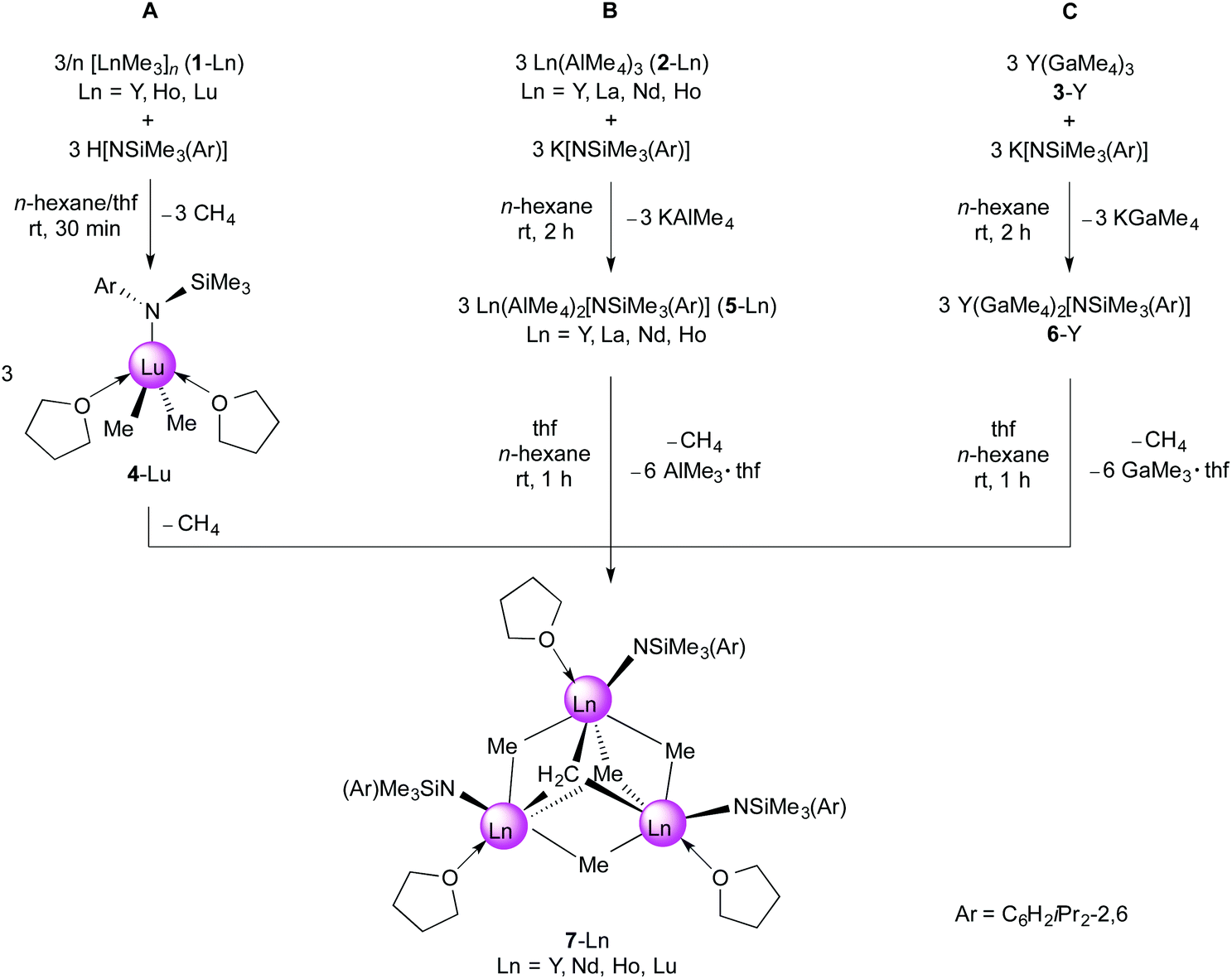 | ||
| Scheme 2 Syntheses of trinuclear rare-earth metal methylidene complexes 7-Ln supported by a bulky amido ligand. | ||
Single-crystal X-ray diffraction of 4-Lu revealed a geometry about the five-coordinate lutetium metal centre which can best be described as distorted trigonal bipyramidal with C1, C2, and N1 occupying the positions in the equatorial plane and O1 and O2 in the apical positions (see Fig. 1). The Lu–C(CH3) bond lengths of 2.366(4) and 2.378(5) Å of complex 4-Lu are in the same range as in dimethyl complex (TptBu,Me)LuMe2 (avg. 2.370 Å)39 and ate complex [NSiMe3(Ar)]2YbMe(μ-Me)Li(thf)3 (Ar = C6H3iPr2-2,6; Ln–C 2.349(8) Å, 2.382(7) Å; Ln–N avg. 2.226 Å),40 but the Ln–C/N bond lengths of 4-Lu are slightly longer than in 4-coordinate [NSiMe3(Ar)]Lu(CH2SiMe3)2(thf) (Ar = C6H3iPr2-2,6; Lu–C avg. 2.318 Å; Ln–N 2.153(2) Å).41 The sterically demanding amido ligand seems suitable for protecting the methyl groups kinetically. However, decomposition of compound 4-Lu starts at ambient temperature within just a few hours, which is consistent with the hypothesis that 4-Lu is a crucial intermediate in the synthesis of 7-Lu. Although the exact mechanism for the formation of multinuclear methylidene complexes is unclear, we assume that species like [NSiMe3(Ar)]LnMe2(thf)x play a pivotal role.
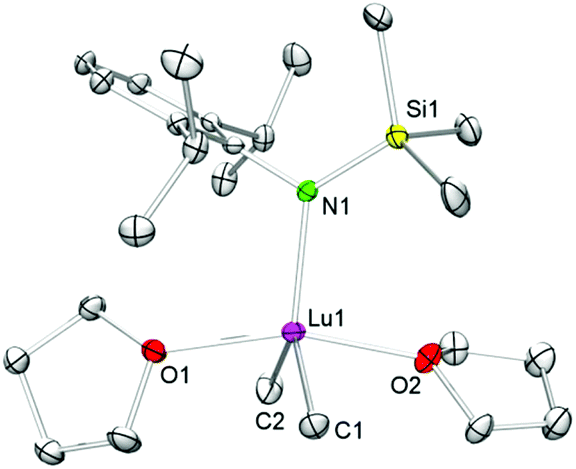 | ||
| Fig. 1 Molecular structure of 4-Lu (atomic displacement parameters are set at the 30% level). Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Lu1–C1 2.366(4), Lu1–C2 2.378(5), Lu1–O1 2.312(3), Lu1–O2 2.333(3), Lu1–N1 2.203(3), C6–N1–Lu1 112.8(2), O1–Lu1–O2 161.6(1), O1–Lu1–C1 88.1(1), O2–Lu1–C1 86.6(1), O1–Lu1–C2 83.1(1), O2–Lu1–C2 84.8(1), C1–Lu1–C2 122.3(2). | ||
The isolation of dimethyl complex [NSiMe3(Ar)]LuMe2(thf)2 (4-Lu) as a plausible intermediate in the formation of the trinuclear complex 7-Lu tempted us to speculate about a possible mechanism (see Scheme 3). The reaction proceeds presumably via a sequence of methane elimination (a) or donor-induced tetramethylaluminate/gallate cleavage (vide infra) (b) to yield dimethyl complexes (I1 or 4-Lu), C–H bond activation under formation of Ln(III)CH2 (I2) and methane (c) and subsequent agglomeration (d) affording trinuclear compounds 7-Ln, which consist of a core with three rare-earth metal centres bridged by μ2-Me, μ3-Me and μ3-CH2 moieties.
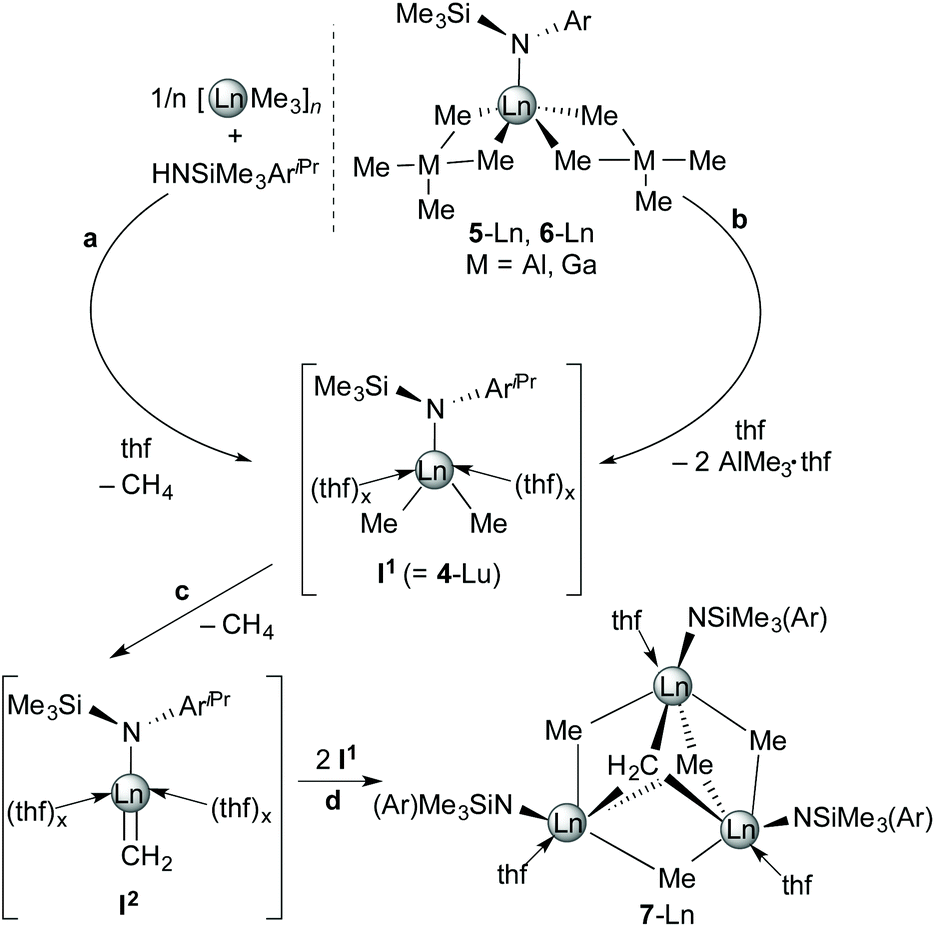 | ||
| Scheme 3 Proposed reaction mechanism for the formation of multinuclear methylidene complexes 7-Ln. | ||
The formation of 7-Y by donor-induced cleavage of bis(aluminate) complex 5-Y (Scheme 2, route B; Scheme 3, b–d) was also previously communicated by us.31 We now found that route B can be adopted for the larger rare-earth metal centres, which is infeasible for route A. Reaction of homoleptic Ln(AlMe4)3 (3-Ln, Ln = Y, La, Nd, Ho)42,43 with one equivalent of potassium amide K[NSiMe3(Ar)]44,45 in n-hexane at ambient temperature cleanly formed the respective rare-earth metal mono(amide) complexes [NSiMe3(Ar)]Ln(AlMe4)2 (5-Ln) in high yields (Scheme 2). The molecular composition of complexes 5-Ln was confirmed by elemental analyses, infrared spectroscopy, single-crystal X-ray diffraction and 1H/13C NMR spectroscopy (except 5-Ho).
The 1H NMR spectra of the diamagnetic complexes 5-Ln (Ln = Y, La) showed the expected resonances for the NSiMe3(Ar) ligand. The methyl groups of the Al(μ-Me)2Me2 moieties appeared as one narrow signal indicating a rapid exchange of bridging and terminal methyl groups (5-Y δ −0.24 ppm, 2JYH = 2.5 Hz; 5-La δ −0.19 ppm). Good quality 1H and 13C NMR spectra could also be obtained for the paramagnetic neodymium compound 5-Nd. However, due to significant paramagnetic shifts and broadening effects, assignment of the signals – except for the AlMe4 resonance at 11.27 ppm in the 1H NMR spectrum – appeared to be difficult.
The X-ray crystallographic analyses of [NSiMe3(Ar)]Ln(AlMe4)2 (5-Ln) revealed structural motifs as found in the solid-state structures of half-sandwich bis(tetramethylaluminate) complexes CpRLn(AlMe4)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46–48 with one AlMe4 ligand coordinating in the routinely observed planar η2 fashion, and the second one showing a bent η2 coordination (Fig. 2 and S1–S3†). The geometry about the five-coordinate Ln(III) centre can best be described as distorted square pyramidal, with the four bridging methyl groups in the corner of the basal plane and the amido nitrogen atom in the apical position. As detected for CpRLn(AlMe4)246–48 the Ln–C(μ-Me) bond lengths increase with increasing Ln(III) size, the bonds in the bent AlMe4 ligand being significantly elongated compared to those in the planar tetramethylaluminate ligand, and C8 is tilted toward the rare-earth metal centre. The solid-state structures of 5-Ln feature additional short contacts between the metal centres and the ipso carbon atoms of the aryl rings. As a consequence, the aryl ring lies almost orthogonally to the Ln–N bond as evidenced by the acute C9–N1–Ln1 bond angles (Table 1). Interestingly, the feasibility of complexes 5-La and 5-Nd might provide access to methylidene complexes of type 7-Ln with large rare-earth metal centres.
46–48 with one AlMe4 ligand coordinating in the routinely observed planar η2 fashion, and the second one showing a bent η2 coordination (Fig. 2 and S1–S3†). The geometry about the five-coordinate Ln(III) centre can best be described as distorted square pyramidal, with the four bridging methyl groups in the corner of the basal plane and the amido nitrogen atom in the apical position. As detected for CpRLn(AlMe4)246–48 the Ln–C(μ-Me) bond lengths increase with increasing Ln(III) size, the bonds in the bent AlMe4 ligand being significantly elongated compared to those in the planar tetramethylaluminate ligand, and C8 is tilted toward the rare-earth metal centre. The solid-state structures of 5-Ln feature additional short contacts between the metal centres and the ipso carbon atoms of the aryl rings. As a consequence, the aryl ring lies almost orthogonally to the Ln–N bond as evidenced by the acute C9–N1–Ln1 bond angles (Table 1). Interestingly, the feasibility of complexes 5-La and 5-Nd might provide access to methylidene complexes of type 7-Ln with large rare-earth metal centres.
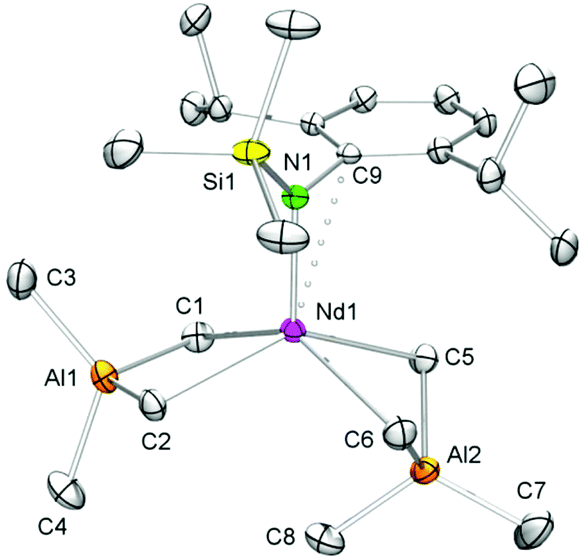 | ||
| Fig. 2 Molecular structure of [NSiMe3(Ar)]Nd(AlMe4)2 (5-Nd), representative of the isostructural complexes 5-La, 5-Nd and 5-Ho. Atomic displacement parameters are set at the 30% level, hydrogen atoms have been omitted for clarity. The asymmetric unit contains two independent molecules with similar structural data. | ||
| 5-La | 5-Nda | 5-Hoa | 6-Y | |
|---|---|---|---|---|
| a Metric parameters of molecule 2 of 5-Nd and 5-Ho are listed in the ESI. | ||||
| Ln–N1 | 2.295(4) | 2.222(2) | 2.160(3) | 2.172(4) |
| Ln–C1 | 2.712(5) | 2.617(3) | 2.524(4) | 2.522(5) |
| Ln–C2 | 2.716(5) | 2.661(4) | 2.525(4) | 2.520(5) |
| Ln–C5 | 2.837(5) | 2.661(4) | 2.631(4) | 2.653(5) |
| Ln–C6 | 2.717(5) | 2.718(3) | 2.585(4) | 2.585(5) |
| Ln⋯M1 | 3.278(2) | 3.194(1) | 3.078(1) | 3.0550(7) |
| Ln⋯M2 | 3.013(1) | 2.976(1) | 2.882(1) | 2.8711(7) |
| Ln⋯C9 | 2.779(4) | 2.755(3) | 2.694(3) | 2.770(4) |
| Ln–N1–C9 | 93.8(2) | 95.7(2) | 95.4(2) | 98.5(2) |
| Ln–C1–M1–C2 | −0.4(2) | 9.6(2) | 7.7(2) | 42.1(2) |
| Ln–C5–M2–C6 | 45.5(2) | −40.0(1) | −40.4(2) | 9.5(2) |
The anticipated formation of a transient [NSiMe3(Ar)]LnMe2(thf)x species is substantiated by synthesis approach B (Scheme 2), since addition of excess thf to n-hexane solutions of [NSiMe3(Ar)]Ln(AlMe4)2 (5-Ln) afforded complexes 7-Ln in high yields. Single-crystal X-ray diffraction revealed the formation of trinuclear rare-earth metal tetramethyl methylidene complexes [NSiMe3(Ar)]3Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3(thf)3 (7-Ln, Ln = Y,31 Nd, Ho) as the product of sequential donor-induced cleavage of Ln(μ2-Me)2(AlMe2) moieties,49 C–H bond activation, and agglomeration (Schemes 2 and 3). Note that, treatment of 5-La with excess thf led to intractable, alkylated species.24 Comparative studies on half-sandwich dialkyl complexes revealed that the capability of alkyl complexes to engage in C–H bond activation reactions strongly depends on the size of the rare-earth metal centre, the amount of Lewis base as well as the steric demand of the ancillary ligand. For example, Hou and coworkers showed that the reaction of Cp′Ln(CH2SiMe3)2(thf) (Cp′ = C5Me4SiMe3) with AlMe3 in diethyl ether at ambient temperature yielded [Cp′LnMe2]3 for lutetium and Cp′3Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3 for thulium.34 We found that the donor-induced cleavage of Cp*Ln(AlMe4)2 (Ln = Y, Lu) with two equivalents of tetrahydrofuran led to trimeric [Cp*LnMe2]3, whereas the reaction with one equivalent thf produced methylidyne complex [Cp*4Y4(μ2-CH3)2{(CH3)Al(μ2-CH3)2}4(μ4-CH)2].47,50
Organoaluminum moieties are well-known to promote C–H bond activation reactions at transition metals.19,50–52 As such, we were interested to elucidate the influence of any coordinated alkylaluminate moieties on the formation of 7-Ln along pathway B. According to Pearson's HSAB concept the methyl groups in GaMe4 moieties should behave less basic than in AlMe4. This is also in accord with the tendency of the tetramethylgallato ligand to separate off GaMe3. Hence we tackled the synthesis of [NSiMe3(Ar)]Y(GaMe4)2 (6-Y) from homoleptic Y(GaMe4)3 (2-Y)53,54 which was obtained aluminium-free from [Y(NMe2)3(LiCl)3] and GaMe3. Reaction of 2-Y with one equivalent of potassium amide K[NSiMe3(Ar)]44,45 in n-hexane at ambient temperature gave exclusively compound 6-Y in high yields (Scheme 2, route C). Comparable to aluminate complexes 5-Ln, 6-Y features a highly fluxional Ga(μ-Me)2Me2 coordination as evidenced by one narrow signal for the methyl groups at δ −0.03 ppm. The 89Y NMR resonance of 6-Y (δ 477.2 ppm) is in the same range as for 7-Y (δ 498.0 ppm).31 The molecular structure of complex 6-Y is isomorphous to those of 5-Ln and similar to Cp*Y(GaMe4)2 (Cp* = C5Me5) regarding the coordination of the GaMe4 ligands (Fig. 3).55
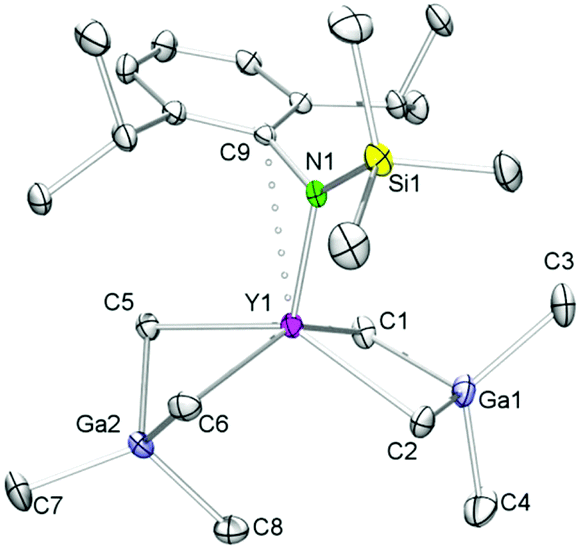 | ||
| Fig. 3 Molecular structure of [NSiMe3(Ar)]Y(GaMe4)2 (6-Y). Atomic displacement parameters are set at the 30% level, hydrogen atoms have been omitted for clarity. | ||
The metrical parameters of 6-Y are comparable to Cp*Y(GaMe4)2 with average Y–C(μ-CH3) bond lengths of 2.616 Å and 2.520 Å for the bent and planar Ga(μ-Me)2Me2 moieties, respectively, being slightly shorter than in the respective half-sandwich complex (average 2.679 and 2.555 Å).55 The Ln–C bond lengths of the related yttrium neosilyl complexes [NSiMe3(Ar)]Y(CH2SiMe3)2(thf) (Ar = C6H3iPr2-2,6; Y–C avg. 2.367 Å)41 and [NSiiPr3(Ar)]Y(CH2SiMe3)3Li(thf)2 (Ar = C6H3iPr2-2,6; Y–C 2.373(3)–2.490(4) Å)56 are as expected shorter than in 6-Y, but the Ln–N(amido) bond lengths compare well (2.190(2) Å;41 2.259(3) Å;566-Y, 2.172(4) Å).
If the organoaluminum moieties in [NSiMe3(Ar)]Ln(AlMe4)2 (5-Ln) would be crucial for the formation of methylidene species, the reaction of the respective methylgallate [NSiMe3(Ar)]Y(GaMe4)2 (6-Y) with a Lewis base might yield a species like {[NSiMe3(Ar)]YMe2}x. However, upon treatment of 6-Y with thf we could isolate [NSiMe3(Ar)]3Y3(μ3-CH2)(μ3-Me)(μ2-Me)3(thf)3 (7-Y) in almost quantitative yield.
The rare-earth metal tetramethyl methylidene complexes 7-Ln are sparingly soluble in n-hexane, but readily dissolve in aromatic solvents and thf, which allowed for elaborate NMR spectroscopic investigations of the diamagnetic representatives 7-Y and 7-Lu.31 The interpretation of the 1H NMR spectra of compounds 7-Ln (Ln = Nd, Ho) is affected by paramagnetic shifts and line broadening.
Representatively for the isostructural complexes 7-Ln, Fig. 4 and 5 illustrate the molecular and core structure of the neodymium derivative 7-Nd (7-Ho: Fig. S5†). Complexes 7-Nd and 7-Ho are isomorphous to the previously reported 7-Y and 7-Lu,31 crystallizing in the monoclinic space group P21![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) . In the solid state, each Ln is six-coordinate by one amido ligand, one thf, three bridging methyl groups, and one μ3-bridging methylidene group. The core atoms of the complex (Fig. 5, left) adopt a distorted hexagonal bipyramid with alternating Ln and μ2-bridging methyl groups in the equatorial, and the μ3-methyl and the methylidene groups in the apical positions. Together with the methylidene group all three amido ligands are residing on one side of the plane spanned by the Ln metal centres. It seems that the amido substituents provide a protective enclosure for the methylidene moiety reminiscent of a picket fence, thus impeding intermolecular deactivation.57 The Ln3(μ3-CH2) unit in 7-Ln resembles those previously found in complexes L3Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3(thf)x (Ln = Sc, Y, Ho, Tm, Lu; x = 0, 1).31,32,34 The Ln–C(CH2) bond lengths of 2.356(10)–2.428(10) Å in 7-Ho (7-Nd: 2.425(7)–2.505(8) Å) are in accord with the distances reported for 7-Y (2.345(5)–2.424(4) Å),31 but are significantly shorter than the values for Cp*3Ln3(μ3-CH2)(μ3-Cl)(μ2-Cl)3 (Y: 2.424(2)–2.450(2) Å, La: 2.537(3)–2.635(3) Å)30 considering the differences of the ionic radii. As expected, the Ln–C bond lengths of the μ2-CH3 moieties are between the values for Ln–C(μ3-CH2) and Ln–C(μ3-CH3) (Table 2).
. In the solid state, each Ln is six-coordinate by one amido ligand, one thf, three bridging methyl groups, and one μ3-bridging methylidene group. The core atoms of the complex (Fig. 5, left) adopt a distorted hexagonal bipyramid with alternating Ln and μ2-bridging methyl groups in the equatorial, and the μ3-methyl and the methylidene groups in the apical positions. Together with the methylidene group all three amido ligands are residing on one side of the plane spanned by the Ln metal centres. It seems that the amido substituents provide a protective enclosure for the methylidene moiety reminiscent of a picket fence, thus impeding intermolecular deactivation.57 The Ln3(μ3-CH2) unit in 7-Ln resembles those previously found in complexes L3Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3(thf)x (Ln = Sc, Y, Ho, Tm, Lu; x = 0, 1).31,32,34 The Ln–C(CH2) bond lengths of 2.356(10)–2.428(10) Å in 7-Ho (7-Nd: 2.425(7)–2.505(8) Å) are in accord with the distances reported for 7-Y (2.345(5)–2.424(4) Å),31 but are significantly shorter than the values for Cp*3Ln3(μ3-CH2)(μ3-Cl)(μ2-Cl)3 (Y: 2.424(2)–2.450(2) Å, La: 2.537(3)–2.635(3) Å)30 considering the differences of the ionic radii. As expected, the Ln–C bond lengths of the μ2-CH3 moieties are between the values for Ln–C(μ3-CH2) and Ln–C(μ3-CH3) (Table 2).
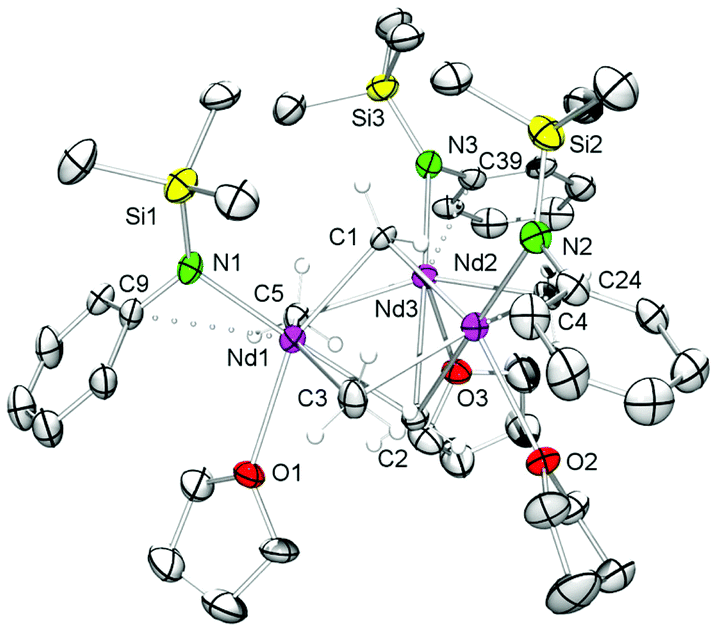 | ||
| Fig. 4 Molecular structure of 7-Nd (atomic displacement parameters are set at the 30% level). Solvent molecules, isopropyl groups and hydrogen atoms, except for Nd–CH2 and Nd–CH3 moieties, have been omitted for clarity. | ||
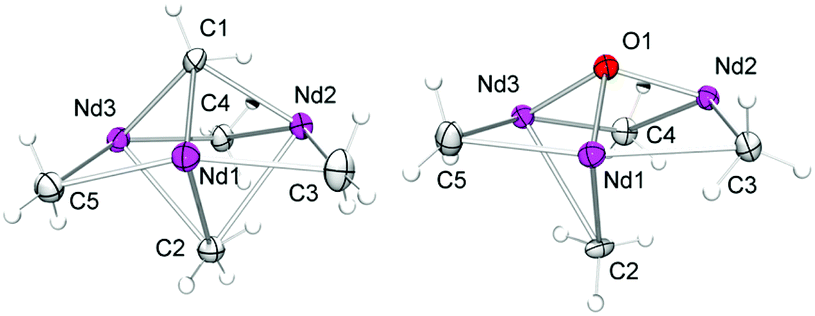 | ||
| Fig. 5 Molecular core structure of 7-Nd (left) and 8-Nd (right). Atomic displacement parameters are set at the 30% level. | ||
| 7-Nd | 7-Ho | 8-Nd | |
|---|---|---|---|
| Ln–C(μ3-CH2) | 2.475 | 2.391 | |
| Ln–C(μ3-CH3) | 2.817 | 2.737 | |
| Ln–C(μ2-CH3) | 2.635 | 2.547 | 2.676 |
| Ln–O(oxo) | 2.239 | ||
| Ln–N | 2.355 | 2.279 | 2.369 |
| Ln–O(thf) | 2.568 | 2.466 | 2.543 |
| Ln–Cipso(Ar) | 3.021 | 3.086 | 3.076 |
Reaction of 7-Ln with ketones
In the presence of carbonylic substrates, methylidene complexes 7-Ln act as Schrock-type nucleophilic carbenes. Monitoring the reactions of 7-Ln with 1 equivalent of 9-fluorenone, benzophenone, and cyclohexanone, respectively, by 1H NMR spectroscopy, clearly revealed the formation of the expected methylenated products (see Scheme 4) and concomitantly led to methylation products.31 If compared to the original Tebbe reagent, which allows a variety of functional substrates (esters, lactones) suppressing the formation of undesired side-products,58 complexes of type 7-Ln are certainly less efficient and selective.30 Theoretical studies on [PhC(NC6H3iPr2-2,6)2]3Sc3(μ3-CH2)(μ3-Me)(μ2-Me)3, however, indicated that methylidene transfer rather than methyl transfer occurs.33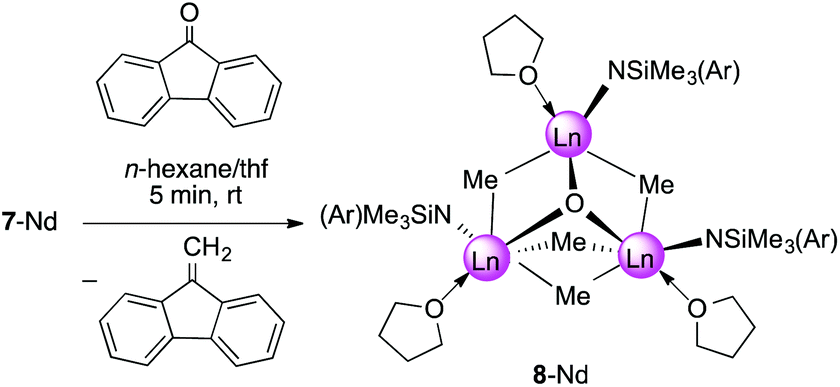 | ||
| Scheme 4 Synthesis of [NSiMe3(Ar)]3Nd3(μ3-O)(μ2-Me)4(thf)3 (8-Nd). | ||
Upscaling of the reaction between 7-Nd and 9-fluorenone (1:1 ratio) in n-hexane/thf led to the formation of 9-methylidene-fluorene and [NSiMe3(Ar)]3Nd3(μ3-O)(μ2-Me)4(thf)3 (8-Nd) as indicated by an immediate colour change from blue to green (Fig. S21†). NMR-scale reactions using 1–5 equivalents of the carbonylic reagent in benzene-d6, revealed that after addition of the third equivalent of substrate the mixture kept a brown colour. Additional signals assignable to a paramagnetically shifted 9-methyl-9H-fluoren-9-yloxy moiety were observed as well, however, due to the existing paramagnetism, a quantification of methylenation versus methylation was infeasible. Single-crystal X-ray diffraction of complex 8-Nd revealed that the core structure of 7-Nd (Fig. 5, left) was replaced by Nd3(μ3-O)(μ2-Me)4 with the NdIII metal centres being bridged by one μ3-oxo moiety and four μ2-Me groups (Fig. 5, right). In contrast to 7-Nd, two of the amido ligands and the oxo are now located on the same side of the plane spanned by the Ln3 metal centres. The coordination of the thf donor molecules is opposite to the amido ligands (Fig. 6). The Nd–O bond lengths (avg. 2.239 Å) are slightly longer than in [Cp*Nd(NC5H4NC4H8)]2(μ-O) (2.157(2) Å).59 Similarly, the benzamidinato-supported complexes [PhC(NC6H3iPr2-2,6)2]3Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3 (Ln = Sc, Lu) could be selectively converted into the oxo derivatives.32
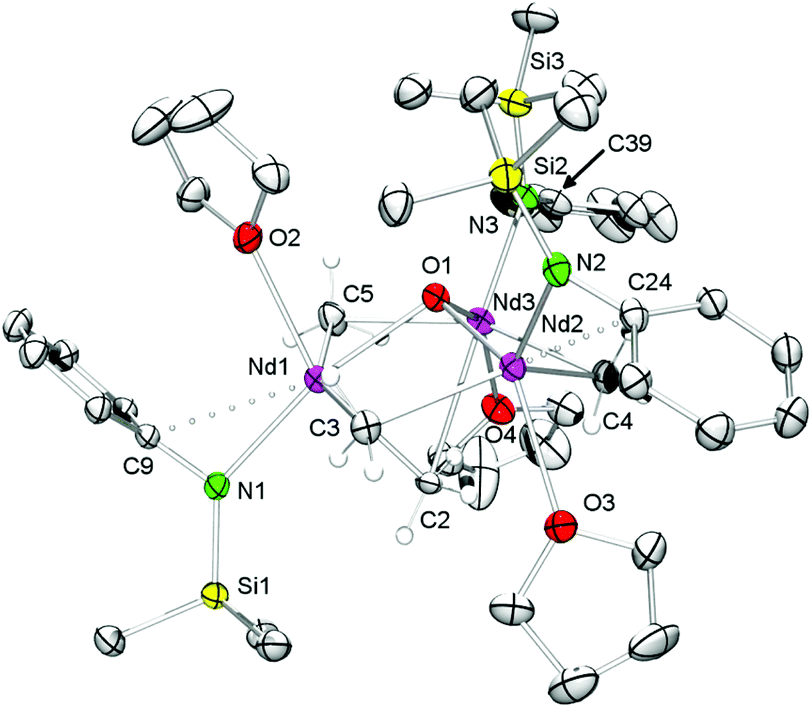 | ||
| Fig. 6 Molecular structure of 8-Nd (atomic displacement parameters are set at the 30% level). Isopropyl groups and hydrogen atoms, except for Nd–CH3 moieties, have been omitted for clarity. | ||
Synthesis and characterization of [NSiMe3(Ar)]2Ln(AlMe4) (9-Ln)
As shown in Scheme 2/route B, reaction of Ln(AlMe4)3 (2-Ln, Ln = La, Nd) with K[NSiMe3(Ar)] in n-hexane for 2 h and separation of precipitated KAlMe4 gave [NSiMe3(Ar)]Ln(AlMe4)2 (5-Ln), however, contaminated by small amounts of the bis(amide) complex [NSiMe3(Ar)]2Ln(AlMe4) (9-Ln, Ln = La, Nd), which were separated by fractional crystallization. Complexes 9-Ln could be obtained in moderate yields via salt metathesis of Ln(AlMe4)3 (2-Ln) with two equivalents of K[NSiMe3(Ar)] in n-hexane at ambient temperature (Scheme 5).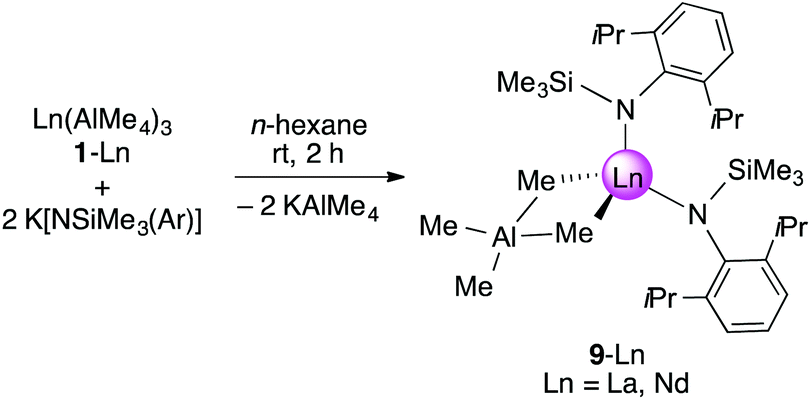 | ||
| Scheme 5 Synthesis of [NSiMe3(Ar)]2Ln(AlMe4) (9-Ln, Ln = La, Nd). | ||
The 1H and 13C{1H} NMR spectra of complex 9-La revealed one set of signals for the amido ligands and the AlMe4 moiety, except for the diastereotopic aryl isopropyl groups. While the 1H NMR resonances for the NSiMe3(Ar) ligand are shifted to lower fields compared to 5-La, a high-field shift was observed for the signal of the AlMe4 ligand (δ −0.47 ppm, 12H; 5-La: δ −0.19 ppm). The resonance of the AlMe4 ligand in paramagnetic 9-Nd appeared at 10.73 ppm. Blue (9-Nd) and colourless (9-La) single crystals were obtained from saturated n-hexane solutions. Previously reported Nd[NSiMe3(Ar)]2Cl(thf)60 is somewhat comparable to isomorphous 9-Ln featuring also a four-coordinate Ln metal centre (Fig. 7). Such simple complexes (NRR′)2Ln(alkyl) and (NRR′)Ln(alkyl)2 coordinated by non-chelating alkyl and amido ligands, however, seem to be rather scarce.40,41,56 Complexes 9-Ln display distorted tetrahedral coordination geometries as evidenced by large N1–Ln–C(μ2-CH3) angles e.g., 9-La, 122.5(2)°; 9-Nd, 123.33(3)° (Table 3). Pronounced interactions of the LnIII metal centre with the amido ligand is also evidenced by close contacts to the ipso carbon atoms of the aryl rings (9-La, 2.791(7) Å; 9-Nd, 2.7502(9) Å). The average Ln–C(μ2-CH3) bond lengths of 2.721 Å (9-La, 2.650 Å (9-Nd)) are shorter than in the respective metallocene complex Cp*2La(AlMe4) (avg. 2.849 Å), which is dimeric in the solid-state.55
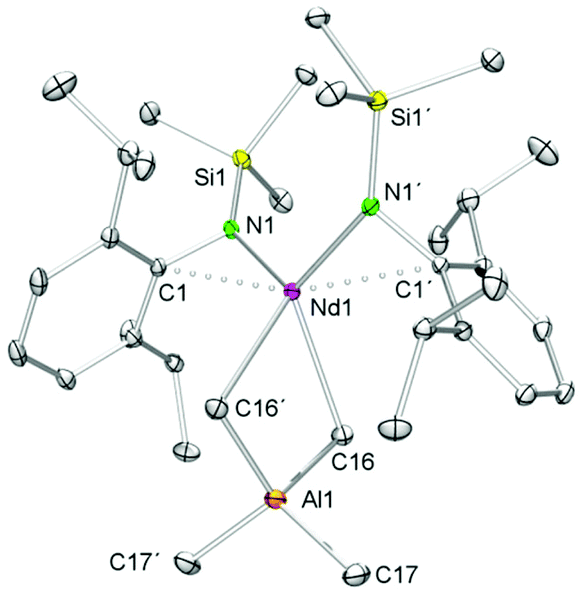 | ||
| Fig. 7 Molecular structure of [NSiMe3(Ar)]2Nd(AlMe4) (9-Nd), representative of the isomorphous complexes 9-La and 9-Nd. Atomic displacement parameters are set at the 30% level, hydrogen atoms have been omitted for clarity. | ||
| 9-La | 9-Nd | |
|---|---|---|
| Ln1–N1 | 2.356(3) | 2.2968(8) |
| Ln1–C16 | 2.719(4) | 2.650(1) |
| Al1–C16 | 2.072(4) | 2.073(1) |
| Al1–C17 | 1.979(4) | 1.980(1) |
| Ln1⋯Al1 | 3.296(2) | 3.2302(4) |
| Ln1⋯C1 | 2.791(3) | 2.7502(9) |
| N1–Ln1–C16 | 103.73(11) | 103.10(3) |
| Ln1–N1–C1 | 91.95(18) | 92.29(5) |
Conclusions
The unique [Ln3(μ3-CH2)(μ3-CH3)(μ2-CH3)3] core structure has been achieved via three synthesis pathways. Such rare-earth metal methylidene complexes do form in the absence of any organoaluminium components as proven by applying alkylaluminium-free synthesis protocols. The generation of [NSiMe3(Ar)]3Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3(thf)3 seems to be rather driven by steric constraints. A possible mechanism involves the formation of intermediate [NSiMe3(Ar)]LnMe2(thf)x species, which upon C–H bond activation agglomerate to the target compound. The dianionic CH22− moiety seems to be preferentially stabilized by Ln3 “cluster-like” entities being protected from intermolecular deactivation by a picket-fence arrangement of the amido ligands. Nevertheless the methylidene ligand retains its nucleophilic character as revealed by Tebbe-like methylenation reactions with carbonylic substrates, which concomitantly form isolable rare-earth metal oxo clusters.Experimental section
General procedures
All operations were performed with rigorous exclusion of air and water, using standard Schlenk, high-vacuum, and glovebox techniques (MBraun MBLab; <1 ppm O2, <1 ppm H2O). Toluene, n-hexane, and tetrahydrofuran were purified by using Grubbs columns (MBraun SPS, solvent purification system) and stored in a glovebox. Benzene-d6 and toluene-d8 were obtained from Aldrich, degassed, dried over Na for 24 h, and filtered. Tetrahydrofuran-d8 was obtained from Aldrich, degassed, dried over CaH2 for four days and vacuum transferred. 9-Fluorenone was obtained from Fluka and sublimed prior to use. [LuMe3]n (1-Lu),38 homoleptic Ln(AlMe4)3 (2-Ln) (Ln = Y, Nd, Ho, Lu)42,43 and Y(GaMe4)353 were prepared according to literature methods. H[NSiMe3(Ar)] and K[NSiMe3(Ar)] were synthesized by a modification of the published procedure.44,45 The NMR spectra of air and moisture sensitive compounds were recorded by using J. Young valve NMR tubes at 26 °C on a Bruker-Avance II 400 (1H: 400.13 MHz; 13C: 100.61 MHz) and a Bruker-Avance II 500 (1H: 500.13 MHz; 13C: 125.76 MHz). 1H and 13C shifts are referenced to internal solvent resonances and reported in parts per million relative to TMS. Coupling constants are given in Hertz. IR and DRIFT spectra were recorded on a NICOLET 6700 FTIR spectrometer using dried KBr and KBr windows or as Nujol mulls sandwiched between CsI plates. Elemental analyses were performed on an Elementar Vario EL III and an Elementar Vario Micro Cube.
X-ray crystallography and crystal structure determination of 4-Lu, 5-Ln, 6-Y, 7-Ln, 8-Nd, and 9-Ln
Crystals of 5-Ln, 6-Y and 9-Ln were grown by standard techniques from saturated solutions using n-hexane or n-hexane/thf (4-Lu, 7-Ln and 8-Nd) at −40 °C. Suitable single crystals for X-ray structure analyses were selected in a glovebox and coated with Parabar 10312 and fixed on a nylon loop/glass fiber. Data for 4-Lu, 5-La, 5-Nd, 5-Ho, 6-Y, 7-Nd, 7-Ho and 8-Nd were collected on a Stoe IPDS 2T instrument equipped with a fine focus sealed tube and graphite monochromator using MoKα radiation (λ = 0.71073 Å) performing ω scans. Raw data were collected and integrated using Stoe's X-Area software package.61 A numerical absorption correction based on crystal shape optimization was applied using Stoe's X-Red62 and X-Shape.63 X-ray data for 9-Ln were collected on a Bruker AXS, TXS rotating anode instrument using a Pt135 CCD detector, and graphite monochromator using MoKα radiation (λ = 0.71073 Å). The Data collection strategy was determined using COSMO64 employing ω- and ϕ scans. Raw data were processed using APEX64 and SAINT,64 corrections for absorption effects were applied using SADABS.64 The structure was solved by direct methods and refined against all data by full-matrix least-squares methods on F2 using SHELXTL64 and ShelXle.65 All Graphics were produced employing ORTEP-366 and POV-Ray.67 Further details of the refinement and crystallographic data are listed in Table S1† and in the CIF files. CCDCs 1410385–1410394 contain all the supplementary crystallographic data for this paper.
Acknowledgements
We are grateful to the German Science Foundation for support (Grant: AN 238/15-1).References
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS PubMed.
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760 CrossRef CAS PubMed.
- P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862 CrossRef PubMed.
- Z.-L. Xue and L. A. Morton, J. Organomet. Chem., 2011, 696, 3924 CrossRef CAS PubMed.
- D. J. Mindiola and J. Scott, Nat. Chem., 2011, 3, 15 CrossRef CAS PubMed.
- O. T. Summerscales and J. C. Gordon, RSC Adv., 2013, 3, 6682 RSC.
- J. Kratsch and P. W. Roesky, Angew. Chem., Int. Ed., 2014, 53, 376 CrossRef CAS PubMed.
- J. W. Herndon, Coord. Chem. Rev., 2015, 286, 30 CrossRef CAS PubMed.
- R. R. Schrock, J. Am. Chem. Soc., 1974, 96, 6796 CrossRef CAS.
- R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748 CrossRef CAS PubMed.
- L. J. Guggenberger and R. R. Schrock, J. Am. Chem. Soc., 1975, 97, 6578 CrossRef CAS.
- R. R. Schrock, J. Am. Chem. Soc., 1975, 97, 6577 CrossRef CAS.
- M. Kamitani, B. Pinter, C.-H. Chen, M. Pink and D. J. Mindiola, Angew. Chem., Int. Ed., 2014, 53, 10913 CrossRef CAS PubMed.
- K. Searles, K. Keijzer, C.-H. Chen, M.-H. Baik and D. J. Mindiola, Chem. Commun., 2014, 50, 6267 RSC.
- J. E. Kickham, F. Guérin and D. W. Stephan, J. Am. Chem. Soc., 2002, 124, 11486 CrossRef CAS PubMed.
- P. Wei and D. W. Stephan, Organometallics, 2003, 22, 1992 CrossRef CAS.
- G. Theurkauff, A. Bondon, V. Dorcet, J.-F. Carpentier and E. Kirillov, Angew. Chem., Int. Ed., 2015, 54, 6343 CrossRef CAS PubMed.
- R. E. von H. Spence, D. J. Parks, W. E. Piers, M.-A. MacDonald, M. J. Zaworotko and S. J. Rettig, Angew. Chem., Int. Ed. Engl., 1995, 34, 1230 CrossRef PubMed.
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611 CrossRef CAS.
- R. Thompson, E. Nakamaru-Ogiso, C.-H. Chen, M. Pink and D. J. Mindiola, Organometallics, 2013, 33, 429 CrossRef.
- J. Scott, H. Fan, B. F. Wicker, A. R. Fout, M.-H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2008, 130, 14438 CrossRef CAS PubMed.
- R. Litlabø, M. Zimmermann, K. Saliu, J. Takats, K. W. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2008, 47, 9560 CrossRef PubMed.
- M. Zimmermann, J. Takats, G. Kiel, K. W. Törnroos and R. Anwander, Chem. Commun., 2008, 612 RSC.
- L. C. H. Gerber, E. Le Roux, K. W. Törnroos and R. Anwander, Chem. – Eur. J., 2008, 14, 9555 CrossRef CAS PubMed.
- W. Huang, C. T. Carver and P. L. Diaconescu, Inorg. Chem., 2011, 50, 978 CrossRef CAS PubMed.
- A. Venugopal, I. Kamps, D. Bojer, R. J. F. Berger, A. Mix, A. Willner, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2009, 5755 RSC.
- D. Bojer, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2011, 17, 6239 CrossRef CAS PubMed.
- D. Bojer, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2011, 2011, 3791 CAS.
- D. Bojer, A. Venugopal, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2011, 17, 6248 CrossRef CAS PubMed.
- H. M. Dietrich, K. W. Törnroos and R. Anwander, J. Am. Chem. Soc., 2006, 128, 9298 CrossRef CAS PubMed.
- M. Zimmermann, D. Rauschmaier, K. Eichele, K. W. Törnroos and R. Anwander, Chem. Commun., 2010, 46, 5346 RSC.
- J. Hong, L. Zhang, X. Yu, M. Li, Z. Zhang, P. Zheng, M. Nishiura, Z. Hou and X. Zhou, Chem. – Eur. J., 2011, 17, 2130 CrossRef CAS PubMed.
- G. Luo, Y. Luo, J. Qu and Z. Hou, Organometallics, 2015, 34, 366 CrossRef CAS.
- W.-X. Zhang, Z. Wang, M. Nishiura, Z. Xi and Z. Hou, J. Am. Chem. Soc., 2011, 133, 5712 CrossRef CAS PubMed.
- T. Li, M. Nishiura, J. Cheng, Y. Li and Z. Hou, Chem. – Eur. J., 2012, 18, 15079 CrossRef CAS PubMed.
- J. Hong, L. Zhang, K. Wang, Y. Zhang, L. Weng and X. Zhou, Chem. – Eur. J., 2013, 19, 7865 CrossRef CAS PubMed.
- K. Wang, G. Luo, J. Hong, X. Zhou, L. Weng, Y. Luo and L. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1053 CrossRef CAS PubMed.
- H. M. Dietrich, G. Raudaschl-Sieber and R. Anwander, Angew. Chem., Int. Ed., 2005, 44, 5303 CrossRef CAS PubMed.
- M. Zimmermann, R. Litlabø, K. W. Törnroos and R. Anwander, Organometallics, 2009, 28, 6646 CrossRef CAS.
- Y. Luo, Y. Yao, W. Li, J. Chen, Z. Zhang, Y. Zhang and Q. Shen, J. Organomet. Chem., 2003, 679, 125 CrossRef CAS.
- Y. Luo, M. Nishiura and Z. Hou, J. Organomet. Chem., 2007, 692, 536 CrossRef CAS PubMed.
- W. J. Evans, R. Anwander and J. W. Ziller, Organometallics, 1995, 14, 1107 CrossRef CAS.
- M. Zimmermann, N. Å. Frøystein, A. Fischbach, P. Sirsch, H. M. Dietrich, K. W. Törnroos, E. Herdtweck and R. Anwander, Chem. – Eur. J., 2007, 13, 8784 CrossRef CAS PubMed.
- Y. W. Chao, P. A. Wexler and D. E. Wigley, Inorg. Chem., 1989, 28, 3860 CrossRef CAS.
- C. Schädle, C. Meermann, K. W. Törnroos and R. Anwander, Eur. J. Inorg. Chem., 2010, 2010, 2841 CrossRef PubMed.
- R. Anwander, M. G. Klimpel, H. M. Dietrich, D. J. Shorokhov and W. Scherer, Chem. Commun., 2003, 1008 RSC.
- H. M. Dietrich, C. Zapilko, E. Herdtweck and R. Anwander, Organometallics, 2005, 24, 5767 CrossRef CAS.
- M. Zimmermann, K. W. Törnroos, H. Sitzmann and R. Anwander, Chem. – Eur. J., 2008, 14, 7266 CrossRef CAS PubMed.
- J. Holton, M. F. Lappert, D. G. H. Ballard, R. Pearce, J. L. Atwood and W. E. Hunter, J. Chem. Soc., Chem. Commun., 1976, 480 RSC.
- H. M. Dietrich, H. Grove, K. W. Törnroos and R. Anwander, J. Am. Chem. Soc., 2006, 128, 1458 CrossRef CAS PubMed.
- R. R. Schrock and P. R. Sharp, J. Am. Chem. Soc., 1978, 100, 2389 CrossRef CAS.
- M. Zimmermann, F. Estler, E. Herdtweck, K. W. Törnroos and R. Anwander, Organometallics, 2007, 26, 6029 CrossRef CAS.
- W. J. Evans, R. Anwander, R. J. Doedens and J. W. Ziller, Angew. Chem., Int. Ed. Engl., 1994, 33, 1641 CrossRef PubMed.
- H. M. Dietrich, C. Meermann, K. W. Törnroos and R. Anwander, Organometallics, 2006, 25, 4316 CrossRef CAS.
- H. M. Dietrich, K. W. Törnroos, E. Herdtweck and R. Anwander, Organometallics, 2009, 28, 6739 CrossRef CAS.
- B. Shen, L. Ying, J. Chen and Y. Luo, Inorg. Chim. Acta, 2008, 361, 1255 CrossRef CAS PubMed.
- J. P. Collman, Acc. Chem. Res., 1977, 10, 265 CrossRef CAS.
- R. C. Hartley, J. Li, C. A. Main and G. J. McKiernan, Tetrahedron, 2007, 63, 4825 CrossRef CAS PubMed.
- W. J. Evans, B. L. Davis, G. W. Nyce, J. M. Perotti and J. W. Ziller, J. Organomet. Chem., 2003, 677, 89 CrossRef CAS.
- H. Schumann, J. Winterfeld, E. C. E. Rosenthal, H. Hemling and L. Esser, Z. Anorg. Allg. Chem., 1995, 621, 122 CrossRef CAS PubMed.
- X-Area v. 1.55, Stoe & Cie GmbH, Darmstadt, Germany, 2009 Search PubMed.
- X-Red 32 v. 1.53, Stoe & Cie GmbH, Darmstad, Germany, 2009 Search PubMed.
- X-Shape v.2.12.2, Stoe & Cie GmbH, Darmstadt, Germany, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- POV-Ray v. 3.6, Persistence of Vision Pty. Ltd., Williamstown, Victoria, Australia, 2004. http://www.povray.org/ Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic and crystallographic data. CCDC 1410385–1410394. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02936h |
| This journal is © The Royal Society of Chemistry 2015 |