
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination polymers of ZnII and 5-methoxy isophthalate†
Laura J.
McCormick
a,
Samuel A.
Morris
a,
Simon J.
Teat
b,
Matthew J.
McPherson
 a,
Alexandra M. Z.
Slawin
a and
Russell E.
Morris
*a
a,
Alexandra M. Z.
Slawin
a and
Russell E.
Morris
*a
aEaSTCHEM School of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife, Scotland, KY16 9ST, UK. E-mail: rem1@st-andrews.ac.uk; Fax: +44 (0)1334 463808; Tel: +44 (0)1334 463800
bAdvanced Light Source, Berkeley Laboratory, 1 Cyclotron Road, Berkeley, California 94720, USA
First published on 21st September 2015
Abstract
Solvothermal reaction of Zn(OAc)2 and 5-methoxy isophthalic acid (H2MeOip) in aqueous alcohols ROH (R = H, Me, Et, or iPr) affords four different novel coordination polymers. Zn2(HMeOip)(MeOip)(OAc) (1) forms as a 1D ‘ribbon of rings’ polymer. Zn6(MeOip)4.5(HMeOip)(OH)2(H2O)2·5.5H2O (2) crystallises as a complex 3D framework. Zn(MeOip)(H2O)2 H2O (3) is a 1D coordination polymer that contains almost planar strips of Zn(MeOip). compound 4, Zn5(MeOip)4(OH)2(H2O)4·H2O, obtained from aqueous iPrOH, crystallises as a 2D polymer containing two crystallographically distinct Zn5(OH)2 clusters. Preliminary nitric oxide release experiments have been conducted.
Introduction
Coordination polymers derived from 5-substituted isophthalic acids have been shown to exhibit interesting gated gas sorption,1 catalytic,2 magnetic properties,3 as well as structural flexibility.4 One of the challenges to be faced in preparing coordination frameworks targeted toward a specific application is that the ability to accurately predict the exact way in which the metal centre and the ligand will be arranged within the framework is still lacking, even if both ligand and metal centre adopt the expected binding and coordination modes.5 In certain systems, changes in reaction conditions, including temperature6 and solvent system,7 have been shown to generate frameworks of different topologies, and in some cases of differing dimensionalities. One example is the similarity in the preparation of HKUST-1 and STAM-1, both of which involve reaction of copper nitrate and trimesic acid (H3btc) in aqueous alcohol, and both of which afford a coordination framework in which the benzene tricarboxylate ligands and CuII centres form copper acetate lantern synthons. HKUST-1 is commonly prepared using a water–ethanol mixture, and all three of the carboxylate groups of the btc3− ligand are involved in the copper acetate lantern formation giving rise to a three-dimensional framework of twisted boracite topology.8 The use of methanol in the synthesis of STAM-1, however, results in the formation of the mono-methyl ester of btc, which acts as a two-connecting ligand within the two-dimensional Kagome lattice of STAM-1.1aWithin the last decade, interest has been growing in the use of porous materials (zeolites,9 coordination polymers1a,10 and polyoxometallates11) to store and deliver biologically active gases, such as nitric oxide (NO). Nitric oxide is of interest due to its in vivo roles in wound healing and preventing thrombosis formation, as well as its antibacterial properties.12 Introducing exogenous NO into the body by means of a porous delivery framework allows the release of the NO to be targeted to a specific and localised area, as opposed to the systemic exposure that occurs when solution (NO from glyceryl trinitrate) or gas phase (NO from a cylinder) delivery is used.13
Given the similarity of isophthalic acid to the common MOF ligands terephthalic acid and trimesic acid, simple 5-substituted isophthalic acids (such as the commercially available 5-methyl,14 bromo,15 and tert-butyl,16 or readily synthesised 5-alkoxy17 or 5-alkylamino18 derivatives) have been used in only a modest number of structural investigations in the absence of a bridging co-ligand. When non-coordinating substituents are used at the 5-position, the relative orientation of the carboxylic acid groups and the 5-substituent give this family of ligands a hydrophilic ‘side’ which can bind to metal centres, and a second side whose polarity and steric bulk may be tuned without directly influencing the binding mode of the isophthalate moiety.
Herein we report the synthesis and structure determinations of four coordination polymers prepared by reaction of zinc acetate and 5-methoxy isophthalic acid (I) under solvothermal conditions. Under most conditions, a mixture of products is formed, from which we were able to identify four different coordination polymers. Preliminary nitric oxide release data are reported for compounds 2 – Zn6(MeOip)4.5(HMeOip)(OH)2(H2O)2·5.5H2O – and 4 Zn5(MeOip)4(OH)2(H2O)4·H2O.

Results and discussion
Structural investigations
Compounds 1–4 were prepared solvothermally by reaction of Zn(OAc)2·2H2O and H2MeOip in water or aqueous alcohols (methanol, ethanol and isopropanol). Details of crystal structure determinations may be found in the Experimental section and in ESI.† Reactions in isopropanol afforded compound 4 as a phase pure product, however, reactions in water, aqueous methanol and aqueous ethanol gave a range of products, of which three coordination polymers in addition to compound 4 could be identified crystallographically. Powder X-ray diffraction experiments show that the pattern produced by these crystalline products vary from reaction to reaction. Fortuitously, during the series of repeat syntheses, compounds 1 and 2 were each prepared once as a phase pure product from reactions in water and aqueous methanol, respectively. Subsequent reactions in aqueous methanol or ethanol afforded compound 2 that was contaminated with other products, whilst every subsequent reaction in water afforded compound 3 either as a pure solid or with contamination of compound 1. Reliable syntheses of compounds 1 and 2 remain elusive, despite attempts to control the products by varying the alcohol to water ratio (for the methanol syntheses) or the reaction time (for water syntheses). Full powder diffraction comparisons on each attempted synthesis are presented in ESI.†Compound 1 forms as a one dimensional ‘ribbon of rings’ polymer of composition Zn2(HMeOip)(MeOip)(OAc), in which all ZnII centres and all 5-methoxy isophthalate ligands are crystallographically equivalent. Pairs of tetrahedral ZnII centres are connected by three μ2 carboxylate groups into dimers that are reminiscent of copper acetate lanterns (Fig. 1).
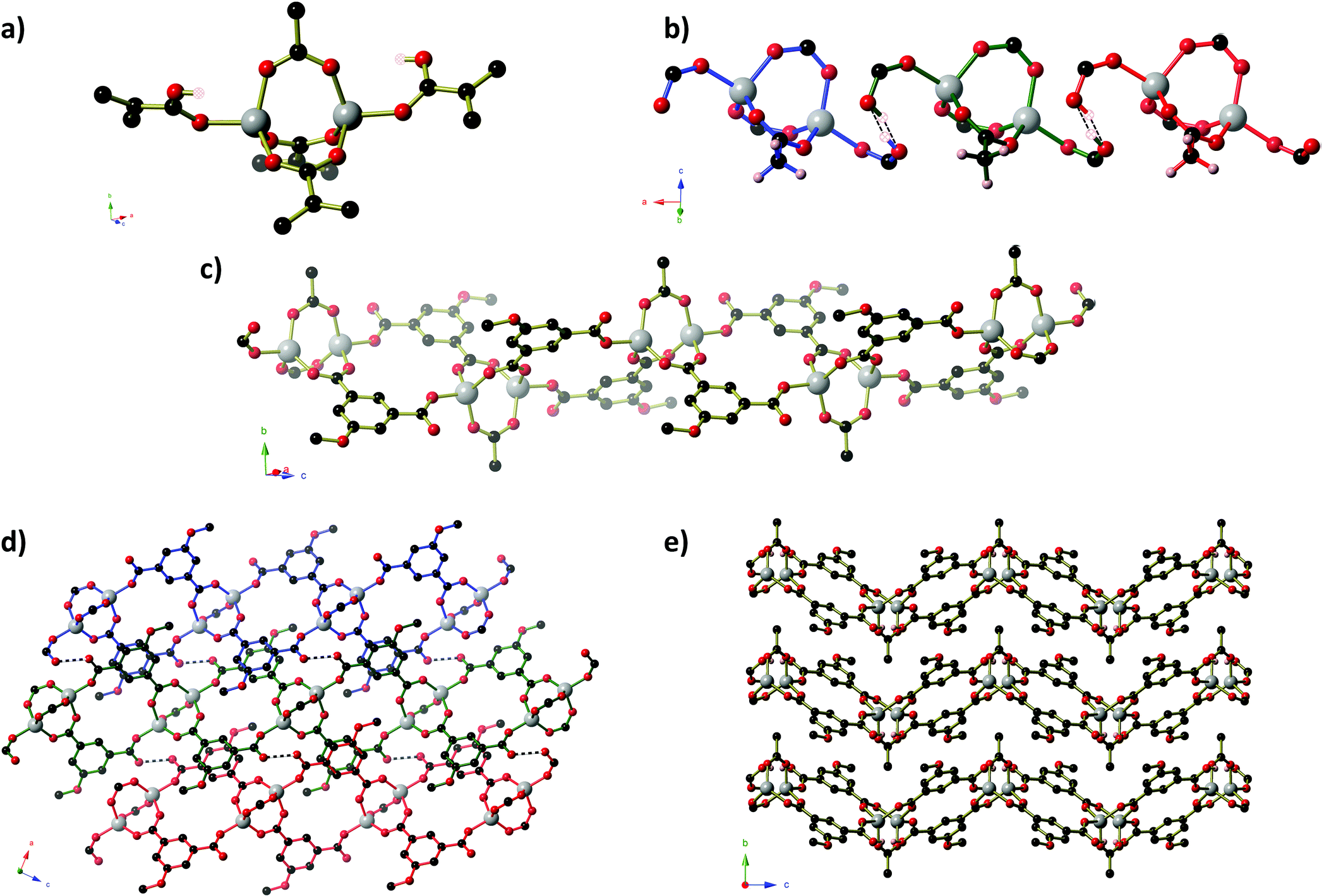 | ||
| Fig. 1 The structure of Zn2(MeOip)(HMeOip)(OAc). (a) One dinuclear Zn2(CO2)3 paddlewheel unit. For clarity, segments of the aromatic ring from the MeOip ligands are shown. The partial occupancy carboxylic hydrogen atoms are shown as hatched spheres. (b) The short hydrogen bonds formed by sharing a proton between two carboxylate groups from neighbouring chains. Short O–H⋯O bonds are shown as dashed lines. (c) A single ribbon-of-rings chain of Zn2(MeOip)(HMeOip)(OAc). (d) Three chains from one hydrogen-bonded sheet of Zn2(MeOip)(HMeOip)(OAc) showing the offset aromatic interactions between MeOip ligands. Short hydrogen bonds are shown as black and white striped bonds. (e) Three parallel hydrogen-bonded sheets. Colour key C: black, O: red, Zn: grey, H: pink. | ||
Two of these acetate groups are symmetry-related and belong to methoxy isophthalate ligands whilst the remaining one belongs to an acetate mono-anion (Fig. 1). The apical coordination sites on the Zn dimers are occupied by monodentate carboxylate groups from MeOip ligands. The non-coordinating oxygen atom of the monodentate carboxylate groups are protonated 50% of the time, and participate in short hydrogen bonds (O⋯O separation 2.435(3) Å) to symmetry-related oxygen atoms on parallel chains. The short oxygen–oxygen distance in these hydrogen bonds is the result of the proton being ‘shared’ between the two carboxylate groups.
Chains of 1 extend parallel to the [1 0 1] direction. Each ‘ring’ within the chain contains two co-planar MeOip ligands that are related by a two-fold axis that passes through the centre of the ‘ring’. Parallel chains are connected by the short hydrogen bonds into layers that extend in the ac-plane. Within a given sheet, the MeOip ligands are aligned such that offset face-to-face π-interactions can occur between the central rings and the bridging carboxylate group (close contact C⋯C separation ∼3.3 Å). The methyl groups of the acetate anion project above and below the plane of the hydrogen-bonded sheets. These methyl groups are directed toward the Zn dimers, where there appear to be weak C–H⋯O hydrogen bonds between the acetate methyl group and the μ2-MeOip carboxylate groups (C⋯O separations of 3.565(4) Å and 3.601(4) Å).
Compound 2 forms as a three-dimensional framework of composition Zn6(MeOip)4.5(HMeOip)(OH)2(H2O)2·5.5H2O, which contains of Zn6(OH)2(RCO2)10(RCO2H)(H2O)2 clusters bridged by methoxy isophthalate ligands (Fig. 2). Within each Zn6 cluster, every atom is crystallographically unique and the Zn centres adopt tetrahedral, octahedral, square pyramidal and distorted trigonal bipyramidal coordination geometries (Fig. 2). Eleven methoxy isophthalate ligands connect each Zn6 cluster to seven symmetry-related clusters (3 via single MeOip ligands, and 4 via pairs of MeOip ligands). The Zn6 clusters may be considered as 7-connecting nodes bridged by MeOip rods, resulting in a uninodal 7-connected framework, whose 33·412·55·6 topology is shown in Fig. 2b. Seven connected frameworks are rare and a search of the literature revealed only 4 other coordination networks with 7-connected nodes,19 although the 33·412·55·6 net has been reported previously in the potassium and rubidium salts of 4-chloro-2,6-dimethylphenolate,20 and the barium salt of terephthalic acid.21
 | ||
| Fig. 2 (a) The Zn6 cluster that occurs within compound 2, Zn6(MeOip)4.5(HMeOip)(OH)2(H2O)2·5.5H2O. Coordinated water molecules are shown in blue. (b) A schematic representation of the 7-connected framework of compound 2. The Zn6 clusters have been simplified to green spheres and the bridging MeOip ligands are shown as long gold connections. | ||
Compound 3, Zn(MeOip)(H2O)2·H2O, like compound 1, exists as a ‘ribbon of rings’ coordination polymer containing Zn2 units. In compound 3, however, the ZnII centres adopt trigonal bipyramidal coordination geometries and are bridged by methoxy isophthalate carboxylate groups to form planar 8-membered ZnOCOZnOCO rings (Fig. 3). Monodentate carboxylate groups occupy the remaining coordination site in the trigonal plane, and the coordination environment of the Zn centres is completed by two axial water molecules. Adjacent pairs of Zn2 units are connected by a pair of MeOip ligands, forming rings similar to those seen compound 1. Along the chain, all of the rings are very close to coplanar, lying either slightly above or below the average plane of the Zn–MeOip strip. The chains of compound 3 propagate parallel to the a-axis. Extensive hydrogen bonds between the coordinated water molecules and the non-coordinated oxygen atoms of the carboxylate and methoxy ether groups connect the chains into sheets. Within the sheets, π-stacking of the MeOip ligands occurs, with close contact C⋯C separations of ∼3.4 Å. Non-coordinated water molecules occupy the small channels between sheets, and further connect the chains into a three-dimensional hydrogen-bonded network. Thermogravimetric analysis shows that both coordinated and guest water molecules may be removed by heating to 100 °C (see ESI† for TGA trace).
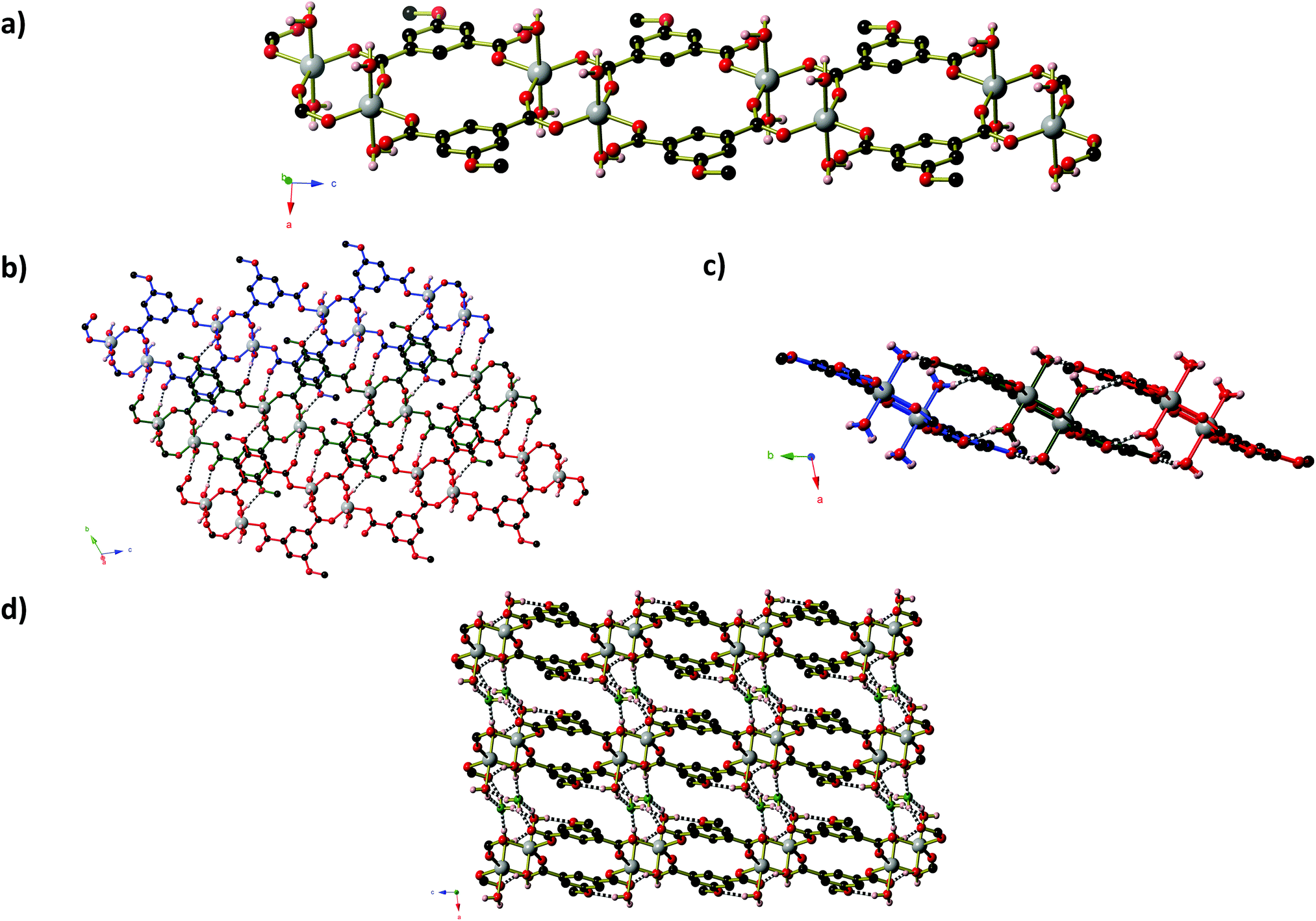 | ||
| Fig. 3 The structure of Zn(MeOip)(H2O)2·H2O (compound 3). (a) One chain of Zn(MeOip)(H2O)2. (b) and c) Views of the hydrogen-bonded sheets of Zn(MeOip)(H2O)2 from (b) almost perpendicular to and (c) along the plane of the sheet. Three chains are shown, coloured in blue, red and green. Hydrogen bonds are shown as black and white striped bonds. (d) The hydrogen-bonded network of Zn(MeOip)(H2O)2·H2O. Oxygen atoms of the guest water molecules are shown in green. Aliphatic and aromatic hydrogen atoms have been omitted for clarity. | ||
Like compound 2, compound 4 also contains clusters of Zn centres bridged by μ3-OH anions (Fig. 4). The two-dimensional framework of 4 has composition Zn5(MeOip)4(OH)2(H2O)4·H2O and contains two distinct penta-nuclear Zn5(OH)2 clusters, which are shown in Fig. 4a and b. These clusters are comprised of four Zn centres in a square planar arrangement about a central octahedral Zn centre. In the first cluster (Type 1), centred around Zn(1), the four peripheral Zn centres (Zn(2) and Zn(3)) have a tetrahedral coordination environment consisting of two carboxylate groups from methoxy isophthalate ligands, one hydroxide anion and one water molecule. Zn(2) centres have an additional long interaction of 2.587(3) Å with a second oxygen of one of the carboxylate groups. In the second cluster (Type 2), centred on Zn(4), two of the peripheral Zn centres have a tetrahedral coordination environment (with the same combination of ligands as Zn(2) and Zn(3)) with an additional long interaction between Zn(5) and O(1) of 2.629(4) Å that we deem too long to be a coordination bond, whilst the remaining two have an octahedral environment, in which a third, bidentate carboxylate group is bound to the Zn(6) centre. In all cases, Zn–O distances lie in the range 1.92 to 2.45 Å. Three of the four 5-methoxyisophthalate ligands play essentially the same role, with one carboxylate arm binding to two Zn centres from one Zn5 cluster (the central Zn centre plus one on the periphery) whilst the second arm binds in a monodentate fashion to one Zn centre on the periphery of an adjacent cluster of the opposite type. These isophthalate ligands connect the Zn5 clusters into strips that extend parallel to the [−1 0 1] direction (Fig. 4c). Inside each strip, non-coordinated water molecules are enclathrated in the space between Type 1 and Type 2 Zn5 clusters, held in place by hydrogen bonds to non-coordinated carboxylate oxygen atoms and μ3-OH anions. One arm of the remaining isophthalate ligand also binds in a μ2 fashion within a Type 2 cluster, but the second carboxylate arm is twisted out of the plane of the phenyl ring −36.88(56)° – such that the second carboxylate oxygen atom can bind to a Zn(6) centre on a neighbouring Type 2 cluster. This connects the strips into sheets that extend parallel to the ac-plane (Fig. 4d). The Zn5 clusters lie in the mid-section of the sheets, whilst the methoxy groups of the ligands project outwards from the sheet, giving the sheets a corrugated surface with hydrophobic peaks. The isophthalate ligands are aligned within the sheets such that their phenyl rings are able to participate in offset π-stacking interactions, with close-contact C⋯C separations of 3.50 to 4.00 Å. Sheets pack along the b-axis in such a way that allows the methoxy groups of one sheet to interdigitate with those belonging to the sheets directly above and below (Fig. 4e).
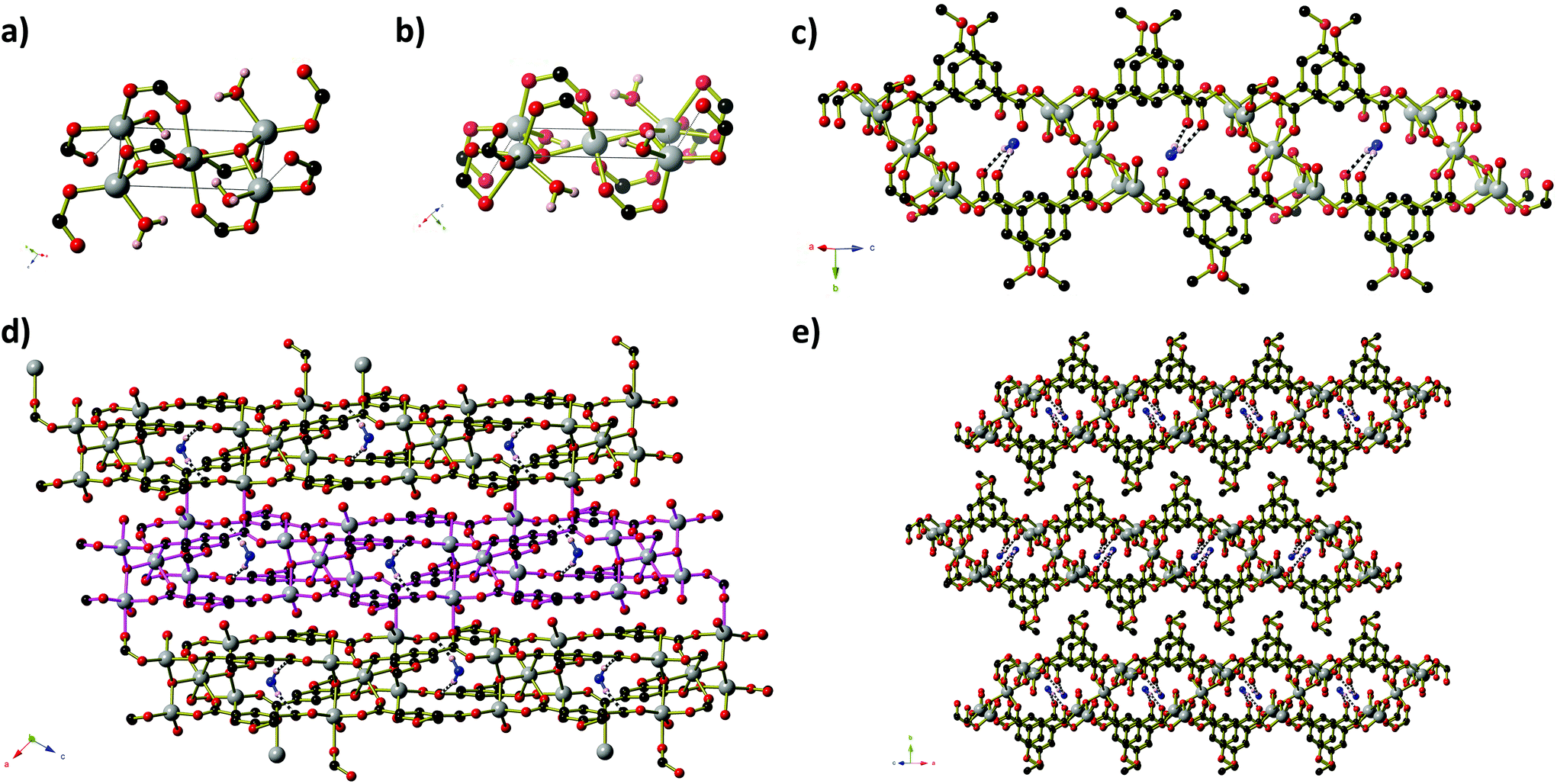 | ||
| Fig. 4 The structure of Zn5(MeOip)4(OH)2(H2O)4·H2O. (a) Type 1 and (b) Type 2 Zn5 clusters. The long Zn⋯O interactions are shown as dotted black lines. (c) A side-on view of one ‘strip’ of Zn5(MeOip)4(OH)2(H2O)4. Water molecules are shown in blue, and hydrogen bonds are depicted as black and white striped bonds. Hydrogen bonds involving OH anion have been omitted for clarity. (d) A view perpendicular to the sheet. The ‘strip’ from (c) has been highlighted using pink bonds. (e) The stacking of the sheets along the b-axis. | ||
The similar makeup of the two types of cluster leads to some pseudo symmetry within the structure. It is possible to obtain a structural solution in which this higher symmetry is taken into account. The ‘higher symmetry’ structure has cell lengths a = 7.8173(16) Å, b = 27.317(5) Å, c = 10.203(2) Å, β = 110.7028(10)°. Apart from the re-ordering of the a and c axes, one cell length has been significantly reduced, from 14.9 Å to 7.8 Å. This solution contains only one Zn5 cluster, however the carboxylate group that cross-links parallel strips is now disordered over two orientations – one where it crosslinks the strips, and one where both oxygen atoms are bound to a single cluster. Given that this disorder is local and does not provide information about the longer range order, the ‘lower’ symmetry solution has been selected as the correct one. PLATON analysis of the lower symmetry solution does not find additional higher symmetry.
Nitric oxide release from compounds 2 and 4
Of the four compounds prepared as part of this work, compounds 2 and 4 were selected for investigations involving nitric oxide uptake and release as they both contain pendant water molecules that may potentially be removed to produce vacant coordination sites on the Zn centres to which the NO molecules can bind. Thermogravimetric analyses showed that, for both compounds, the preliminary mass loss step had occurred before 200 °C under atmospheric pressure and no further mass loss steps occur until approximately 350 °C. There was insufficient solid to run a surface area measurement on compound 2 after NO release experiments were conducted, however compound 4 was found to have a BET surface area of 29 m2 g−1. The two compounds were dehydrated at 200 °C under dynamic vacuum for approximately 16 hours. After cooling, the dehydrated solid was exposed to 1 atm NO for one hour. Typical release plots are shown in Fig. 5 and 6 (further details are included in ESI†). Both compounds show a very small release (∼20 nmol g−1) which occurs in one quick burst. This is indicative of nitric oxide interacting with only the surface of the powder and not with the vacant coordination sites generated by dehydration of the compounds. This is approximately 5 orders of magnitude lower than the NO release from Co- or Ni-CPO-27, approximately 6 mmol per gram of loaded material, which have the best release of MOF-stored NO known to date.22 Other MOFs whose nitric oxide storage and release properties have been investigated lie between these two values: HKUST-1 adsorbs approx 3 mmol g−1, but releases only approx 2 μmol g−1,10a and STAM-1 adsorbs approx 600 μmol g−1.1a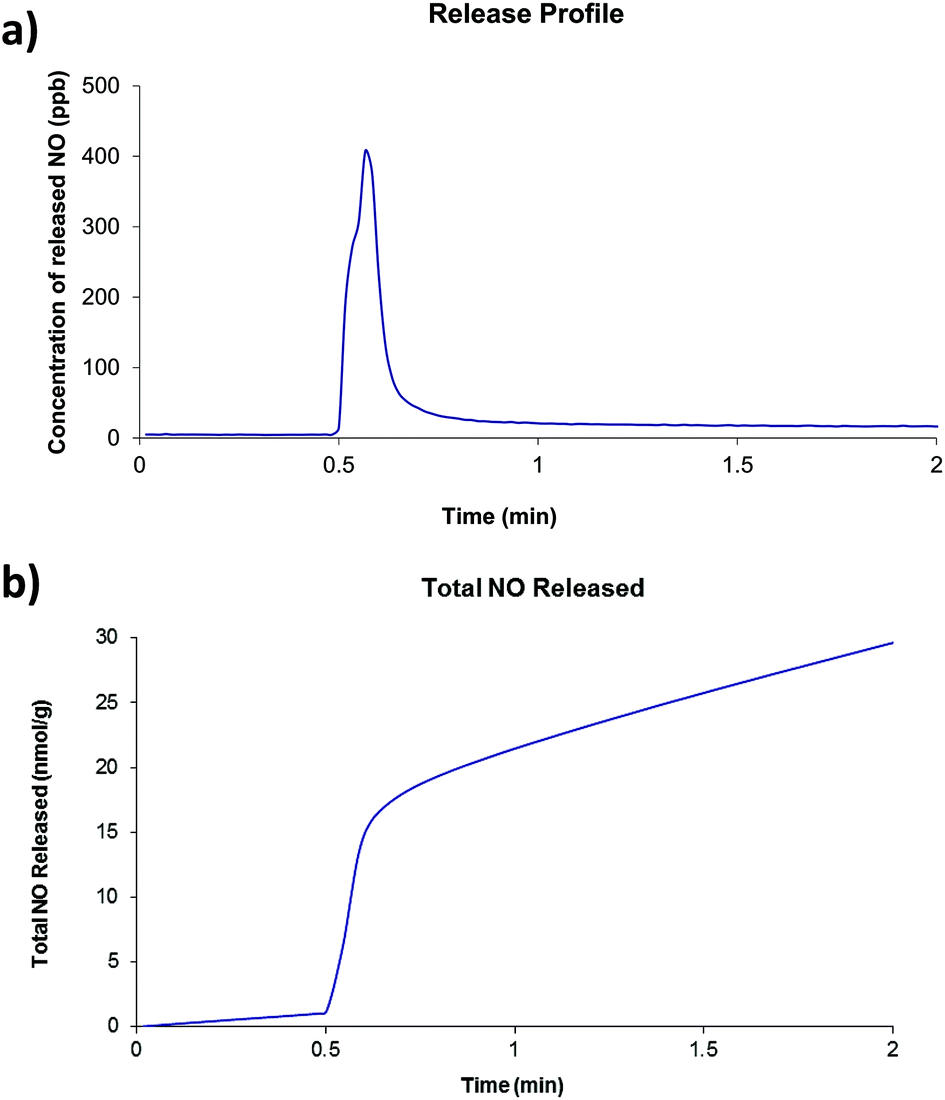 | ||
| Fig. 5 (a) Typical nitric oxide release profile and (b) total nitric oxide released for compound 2. | ||
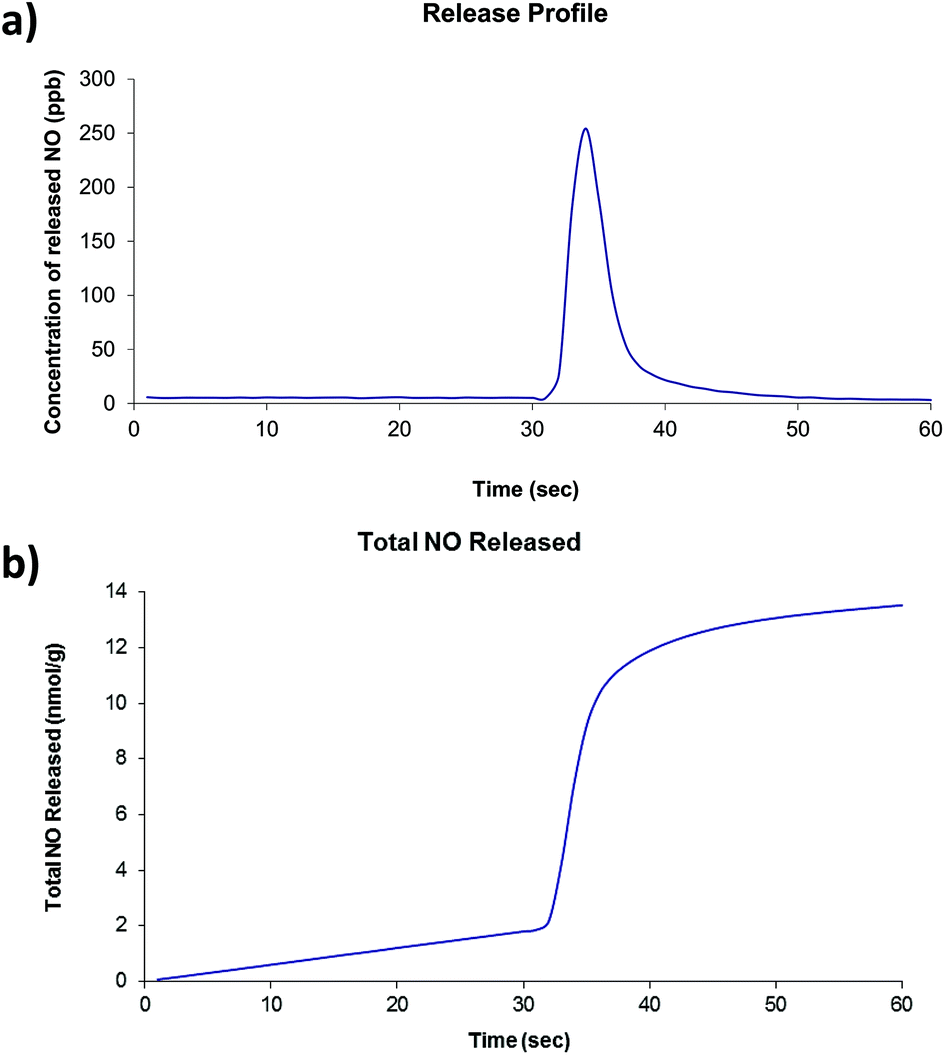 | ||
| Fig. 6 (a) Typical nitric oxide release profile and (b) total nitric oxide released for compound 4. | ||
Some variation in the amount of NO released per gram of material is observed, however, this a common occurrence in our experience. Given the very small total amount of NO release (nmol g−1 released by ∼ 12 mg), very small changes in ambient NO at the beginning of the measurements, and the arbitrary collection time have a large knock-on effect. Powder diffraction patterns collected on both compounds after the NO release (see ESI†) show that compound 2 remains virtually unchanged but compound 4 displays a loss of crystallinity which is consistent with significant decomposition of the framework. A summary of the NO release experiments is presented in Tables 1 and 2. Given the poor NO release observed in these initial tests, further investigations are considered to be unwarranted.
| Vial | Loaded weight | Total NO released per gram of loaded material after 120 s release time |
|---|---|---|
| 1 | 17.8 mg | 29.6 nmol g−1 |
| 2 | 18.4 mg | 22.3 nmol g−1 |
| 3 | 19.3 mg | 18.7 nmol g−1 |
| 4 | 17.7 mg | 16.6 nmol g−1 |
| Vial | Loaded weight | Total NO released per gram of material after 60 s release time |
|---|---|---|
| 1 | 12.3 mg | 17.4 nmol g−1 |
| 2 | 9.2 mg | 13.5 nmol g−1 |
| 3 | 12.2 mg | 13.5 nmol g−1 |
| 4 | 11.1 mg | 22.3 nmol g−1 |
| 5 | 12.6 mg | 23.1 nmol g−1 |
Conclusions
We have prepared 4 novel coordination polymers by adjusting the solvent system of reactions between zinc acetate and 5-methoxy isophthalic acid. In each case, poly-nuclear Zn units are bridged by methoxy isophthalate anions to form 1-, 2- or 3-dimensional coordination polymers. Nitric oxide release experiments on compounds 2 and 4 show very poor release of nitric oxide.Experimental
General
All reagents were obtained from commercial sources and were used without further purification. 5-Methoxy isophthalic acid was prepared according to literature procedure.23 NMR data were recorded using automated procedures on a Bruker Avance II 400 Hz spectrometer. Elemental analyses were conducted by London Metropolitan University. Powder X-ray diffraction patterns were collected using Cu Kα1 radiation on a PANalytical Empyrean diffractometers radiation (compounds 2 to 4) or a STOE STADIP diffractometer operating in Debye–Scherrer mode.Syntheses
5-Methoxy isophthalic acid,23 5-hydroxy isophthalic acid (18.2 g, 0.10 mol), K2CO3 (40.73 g, 0.34 mol) and dimethyl sulphate (52 mL, 0.55 mol) were refluxed for 18 hours in acetone (300 mL). The mixture was allowed to cool, and was poured onto 1.3 L of water. The resulting white powder was collected on a glass frit. Dimethyl 5-methoxyisophthalate was recrystallised from cyclohexane. Yield: 17.47 g, 0.078 mol, 78% yield. 1H NMR (400 MHz, d4-MeOD) δ 3.89 (s, 3H, –OCH3), 3.93 (s, 6H, –CO2CH3), 7.73 (d, 2H, aromatic CH, J = 1.44 Hz), 8.19 (t, 1H, aromatic CH, J = 1.46 Hz) ppm. 13C NMR (101 MHz, d4-MeOH) δ 52.95 (–OCH3), 56.28 (–CO2CH3), 120.06 (aromatic), 123.53 (aromatic), 133.14 (C–CO2Me), 161.34 (C–OMe), 167.37 (–CO2Me) ppm.Aqueous potassium hydroxide (5.72 M, 40 mL, 0.23 mol) was added to a methanolic solution (150 mL) of dimethyl 5-methoxyisophthalate (15.35 g, 0.068 mol) and heated to reflux for 7 hours. The cooled mixture was then poured onto distilled water (375 mL), and the solution acidified to pH 1 using conc. HCl. The crude 5-methoxy isophthalic acid was recrystallised from glacial acetic acid. Yield: 12.87 g, 0.065 mol, 84%. 1H NMR (400 MHz, d4-MeOD) δ 3.89 (s, 3H, –OCH3), 7.74 (d, 2H, aromatic CH, J = 1.44 Hz), 8.23 (t, 1H, aromatic CH, J = 1.44 Hz) ppm. 13C NMR (101 MHz, d4-MeOH) δ 56.19 (–OCH3), 120.12 (aromatic), 124.10 (aromatic), 133.70 (C–CO2H), 161.22 (C–OMe), 168.68 (–CO2H) ppm.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 water:iso-propanol (30 mL) to 110 °C in a 50 mL teflon-lined steel autoclave for 3 days. Yield: 357 mg, 0.29 mmol, 73%. Elemental analysis Calcd for Zn5(MeOip)4(OH)2(H2O)4·H2O C: 35.22, H: 2.96, Found C: 35.34, H: 3.03%.
:2 water:iso-propanol (30 mL) to 110 °C in a 50 mL teflon-lined steel autoclave for 3 days. Yield: 357 mg, 0.29 mmol, 73%. Elemental analysis Calcd for Zn5(MeOip)4(OH)2(H2O)4·H2O C: 35.22, H: 2.96, Found C: 35.34, H: 3.03%.
Crystallography
Crystals were coated in a protective oil and mounted on a MiTeGen fibre tip (2) or a Molecular dimensions mounted 0.1 mm litholoop (1, 3 and 4). Data were collected on a Rigaku Mercury2 SCXMini diffractor (λ = 0.71075 Å) or on a Bruker APEXII diffractometer (λ = 0.77490 Å) at station 11.3.1 of the Advanced Light Source. For data collected at beamline 11.3.1, appropriate scattering factors were applied using XDISP.24 Absorption corrections were applied using multi-scan25 or numerical methods.25b Structure solutions were obtained using either SHELXS-97 or SHELXT (beta) and refined by full matrix on F2 using SHELXL-97.26 All full occupancy non-hydrogen atoms were refined with anisotropic thermal displacement parameters. Aromatic and aliphatic hydrogen atoms were included at their geometrically estimated positions as were hydrogen atoms bound to μ3-OH− anions in compound 4 and non-coordinated carboxylic OH groups. Hydrogen atoms belonging to free or coordinated water molecules were assigned, fixed at a distance of 0.9 Å from the oxygen atom and 1.47 Å from the second hydrogen atom of the water molecule, and their thermal parameters linked to those of the oxygen atom to which they are bound.080(3) Å3, space group Pbcn, Z = 8, θmax = 26.19°, T = 150(2) K, 836 parameters, 3 restraints, R1 = 0.0769 (for 8556 data I > 2σ(I)), wR2 = 0.2056 (all 10004 data).
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, θmax = 27.49°, T = 173(2) K, 182 parameters, 9 restraints, R1 = 0.0451 (for 1947 data I > 2σ(I)), wR2 = 0.0921 (all 2600 data).
, Z = 2, θmax = 27.49°, T = 173(2) K, 182 parameters, 9 restraints, R1 = 0.0451 (for 1947 data I > 2σ(I)), wR2 = 0.0921 (all 2600 data).
Acknowledgements
This work was funded by the British Heart Foundation (NH/11/8/29253) and the EPSRC (EP/K005499/1). Crystallographic data for compound 2 were collected at station 11.3.1 at the Advanced Light Source, Berkeley, CA, USA. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.Notes and references
- (a) M. I. H. Mohideen, B. Xiao, P. S. Wheatley, A. C. McKinlay, Y. Li, A. M. Z. Slawin, D. W. Aldous, N. F. Cessford, T. Düren, X. Zhao, R. Gill, K. M. Thomas, J. M. Griffin, S. E. Ashbrook and R. E. Morris, Nat. Chem., 2011, 3, 304 CrossRef CAS PubMed; (b) H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takaa and S. Kitagawa, Science, 2014, 343, 167 CrossRef CAS PubMed.
- R.-Q. Zou, H. Sakurai, S. Han, R.-Q. Zhong and Q. Xu, J. Am. Chem. Soc., 2007, 129, 8402 CrossRef CAS PubMed.
- (a) X.-F. Wang, L. Li, Y.-M. Kong and Y. Liu, Inorg. Chem. Commun., 2012, 21, 72 CrossRef CAS; (b) L.-F. Ma, L.-Y. Wang, D.-H. Lu, S. R. Batten and J.-G. Wang, Cryst. Growth Des., 2009, 9, 1741 CrossRef CAS.
- P. K. Allan, K. W. Chapman, P. J. Chupas, J. A. Hriljac, C. L. Renouf, T. C. A. Lucas and R. E. Morris, Chem. Sci., 2012, 3, 2559 RSC.
- M. G. Goesten, F. Kapteijn and J. Gascon, CrystEngComm, 2013, 15, 9249 RSC.
- (a) M. Dai, Z. Yang, C.-Y. Ni, H. Yu, Y. Chen, H.-X. Li, Z.-G. Ren and J.-P. Lang, Inorg. Chem. Commun., 2014, 40, p205 CrossRef; (b) H.-L. Jiang, Y. Tatsu, Z.-H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586 CrossRef CAS PubMed.
- (a) D. Frahm, F. Hoffmann and M. Fröba, Cryst. Growth Des., 2014, 14, 1719 CrossRef CAS; (b) W.-H. Huang, X.-J. Luan, X. Zhou, J. Chen, Y.-Y. Wang and Q.-Z. Shi, CrystEngComm, 2013, 15, 10398 Search PubMed; (c) X. Zhou, P. Liu, W.-H. Huang, M. Kang, Y.-Y. Wang and Q.-Z. Shi, CrystEngComm, 2013, 15, 8125 RSC.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS PubMed.
- (a) P. S. Wheatley, A. R. Butler, M. S. Crane, S. Fox, B. Xiao, A. G. Rosse, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2006, 128, 502 CrossRef CAS PubMed; (b) B. Xiao, P. S. Wheatley and R. E. Morris, Studies Surf. Sci. Cat., 2007, 170A, 902 CAS; (c) M. Mowbray, X. J. Tan, P. S. Wheatley, A. G. Rossi, R. E. Morris and R. B. Weller, J. Invest. Dermatol., 2008, 128, 352 CrossRef CAS PubMed; (d) H. A. Liu and K. J. Balkus, Chem. Mater., 2009, 21, 5032 CrossRef CAS; (e) G. Narin, C. B. Albayrak and S. Ulku, Appl. Clay Sci., 2010, 50, 560 CrossRef CAS; (f) M. L. Pinto, A. C. Fernandes, J. Rocha, A. Ferreira, F. Antunes and J. Pires, J. Mater. Chem. B, 2014, 2, 224 RSC.
- (a) B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203 CrossRef CAS PubMed; (b) A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440 CrossRef CAS PubMed; (c) J. G. Nguyen, K. K. Tanabe and S. M. Cohen, CrystEngComm, 2010, 12, 2335 RSC; A. C. McKinlay, J. F. Eubank, S. Wuttke, B. Xiao, P. S. Wheatley, P. Bazin, J.-C. Lavalley, M. Daturi, A. Vimont, G. de Weireld, P. Horcajada, C. Serre and R. E. Morris, Chem. Mater., 2013, 25, 1592 Search PubMed; (d) F. R. S. Lucena, L. C. C. de Araujo, M. do D. Rodrigues, T. G. da Silva, V. R. A. Pereira, G. C. G. Militao, D. A. F. Fontes, P. J. Rolim-Neto, F. F. da Silva and S. C. Nascimento, Biomed. Pharmacother., 2013, 67, 707 CrossRef CAS PubMed; (e) S. R. Miller, E. Alvarez, L. Fradcourt, T. Devic, S. Wuttke, P. S. Wheatley, N. Steunou, C. Bonhomme, C. Gervais, D. Laurencin, R. E. Morris, A. Vimont, M. Daturi, P. Horcajada and C. Serre, Chem. Commun., 2013, 49, 7773 RSC; (f) J. L. Harding and M. M. Reynolds, J. Mater. Chem. B, 2014, 2, 2530 RSC.
- J. Zhang, J. Hao, R. N. N. Khan, J. Zhang and Y. Wei, Eur. J. Inorg. Chem., 2013, 1664 CrossRef.
- (a) H. F. Zhu, B. Ka and F. Murad, World J. Surg., 2007, 31, 624 CrossRef PubMed; (b) M. R. Miller and I. L. Megson, Br. J. Pharmacol., 2007, 151, 305 CrossRef CAS PubMed.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966 CrossRef CAS PubMed.
- (a) R.-Q. Zou, H. Sakurai, S. Han, R.-Q. Zhong and Q. Xu, J. Am. Chem. Soc., 2007, 129, 8402 CrossRef CAS PubMed; (b) Z.-L. Yu and J.-L. Hu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1522 CrossRef CAS PubMed; (c) H. Chun and H. Jung, Inorg. Chem., 2009, 48, 417 CrossRef CAS PubMed; (d) W.-X. Chen, Y.-P. Ren, L.-S. Long, R.-B. Huang and L.-S. Zheng, CrystEngComm, 2009, 11, 1522 RSC; (e) L. Wang, J. Liu, C.-Z. Zheng and Y.-J. Wang, Huaxue Yu Shengwu Gongcheng, 2011, 28, 17 CAS; (f) Q.-B. Bo, Z.-W. Zhang, J.-L. Miao, D.-Q. Wang and G.-X. Sun, CrystEngComm, 2011, 13, 1765 RSC; (g) Q.-B. Bo, H.-Y. Wang, D.-Q. Wang, Z.-W. Zhang, J.-L. Miao and G.-X. Sun, Inorg. Chem., 2011, 50, 10163 CrossRef CAS PubMed; (h) H. Y. Zhu, J. W. Ji, Y. W. Li, C. W. Li and K. Z. Zhu, Koord. Khim., 2012, 38, 157 Search PubMed; (i) J.-W. Ji, X.-F. Wang, B.-Y. Li and Z.-B. Han, Solid State Sci., 2012, 14, 335 CrossRef CAS; (j) S. Du, C. Hu, J.-C. Xiao, H. Tan and W. Liao, Chem. Commun., 2012, 48, 9177 RSC; (k) Q.-B. Bo, H.-Y. Wang, J.-L. Miao and D.-Q. Wang, RSC Adv., 2012, 2, 11650 RSC; (l) X.-L. Deng, S.-Y. Yang, R.-F. Jin, J. Tao, C.-Q. Wu, Z.-L. Li, L.-S. Long, R.-B. Huang and L.-S. Zheng, Polyhedron, 2013, 50, 219 CrossRef CAS; (m) Q.-B. Bo, H.-Y. Wang and D.-Q. Wang, New J. Chem., 2013, 37, 380 RSC; (n) L.-P. Xue, Y.-C. Liu, Y.-H. Han, C.-B. Tain, Q.-P. Li, P. Lin and S.-W. Du, Jiegou Huaxue, 2014, 33, 1191 CAS; (o) C.-B. Tian, R.-P. Chen, C. He, W.-J. Li, Q. Wei, X.-D. Zhang and S.-W. Du, Chem. Commun., 2014, 50, 1915 RSC; (p) L. Jin, L.-L. Zha, S. Gao, S.-Y. Yang and R.-B. Huang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2015, 71, m1 CrossRef CAS PubMed.
- (a) G.-X. Liu, Y. Wang, L.-F. Huang, X.-J. Kong and H. Chen, J. Inorg. Organomet. Polym. Mater., 2008, 18, 358 CrossRef CAS; (b) L. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m974 CrossRef CAS PubMed; (c) G.-X. Liu, R.-Y. Huang, H. Xu, X.-J. Kong, L.-F. Huang, K. Zhu and X.-M. Ren, Polyhedron, 2008, 27, 2327 CrossRef CAS; (d) H. Furukawa, J. Kim, N. W. Ockwig, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11650 CrossRef CAS PubMed; (e) Y.-P. Li, Z. Kristallogr. - New Cryst. Struct., 2009, 224, 523 CAS; (f) G.-X. Liu, Y.-Y. Xu and X.-M. Ren, Wuji Huaxue Xuebao, 2010, 26, 1885 CAS; (g) P.-F. Wang, M.-G. Sheng, X.-S. Wu and X. Wang, Inorg. Chim. Acta, 2011, 379, 135 CrossRef CAS; (h) R. Zhang, Q. Gong, T. J. Emge, D. Banerjee and J. Li, Chimia, 2013, 67, 393 CrossRef CAS PubMed; (i) D. Feng, K. Wang, Z. Wei, Y.-P. Chen, C. M. Simon, R. Arvapally, R. L. Martin, M. Bosch, T.-F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H.-C. Zhou, Nat. Commun., 2014, 5, 5723 CrossRef CAS PubMed.
- (a) L. Pan, B. Parker, X. Huang, D. H. Olson, J. Y. Lee and J. Li, J. Am. Chem. Soc., 2006, 128, 4180 CrossRef CAS PubMed; (b) S. Ma, D. Sun, X.-S. Wang and H.-C. Zhou, Angew. Chem., Int. Ed., 2007, 46, 2458 CrossRef CAS PubMed; (c) X.-L. Li and M.-L. Huang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1501 CrossRef CAS PubMed; (d) X.-L. Li and M.-L. Huang, Z. Kristallogr. - New Cryst. Struct., 2009, 224, 127 CAS; (e) Y.-T. Fan, B. Li, L.-F. Ma and L.-Y. Wang, Jiegou Huaxue, 2009, 28, 1061 CAS; (f) Z.-X. Du, L.-Y. Wang and H.-W. Hou, Z. Anorg. Allg. Chem., 2009, 635, 1659 CrossRef CAS; (g) L.-F. Ma, L.-Y. Wang, D.-H. Lu, S. R. Batten and J.-G. Wang, Cryst. Growth Des., 2009, 9, 1741 CrossRef CAS; (h) D.-S. Zhou, F.-K. Wang, S.-Y. Yang, Z.-X. Xie and R.-B. Huang, CrystEngComm, 2009, 11, 2548 RSC; (i) J.-D. Lin, X.-F. Long, P. Lin and S.-W. Du, Cryst. Growth Des., 2010, 10, 146 CrossRef CAS; (j) J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893 CrossRef CAS PubMed; (k) J.-D. Lin, S.-T. Wu, Z.-H. Li and S.-W. Du, Dalton Trans., 2010, 39, 10719 RSC; (l) C.-B. Tian, Z.-Z. He, Z.-H. Li, P. Lin and S.-W. Du, CrystEngComm, 2011, 13, 3080 RSC; (m) B. Kilbas, S. Mirtschin, T. Riis-Johannessen, R. Scopelliti and K. Severin, Inorg. Chem., 2012, 51, 5795 CrossRef CAS PubMed; (n) L.-J. Chen, J.-B. Su, R.-B. Huang, S. Lin, M.-X. Yang and H. Huang, Dalton Trans., 2011, 40, 9731 RSC; (o) X.-F. Wang, L. Li, Y.-M. Kong and Y. Liu, Inorg. Chem. Commun., 2012, 21, 72 CrossRef CAS; (p) S.-H. Li and J.-B. Guo, Z. Kristallogr .- New Cryst. Struct., 2012, 227, 389 CrossRef CAS; (q) Y.-F. Liang, Y.-R. Zhang and Z.-B. Han, Z. Anorg. Allg. Chem., 2012, 638, 423 CrossRef CAS; (r) R.-F. Jin, S.-Y. Yang, H.-M. Li, L.-S. Long, R.-B. Huang and L.-S. Zheng, CrystEngComm, 2012, 14, 1301 RSC; (s) Q.-B. Bo, H.-Y. Wang, J.-L. Miao and D.-Q. Wang, RSC Adv., 2012, 2, 11650 RSC; (t) S.-Y. Yang, H.-B. Yuan, X.-B. Xu and R.-B. Huang, Inorg. Chim. Acta, 2013, 403, 53 CrossRef CAS; (u) Q.-B. Bo, H.-Y. Wang and D.-Q. Wang, New J. Chem., 2013, 37, 380 RSC; (v) K. Noh and J. Kim, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m579 CrossRef CAS PubMed; (w) J. Wang, J.-Q. Tao, X.-J. Xu and C.-Y. Tan, Zh. Strukt. Khim., 2013, 54, 759 CAS; (x) C. Tian, Z. Lin and S. Du, Cryst. Growth Des., 2013, 13, 3746 CrossRef CAS; (y) A. K. Chaudhari, S. Mukherjee, S. S. Nagarkar, B. Joarder and S. K. Ghosh, CrystEngComm, 2013, 15, 9465 RSC.
- (a) G.-L. Wen, Z. Kristallogr. - New Cryst. Struct., 2009, 224, 497 CAS; (b) S.-M. Shen, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1173 CrossRef CAS PubMed; (c) F.-P. Liang, Q.-L. Meng, L.-F. Ma and L.-Y. Wang, Jiegou Huaxue, 2010, 29, 157 CAS; (d) H.-J. Zhang, Z. Kristallogr. - New Cryst. Struct., 2011, 226, 629 CAS; (e) X.-H. Huang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 483 CrossRef CAS PubMed; (f) C. Tian, Z. Lin and S. Du, Cryst. Growth Des., 2013, 13, 3746 CrossRef CAS; (g) A. Dorazco-Gonzalez, S. Martinez-Vargas, S. Hernandez-Ortega and J. Valdes-Martinez, CrystEngComm, 2013, 15, 5961 RSC; (h) X.-H. Chang, J.-H. Qin, M.-L. Han, L.-F. Ma and L.-Y. Wang, CrystEngComm, 2014, 16, 870 RSC; (i) H. Abourahma, G. J. Bodwell, J. Lu, B. Moulton, I. R. Pottie, R. B. Walsh and M. J. Zaworotko, Cryst. Growth Des., 2003, 3, 513 CrossRef CAS; (j) J. J. Perry, G. J. McManus and M. J. Zaworotko, Chem. Commun., 2004, 2534 RSC; (k) X. Zhang, J.-X. Yang and Y.-G. Yao, J. Inorg. Organomet. Polym. Mater., 2012, 22, 1189 CrossRef CAS; (l) X. Li, J. Li, M.-K. Li and Z. Fei, J. Mol. Struct., 2014, 1059, 294 CrossRef CAS; (m) D.-W. Zhang and G.-Y. Zhao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2015, 45, 524 CrossRef CAS.
- (a) S. Xiaofei, Y. J. X. X. Hong and L. Xiaoju, Jiegou Huaxue, 2015, 34, 562 Search PubMed; (b) J. Xu, Z.-S. Bai, T. Okamura, M.-S. Chen, W.-Y. Sun and N. Ueyama, Polyhedron, 2009, 28, 2480 CrossRef CAS; (c) J. Xu, Z. Su, M.-S. Chen, S.-S. Chen and W.-Y. Sun, Inorg. Chim. Acta, 2009, 362, 4002 CrossRef CAS; (d) X.-C. Cheng, X.-H. Zhu and H.-W. Kuai, Z. Naturforsch., B: Chem. Sci., 2013, 68, 1007 CAS; (e) H.-W. Kuai, X.-C. Cheng, L.-D. Feng and X.-H. Zhu, Z. Anorg. Allg. Chem., 2011, 637, 156 CrossRef; (f) X.-H. Zhu and X.-C. Cheng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1862 CrossRef CAS PubMed; (g) X.-J. Li, X.-H. Xu and X.-F. Guo, Jiegou Huaxue, 2014, 33, 721 CrossRef CAS PubMed; (h) Y. Wang, T. Wang, G.-X. Liu and Y.-C. Chen, Wuji Huaxue Xuebao, 2010, 26, 1467 CAS; (i) X. Liu, M. Oh and M. S. Lah, Cryst. Growth Des., 2011, 11, 5064 CrossRef CAS; (j) Y. Wang, Y.-J. Dong, J.-H. Zhou, S.-N. Wang, G.-X. Liu and Y.-C. Chen, Wuji Huaxue Xuebao, 2012, 28, 2407 CAS; (k) M. C. Das and P. K. Bharadwaj, J. Am. Chem. Soc., 2009, 131, 10942 CrossRef CAS PubMed; (l) M. C. Das and P. K. Bharadwaj, Chem. – Eur. J., 2010, 16, 5070 CrossRef CAS PubMed.
- (a) D.-L. Long, A. J. Blake, N. R. Champness, C. Wilson and M. Schröder, Angew. Chem., Int. Ed., 2001, 40, 2443 CrossRef; (b) P. Ren, W. Shi and P. Cheng, Inorg. Chem. Commun., 2008, 11, 125 CrossRef CAS; (c) W.-P. Xie, N. Wang, Y. Long, X.-R. Ran, J.-Y. Gao, C.-J. Chen, S.-T. Yue and Y.-P. Cai, Inorg. Chem. Commun., 2014, 40, 151 CrossRef CAS; (d) Q. Chen, W. Xue, J.-B. Lin, R.-B. Lin, M.-H. Zeng and X.-M. Chen, Dalton Trans., 2012, 41, 4199 RSC.
- J. J. Morris, B. C. Noll and K. W. Henderson, Chem. Commun., 2007, 5191 RSC.
- S. M. F. Lo, S. S. Y. Chiu and I. D. Williams, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, C54, 1846 CrossRef CAS.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440 CrossRef CAS PubMed.
- A. J. Guenthner, M. C. Davis, M. D. Ford, J. T. Reams, T. J. Groshens, L. C. Baldwin, L. M. Lubin and J. M. Mabry, Macromolecules, 2012, 45, 9707 CrossRef CAS.
- XDISP Program in WinGX, L. Kissel and R. H. Pratt, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 170 CrossRef.
- (a) SADABS: Area-Detector Absorption Correction, Siemens Industrial Automation, Inc., Madison, WI, 1996 Search PubMed; (b) CrystalClear 2.1, CrystalClear Software User's Guide, Molecular Structure Corporation, Rigaku Corporation, 2014 Search PubMed; (c) J. W. P. Flugrath, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1718 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and calculated PXRD patterns, TGA traces, details of NO release experiments, additional crystallographic information on compound 2 and selected bond lengths and angles on compounds 1–4. CCDC 1039561–1039564. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02924d |
| This journal is © The Royal Society of Chemistry 2015 |