
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild partial deoxygenation of esters catalyzed by an oxazolinylborate-coordinated rhodium silylene†
Songchen
Xu
,
Jeffery S.
Boschen
,
Abhranil
Biswas
,
Takeshi
Kobayashi
,
Marek
Pruski
,
Theresa L.
Windus
and
Aaron D.
Sadow
*
U.S. Department of Energy Ames Laboratory and Department of Chemistry, Iowa State University, 1605 Gilman Hall, Ames, Iowa 50011, USA
First published on 17th August 2015
Abstract
An electrophilic, coordinatively unsaturated rhodium complex supported by borate-linked oxazoline, oxazoline-coordinated silylene, and N-heterocyclic carbene donors [{κ3-N,Si,C-PhB(OxMe2)(OxMe2SiHPh)ImMes}Rh(H)CO][HB(C6F5)3] (2, OxMe2 = 4,4-dimethyl-2-oxazoline; ImMes = 1-mesitylimidazole) is synthesized from the neutral rhodium silyl {PhB(OxMe2)2ImMes}RhH(SiH2Ph)CO (1) and B(C6F5)3. The unusual oxazoline-coordinated silylene structure in 2 is proposed to form by rearrangement of an unobserved isomeric cationic rhodium silylene species [{PhB(OxMe2)2ImMes}RhH(SiHPh)CO][HB(C6F5)3] generated by H abstraction. Complex 2 catalyzes reductions of organic carbonyl compounds with silanes to give hydrosilylation products or deoxygenation products. The pathway to these reactions is primarily influenced by the degree of substitution of the organosilane. Reactions with primary silanes give deoxygenation of esters to ethers, amides to amines, and ketones and aldehydes to hydrocarbons, whereas tertiary silanes react to give 1,2-hydrosilylation of the carbonyl functionality. In contrast, the strong Lewis acid B(C6F5)3 catalyzes the complete deoxygenation of carbonyl compounds to hydrocarbons with PhSiH3 as the reducing agent.
Introduction
Partial reductions are an important part of an overall strategy for conversions of oxygenated materials and are germane to conversions of biorenewables. Such transformations can be considered the counterpart to selective partial oxidation of hydrocarbons typically sought for petrochemical conversions. Selectivity for partial oxidation or partial reduction products is a key longstanding challenge in catalysis. Organometallic complexes and homogeneous catalytic systems can provide insight into active sites to guide downstream applications and strategies for the development of mild conversions. In this regard, complete deoxygenation vs. partial deoxygenation of sugars and protected sugars into hydrocarbons may be affected by secondary vs. tertiary silanes, respectively, in the presences of strong Lewis acid catalysts.1 Similar control in metal-catalyzed hydrosilylation and hydrodeoxygenation catalysis has focused on catalyst site design2–5 or tailored oxygenated substrates.6The ester group, as a common functionality in biorenewable feedstocks, fuels, chemicals, and materials, typically reacts under reducing conditions to give alcohols or protected alcohols through C–O bond cleavage, whereas hydrodeoxygenation to alkanes through complete C–O hydrogenolysis is obtained under more forcing conditions. Alternatively, the conversion of esters to ethers through selective partial reduction could be valuable in synthetic schemes and in biorenewable conversions.
The conversion of cyclic esters to ethers may be accomplished through stoichiometric or multistep methods, while transformations of acyclic esters have been more challenging.7–9 The seminal report of mid-transition-metal-catalyzed partial ester reduction involves hydrosilylation and gives a mixture of ethers and alkoxysilanes.10 Group 8-based multimetallic carbonyl compounds were more recently reported to catalyze ether formation using trialkylsilanes (Ru) or tetramethyldisiloxane (Fe).11 These catalysts require heat or photochemical activation for catalytic conversion. Other metal precursors divert the products to the more common ester reductive cleavage12 or partial reduction to aldehydes.13 Titanium complexes also catalyze hydrosilylations to give ester cleavage or partial reduction of lactones to lactols, depending on catalytic conditions and silane reductant.14 Alternatively, gallium bromide15 and indium bromide16 Lewis acid catalysts employ tertiary silanes or disiloxanes for conversions of esters to ethers, whereas B(C6F5)3 catalyzes hydrosilylation to give silyl acetals.17
Catalytic intermediates in the above transition-metal systems are ambiguous, whereas the Lewis acid-based silane activations have been established by kinetics and isolation of models of proposed intermediates.17b,18 Organometallic-like intermediates in the above pathways may include metal silane compounds19 and metal silylene compounds.20 Metal-coordinated silylenes are known to have Lewis acid character21 and are proposed as catalytic intermediates in the hydrosilylation of carbonyls and olefins.20,22 In addition, iridium compounds with bridging silylenes [Ir(μ-SiEt2)(H)2SiHEt2]2,23 and iridium-coordinated N-heterocyclic silylenes24 are catalysts for silane-mediated reduction of amides to amines. The related catalytic reactions were not reported with rhodium. Migrations of chloride from rhodium to a transient unsaturated silicon center (as well as hydride and methyl migrations from silicon to rhodium) are proposed in formation of rhodium disilacyclobutanes.25 However, dirhodium silylene dimers were ruled out as intermediates in silane redistribution and dehydrocoupling reactions,26 and the presence of a donor as a base-stabilized silylene or in the form of a bimetallic compound can diminish reactivity. Nonetheless, the Lewis acid character of silylenes may provide a mechanistic connection to the B(C6F5)3, GaBr3, and InBr3 conversions. For example, rhodium-coordinated silylenes are invoked as intermediates in a cationic carbene-oxazoline rhodium(I)-catalyzed carbonyl hydrosilylation.20a,27,28 However, the dearth of isolated rhodium-coordinated silylenes is notable.24,29
We recently synthesized a zwitterionic rhodium(III) silyl hydride containing the mixed carbene–oxazoline borate ligand {PhB(OxMe2)2ImMes}RhH(SiH2Ph)CO (1, OxMe2 = 4,4-dimethyl-2-oxazoline; ImMes = 1-mesitylimidazole).30 Neither 1 nor its synthetic precursor {PhB(OxMe2)2ImMes}Rh(CO)2 show any catalytic hydrosilylation activity in the presence of ethyl acetate and phenylsilane. Therefore, we attempted to enhance the reactivity of 1 by reaction with B(C6F5)3 to form a cationic complex. The resulting isolated oxazoline-coordinated rhodium silylene catalyzes the deoxygenation of esters to ethers, amides to amines, and ketones to alkanes in the presence of primary organosilanes as the reducing agent and oxygen-acceptor. These catalytic dexoygenations contrast the expected addition chemistry of silanes and carbonyls typically observed with rhodium complexes.31 Herein we describe the characterization of the internal base-stabilized silylene catalyst and its reactivity in carbonyl deoxygenation.
Results and discussion
The reaction of {PhB(OxMe2)2ImMes}RhH(SiH2Ph)CO (1) and B(C6F5)3 in benzene at room temperature produces a dark oily precipitate 2 (eqn (1)), and this material is purified to a black solid by washing with benzene and pentane. | (1) |
X-ray quality single crystals of 2 are not yet available. In addition, attempts to generate 2 as [B(C6F5)4]− or [B{C6H3(CF3)2}4]−-counterion containing complexes from reactions of 1 and [Ph3C][B(C6F5)4] or [(Et2O)2H][B(C6H3(CF3)2)4] did not provide pure samples. Despite this, the characterization of 2 as an unusual oxazoline-coordinated silylene complex of rhodium is supported by extensive and unambiguous spectroscopic and computational analysis, as described below. The 1H NMR spectra of 2 in bromobenzene-d5, tetrahydrofuran-d8, chloroform-d1 or methylene chloride-d2 provide evidence for the same species, and solid state NMR spectroscopy also supports the assignment. The discussion below focuses on data acquired in bromobenzene-d5.
The 1H NMR spectrum of the isolated product 2 contained resonances assigned to one RhH and one SiH, as well as a pattern of PhB(OxMe2)2ImMes signals that indicated overall C1 symmetry for the product and a set of phenyl resonances from the silyl moiety. In the isolated material, the rhodium hydride resonance was observed as a doublet of doublets at −12.92 ppm (1JRhH = 20.0 Hz, 3JHH = 4.0 Hz). Silicon satellite signals were not detected for the rhodium hydride resonances in either 1 or 2, and from this we rule out Rh↽H–Si or Rh–H⇀Si interactions. A virtual triplet at 5.15 ppm (J = 4.0 Hz) was assigned to the single SiH, with the splitting resulting from 3JHH and 2JRhH coupling of similar magnitude. The assignment of the former coupling is supported by a crosspeak in a COSY experiment. The SiH signal was further downfield and exhibited a greater 1JSiH coupling constant (1JSiH = 234 Hz) than the diastereotopic SiHs in the rhodium starting material 1, which were observed as doublets at 4.23 and 4.68 ppm (2JHH = 6.0 Hz, 1JSiH = 170 and 188 Hz, 1 H each). The increase in one-bond coupling constant is typically attributed to greater s-character in the Si–H bond, which results from the interaction of the silicon with a substituent of greater effective electronegativity than H. An EXSY experiment did not reveal crosspeaks between RhH and SiH signals.
In addition to the hydride, the rhodium coordination sphere of 2 includes carbene and carbonyl ligands. The 13C{1H} NMR spectrum provided unambiguous evidence for coordination of these ligands through two doublets at 186.33 (1JRhC = 51 Hz, NHC) and 169.23 ppm (1JRhC = 37.5 Hz, CO). The rhodium coordination sphere also includes a silicon-based ligand, assigned on the basis of the 1JRhSi value of 42 Hz. The 29Si NMR chemical shift of 6.5 ppm is consistent with a four-coordinate silicon center. To validate the chemical shift measured in solution, we measured a 29Si NMR spectrum of 2 in the solid state, using 1H → 29Si cross-polarization under magic angle spinning (CPMAS), which similarly featured a single resonance centered at 6 ppm (see Fig. S6 in the ESI†). The MAS 1H NMR spectrum also contained an upfield signal at ∼−13 ppm assigned to the RhH.
The fifth group bonded to rhodium is an oxazoline. Correlations between 15N and 1H NMR resonances assigned to 4,4-dimethyl-2-oxazoline provided two 15N NMR chemical shifts of −199 and −167 ppm for the two inequivalent groups. These chemical shifts indicate that both oxazolines are coordinated in 2, because the chemical shift of non-coordinated 4,4-dimethyl-2-oxazoline is −128 ppm. In the precursor 1, 15N NMR chemical shifts of −161 and −172 ppm were observed for rhodium-coordinated oxazolines trans to SiH2Ph and hydride, respectively. One of these signals shifts 30 to 40 ppm upfield in the cationic compound 2. Significantly, a correlation between the upfield 15N signal at −199 ppm and the 1H NMR signal assigned to the SiH indicates that a N–Si bond has formed in 2. The remaining oxazoline that appeared at −167 ppm was assigned to the fifth ligand on rhodium. With the exception of the oxazoline, the coordination of H, SiHPh, NHC, and CO to rhodium is unambiguously established by 1JRhE coupling, and silylene insertion into rhodium-hydride, -carbonyl or -N-heterocyclic carbene donor, rather than the oxazoline, are ruled out. At the same time, these NMR data indicate that the four groups bonded to the silicon center are hydrogen, phenyl, rhodium, and an oxazoline.
Characterization of 2 by 11B and 19F NMR spectroscopy provided additional support for hydride abstraction. Two resonances were observed in the 11B NMR spectrum at −8 and −25 ppm assigned to the PhB(OxMe2)2ImMes ligand and [HB(C6F5)3]−, respectively. The latter signal was broad (400 Hz peak width at half-height), and the 1JBH was not resolved. The chemical shift of −25 ppm is consistent with a four-coordinate anionic borate center.32 Three 19F NMR resonances at −132.08, −162.79, and −165.72 ppm were distinguished from those of B(C6F5)3 in bromobenzene-d5, which appeared at −126.93, −141.38, and −158.88 ppm. Moreover, a similar ortho-F signal for [HB(C6F5)3]− was reported for [Cp*2ZrH][HB(C6F5)3].33 The formation of [HB(C6F5)3]− was further supported by mass spectral analysis in negative ion mode, which provided spectra dominated by a peak at 512.9 m/z in bromobenzene or chloroform.
Mass spectrometry of 2 in bromobenzene, in positive ion mode, provided a spectrum with the major peak at 707.21 m/z, which corresponds to the calculated mass of [{PhB(OxMe2)2ImMes}RhSiH2PhCO]+. In addition, peaks with M+ ± 27.99 m/z were observed, corresponding to [{PhB(OxMe2)2ImMes}RhSiH2Ph(CO)2]+ and [{PhB(OxMe2)2ImMes}RhSiH2Ph]+, which may come from gas-phase reactions involving loss or gain of CO. There was also a peak at 601.18 m/z that corresponds to the mass of [{PhB(OxMe2)2ImMes}RhH(CO)]+, resulting from loss of a SiHPh group.
Some evidence for the appearance of the dicarbonyl species through a gas-phase reaction in the mass spectrometer comes from independent generation of [{PhB(OxMe2)2ImMes}Rh(H)SiHPh(CO)2]+ through the addition of an atmosphere of CO to a solution of 2. [{PhB(OxMe2)2ImMes}Rh(H)SiHPh(CO)2]+ gave an identical m/z for the parent ion as the minor signal observed for 2. The 1H NMR spectrum of this dicarbonyl species contained a new RhH at −13.12 ppm (1 H, doublet of doublets) and a new SiH at 5.53 ppm (1 H). The coordination of CO to 2 indicates that there is an open coordination site in the complex. Unfortunately, [{PhB(OxMe2)2ImMes}Rh(H)SiHPh(CO)2]+ is too short-lived at room temperature for isolation or detailed NMR characterization. Attempts to extend its lifetime through low temperature NMR acquisition gave broad signals, further hindering analysis by 13C, 15N, and 29Si NMR spectroscopic methods. In the absence of added CO, [{PhB(OxMe2)2ImMes}Rh(H)SiHPh(CO)2]+ is not detected in the 1H NMR spectra of 2. Thus, we conclude that the CO transfer in 2 is a feature specific to the mass spectral analytical method.
Contrary to the parent ion signal for 2 in bromobenzene, the mass spectrum of 2 dissolved in chloroform provided a signal at 797.28 m/z. This mass is consistent with the formulation [{PhB(OxMe2)2ImMes}RhSiH2(C6F5)(CO)]+ resulting from C6H5/C6F5 exchange under MS data acquisition conditions. There is no evidence for this rearrangement in 1H, 19F, or 11B NMR spectra of 2 in chloroform-d1, and mass spectrum acquired in negative ion mode contained signals from only [HB(C6F5)3]−. Moreover, the 1H NMR spectra of isolated 2 dissolved in chloroform-d1, methylene chloride-d2, and bromobenzene-d5 contained similar signals in terms of chemical shifts and coupling constants, and all three solvents provided catalytically active species. The NMR signals of a bromobenzene solution of 2 are maintained at 80 °C for 72 h, although a small amount of black precipitate and aliphatic signals associated with decomposition products were observed. In addition, solutions of 2 persist at room temperature for greater than 1 month.
DFT calculations of 15N and 29Si NMR chemical shifts for gas-phase model compounds 1-A, a plausible intermediate [{PhB(OxMe2)2ImMes}Rh(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SiHPh)(H)CO]+ (B), and oxazoline-coordinated silylene (2-A) provide additional support for the proposed structure of 2 and a possible pathway for its formation via the coordinatively unsaturated rhodium silylene (Fig. 1). The calculations also provide additional evidence ruling out an unsaturated silicon center in 2. The calculated atomic coordinates (1-A) and the coordinates of 1 determined by single crystal X-ray diffraction are in good agreement, and the calculated configuration of 1-A maintains the trans disposed carbene and carbonyl ligands. The calculated 15N NMR chemical shifts of 1 are well reproduced in 1-A: −150 (trans to SiH2Ph, cf. experimental −161 ppm) and −158 ppm (trans to H, cf. experimental −172 ppm). The gas phase calculated 29Si NMR chemical shift of 1-A is 14 ppm, in reasonable agreement with the experimental value of −21 ppm (see Tables S1 and S2† for a comparison of calculated and experimental NMR chemical shifts).
SiHPh)(H)CO]+ (B), and oxazoline-coordinated silylene (2-A) provide additional support for the proposed structure of 2 and a possible pathway for its formation via the coordinatively unsaturated rhodium silylene (Fig. 1). The calculations also provide additional evidence ruling out an unsaturated silicon center in 2. The calculated atomic coordinates (1-A) and the coordinates of 1 determined by single crystal X-ray diffraction are in good agreement, and the calculated configuration of 1-A maintains the trans disposed carbene and carbonyl ligands. The calculated 15N NMR chemical shifts of 1 are well reproduced in 1-A: −150 (trans to SiH2Ph, cf. experimental −161 ppm) and −158 ppm (trans to H, cf. experimental −172 ppm). The gas phase calculated 29Si NMR chemical shift of 1-A is 14 ppm, in reasonable agreement with the experimental value of −21 ppm (see Tables S1 and S2† for a comparison of calculated and experimental NMR chemical shifts).
 | ||
| Fig. 1 Calculated structures of [{PhB(OxMe2)2ImMes}Rh(SiH2Ph)(H)CO] (1-A), [{PhB(OxMe2)2ImMes}Rh(SiHPh)(H)CO]+ (B), and [{κ3-N,Si,C-PhB(OxMe2)(OxMe2SiHPh)ImMes}Rh(H)CO]+ (2-A). H atoms on Si and Rh are illustrated, and all other H atoms are not included in the rendered depiction for clarity. | ||
[{κ3-N,Si,C-PhB(OxMe2)(OxMe2SiHPh)ImMes}Rh(H)CO]+ (2-A) was calculated as a gas-phase model for 2. Satisfyingly, the calculated 15N NMR chemical shifts of the oxazolines in 2-A mirror the experimental pattern, with the chemical shifts of the oxazoline N bonded to Si calculated to be −191 ppm and the N coordinated to Rh at −167 ppm. The calculated 29Si NMR chemical shift of 2-A is 73 ppm, which is ∼65 ppm downfield compared to the experimental value. The difference in calculated vs. experimental 29Si NMR chemical shifts may be related to the cationic charge in the gas phase vs. a solvated ionic species.
For comparison, the calculated geometry of a putative rhodium silylene [{PhB(OxMe2)2ImMes}Rh(SiHPh)(H)CO]+ (B) was optimized from a few starting configurations to the stationary point with carbene and carbonyl ligands trans, a hydride and oxazoline trans, and the SiHPh and oxazoline trans. Moreover, the calculated 29Si NMR chemical shift for B is 370 ppm as expected for a coordinatively unsaturated silicon center, and this is 364 ppm downfield from the experimental value for 2 and 280 ppm shift downfield from 2-A. Thus, the terminal silylene structure is unambiguously ruled out by experimental and computed 29Si NMR chemical shift values. The optimized structure of 2-A is 17 kcal mol−1 lower in energy than the silylene isomer B, suggesting that the terminal unsaturated silylene isomer is unlikely to be accessed. In line with this, compound 2 is also formed from reactions of 1 and B(C6F5)3 in THF as a competitive donor rather than the THF-stabilized silylene. In addition, identical chemical shifts for 2 were observed upon treatment with carbonyl compounds such as acetophenone and ethyl acetate. That is, there is no apparent substitution of the oxazoline for other 2-electron donor groups. In addition, there is no detectable interaction between 2 and an added equivalent of B(C6F5)3.
Despite the lack of observable interaction between 2 and carbonyls, complex 2 is a catalyst for the selective partial reduction of esters to ethers (see Table 1), silyl-protected acetals, or silyl-protected alcohols. Notably, the pathway for conversion of the ester to ether or to hydrosilylation products depends on the silane, with primary silanes giving selective deoxygenation and tertiary silanes giving 1,2-addition. In an initial set of experiments, phenylsilane, methylphenylsilane, and benzyldimethylsilane were tested as representative primary, secondary, and tertiary silanes in the partial reduction of ethyl acetate. The primary silane PhSiH3 gives diethyl ether as the partial deoxygenation product. Compound 2 is highly reactive and selective for this transformation, giving full conversion and similar yields with 0.1–1 mol% catalyst in 30 min.
| Conversion | mol% cat. | Solvent | Time (h) | Yield (%) | TOF (h−1) |
|---|---|---|---|---|---|

|
1 | C6D5Br | 24 | 55 | 2.3 |
| 1 | CDCl3 | 0.5 | 75 | 150 | |
| 0.2 | CDCl3 | 0.5 | 70 | 700 | |

|
1 | C6D5Br | 12 | 77 | 6.4 |
| 1 | CDCl3 | 24 | 0 | n.a | |

|
1 | C6D5Br | 12 | 88 | 7.3 |
In contrast, the tertiary silane BnMe2SiH gives the hydrosilylation product and a small amount of ester cleavage product benzyl(ethoxy)dimethylsilane. The yield of ester cleavage increases with 8 equiv. of BnMe2SiH. Interestingly, the secondary silane reagent PhMeSiH2 gives a mixture of both products (eqn (2)). In this case, the ester cleavage product is not formed, as determined by comparison of the 1H NMR spectrum of the reaction to an authentic sample prepared by zinc-catalyzed dehydrocoupling of PhMeSiH2 and EtOH.34 The relative amounts of diethyl ether and disiloxane byproduct from reduction with PhMeSiH2 are ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as expected by stoichiometry, while PhSiH3 gives unquantified, presumably oligomeric, siloxanes as byproducts.
:1 as expected by stoichiometry, while PhSiH3 gives unquantified, presumably oligomeric, siloxanes as byproducts.
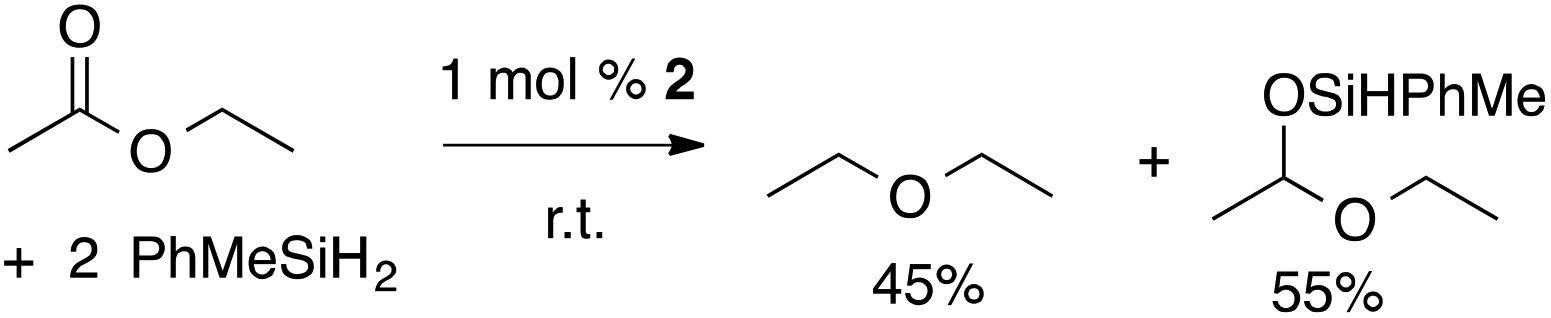 | (2) |
Although the deoxygenation reaction occurs both in bromobenzene-d5 and chloroform-d1, the reaction is significantly faster in chloroform. However, all scaled-up reactions were performed in methylene chloride and equivalent yields are obtained in the two chlorinated solvents. There is no evidence that 2 and methylene chloride react, and chloroform and methylene chloride may provide identical active catalytic sites. However, we cannot rule out the possible involvement of solvent, especially one as reactive as chloroform, in the catalytic cycle. Indeed, solvent effects on catalysis are observed. For example, bromobenzene is a superior solvent to chloroform for hydrosilylation using the tertiary silane BnMe2SiH, as the latter solvent gives no detectable conversion. Interestingly both chloroform-d1 and bromobenzene-d5 are equally effective solvents for conversions involving the secondary silane PhMeSiH2, which gives a mixture of both hydrosilylation and deoxygenation products as noted above.
These optimized conditions for diethyl ether synthesis were applied to the deoxygenation of a variety of ester substrates in the presence of 2.0 equiv. of PhSiH3 in chloroform or methylene chloride, and the results are summarized in Table 2. Typically, 1 mol% catalyst is required for conversion of most esters to ethers in Table 2, and the lower catalyst loading (0.1 mol%) determined for ethyl acetate above is effective for less sterically-demanding substrates such as ethyl formate and exo-2-norbornyl formate. Substrates with long alkyl chains such as methyl palmitate, or substituents with heteroatoms, such as 6-bromohexanoate and 2-thiopheneacetate, require 1 mol% catalyst loading and elevated temperature to give the ether products. The low yield for the deoxygenation of ethyl formate is probably due to the volatility of the gaseous methyl ethyl ether product.
| Ester | Cat. (mol%) | °C | h | Products | Yield (%)b |
|---|---|---|---|---|---|
| a See ESI for experimental conditions for catalytic reactions and product characterization. b Spectroscopic yields from micromolar scale experiments, isolated yields given in parenthesis. c Substrate conversion. d Trace 2-ethylthiophene observed. | |||||
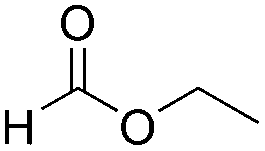
|
2 (0.1) | 25 | 2.5 | MeOEt | 54 |
| B(C6F5)3 (1.0) | 25 | 0.5 | C2H6 + CH4 | 100c | |

|
2 (0.1) | 25 | 25 |

|
56 (49) |
| B(C6F5)3 (1.0) | 25 | 0.5 |

|
95 | |
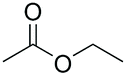
|
2 (0.1) | 25 | 0.5 | Et2O | 70 |
| B(C6F5)3 (1.0) | 25 | 0.5 | C2H6 | 100c | |

|
2 (1.0) | 25 | 6 |

|
87 (62) |
| B(C6F5)3 (1.0) | 25 | 0.5 | C16H34 + CH4 | 94 | |

|
2 (1.0) | 80 | 72 |

|
82 (75) |
| B(C6F5)3 (1.0) | 25 | 0.5 | C6H13Br + C2H6 | 95 | |

|
2 (1.0) | 80 | 24 |

|
54 (50) |
| B(C6F5)3 (1.0) | 25 | 0.5 |

|
93d | |

|
2 (1.0) | 60 | 72 |

|
86 (62) |

|
2 (1.0) | 80 | 24 |
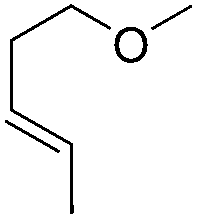
|
65 |

|
2 (1.0) | 25 | 8 |

|
60 (55) |

|
2 (2.0) | 25 | 6 |
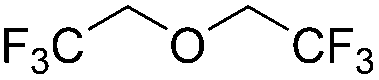
|
50 |
The catalysis product of exo-2-norbornyl formate is exo-2-methoxybicyclo[2.2.1]heptane (Table 2), indicating that the R–O bond in R′CO2R is not broken during reduction. The isolated yield of the deoxygenation is comparable to the yield from exo-norborenol and iodomethane using sodium hydride via the Williamson ether method,35 suggesting this deoxygenation method has potential synthetic utility. Olefins are unaffected by catalytic ester deoxygenation conditions. Methyl oleate and methyl trans-3-pentenoate are effectively and selectively deoxygenated to their corresponding ethers in good yield, although elevated temperature is required. The stereochemistry of the CC double bond is maintained as suggested by the 13C{1H} NMR spectrum of the isolated products. The cyclic ester γ-butyrolactone is deoxygenated quickly at room temperature with moderate yield of the product, while elevated temperatures result in ring-opening. Both carbonyl groups in trifluoroacetic anhydride are deoxygenated with 2.0 mol% of the catalyst loading at room temperature.
Tris(perfluorophenyl)borane is a catalyst for carbonyl hydrosilylation and C–O bond cleavage in the presence of tertiary and secondary silanes,1 and might be involved in the present deoxygenation since InBr3 and GaBr3 are known catalysts with tertiary silanes.15,16 However to the best of our knowledge, B(C6F5)3-catalyzed reactions of esters and primary silanes are not yet reported,17a and the above studies demonstrate a significant effect of silane upon the product of ester conversion. Although the 11B NMR and mass spectrometry results indicated that the precatalyst contains [HB(C6F5)3]−, and no B(C6F5)3 was detected by 11B or 19F NMR spectroscopy, a small amount of that Lewis acid could form during catalysis. For these reasons, the interaction of PhSiH3 and esters in the presence of catalytic B(C6F5)3 was tested as a possible background reaction, and the results are given in Table 2 for comparison to the catalytic action of 2. Interestingly, the combination of PhSiH3 and B(C6F5)3 with esters provides hydrocarbons (complete deoxygenation) through C–O bond cleavage rather than the selective deoxygenation to ethers observed with 2. These experiments indicate that trace B(C6F5)3, as a unilateral catalytic actor, is not responsible for selective conversion of esters to ethers.
As noted above, the common pathway for reactions of esters and hydridic reagents involves reductive cleavage of the C–O linkage. In contrast, deoxygenation of amides to amines is the typical pathway, observed with metal hydride reagents.36 Catalytic hydrosilylation reactions of esters and amides also tend to follow the pathways established for metal hydrides, with ester hydrosilylation giving reductive cleavage and amide hydrosilylation providing amines. Thus, catalytic reactions of phenylsilane with amides, employing 2 or B(C6F5)3 as catalysts, provide further insight into the pathway for ester deoxygenation.
Interestingly, both catalysts mediate amide deoxygenation to tertiary amines (Table 3). These experiments were then extended to ketones and aldehydes, which generally give complete deoxygenation to alkanes. Both 2 and B(C6F5)3 catalyze the conversion of acetophenone to ethylbenzene, with the B(C6F5)3-catalyzed reaction occurring in a shorter time. However, the dimethoxy-substituted electron-rich ketone is deoxygenated with similar reaction times with 2 or B(C6F5)3, while the 2,4,6-trimethoxybenzaldehyde is selectively deoxygenated by 2 but is unreactive under B(C6F5)3-catalyzed conditions at room temperature. Furthermore, the dimethoxy-substituted aldehyde decomposes under the B(C6F5)3-catalyzed conditions, but is selectively deoxygenated in the presence of 2. The neutral hydrosilyl hydridorhodium compound, which might be envisioned to catalyze ketone hydrosilylation via an oxidative addition and insertion pathway,31 is not reactive under the tested catalytic conditions.
| Reaction | Cat. | °C | h | Yieldb (%) |
|---|---|---|---|---|
| a See ESI for experimental condition for catalytic reactions and product characterization. b Spectroscopic yields from micromolar scale experiments, isolated yields given in parenthesis. c Not detected (starting material recovered). d Unidentified product mixture. | ||||

|
2 | 80 | 3 | 85 |
| B(C6F5)3 | 80 | 3 | 88 | |

|
2 | 80 | 24 | 86 (80) |
| B(C6F5)3 | 80 | 18 | 84 | |

|
2 | 25 | 7 | 65 (58) |
| B(C6F5)3 | 25 | 0.5 | 87 | |
| 1 | 25 | 18 | n.d.c | |

|
2 | 25 | 24 | 57 (55) |
| B(C6F5)3 | 25 | 18 | 65 | |

|
2 | 25 | 7 | 65 (48) |
| B(C6F5)3 | 25 | 18 | n.d.c | |

|
2 | 25 | 4 | 77 |
| B(C6F5)3 | 25 | 4 | n.d.d | |
The combined results from the catalytic reactions using B(C6F5)3 or 2 as catalysts (Tables 2 and 3) shed some light on the pathways and active species involved in the transformations. First, B(C6F5)3 catalyzes the C–O bond cleavage reactions of ketones and amides in the presence of phenylsilane to give the same products that are observed with 2. However, the relative rates observed from the two catalysts vary significantly with the substrates, indicating that the reactive species in the two systems are not equivalent. That is, compound 2 is not providing free B(C6F5)3 as the catalyst, nor is the reaction of PhSiH3 with B(C6F5)3 or 2 affording an equivalent activated organosilane species that interacts with the carbonyl substrate. This point is further emphasized by the distinct products observed from reaction of PhSiH3 and esters or aldehydes, as catalyzed by 2 or B(C6F5)3.
A possible pathway for the catalytic processes involves stepwise carbonyl hydrosilylation to give a silyl-protected acetal followed by C–O bond cleavage to provide either two silyl ethers or the ether/disiloxane products (Scheme 1).
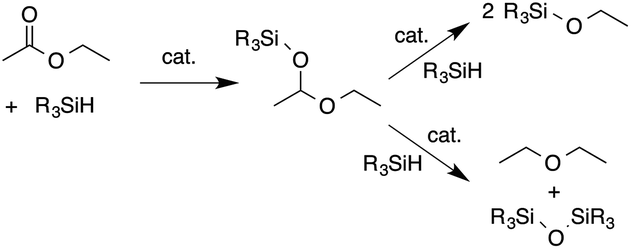 | ||
| Scheme 1 Two C–O bond cleavage pathways for the carbonyl hydrosilylation product. | ||
A few points emerge from the effect of the silane the product selectivity. First, the addition of PhSiH3 to a mixture of the silyl-protected acetal BnMe2SiOCHMeOEt and BnMe2SiH increases the yield of the ester cleavage product BnMe2SiOEt. This likely occurs through formation of PhH2SiOEt-type intermediates that undergo silyl exchange with BnMe2SiH, as suggested by transient OEt resonances in the 1H NMR spectrum of the reaction mixture. This ester cleavage reaction may be responsible for the difference between conversion and yield of ethers from deoxygenation. In addition, the silyl protected acetal is also an intermediate that leads to diethyl ether. There is no ethyl acetate remaining in the in situ reaction mixture of Scheme 2, yet diethyl ether is observed upon addition of PhSiH3. Thus, 1,2-hydrosilylation is the first step of both deoxygenation and ester cleavage pathways. However, the ether yield is lower when BnMe2SiOCHMeOEt is allowed to react with PhSiH3 in comparison to the reaction of EtOAc and PhSiH3 described in Table 1 (24 vs. 75%). The difference in ether yield may result from steric effects due to the large BnMe2Si group, or the effect of added PhSiH3 also may reflect a different pathway accessible to SiH-substituted acetals vs. tertiary silyl substituted acetals. A related experiment with acetophenone shows that the hydrosilylation product PhMeHCOSiH2Ph reacts to give ethylbenzene in the presence of excess PhSiH3 and catalytic quantities of 2.
 | ||
| Scheme 2 Effect of PhSiH3 on ester cleavage vs. deoxygenation pathways. | ||
Conclusions
The reaction of rhodium silyl {PhB(OxMe2)2ImMes}RhH(SiH2Ph)CO (1) and B(C6F5)3 affords the zwitterionic complex 2, which contains a SiHPh group bonded to both rhodium and the nitrogen of an oxazoline donor. This species is formulated as a rhodium-bonded silylene that is coordinated by a two-electron donor (a donor-stabilized silylene), leaving a coordinatively unsaturated rhodium site. The preference for oxazoline-silicon vs. oxazoline-rhodium coordination suggests that the putative silylene is more electrophilic than the rhodium center in 2. Also, note that the five-coordinate rhodium silyl [{κ3-N,N,C-PhB(OxMe2)2ImMes}Rh(SiH2Ph)CO]+ isomer, which might be anticipated based on established α-H-migration chemistry,21,37 is not observed. The isomer resulting from silylene insertion into the Rh–C of the more nucleophilic N-heterocyclic carbene is also not observed. NHC-coordination to an unsaturated silicon center might also be expected based on the stabilization of silanone derivatives and zero-valent silicon centers with NHCs.29,38 In addition, a series of dichlorosilylene compounds coordinated by metal centers (V, Fe, Co, Ni, Rh) and N-heterocyclic carbenes have related connectivity ([M]–SiX2–NHC) to the unobserved isomer of 2.39 Despite this precedent, compound 2 apparently does not isomerize to the carbene-coordinated rhodium silylene, likely as a result of the inert rhodium–carbene interaction.Compound 2 is an excellent catalyst for the deoxygenation of carbonyl-containing compounds with organosilanes. The deoxygenation pathway is unusual for esters, which are more commonly cleaved under reductive conditions. For most substrates, less than 1 mol% catalyst is needed, many conversions occur at room temperature, acyclic esters are deoxygenated, and the chemistry may be tuned for hydrosilylation vs. deoxygenation by the organosilane. Moreover, partial deoxygenation of fatty esters is readily accomplished with this catalyst. Unlike many coordinatively unsaturated late-metal catalysts for hydrosilylation, redistribution of organosilanes is not observed with 2. We note, however, that an iridium catalyst for amide deoxygenation includes a silylene bridging between two iridium centers,23 while a related catalytic system mediates the partial reduction of esters to aldehydes.40
The selective conversion of substrates through hydrosilylation or deoxygenation is systematically affected by the degree of substitution of the silane. Primary silanes give deoxygenation and tertiary silanes give hydrosilylation products, while secondary silanes give mixtures of both processes. This reactivity pattern is also followed with B(C6F5)3-catalyzed reactions of silanes and carbonyls, with primary silanes giving complete deoxygenation and tertiary silanes giving addition chemistry.17 However, the selectivity, rates, and in many cases, products of 2-catalyzed conversions are inequivalent from those of B(C6F5)3, suggesting that the unsaturated rhodium-silyl moiety is central to the conversions observed here. Catalytic hydrosilylation chemistry involving tertiary silanes does not apparently involve Si–C bond cleavage because the tertiary silane is incorporated into the product. This observation rules out silylene transfer as the mode of action for the hydrosilylation of esters and other carbonyls by tertiary silanes. Currently, we are studying related systems with kinetic and isotopic experiments to understand the effects of silane and catalyst on selective partial deoxygenation pathways. The tunability of the latter, through systematic ligand variation, will be key to controlling selectivity in both activation reactions involving metal-silicon and metal-hydrogen bonds or Lewis acid sites.
Acknowledgements
Dr Bruce Fulton is thanked for assistance with solution-phase 15N NMR measurements. This research was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences through the Ames Laboratory Catalysis and Chemical Physics projects (Contract no. DE-AC02-07CH11358).Notes and references
- (a) L. L. Adduci, M. P. McLaughlin, T. A. Bender, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2014, 53, 1646–1649 CrossRef CAS PubMed; (b) T. Robert and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 5216–5218 CrossRef CAS PubMed.
- (a) E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408–11409 CrossRef CAS PubMed; (b) E. Arceo, P. Marsden, R. G. Bergman and J. A. Ellman, Chem. Commun., 2009, 3357–3359 RSC.
- (a) M. Schlaf, P. Ghosh, P. J. Fagan, E. Hauptman and R. M. Bullock, Angew. Chem., Int. Ed., 2001, 40, 3887–3890 CrossRef CAS; (b) M. Schlaf, P. Ghosh, P. J. Fagan, E. Hauptman and R. M. Bullock, Adv. Synth. Catal., 2009, 351, 789–800 CrossRef CAS.
- (a) S. Stanowski, K. M. Nicholas and R. S. Srivastava, Organometallics, 2012, 31, 515–518 CrossRef CAS; (b) I. Ahmad, G. Chapman and K. M. Nicholas, Organometallics, 2011, 30, 2810–2818 CrossRef CAS; (c) S. Vkuturi, G. Chapman, I. Ahmad and K. M. Nicholas, Inorg. Chem., 2010, 49, 4744–4746 CrossRef CAS PubMed.
- (a) B. L. Wegenhart and M. M. Abu-Omar, Inorg. Chem., 2010, 49, 4741–4743 CrossRef CAS PubMed; (b) J. E. Ziegler, M. J. Zdilla, A. J. Evans and M. M. Abu-Omar, Inorg. Chem., 2009, 48, 9998–10000 CrossRef CAS PubMed.
- A. D. Sutton, F. D. Waldie, R. Wu, M. Schlaf, L. A. ‘Pete’ Silks and J. C. Gordon, Nat. Chem., 2013, 5, 428–432 CrossRef CAS PubMed.
- (a) G. R. Pettit and T. Kasturi, J. Org. Chem., 1960, 25, 875–876 CrossRef CAS; (b) G. R. Pettit, U. Ghatak, B. Green, T. Kasturi and D. Piatak, J. Org. Chem., 1961, 26, 1685–1686 CrossRef CAS; (c) G. R. Pettit and D. M. Piatak, J. Org. Chem., 1962, 27, 2127–2130 CrossRef CAS.
- G. A. Kraus, K. A. Frazier, B. D. Roth, M. J. Taschner and K. Neuenschwander, J. Org. Chem., 1981, 46, 2417–2419 CrossRef CAS.
- S. L. Baxter and J. S. Bradshaw, J. Org. Chem., 1981, 46, 831–832 CrossRef CAS.
- Z. Mao, B. T. Gregg and A. R. Cutler, J. Am. Chem. Soc., 1995, 117, 10139–10140 CrossRef CAS.
- S. Das, Y. Li, K. Junge and M. Beller, Chem. Commun., 2012, 48, 10742–10744 RSC.
- (a) K. Junge, B. Wendt, S. Zhou and M. Beller, Eur. J. Org. Chem., 2013, 2061–2065 CrossRef CAS; (b) A. J. Ruddy, C. M. Kelly, S. M. Crawford, C. A. Wheaton, O. L. Sydora, B. L. Small, M. Stradiotto and L. Turculet, Organometallics, 2013, 32, 5581–5588 CrossRef CAS.
- H. Li, L. C. Misal Castro, J. Zheng, T. Roisnel, V. Dorcet, J.-B. Sortais and C. Darcel, Angew. Chem., Int. Ed., 2013, 52, 8045–8049 CrossRef CAS PubMed.
- (a) S. C. Berk, K. A. Kreutzer and S. L. Buchwald, J. Am. Chem. Soc., 1991, 113, 5093–5095 CrossRef CAS; (b) X. Verdaguer, S. C. Berk and S. L. Buchwald, J. Am. Chem. Soc., 1995, 117, 12641–12642 CrossRef CAS.
- U. Biermann and J. O. Metzger, ChemSusChem, 2014, 7, 644–649 CrossRef CAS PubMed.
- N. Sakai, T. Moriya and T. Konakahara, J. Org. Chem., 2007, 72, 5920–5922 CrossRef CAS PubMed.
- (a) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS; (b) D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed; (c) J. M. Blackwell, D. J. Morrison and W. E. Piers, Tetrahedron, 2002, 58, 8247–8254 CrossRef CAS.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- (a) M. C. Lipke and T. D. Tilley, J. Am. Chem. Soc., 2014, 136, 16387–16398 CrossRef CAS PubMed; (b) S. Park and M. Brookhart, Organometallics, 2010, 29, 6057–6064 CrossRef CAS PubMed.
- (a) N. Schneider, M. Finger, H. Christian, S. Bellemin-Laponnaz, P. Hofmann and L. H. Gade, Angew. Chem., Int. Ed., 2009, 48, 1609–1613 CrossRef CAS PubMed; (b) E. Calimano and T. D. Tilley, Organometallics, 2010, 29, 1680–1692 CrossRef CAS.
- R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed.
- (a) P. B. Glaser and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 13640–13641 CrossRef CAS PubMed; (b) P. G. Hayes, C. Beddie, M. B. Hall, R. Waterman and T. D. Tilley, J. Am. Chem. Soc., 2006, 128, 428–429 CrossRef CAS PubMed; (c) E. Calimano and T. D. Tilley, J. Am. Chem. Soc., 2008, 130, 9226–9227 CrossRef CAS PubMed; (d) E. Calimano and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 11161–11173 CrossRef CAS PubMed; (e) M. E. Fasulo, M. C. Lipke and T. D. Tilley, Chem. Sci., 2013, 4, 3882–3887 RSC.
- C. Cheng and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11304–11307 CrossRef CAS PubMed.
- M. Stoelzel, C. Präsang, B. Blom and M. Driess, Aust. J. Chem., 2013, 66, 1163–1170 CAS.
- H. Hashimoto, T. Suzuki and H. Tobita, Dalton Trans., 2010, 39, 9386–9400 RSC.
- L. Rosenberg, M. D. Fryzuk and S. J. Rettig, Organometallics, 1999, 18, 958–969 CrossRef CAS.
- (a) L. H. Gade, V. Cesar and S. Bellemin-Laponnaz, Angew. Chem., Int. Ed., 2004, 43, 1014–1017 CrossRef CAS PubMed; (b) V. César, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2005, 11, 2862–2873 CrossRef PubMed.
- P. Gigler, B. Bechlars, W. A. Herrmann and F. E. Kühn, J. Am. Chem. Soc., 2011, 133, 1589–1596 CrossRef CAS PubMed.
- R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444–456 CrossRef CAS PubMed.
- S. Xu, K. Manna, A. Ellern and A. D. Sadow, Organometallics, 2014, 33, 6840–6860 CrossRef CAS.
- I. Ojima, T. Kogure and M. Kumagai, J. Org. Chem., 1977, 42, 1671–1679 CrossRef CAS.
- R. G. Kidd, Boron-11, Academic Press, New York, 1983 Search PubMed.
- X. Yang, C. L. Stern and T. J. Marks, Angew. Chem., Int. Ed. Engl., 1992, 31, 1375–1377 CrossRef.
- D. Mukherjee, R. R. Thompson, A. Ellern and A. D. Sadow, ACS Catal., 2011, 698–702 CrossRef CAS.
- T. Hirai, A. Hamasaki, A. Nakamura and M. Tokunaga, Org. Lett., 2009, 11, 5510–5513 CrossRef CAS PubMed.
- D. L. Dodds and D. J. Cole-Hamilton, in Sustainable catalysis: challenges and practices for the pharmaceutical and fine chemical industries, ed. P. J. Dunn, K. K. Hii, M. J. Krische and M. T. Williams, Wiley, Hoboken NJ, 2013, ch. 1, pp. 1–36 Search PubMed.
- G. P. Mitchell and T. D. Tilley, Organometallics, 1998, 17, 2912–2916 CrossRef CAS.
- (a) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, P. von R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 CrossRef CAS PubMed; (b) Y. Xiong, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 4302–4311 CrossRef CAS PubMed; (c) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815–11832 CrossRef CAS PubMed.
- (a) J. Li, S. Merkel, J. Henn, K. Meindl, A. Döring, H. W. Roesky, R. S. Ghadwal and D. Stalke, Inorg. Chem., 2010, 49, 775–777 CrossRef CAS PubMed; (b) G. Tavčar, S. S. Sen, R. Azhakar, A. Thorn and H. W. Roesky, Inorg. Chem., 2010, 49, 10199–10202 CrossRef PubMed; (c) R. S. Ghadwal, R. Azhakar, K. Pröpper, J. J. Holstein, B. Dittrich and H. W. Roesky, Inorg. Chem., 2011, 50, 8502–8508 CrossRef CAS PubMed.
- C. Cheng and M. Brookhart, Angew. Chem., Int. Ed., 2012, 51, 9422–9424 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental description of catalysis, solution-phase and solid-state NMR spectra, and computational results. See DOI: 10.1039/c5dt02844b |
| This journal is © The Royal Society of Chemistry 2015 |