
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoretentive formylation of (S)-proline: new application of the self-regeneration of stereo-centres (SRS) principle via chelation to cobalt(III)†
Mehmet
Temizsoy
,
Waqas
Sethi
,
Anders
Reinholdt
,
Magnus
Schau-Magnussen
,
Jesper
Bendix
and
Anders
Hammershøi
*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark. E-mail: anders@kiku.dk; Tel: +45 3532 0110
First published on 29th September 2015
Abstract
In a Vilsmeier–Haack-type formylation reaction the α-(dihydroxymethyl)-(S)-prolinato complex (+)578-p-[Co(tren){(RC,SN)-Pro[CH(OH)2]O}]Cl2·2H2O (22) was produced stereoselectively (85% ee) from the (S)-prolinato complex, (+)578-p-[Co(tren){(SC,SN)-ProO}]2(H3O)2(HOEt2)(O3SCF3)7 (18). Similar reaction of the (S)-alaninato complex, (−)578-p-[Co(tren)(S-AlaO)](H3O)(O3SCF3)3 (13), produced the racemate, rac-p-[Co(tren)(Ala{CH(OH)2}O)]SO4·2H2O (17). The contrasting stereochemical outcomes of the formylation reaction with 18versus13 were ascribed to the stereogenic character of the coordinated sec. amine of the (S)-prolinate chelate in 18, which serves to uphold a chiral environment during reaction, whereas reaction intermediates derived from 13 lack this stereochemical feature. The stereoselective formylation of (S)-proline, relying on coordination to an inert metal centre, as conducted here, constitutes a novel application of the concept of Self-Regeneration of Stereocentres (SRS). The α-(hydroxymethyl)-(S)-prolinato complex, (+)578-p-[Co(tren){(RC,SN)-Pro(CH2OH)O}]Cl2·2H2O (23) resulted from borohydride reduction of 22. The molecular structures of (+)578-p-[Co(tren){(RC,SN)-Pro[CH(OH)2]O}]Cl2·2H2O (22), rac-p-[Co(tren)-(Ala{CH(OH)2}O)]I2·H2O (17) and (+)578-p-[Co(tren){(RC,SN)-Pro(CH2OH)O}]Cl2·2H2O (23) were established by X-ray crystallography.
Introduction
Proline (1) and derivatives play essential roles in chemistry and biology.1 Chiral proline and analogues constitute archetypal agents in enantioselective organocatalysis.2,3 Prolyl segments within peptide strands impose rigid constraints on the peptide secondary structure, and in this respect proline is unique among the proteinogenic amino acids.1 Synthetic studies have focussed on extending proline entities in order to create “proline chimeras” that mimic other peptidal amino acids with respect to side chain while retaining the conformational rigidity of the prolyl segment.4 This prospect has sparked interest in producing proline derivatives stereoselectively by elaboration from the α-C centre. However, modification at this, the sole stereogenic centre of proline, without loss of chirality is not straightforward.4 In 1981, Seebach et al. devised an elegant strategy which permitted α-alkylation of (S)-proline (1) to be conducted enantioselectively with a number of electrophilic reactants (Scheme 1).5,6 The strategy relied on temporary incorporation of an auxiliary, stereogenic centre via formation of a bicyclic acetal (2) by condensation with pivaldehyde. The auxiliary stereogenic centre arose diastereoselectively at C-2 of the so formed 2-(tert-butyl)oxazolidin-5-one ring. Alkylation at the α-C centre of the lithium enolate intermediate 3 to give 4 also proceeded diastereoselectively. The inherent stereogenicity of the α-C centre in 1 was lost in the deprotonation step providing 3, but the auxiliary stereocentre upheld an asymmetric environment for the alkylation step to yield 4 stereoselectively. Hydrolytic removal of the auxiliary centre provided 5.6 This, overall, stereoretentive sequence (Scheme 1) constitutes a seminal example of the application of the principle of Self-Regeneration of Stereocentres (SRS), due to Seebach.4,7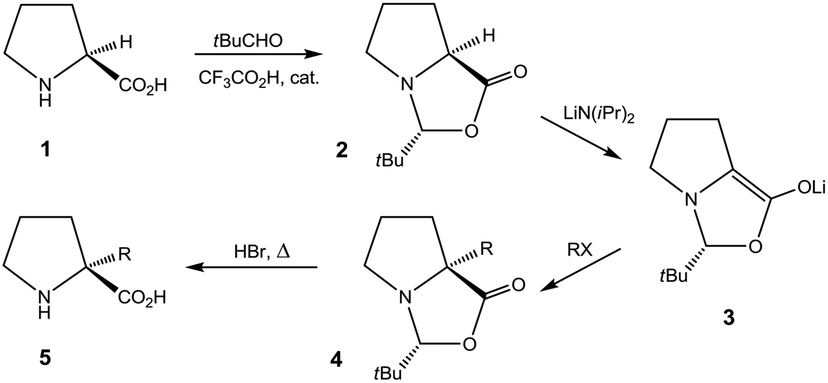 | ||
| Scheme 1 | ||
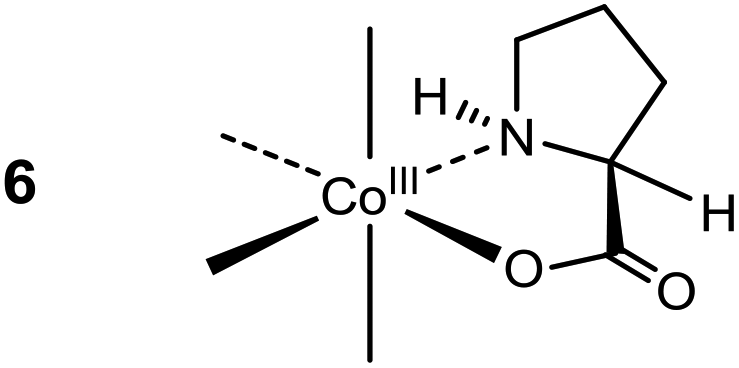
The creation of a quaternary α-C centre by introduction of a formyl group is an obvious goal, generating the basis for further elaboration. So far, successful introduction of the formyl group at the α-C centre of a cobalt(III)-bound amino acidate is only reported with glycinate (7). This was achieved by reaction with POCl3 in DMF via a Vilsmeier–Haack-type reaction,26 and results are summarised in Scheme 2 {L4Co = p-Co(tren), t-Co(tren), Co(en)2, cis-β2-Co(trien) }.14–17,27 The argued mechanism assumes initial formation of a glycyl chloride (8) which upon deprotonation at the α-C site is added to the N,N-dimethyl-chloromethyliminium ion (“Vilsmeier reagent”, generated in situ from reaction of POCl3 with DMF) followed by elimination of HCl to produce the crystallographically verified 3-(dimethylamino)-2-aminoacrylyl chloride complex, 9[L4Co = p-Co(tren)].14 The conjugationally stabilized ligand of 9 yielded to hydrolysis in strong aqueous acid, providing15,16 the aldehyde complex 11, which was distributed between its hydrated (12) and enol (10) forms in water in ca. 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (L4 = (en)2).17 A published solid state structure has the aldehyde in the enol form [10, L4Co = cis-β2-Co(trien)].27
:1 ratio (L4 = (en)2).17 A published solid state structure has the aldehyde in the enol form [10, L4Co = cis-β2-Co(trien)].27
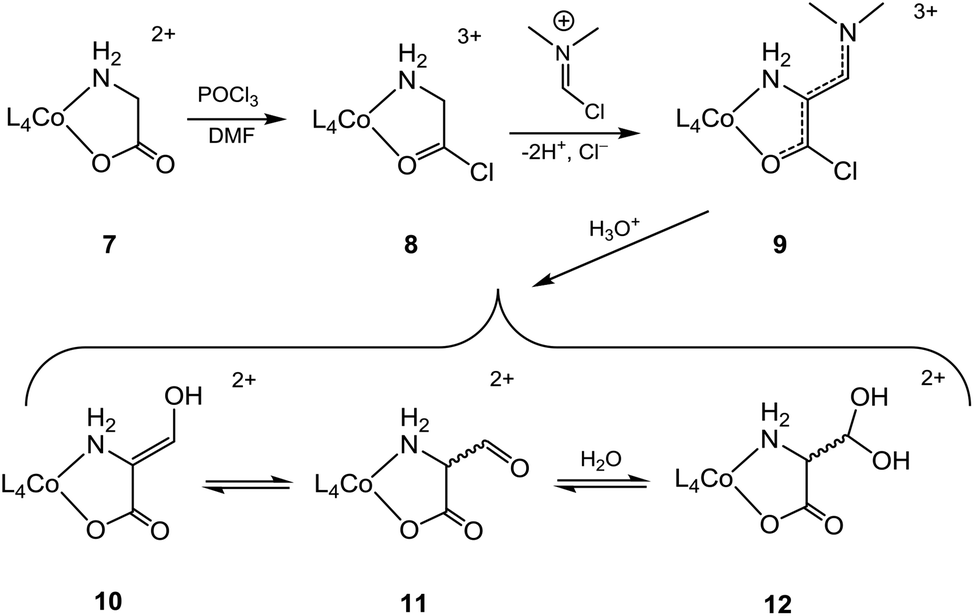 | ||
| Scheme 2 | ||
In the sequence of Scheme 2 the site of reaction was the glycinate α-CH2 group. However, if parallel chemistry was to be conducted with other α-amino acidates (different from GlyO−) with the aim of converting an α-CH group into a quaternary centre, the extensive conjugation noted for 9 would not apply. Therefore, such parallel chemistry was intuitively viewed less feasible. However, in this paper we report the successful formylation of the α-CH centres of the amino acidate ligands in (−)578-p-[Co(tren)(S-AlaO)]2+ and (+)578-p-[Co(tren)(S-ProO)]2+ producing the derivative aldehydes (hydrated form). While the (S)-alaninato complex resulted in a racemized product the reaction of the (S)-prolinato complex proceeded stereoselectively.
Experimental
General
Absorption spectra and optical rotations were monitored in water with a Lambda 17 spectrophotometer and a Perkin-Elmer P22 polarimeter (±0.002°), respectively; for the latter in 1 dm quartz cells at 25 °C. Within experimental error all listed values for specific rotations ([α], in units of 10−1 deg cm2 g−1) of chiral products did not change on further recrystallization of the product, and this was taken as evidence of optical purity. 13C{1H} NMR spectra were recorded in D2O on a (500 MHz) Bruker spectrometer (with cryoprobe) using 1,4-dioxane (13C, δ = 69.14 ppm relative to Me4Si) as internal standard. When applicable, assignments of 13C resonances were made on the basis of the APT technique. The cation exchange resin AG 50W-X2 (Bio-Rad, 200–400 mesh) was used throughout and resin column dimensions are given as diameter × length. Routine concentration of solutions by removal of solvent was carried out at reduced pressure (ca. 20 Torr) in a Büchi rotary evaporator using a water aspirator and water bath (ca. 60 °C). Drying “in vacuo” was accomplished over P4O10. Anhydrous CF3SO3H (3M Comp.) was used as supplied.Crystallography
Adequate crystals of rac-p-[Co(tren)(Ala{CH(OH)2}O)]I2·H2O ([17]I2·H2O), (+)578-p-[Co(tren){(RC,SN)-Pro[CH(OH)2]O}]Cl2·2H2O ([22]Cl2·2H2O) and (+)578-p-[Co(tren){(RC,SN)-Pro(CH2OH)O}]Cl2·2H2O ([23]Cl2·2H2O) were selected and each mounted on a glass fiber or nylon loop attached to a copper pin and placed in the N2 stream of a Bruker D8 Venture diffractometer. Data were collected using Mo-Kα radiation (λ = 0.71073 Å). Details of the crystal structure determination and refinement are given in Table 1. The structures were solved using the charge-flipping method (olex2.solve)28 and refined using the program packages SHELXL29 ([17]I2·H2O and [23]Cl2·2H2O) or Olex230 ([22]Cl2·2H2O).| Compound reference | [17]I2·H2O | [22]Cl2·2H2O | [23]Cl2·2H2O |
|---|---|---|---|
| Emperical formula | C10H28CoI2N5O5 | C12H32Cl2CoN5O6 | C12H32Cl2CoN5O5 |
| M | 611.11 | 472.26 | 456.25 |
| Crystal system | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | P21/n | P212121 | P212121 |
| a/Å | 21.619(3) | 9.6314(6) | 8.9450(3) |
| b/Å | 9.0014(10) | 13.5664(6) | 12.4236(4) |
| c/Å | 22.283(3) | 14.7998(8) | 17.4130(6) |
| α/° | 90 | 90 | 90 |
| β/° | 117.732(6) | 90 | 90 |
| γ/° | 90 | 90 | 90 |
| Unit cell volume/Å3 | 3838.1(8) | 1933.8(2) | 1935.1(1) |
| Temperature/K | 298(1) | 122(1) | 122(1) |
| Z | 8 | 4 | 4 |
| Refl. measured | 35227 |
14308 |
30215 |
| Independ. refl. | 6799 | 4259 | 5425 |
| Observed refl. (I > 2σ(I)) | 5152 | 3991 | 4854 |
| Parameters | 427 | 281 | 233 |
| R int | 0.051 | 0.0366 | 0.050 |
| R 1 (I > 2σ(I)) | 0.0550 | 0.0269 | 0.0271 |
| wR (F2) (I > 2σ(I)) | 0.1316 | 0.0617 | 0.0556 |
| R 1 (all data) | 0.0765 | 0.0309 | 0.0356 |
| wR (F2) (all data) | 0.1443 | 0.0662 | 0.0585 |
| S | 1.084 | 1.013 | 1.043 |
| Δρmax, Δρmax (e Å−3) | 1.31, −0.71 | 0.63, −0.37 | 0.63, −0.37 |
| Flack parameter | — | 0.010(13) | 0.006(6) |
| CCDC number | 1411924 | 1411925 | 1411926 |
Results and discussion
The chiral (S)-alaninato- and (S)-prolinato cobalt(III) complexes were each formylated at their α-methine centres in a Vilsmeier–Haack-type reaction, essentially as done before at the α-methylene group of the analogous glycinato complexes.14–17 However, in the present study quaternary carbon centres were created, and the products, rac-p-[Co(tren)(Ala{CH(OH)2}O)]I2·H2O ([17]I2·H2O) and (+)578-p-[Co(tren){(RC,SN)-Pro[CH(OH)2]O}]Cl2·2H2O ([22]Cl2·2H2O), were each isolated in their hydrated, i.e. α-(dihydroxymethyl) forms, and their structures verified by X-ray crystallography. While the formylation of the chiral alaninato complex (13) gave racemic 17, the formylation of the chiral prolinato complex (18) produced the chiral α-formyl (R)-prolinato complex (22) in 85% enantiomeric excess (ee).Synthesis and characterisation
The two precursor complexes were readily produced in a “one-pot” procedure comprising aeration of a solution of cobalt(II) salt, tris(2-aminoethyl)amine (tren) and S-amino acid in water in the presence of activated-charcoal catalyst. Enantiopure products, (−)578-p-[Co(tren)(S-AlaO)]Cl2·5H2O and (+)578-p-[Co(tren){(SC,SN)-ProO}]Cl2·1.5H2O, respectively, were isolated in good yields. Usual cobalt(III) behaviour predicts activated charcoal to catalyse the equilibration of coordination isomers.31,32 Here, the p isomer was the sole geometrical isomer observed in both instances.33,34 This isomer has the carboxylate group coordinated trans to a primary amine of the tren ligand in the complex,35 and the same single product geometry was observed in similar syntheses of the homologous glycinato-, ornithinato- and sarcosinato complexes.33,36,37 These concurring results corroborate the suggestion that this facile strategy is generally applicable for the directed, stereoselective synthesis of p-[CoIII(tren)(amino acidato)]n+ complexes, obviating earlier (non-stereoselective) methods based on substitution of ligands of a suitable cobalt(III) complex {[Co(tren)Cl2]Cl,38,39 [Co(tren)Cl2](ClO4)40 or [Co(tren)(OH2)(OH)](ClO4)214,41,42}.The precursor complex chlorides were converted to their DMF-soluble triflate salts, (−)578-p-[Co(tren)(S-AlaO)](H3O)(O3SCF3)3 and (+)578-p-[Co(tren){(SC,SN)-ProO}]2(H3O)2(HOEt2)(O3SCF3)7, and each subjected to Vilsmeier–Haack-formylation in DMF followed by chromatographic workup and crystallization of products. Thereby, the chiral (S)-alaninato complex (13) was converted to the racemic α-(dihydroxymethyl) product, rac-p-[Co(tren)(Ala{CH(OH)2}O)]SO4·2H2O (17), whereas the chiral (S)-prolinato complex (18) gave the product enantiomer (+)578-p-[Co(tren){(RC,SN)-Pro[CH(OH)2]O}]Cl2·2H2O (22) in 85% ee (in eluate after chromatography), but recrystallization afforded enantiopure product in 80% yield. The 13C NMR spectra (in D2O) of both formyl complexes (17, 22) revealed the hydrated, α-(dihydroxymethyl) forms of the crystallized products to also persist in aqueous solution. With NaBH4 in water (pH 6.9) the α-formyl-prolinato complex (22) was selectively reduced to the derivative alcohol complex (+)578-p-[Co(tren){(RC,SN)-Pro(CH2OH)O}]Cl2·2H2O ([23]Cl2·2H2O).
Structures
A number of prolinato cobalt(III) complexes are reported before.8–12,43,44 In all instances for which a crystal structure was also reported, the five-membered pyrrolidine ring was found to reside in a cisoid fashion on the same side of the plane defined by the prolinate chelate ring.8–12 Thereby, the configurations of the α-C- and sec.-amine-N atoms of chelated prolinate are mutually interlocked. Thus, in the published12 structure of chiral p-[Co(tren){(SC,SN)-ProO}]I2·H2O ([18]I2·H2O) the absolute configurations of these centres are SC,SN, and inspection of a molecular model reveals the alternative transoid disposition (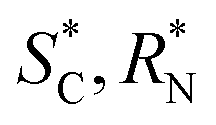 , relative configuration) to be more strained, relatively.
, relative configuration) to be more strained, relatively.
Here, crystal structure determinations were undertaken of product complexes rac-p-[Co(tren)(Ala{CH(OH)2}O)]I2·H2O ([17]I2·H2O), (+)578-p-[Co(tren){(RC,SN)-Pro[CH(OH)2]O}]Cl2·2H2O ([22]Cl2·2H2O) and (+)578-p-[Co(tren){(RC,SN)-Pro(CH2OH)O}]Cl2·2H2O ([23]Cl2·2H2O), and the complex cations of each structure are depicted in Fig. 1–3. The monoclinic crystal of racemic [17]I2·H2O contains two formula units per asymmetric unit, but since the two crystallographically independent complex ions are structurally very similar, only one of these is depicted in Fig. 1. Selected bonding parameters for all three structures appear in Table 2. Since all corresponding values of the two crystallographically independent complex cations of [17]I2·H2O are similar within a few standard deviations, only averaged values are given in Table 2 for this compound.
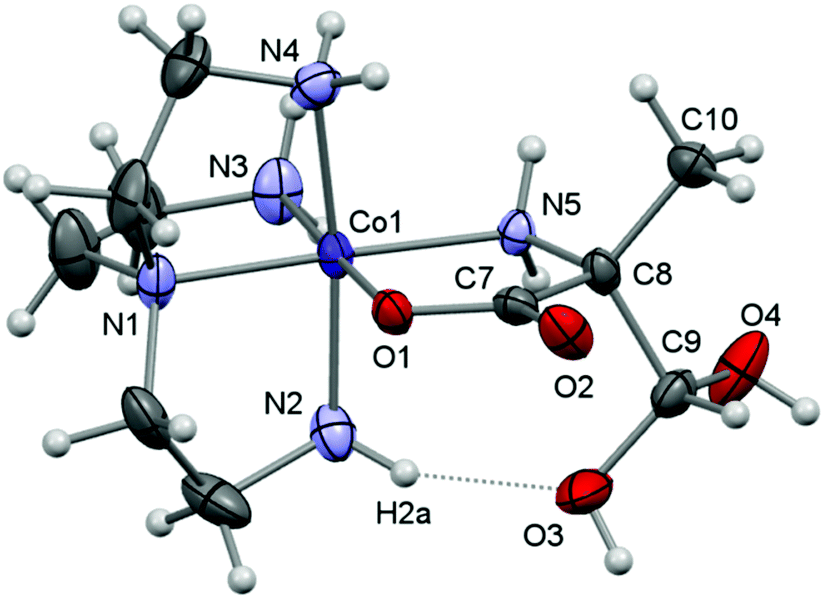 | ||
| Fig. 1 A view of the molecular structure of one of the two crystallographically independent but structurally similar complex ions of [17]I2·H2O showing the atom-labelling scheme for selected atoms. Displacement ellipsoids are drawn at the 30% probability level and H atoms are shown as small spheres of arbitrary radii. | ||
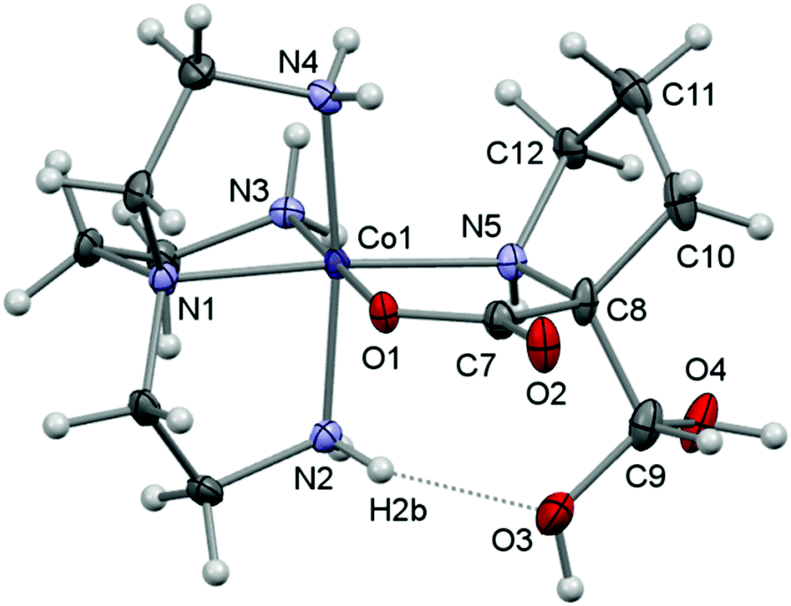 | ||
| Fig. 2 A view of the molecular structure of the complex cation of [22]Cl2·2H2O showing the atom-labelling scheme for selected atoms. Displacement ellipsoids are drawn at the 50% probability level and H atoms are shown as small spheres of arbitrary radii. | ||
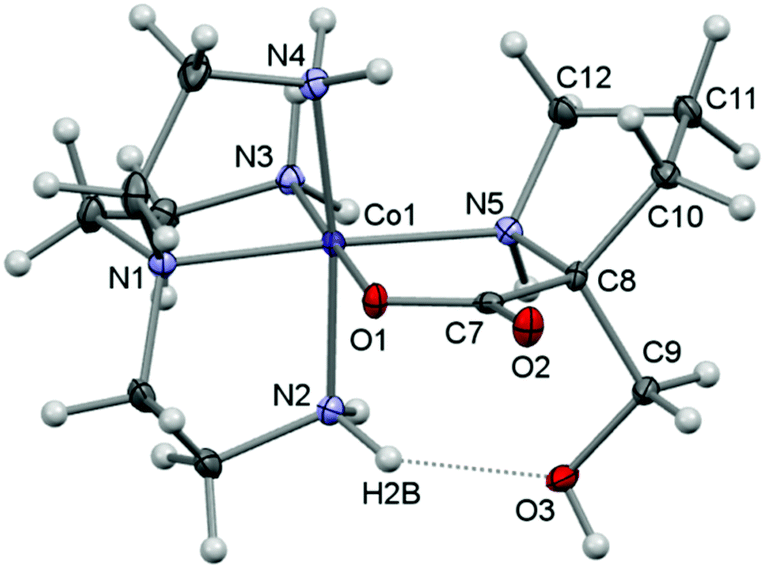 | ||
| Fig. 3 A view of the molecular structure of the complex cation of [23]Cl2·2H2O showing the atom-labelling scheme for selected atoms. Displacement ellipsoids are drawn at the 50% probability level and H atoms are shown as small spheres of arbitrary radii. | ||
| Compound | [17]I2·H2Oa | [22]Cl2·2H2O | [23]Cl2·2H2O |
|---|---|---|---|
| a Averages. | |||
| Co–N1 | 1.938 | 1.956(2) | 1.945(2) |
| Co–N2 | 1.948 | 1.969(2) | 1.971(2) |
| Co–N3 | 1.934 | 1.940(2) | 1.943(2) |
| Co–N4 | 1.944 | 1.971(2) | 1.957(2) |
| Co–N5 | 1.952 | 1.973(2) | 1.977(2) |
| Co–O1 | 1.885 | 1.902(2) | 1.906(2) |
| N1–Co–N2 | 86.1 | 85.0(1) | 86.2(1) |
| N1–Co–N3 | 87.9 | 86.9(1) | 87.9(1) |
| N1–Co–N4 | 86.2 | 86.5(1) | 86.5(1) |
| O1–Co–N5 | 85.3 | 86.0(1) | 85.9(1) |
| N2–Co–N3 | 92.5 | 91.7(1) | 92.0(1) |
| N3–Co–N4 | 91.8 | 92.3(1) | 91.6(1) |
| N4–Co–O1 | 86.7 | 87.1(1) | 87.0(1) |
| O1–Co–N2 | 89.1 | 88.8(1) | 89.3(1) |
| N4–Co–N5 | 94.8 | 97.6(1) | 96.5(1) |
| N2–Co–N5 | 92.8 | 90.7(1) | 91.3(1) |
| N1–Co–N5 | 177.3 | 175.6(1) | 176.8(1) |
| N2–Co–N4 | 171.1 | 170.4(1) | 171.1(1) |
| N3–Co–O1 | 178.4 | 179.4(1) | 178.6(1) |
| Co–N5–C12 | — | 120.1(2) | 119.8(2) |
| Co–N5–C8 | 111.8 | 110.1(1) | 109.8(1) |
| Co–O1–C7 | 116.6 | 117.1(2) | 116.5(1) |
All three complex cations display very similar overall geometries. A common feature is the reduced N2–Co–N4 angle (ca. 171°) spanning the “meridional” segment of the tren ligand. In the formyl-alaninato complex (17) the corresponding angular increase is distributed almost equally over the angles N4–Co–N5 (94.8°) and N2–Co–N5 (92.8°). However, in the formyl-prolinato- (22) and the hydroxymethyl-prolinato (23) complexes the increase primarily affects the angle N4–Co–N5 [97.7° (22); 96.5° (23)], while the angle N2–Co–N5 [90.7° (22); 91.3° (22)] is close to the ideal octahedral value in both instances. The expanded N4–Co–N5 angles and the also enlarged Co–N5–C12 angles of 120.1° of 22 and 119.8° of 23 (>109.5°) may reflect the result of an alleviating effect arising from repulsive interactions with the pyrrolidine ring within the complex. Nevertheless, in 22 the pyrrolidine ring adopts the envelope conformation with the flap oriented towards the complex core. This orientation contrasts with the conformation in the structure of 23 and in other structures of prolinato cobalt(III) complexes which all have the flap pointing away from the complex centre.8–12
All three structures are dominated by extensive hydrogen-bonding networks. A common feature is a strong intramolecular hydrogen-bond interaction between a coordinated amine and one O atom of the α-(dihydroxymethyl) or α-hydroxymethyl groups [N2⋯O3: 2.799 (13) Å in 17, 2.865 (3) Å in 22 and 2.791 (3) Å in 23], a general feature in related structures.33,37,45
Stereoselectivity
The contrasting stereochemical outcomes of the formylation reactions of the two amino acidate complexes of this study provide important mechanistic insight. A mechanistic proposal for the formylation (accompanied by racemization) of the (S)-alaninato complex (13) is depicted in Scheme 3, and the mechanism is argued on the background of the parallel reaction of the analogous glycinato system (Scheme 2).14–17 Thus, with POCl3 the (S)-alaninato complex 13 is converted to the triply charged acid-chloride complex 14, which after proton loss and electronic readjustment provides enolate complex 15. This would be stabilized, relatively, due to the overall lower charge and electronic delocalization of the ligand with negative charge shifted towards the metal ion. The planarity of the enolate ligand renders 15 achiral, and any reaction which restores sp3-hybridization at the α-C atom would result in racemic product mixtures. Here, addition to the imine of the Vilsmeier reagent followed by elimination of Cl− produces 16, which after hydrolysis of the iminium- and acid chloride functionalities provides the racemic α-(dihydroxymethyl)alaninato complex 17. Once formed, the quaternary α-C centre of this product is configurationally locked. Thus, while enantiomers of the related α-formyl-glycinato system may interconvert via an enol intermediate (10, Scheme 2),17,27 such path is clearly not available to the α-formyl-alaninato complex (17).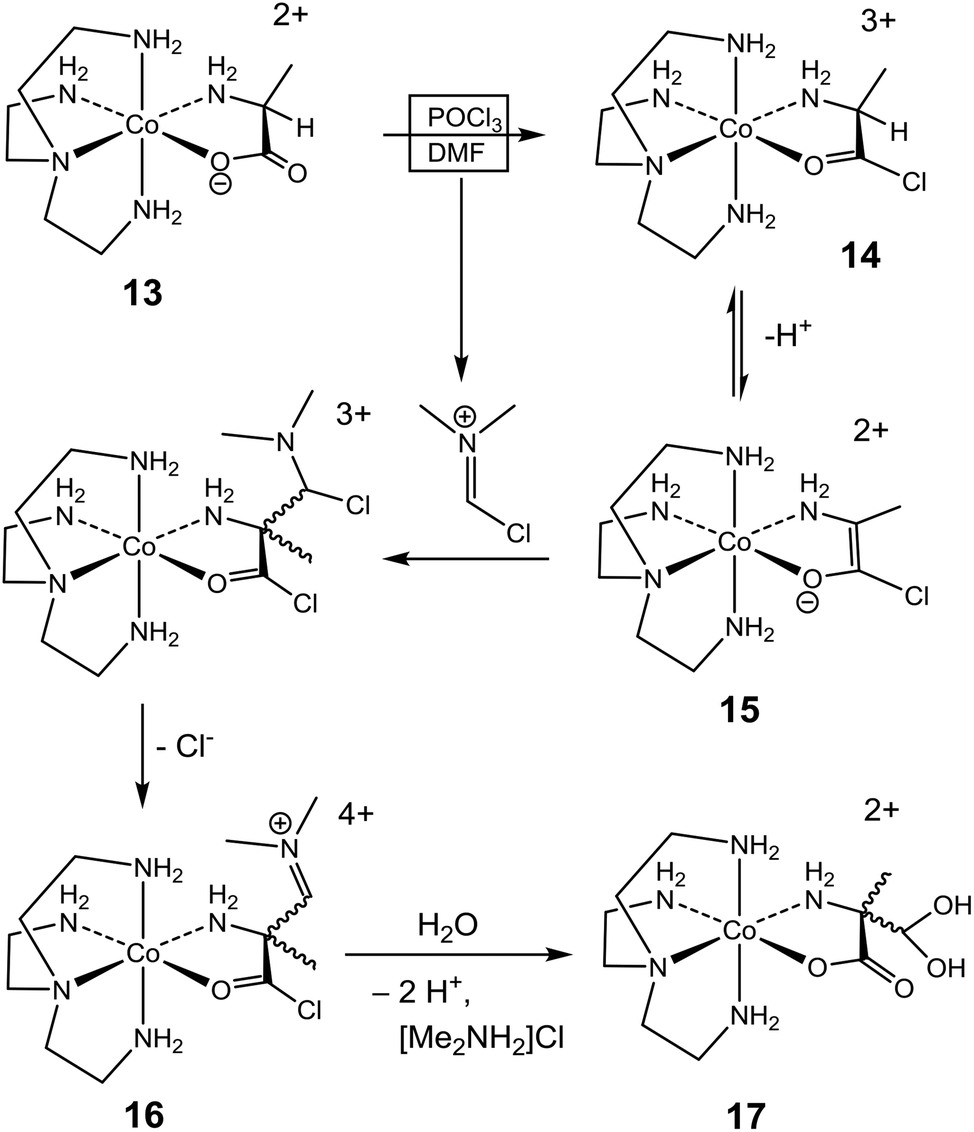 | ||
| Scheme 3 | ||
In the mechanistic proposal above (Scheme 3) the identity of the acid halide 14 was inferred by analogy. Thus, the formation of a chelated acid halide (9, Scheme 2) from p-[Co(tren)GlyO]2+ and POCl3 in DMF has been verified crystallographically.14 Since 15 is achiral, re-protonation yields both enantiomers of 14 in equal proportion, and the reversible inter-conversion between 14 and 15 affects overall racemization of 14, even though this was not verified directly. However, the same effect of activation due to acid halide formation has been noted in a related system. Thus, SOCl2, POCl3 and PBr3 were each shown to induce epimerization at the α-C centre of chelated (S)-valinate in the Δ,Λ-[Co(en)2(S-ValO)]2+ complex in DMF, shifting a non-equilibrium distribution of diastereoisomers ([Δ-S]/[Λ-S] = 50:50) towards equilibrium ([Δ-S, Λ-R]/[Δ-R, Λ-S] = 1.9, in water).18,23 Altogether, this evidence implies that temporary removal of the proton at the α-C centre must occur, since inversion at this centre could otherwise not occur.46,47 However, the fate of the removed proton is uncertain, but general base catalysis may operate, aided by dissolved PO2Cl2− or other base. Alternatively, the proton could reside at the O atom of the chelate, which in the present system (Scheme 3) amounts to the enol tautomer of 14.
The pronounced stereoselectivity associated with the formylation of (+)578-p-[Co(tren){(SC,SN)-ProO}]2+ (18) to give (+)578-p-[Co(tren){(RC,SN)-Pro[CH(OH)2]O}]2+ (22) is remarkable, not least because the “Co(tren)” backbone is achiral and chelated amino acidato ligands typically equilibrate in the conditions applied, vide supra.18,23 Nevertheless, the ee value of 85% testifies that chirality is largely retained in this instance. A mechanistic proposal, depicted in Scheme 4, assumes initial conversion of 18 to form the acid chloride 19, as before (Scheme 3).14–24 The α-C centre of this triply-charged complex (19) is activated towards deprotonation to generate enolate intermediate 20, stabilized by conjugation and the lower charge, but with some strain in the bound pyrrolidine ring. Most importantly, the absolute configuration of the coordinated sec. amine (stereogenic) is retained, and this asymmetry directs stereoselective addition to the imine of the Vilsmeier reagent to occur from the Re face of the enolate ligand. Subsequent loss of Cl− (to yield 21) followed by hydrolysis provide the α-(RC,SN)-di(hydroxy)methyl prolinate complex 22. Effectively, the inserted formyl group occupies the position initially occupied by the α-proton, thus rendering the overall reaction “stereoretentive” even though, formally, the change of absolute configuration of the α-C site is from S (18 and 19) to R (21 and 22). Once formed, the quaternary α-C centre of 22 is configurationally locked.
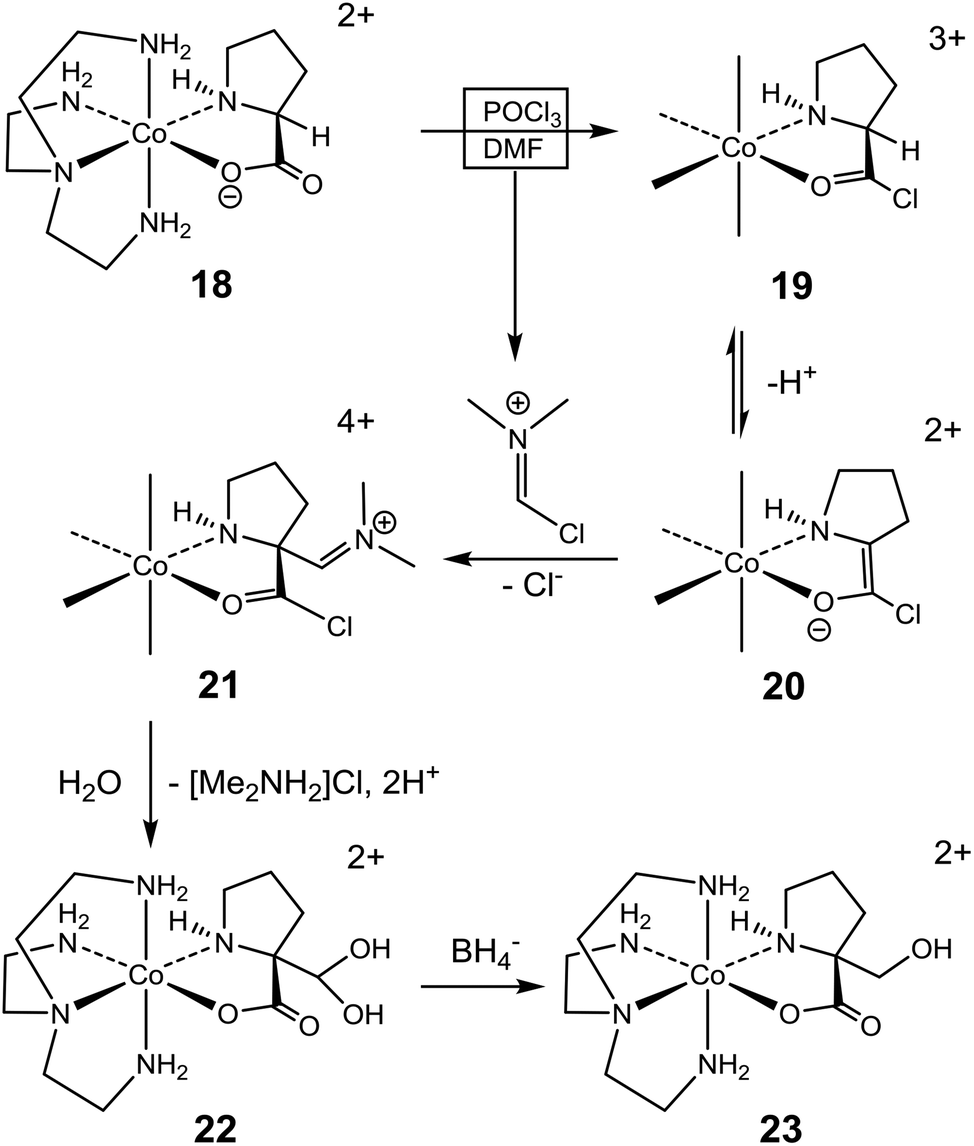 | ||
| Scheme 4 | ||
The 85% ee for 22 implies slight racemization in the overall reaction. Conceivably, this may arise via either of two paths. One possibility is partial racemization of 20 prior to attack by the Vilsmeier reagent. At the molecular level such racemization requires partial inversion of the sec. amine of 20, but at the inert cobalt(III) centre such event is only mechanistically feasible after prior deprotonation of the amine.43,44 However, further deprotonation of 20 (a product of deprotonation, itself!) would seem less probable, and this accords with the actual modest level of racemization. Alternatively, the modest degree of racemization may be explained by less than complete discrimination in the stereoselective reaction of 20 with the Vilsmeier reagent, which attacks preferentially at the Re face of the enolate plane to produce 21. In the rarer event of attack at the Si face, the immediate product (a putative diastereoisomer of 21) would inevitably have the pyrrolidine ring in the more strained transoid arrangement relative to the chelate ring. Subsequently, this situation would have to be alleviated by equilibration (via inversion of the sec. amine) in order to bring the pyrrolidine ring into the more stable cisoid arrangement, which was the only observed (13C NMR and X-ray crystallography) in the isolated product. If such equilibration is operating for the putative diastereoisomer of 21, the mechanistically required deprotonation of the sec. amine would be facilitated by the high ionic charge of this complex.
Conclusions
The feasibility of conducting Vilsmeier–Haack-type formylation reactions at the α-C centre of a chelated amino acidate, hitherto only performed at the glycinate methylene group,14–17,27 has been expanded to also incorporate the methine groups of chelated alaninate and prolinate.The highly stereoselective formylation reaction of chelated (S)-prolinate to give α-(R)-di(hydroxy)methyl prolinate (22) constitutes a novel and highly efficient example of the execution of the SRS principle.7 Here, the prolinato cobalt(III) system is unique in the sense that the combined exploitation of the activating and protecting effects of an inert metal centre, modulating the chemistry while directing the stereochemistry, has not been exemplified in this manner before. The formyl group thus introduced at the quaternary α-C centre of the chiral complex 22 makes this an ideal starting point, easily synthesized, for future controlled elaboration of the proline segment. In the complex, the amine and carboxylate functionalities remain protected by the coordination to the inert metal centre. However, product amino acids may be readily liberated at any time by gentle reduction to the labile cobalt(II) state, as demonstrated before.23,32,33
Further enhancement of the stereoselectivity, and fine-tuning of reactivity, may be envisaged by the introduction of suitable steric demands in the backbone ligand sphere. Investigations along these lines are currently being pursued.
Notes and references
- S. K. Panday, Tetrahedron: Asymmetry, 2011, 22, 1817–1847 CrossRef CAS PubMed.
- Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed.
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138–5175 CrossRef CAS PubMed.
- M. I. Calaza and C. Cativiela, Eur. J. Org. Chem., 2008, 3427–3448 CrossRef CAS PubMed.
- D. Seebach and R. Naef, Helv. Chim. Acta, 1981, 64, 2704–2708 CrossRef CAS PubMed.
- D. Seebach, M. Boes, R. Naef and W. Bernd Schweizer, J. Am. Chem. Soc., 1983, 105, 5390–5398 CrossRef CAS; D. Seebach, A. R. Sting and M. Hoffmann, Angew. Chem., Int. Ed. Engl., 1996, 35, 2708–2748 CrossRef PubMed.
- D. Seebach, A. R. Sting and M. Hoffmann, Angew. Chem., Int. Ed. Engl., 1996, 35, 2708–2748 CrossRef CAS PubMed.
- H. C. Freeman and I. E. Maxwell, Inorg. Chem., 1970, 9, 649–655 CrossRef CAS.
- H. C. Freeman, L. G. Marzilli and I. E. Maxwell, Inorg. Chem., 1970, 9, 2408–2415 CrossRef CAS.
- E. F. Birse, P. A. Williams, F. S. Stephens and R. S. Vagg, Inorg. Chim. Acta, 1988, 148, 63–69 CrossRef CAS.
- R. R. Fenton, F. S. Stephens, R. S. Vagg and P. A. Williams, Inorg. Chim. Acta, 1995, 236, 109–115 CrossRef CAS.
- J. Cai, X. Hu, X. Feng, W. Shao, L. Ji and I. Bernal, Eur. J. Inorg. Chem., 2000, 2199–2205 CrossRef CAS.
- W. Beck, Z. Naturforsch., B: Chem. Sci., 2009, 64, 1221–1245 CAS.
- W. G. Jackson, A. M. Sargeson, P. A. Tucker and A. D. Watson, J. Am. Chem. Soc., 1981, 103, 533–540 CrossRef CAS.
- W. G. Jackson, G. M. McLaughlin, A. M. Sargeson and A. D. Watson, J. Am. Chem. Soc., 1983, 105, 2426–2430 CrossRef CAS.
- N. J. Curtis, A. Hammershøi, L. M. Nicolas, A. M. Sargeson and K. J. Watson, Acta Chem., Scand. Ser. A, 1987, 41, 36–51 CrossRef.
- R. M. Hartshorn, Aust. J. Chem., 1996, 49, 905–909 CAS.
- (a) A. Hammershøi, R. M. Hartshorn and A. M. Sargeson, J. Chem. Soc., Chem. Commun., 1988, 1226–1227 RSC; (b) A. Hammershøi, R. M. Hartshorn and A. M. Sargeson, Inorg. Chem., 1990, 29, 4525–4530 CrossRef.
- L. Grøndahl, A. Hammershøi, R. M. Hartshorn and A. M. Sargeson, Inorg. Chem., 1991, 30, 1800–1805 CrossRef.
- R. M. Hartshorn, A. C. Willis and A. M. Sargeson, J. Chem. Soc., Chem. Commun., 1988, 1269–1271 RSC.
- R. M. Hartshorn and A. M. Sargeson, Aust. J. Chem., 1992, 45, 5–20 CrossRef CAS.
- L. Bendahl, A. Hammershøi, D. Kjærgaard Jensen, E. Kaifer and A. M. Sargeson, Chem. Commun., 1996, 1649–1650 RSC.
- L. Bendahl, A. Hammershøi, D. Kjærgaard Jensen, S. Larsen, A. Riisager, A. M. Sargeson and H. Osholm Sørensen, J. Chem. Soc., Dalton Trans., 2002, 3054–3064 RSC.
- L. Grøndahl, A. Hammershøi, R. M. Hartshorn and A. M. Sargeson, Acta Chem. Scand., 1995, 49, 781–791 CrossRef.
- L. Grøndahl, A. Hammershøi and S. Larsen, Acta Chem. Scand., 1995, 49, 792–799 CrossRef.
- G. Jones and S. P. Stanforth, Org. React., 2000, 56, 355–659 CrossRef CAS PubMed.
- E. E. Eduok, R. P. Kashyap, A. Nagl, S. A. Bourne and W. H. Watson, J. Chem. Crystallogr., 1994, 24, 627–638 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 59–75 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Hammershøi and E. Larsen, Acta Chem. Scand., Ser. A, 1978, 32, 485–491 CrossRef.
- R. Barfod, J. Eriksen, B. T. Golding, A. Hammershøi, T. A. Jacobsen, A. Langkilde, S. Larsen, O. Mønsted, A. M. Sargeson and H. O. Sørensen, Dalton Trans., 2005, 491–500 RSC.
- R. Barfod, L. Bendahl, A. Hammershøi, D. Kjærgaard Jensen, A. M. Sargeson and A. C. Willis, J. Chem. Soc., Dalton Trans., 1999, 449–457 RSC.
- Y. Mitsin, J. Watanabe, Y. Haroda, T. Skamaki, Y. Iitalea, Y. Kuslin and I. Kimura, J. Chem. Soc., Dalton Trans., 1976, 2095–2102 Search PubMed.
- A. G. Blackman, Polyhedron, 2005, 24, 1–39 CrossRef CAS PubMed.
- P. A. Butler, C. G. Crane, B. T. Golding, A. Hammershøi, D. C. Hockless, T. B. Pedersen, A. M. Sargeson and D. C. Ware, Inorg. Chim. Acta, 2002, 331, 318–321 CrossRef CAS.
- S. Oerum, P. Krabbe, M. Schau-Magnussen, S. Furbo, J. Bendix and A. Hammershøi, Dalton Trans., 2013, 292–298 RSC.
- E. Kimura, S. Young and J. P. Collman, Inorg. Chem., 1970, 9, 1183–1191 CrossRef CAS.
- H. Wautier, V. Daffe, M.-N. Smets and J. Fastrez, J. Chem. Soc., Dalton Trans., 1981, 2479–2483 RSC.
- K. Akamatsu, T. Komorita and Y. Shimura, Bull. Chem. Soc. Jpn., 1981, 54, 3000–3005 CrossRef CAS.
- R. M. Hartshorn, J. Chem. Soc., Dalton Trans., 2002, 3214–3218 RSC.
- E. K. Chong, J. M. Harrowfield, W. G. Jackson, A. M. Sargeson and J. Springborg, J. Am. Chem. Soc., 1985, 107, 2015–2022 CrossRef CAS.
- D. A. Buckingham, J. Dekkers, A. M. Sargeson and M. Wein, Inorg. Chem., 1973, 12, 2019–2023 CrossRef CAS.
- P. J. Lawson, M. G. McCarthy and A. M. Sargeson, J. Am. Chem. Soc., 1982, 104, 6710–6716 CrossRef CAS.
- A. Hammershøi, M. Schau-Magnussen, J. Bendix and A. Mølgaard, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, m319–m322 Search PubMed.
- B. Halpern, A. M. Sargeson and K. R. Turnbull, J. Am. Chem. Soc., 1966, 88, 4630–4636 CrossRef CAS.
- D. A. Buckingham, L. G. Marzilli and A. M. Sargeson, J. Am. Chem. Soc., 1967, 89, 825–830 CrossRef CAS.
Footnote |
| † CCDC 1411924–1411926. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02674a |
| This journal is © The Royal Society of Chemistry 2015 |