DOI:
10.1039/C5DT02648B
(Paper)
Dalton Trans., 2015,
44, 19163-19174
Synthesis, crystal structure, optical and thermal properties of lanthanide hydrogen-polyphosphates Ln[H(PO3)4] (Ln = Tb, Dy, Ho)†
Received
13th July 2015
, Accepted 7th October 2015
First published on 7th October 2015
Abstract
Lanthanide hydrogen-polyphosphates Ln[H(PO3)4] (Ln = Tb, Dy, Ho) were synthesised as colourless (Ln = Tb, Dy) and light pink (Ln = Ho) crystalline powders by reaction of Tb4O7/Dy2O3/Ho2O3 with H3PO3 at 380 °C. All compounds crystallise isotypically (P21/c (no. 14), Z = 4, aTb = 1368.24(4) pm, bTb = 710.42(2) pm, cTb = 965.79(3) pm, βTb = 101.200(1)°, 3112 data, 160 parameters, wR2 = 0.062, aHo = 1363.34(5) pm, bHo = 709.24(3) pm, cHo = 959.07(4) pm, βHo = 101.055(1)°, 1607 data, 158 parameters, wR2 = 0.058). The crystal structure comprises two different infinite helical chains of corner-sharing phosphate tetrahedra. In-between these chains the lanthanide ions are located, coordinated by seven oxygen atoms belonging to four different polyphosphate chains. Vibrational, UV/Vis and fluorescence spectra of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) as well as Dy[H(PO3)4]:Ln (Ln = Ce, Eu) and the magnetic and thermal behaviour of Tb[H(PO3)4] are reported.
1 Introduction
In general, phosphates exhibit a large variety of structures and composition. Besides monophosphates, various condensed oligo-, long-chain polyphosphates and cyclophosphates exist.1,2 Phosphates doped with lanthanides or stoichiometric lanthanide phosphates, such as Sr3(PO4)5Cl:Eu3+/Eu2+, LaPO4:Ce3+,Tb3+, (Y,Gd)PO4:Eu3+,3–7 Ln(PO3)3:Eu3+ (Ln = Y, Gd, Lu),8 α-Sr(PO3)2:Eu2+,Mn2+, SrM(P2O7):Eu2+,Mn2+ (M = Zn, Sr)9,10 and BaCa(PO3)4:Eu2+ (ref. 11), reveal great potential for optical applications as luminescent materials. Many transitions between the energy levels of the Ln3+ ions are in the visible range (12500–25000 cm−1).12,13 Due to the strong shielding of the 5s and 5p orbitals, crystal field splitting is very small, and weak 4f–4f transitions occur, leading to the characteristic line emissions.4,7,14 Since these are parity forbidden, they only become allowed by an admixture of states of the opposite parity.12,13 Despite the rather low emission, a good colour rendering can be achieved.4,7
Acid polyphosphates with the sum formula Ln[H(PO3)4] (Ln = Sm, Eu) are known to crystallise isostructurally to Bi[H(PO3)4]15 in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .16 Surprisingly, acid polyphosphates of the elements at the origin of the lanthanide row (La, Pr, Nd) and at the end (Tm, Yb) have not yet been observed. Instead, only non-protonated polyphosphates and ultraphosphates are formed depending on the synthesis temperature.17 Ln[H(PO3)4] (Ln = Tb–Er) crystallises in the monoclinic space group P21/c. Gd[H(PO3)4] represents an exception, which crystallises in both configurations at the same time. Up to now only the crystal structure of Er[H(PO3)4] was solved from single-crystal data.17 With regard to optical applications, both the spectroscopic properties and the thermal stability of the host lattice are important characteristics.
.16 Surprisingly, acid polyphosphates of the elements at the origin of the lanthanide row (La, Pr, Nd) and at the end (Tm, Yb) have not yet been observed. Instead, only non-protonated polyphosphates and ultraphosphates are formed depending on the synthesis temperature.17 Ln[H(PO3)4] (Ln = Tb–Er) crystallises in the monoclinic space group P21/c. Gd[H(PO3)4] represents an exception, which crystallises in both configurations at the same time. Up to now only the crystal structure of Er[H(PO3)4] was solved from single-crystal data.17 With regard to optical applications, both the spectroscopic properties and the thermal stability of the host lattice are important characteristics.
Herein we study the crystal structures of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) determined from single-crystal data (Ln = Tb, Ho) and refined via a Rietveld refinement (Ln = Dy) as well as their optical, magnetic and thermal properties.
2 Crystal structure
Ln[H(PO3)4] (Ln = Tb, Dy, Ho) crystallise isotypically to Er[H(PO3)4]17 in the centrosymmetric monoclinic space group P21/c (no. 14) (Tables 5–9 and 11). The anionic partial structure of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) consists of two different infinite helical corner-sharing phosphate chains arranged parallel to each other along [010] with alternating poly-hydrogenphosphate and polyphosphate chains (Fig. 1).
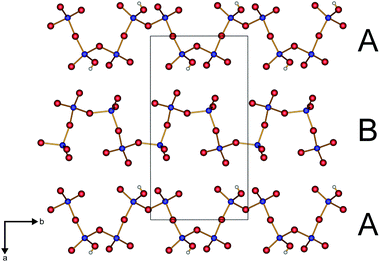 |
| | Fig. 1 Infinite, parallel hydrogen polyphosphate (A) and polyphosphate (B) chains of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) along [010] (viewed along direction [001]); phosphorus blue, oxygen red, hydrogen white. | |
Table 1 MAPLE calculations for Ln[H(PO3)4] (Ln = Tb, Ho)
| Tb[H(PO3)4] |
α-TbP3O9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 + 0.5H2O34 + 0.5P2O535 22 + 0.5H2O34 + 0.5P2O535 |
| MAPLE = 98 590 kJ mol−1 |
MAPLE = 98 466 kJ mol−1 |
| (Δ = 0.1%) |
|
| Ho[H(PO3)4] |
α-HoP3O922 + 0.5H2O34 + 0.5P2O535 |
| MAPLE = 98 811 kJ mol−1 |
MAPLE = 98 326 kJ mol−1 |
| (Δ = 0.5%) |
|
Table 2 Absorption band energies for the strongest transitions in Tb[H(PO3)4] and in free Tb3+ ions between 225 and 800 nm
| No. |
Absorption |
λ/nm |
Tb[H(PO3)4] energy/103 cm−1 |
Free Tb3+ (ref. 42) energy/103 cm−1 |
| 1 |
7F6 → 5H7 |
319 |
31.3 |
31.6 |
| 2 |
7F6 → 5G2 |
336 |
29.8 |
29.6 |
| 3 |
7F6 → 5L9 |
351 |
28.5 |
28.5 |
| 4 |
7F6 → 5G5 |
358 |
27.9 |
27.8 |
| 5 |
7F6 → 5L10 |
369 |
27.1 |
27.1 |
| 6 |
7F6 → 5G6 |
377 |
26.5 |
26.4 |
| 7 |
7F6 → 5D4 |
486 |
20.6 |
20.5 |
Table 3 Absorption band energies for the strongest transitions in Dy[H(PO3)4] and in free Dy3+ ions between 225 and 800 nm
| No. |
Absorption |
λ/nm |
Dy[H(PO3)4] energy/103 cm−1 |
Free Dy3+ (ref. 42) energy/103 cm−1 |
| 1 |
6H15/2 → 4D7/2 |
295 |
33.9 |
33.8 |
| 2 |
6H15/2 → 6P3/2 |
325 |
30.8 |
30.8 |
| 3 |
6H15/2 → 4F5/2 + 4D5/2 |
338 |
29.6 |
29.6 |
| 4 |
6H15/2 → 6P7/2 |
350 |
28.6 |
28.6 |
| 5 |
6H15/2 → 6P5/2 |
364 |
27.5 |
27.5 |
| 6 |
6H15/2 → 4K17/2 |
378 |
26.5 |
26.4 |
| 7 |
6H15/2 → 4I13/2 |
386 |
25.9 |
25.9 |
| 8 |
6H15/2 → 4G11/2 |
425 |
23.5 |
23.3 |
| 9 |
6H15/2 → 4I15/2 |
452 |
22.1 |
22.3 |
| 10 |
6H15/2 → 4F9/2 |
473 |
21.1 |
21.1 |
| 11 |
6H15/2 → 6F3/2 |
753 |
13.3 |
13.2 |
Table 4 Absorption band energies for the strongest transitions in Ho[H(PO3)4] and in free Ho3+ ions between 225 and 800 nm
| No. |
Absorption |
λ/nm |
Ho[H(PO3)4] energy/103 cm−1 |
Free Ho3+ (ref. 41) energy/103 cm−1 |
| 1 |
5I8 → 3H6 + 1I6 |
219 |
45.7 |
45.7 |
| 2 |
5I8 → 3F4 + 5D4 |
241 |
41.6 |
41.5 |
| 3 |
5I8 → 5D3 |
250 |
40.0 |
40.0 |
| 4 |
5I8 → 3I7 |
261 |
38.2 |
38.5 |
| 5 |
5I8 → 3P0 |
278 |
36.0 |
36.0 |
| 6 |
5I8 → 5G4 + 5D4 + 3G4 |
290 |
34.5 |
34.8 |
| 7 |
5I8 → 3F4 + 3H4 + 3G4 |
334 |
29.9 |
30.0 |
| 8 |
5I8 → 3L9 |
346 |
28.9 |
29.0 |
| 9 |
5I8 → 3H6 |
361 |
27.7 |
27.7 |
| 10 |
5I8 → 5G4 |
386 |
25.9 |
25.8 |
| 11 |
5I8 → 5G5 + 3G5 |
417 |
24.0 |
24.0 |
| 12 |
5I8 → 5F1 |
451 |
22.2 |
22.4 |
| 13 |
5I8 → 3K8 |
468 |
21.4 |
21.3 |
| 14 |
5I8 → 5F2 |
473 |
21.1 |
21.1 |
| 15 |
5I8 → 5F3 |
485 |
20.6 |
20.7 |
| 16 |
5I8 → 5F4 |
538 |
18.6 |
18.6 |
| 17 |
5I8 → 5F5 |
648 |
15.4 |
15.5 |
Table 5 Single-crystal data and structure refinements of Ln[H(PO3)4] (Ln = Tb, Ho)
| |
Tb[H(PO3)4] |
Ho[H(PO3)4] |
| Temperature/K |
293(2) |
296(2) |
| Molar weight/g mol−1 |
475.81 |
481.77 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c (no. 14) |
P21/c (no. 14) |
| Crystal shape |
Plate |
Irregular |
| Crystal size/mm3 |
0.047 × 0.018 × 0.019 |
0.050 × 0.018 × 0.020 |
| Colour |
Colourless |
Pink/yellow |
|
a/pm |
1368.24(4) |
1363.34(5) |
|
b/pm |
710.42(2) |
709.24(3) |
|
c/pm |
965.79(3) |
959.07(4) |
|
β/° |
101.200(1) |
101.055(1) |
| Volume/106 pm3 |
920.89(5) |
910.15(6) |
|
Z
|
4 |
4 |
| Calculated density Dx/g cm−3 |
3.432 |
3.499 |
| Absorption coefficient μ/mm−1 |
8.440 |
9.462 |
|
F(000) |
888 |
896 |
| Radiation (λ/Å) |
Mo-Kα (0.7093) |
Mo-Kα (0.7093) |
| Diffractometer |
Bruker D8 Venture |
Bruker D8 Venture |
| Absorption correction |
Multi-scan |
Multi-scan |
| Transmission factor (min./max.) |
0.6359/0.7457 |
0.6029/0.7458 |
| Index range h|k|l (min./max.) |
0/16|−8/0|−11/11 |
−16/16|−8/8|−11/11 |
| Theta range/° |
3.04 ≤ Θ ≤ 25.00 |
3.04 ≤ Θ ≤ 25.00 |
| Reflections collected |
3112 |
27195 |
| Independent reflections |
3111 |
1607 |
| Observed reflections (I > 2σ) |
2790 |
1421 |
|
R
int
|
— |
0.0537 |
| Refined parameters |
160 |
158 |
|
R
1 (all data) |
0.028 |
0.029 |
| wR2 (all data) |
0.062 |
0.058 |
| Weighting scheme |
w−1 = σ2Fo2 + (0.0389P)2 |
w−1 = σ2Fo2 + (0.0423P)2 + 1.5847P |
|
P = (Fo2 + 2Fc2)/3 |
P = (Fo2 + 2Fc2)/3 |
| GooF |
1.131 |
1.030 |
| Residual electron density (min./max.)/e− Å−3 |
−0.499/0.144 |
−0.841/1.981 |
Table 6 Refined atomic coordinates, Wyckoff symbols and isotropic displacement parameters Ueq/Å2 in Tb[H(PO3)4]
| Atom |
Wyckoff symbol |
x
|
y
|
z
|
U
eq
|
| Obr = bridging oxygen atom; Oterm = terminal oxygen atom; OH = hydroxyl group. |
| Tb1 |
4e |
0.729401(15) |
0.05792(3) |
0.05897(2) |
0.00704(9) |
| P1 |
4e |
0.90133(8) |
−0.32485(16) |
0.12591(13) |
0.0083(3) |
| P2 |
4e |
0.90026(9) |
0.15142(17) |
−0.18737(12) |
0.0080(3) |
| P3 |
4e |
0.61157(9) |
−0.20990(17) |
0.29579(12) |
0.0077(2) |
| P4 |
4e |
0.59385(9) |
0.40127(16) |
0.20937(12) |
0.0078(3) |
| Obr1 |
4e |
0.9446(3) |
−0.4771(5) |
0.2377(3) |
0.0135(7) |
| Obr2 |
4e |
0.9990(2) |
−0.2305(4) |
0.0948(3) |
0.0114(7) |
| Obr3 |
4e |
0.5802(2) |
−0.3766(4) |
0.1876(3) |
0.0117(7) |
| Obr4 |
4e |
0.5152(2) |
−0.1724(4) |
0.3594(3) |
0.0098(7) |
| Oterm1 |
4e |
0.8343(3) |
−0.1896(5) |
0.1741(3) |
0.0145(8) |
| Oterm2 |
4e |
0.8414(3) |
0.0764(5) |
−0.0888(3) |
0.0174(8) |
| Oterm3 |
4e |
0.8587(3) |
0.2914(5) |
−0.2952(3) |
0.0156(8) |
| Oterm4 |
4e |
0.6343(3) |
−0.0457(4) |
0.2130(3) |
0.0132(7) |
| Oterm5 |
4e |
0.6902(3) |
−0.2722(4) |
0.4168(3) |
0.0123(7) |
| Oterm6 |
4e |
0.6158(2) |
0.3586(5) |
0.3627(3) |
0.0115(7) |
| Oterm7 |
4e |
0.6615(2) |
0.3364(4) |
0.1191(3) |
0.0114(7) |
| OH1 |
4e |
0.8569(3) |
−0.4300(5) |
−0.0105(4) |
0.0158(8) |
| H1 |
4e |
0.804(3) |
−0.350(8) |
−0.048(5) |
0.05 |
Table 7 Anisotropic displacement parameters Uij/Å2 in Tb[H(PO3)4]
| Atom |
U
11
|
U
22
|
U
33
|
U
12
|
U
13
|
U
23
|
| Tb1 |
0.00640(13) |
0.00779(13) |
0.00678(13) |
−0.00024(9) |
0.00093(10) |
0.00006(9) |
| P1 |
0.0056(6) |
0.0087(6) |
0.0100(6) |
0.0002(5) |
0.0001(5) |
−0.0004(5) |
| P2 |
0.0051(6) |
0.0086(6) |
0.0104(6) |
0.0007(5) |
0.0017(5) |
0.0010(5) |
| P3 |
0.0068(6) |
0.0074(6) |
0.0082(5) |
0.0005(5) |
−0.0005(5) |
−0.0001(5) |
| P4 |
0.0070(6) |
0.0062(6) |
0.0094(6) |
−0.0008(5) |
−0.0001(5) |
−0.0002(5) |
| Obr1 |
0.0099(18) |
0.0108(17) |
0.0184(19) |
0.0068(14) |
−0.0009(14) |
0.0002(15) |
| Obr2 |
0.0129(19) |
0.0104(17) |
0.0091(14) |
0.0030(14) |
−0.0025(13) |
−0.0011(14) |
| Obr3 |
0.0195(19) |
0.0051(16) |
0.0087(16) |
−0.0010(13) |
−0.0021(14) |
0.0022(15) |
| Obr4 |
0.0076(17) |
0.0136(19) |
0.0084(16) |
0.0024(13) |
0.0021(13) |
0.0023(14) |
| Oterm1 |
0.020(2) |
0.0089(18) |
0.0137(17) |
0.0017(14) |
0.0021(15) |
0.0029(16) |
| Oterm2 |
0.0116(18) |
0.027(2) |
0.0156(17) |
−0.0013(16) |
0.0066(15) |
−0.0042(15) |
| Oterm3 |
0.0087(19) |
0.0172(18) |
0.0182(17) |
0.0070(16) |
−0.0042(14) |
0.0015(16) |
| Oterm4 |
0.0148(19) |
0.0093(18) |
0.0171(18) |
0.0040(14) |
0.0068(15) |
0.0012(14) |
| Oterm5 |
0.0082(19) |
0.0111(18) |
0.0155(15) |
0.0013(14) |
−0.0032(13) |
0.0025(13) |
| Oterm6 |
0.012(18) |
0.0126(17) |
0.0075(16) |
0.0000(14) |
−0.0025(14) |
−0.0054(15) |
| Oterm7 |
0.0073(17) |
0.0113(18) |
0.0168(18) |
−0.0026(14) |
0.0054(14) |
−0.0006(14) |
| OH1 |
0.012(2) |
0.0154(19) |
0.0176(18) |
−0.0077(15) |
−0.0019(15) |
−0.0002(14) |
Table 8 Refined atomic coordinates, Wyckoff symbols and isotropic displacement parameters Ueq/Å2 in Ho[H(PO3)4]
| Atom |
Wyckoff symbol |
X
|
y
|
z
|
U
eq
|
| Ho1 |
4e |
0.729532(17) |
0.05657(3) |
0.05931(3) |
0.00637(13) |
| P1 |
4e |
0.90118(10) |
−0.3250(2) |
0.12488(15) |
0.0080(3) |
| P2 |
4e |
0.89935(9) |
0.1530(2) |
−0.18799(15) |
0.0073(3) |
| P3 |
4e |
0.38820(10) |
0.2885(2) |
0.20467(15) |
0.0070(3) |
| P4 |
4e |
0.59443(10) |
0.3988(2) |
0.21039(15) |
0.0070(3) |
| Obr1 |
4e |
0.9438(3) |
−0.0209(6) |
−0.2624(4) |
0.0138(9) |
| Obr2 |
4e |
0.9995(3) |
−0.2308(5) |
0.0942(4) |
0.0101(8) |
| Obr3 |
4e |
0.4186(3) |
0.1212(5) |
0.3132(4) |
0.0111(8) |
| Obr4 |
4e |
0.4853(3) |
0.3257(6) |
0.1412(4) |
0.0107(8) |
| Oterm1 |
4e |
0.8334(3) |
−0.1891(5) |
0.1737(4) |
0.0117(8) |
| Oterm2 |
4e |
0.8395(3) |
0.0788(6) |
−0.0889(5) |
0.0166(9) |
| Oterm3 |
4e |
0.8581(3) |
0.2080(6) |
0.2034(4) |
0.0154(9) |
| Oterm4 |
4e |
0.6349(3) |
−0.0475(5) |
0.2120(4) |
0.0125(9) |
| Oterm5 |
4e |
0.6904(3) |
−0.2267(6) |
−0.0824(4) |
0.0119(8) |
| Oterm6 |
4e |
0.6161(3) |
0.1414(6) |
−0.1351(4) |
0.0132(9) |
| Oterm7 |
4e |
0.6633(3) |
0.3331(5) |
0.1200(4) |
0.0121(9) |
| OH1 |
4e |
0.8568(3) |
−0.4297(6) |
−0.0126(5) |
0.0178(10) |
| H1 |
4e |
0.795(3) |
−0.394(12) |
−0.068(7) |
0.050 |
Table 9 Anisotropic displacement parameters Uij/Å2 in Ho[H(PO3)4]
| Atom |
U
11
|
U
22
|
U
33
|
U
12
|
U
13
|
U
23
|
| Ho1 |
0.00672(16) |
0.00555(17) |
0.00677(17) |
−0.00023(10) |
0.00109(10) |
−0.00002(10) |
| P1 |
0.0072(6) |
0.0059(7) |
0.0103(7) |
0.0007(6) |
0.0004(6) |
−0.0001(6) |
| P2 |
0.0053(6) |
0.0069(7) |
0.0094(7) |
−0.0003(6) |
0.0008(5) |
−0.0011(6) |
| P3 |
0.0066(6) |
0.0054 (7) |
0.0088(7) |
−0.0006(6) |
0.0010(5) |
−0.0006(5) |
| P4 |
0.0069(6) |
0.0048(7) |
0.0090(7) |
−0.0015(6) |
0.0002(6) |
0.0001(5) |
| Obr1 |
0.0094(19) |
0.012(2) |
0.019(2) |
−0.0050(18) |
−0.0009(17) |
0.0009(17) |
| Obr2 |
0.0121(19) |
0.011(2) |
0.0064(18) |
0.0006(17) |
0.0004(15) |
−0.0025(16) |
| Obr3 |
0.018(2) |
0.0037(19) |
0.010(2) |
0.0033(17) |
0.0002(17) |
−0.0011(17) |
| Obr4 |
0.0084(18) |
0.015(2) |
0.0093(19) |
−0.0018(17) |
0.0029(15) |
−0.0026(16) |
| Oterm1 |
0.0139(19) |
0.010(2) |
0.011(2) |
0.0014(17) |
0.0020(16) |
0.0033(16) |
| Oterm2 |
0.014(2) |
0.019 (2) |
0.018(2) |
0.0005(19) |
0.0064(18) |
−0.0033(17) |
| Oterm3 |
0.0119(19) |
0.014(2) |
0.018(2) |
−0.0014(19) |
−0.0037(17) |
−0.0012(17) |
| Oterm4 |
0.014(2) |
0.010(2) |
0.016(2) |
0.0020(17) |
0.0069(17) |
−0.0008(16) |
| Oterm5 |
0.0091(18) |
0.010(2) |
0.0015(2) |
0.0004(18) |
−0.0015(16) |
0.0002(16) |
| Oterm6 |
0.016(2) |
0.010(2) |
0.013(2) |
−0.0019(17) |
0.0000(17) |
0.0043(17) |
| Oterm7 |
0.0105(18) |
0.010(2) |
0.018(2) |
−0.0016(18) |
0.0068(17) |
0.0006(16) |
| OH1 |
0.015(2) |
0.018(2) |
0.017(2) |
−0.0100(19) |
−0.0039(18) |
0.0004(17) |
Table 10 Selected interatomic distances/pm and angles/°
| |
Tb[H(PO3)4] |
Dy[H(PO3)4] |
Ho[H(PO3)4] |
| Ln–O |
228.0(3)–245.1(3) |
224(2)–247(2) |
2.254(4)–2.427(4) |
| P–Obr |
156.1(3)–161.1(3) |
145(3)–188(3) |
1.568(4)–1.601(4) |
| P–Oterm |
146.2(3)–149.4(3) |
130(2)–160(3) |
1.463(4)–1.495(4) |
| P–OH |
153.3(3) |
171 (3) |
1.533(4) |
| POH–Obr–PO |
135.8(2)–135.90(2) |
126.5(18)–134.9(16) |
135.8(2)–136.3(3) |
| PO–Obr–PO |
129.9(2)–132.5(2) |
129.5(19)–141.5(19) |
129.8(3)–132.6(3) |
| Oterm–P–Oterm |
117.4(2)–121.4(2) |
111(2)–118(3) |
117.4(2)–121.2(2) |
| Oterm–P–Obr |
105.9(2)–114.3(2) |
94(2)–130(3) |
105.9(2)–114.1(2) |
| Oterm–P–OH |
115.1(2) |
135(2) |
115.0(2) |
| Obr–P–Obr |
99.4(2)–104.1(2) |
93(2)–111(2) |
99.1(2)–104.2(2) |
| Obr–P–OH |
104.6(2)–106.9(2) |
98.1(20)–108.9(20) |
104.7(2)–106.7(2) |
Table 11 Rietveld refinement parameters of Dy[H(PO3)4]
| Molar weight/g mol−1 |
479.40 |
| Crystal system |
Monoclinic |
| Space group |
P21/c (no. 14) |
| Colour |
Colourless |
|
a/pm |
1365.85(3) |
|
b/pm |
709.74(1) |
|
c/pm |
962.15(2) |
|
β/° |
101.116(1) |
| Volume/106 pm3 |
915.21(3) |
|
Z
|
4 |
| Calculated density Dx/g cm−3 |
3.479 |
| Radiation |
Cu-Kα |
| Wavelength λ/Å |
1.5406 |
| Diffractometer |
Bruker D8 Advance |
| Theta range/° |
5.00 ≤ 2θ ≤ 140.00 |
| Refined parameters |
62 |
|
R
p
|
0.009 |
|
R
wp
|
0.012 |
|
χ
2
|
4.04 |
The protonated chain (A) reveals a larger diameter compared to the non-protonated polyphosphate chain (B) (Fig. 2) due to the widened P–O–P angles (136° in A vs. 130–133° in B).
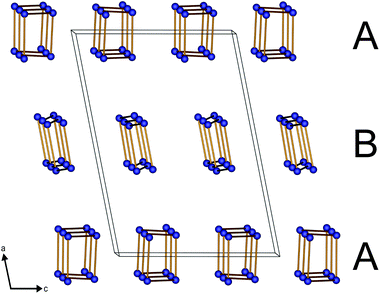 |
| | Fig. 2 Reduced presentation of the infinite helical poly-hydrogenphosphate (A) and polyphosphate (B) chains of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) viewing in the [010] direction. O and H atoms are omitted for clarity; phosphorus blue. | |
The adjacent A and B chains in the a and c directions, respectively, exhibit a phase shift of 180° towards each other (Fig. 3).
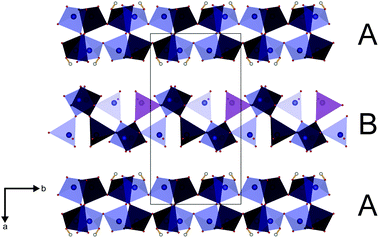 |
| | Fig. 3 Phase-shifted chains of poly-hydrogenphosphate (A) and polyphosphate (B) tetrahedra (blue: front, violet: back) of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) in [001]; phosphate tetrahedra blue, oxygen red, hydrogen white. | |
In the phosphate tetrahedra P–O bond lengths range between 146 and 161 pm. The P–OH bond is 153 pm long. P–Obr distances range between 156 and 161 pm, whereas P–Oterm distances range between 146 and 150 pm. These values correspond very well to the data found for other poly-hydrogenphosphates15–17 or polyphosphates.18–23 In general, P–O distances increase from P–Oterm to P–OH and up to P–Obr due to the decreasing effective negative charge on the involved oxygen atom which leads to weaker electrostatic interactions between P and O. Angles of the POH–Obr–PO and PO–Obr–PO bridges range between 135 and 137° and between 129 and 133°, respectively. Selected bond lengths and angles of Ln[H(PO3)4] (Ln = Tb, Ho) are listed in Table 10.
An appropriate method for the calculation of the deviation of tetrahedra from ideal symmetry was introduced by Balić-Žunić and Makovicky.24,25 We adopted and explained the application of this method with the example of α-BaHPO4;26 in the course of our recent results we assigned deviations of less than 1% to regular tetrahedra. The four crystallographically different phosphate and hydrogenphosphate tetrahedra in Tb[H(PO3)4] and Ho[H(PO3)4] show the values of −0.32% (P1), −0.29 (P2), −0.22 (P3), −0.20 (P4) and −0.32 (P1), −0.29% (P2), −0.21 (P3), −0.21 (P4), respectively. These values are on the typical scale for phosphate tetrahedra.22,26–29 Exhibiting a deviation far below 1%, all tetrahedra are classified as regular.
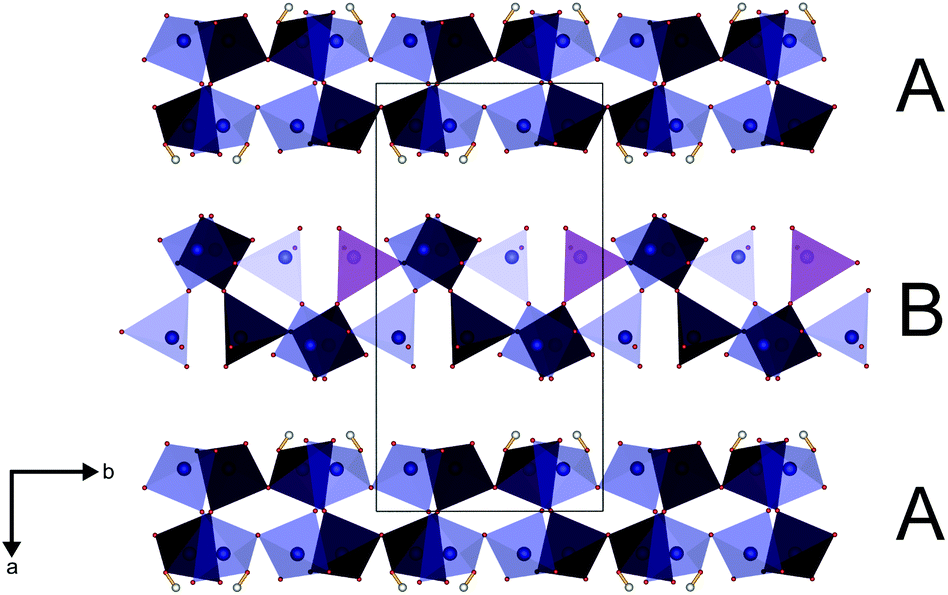
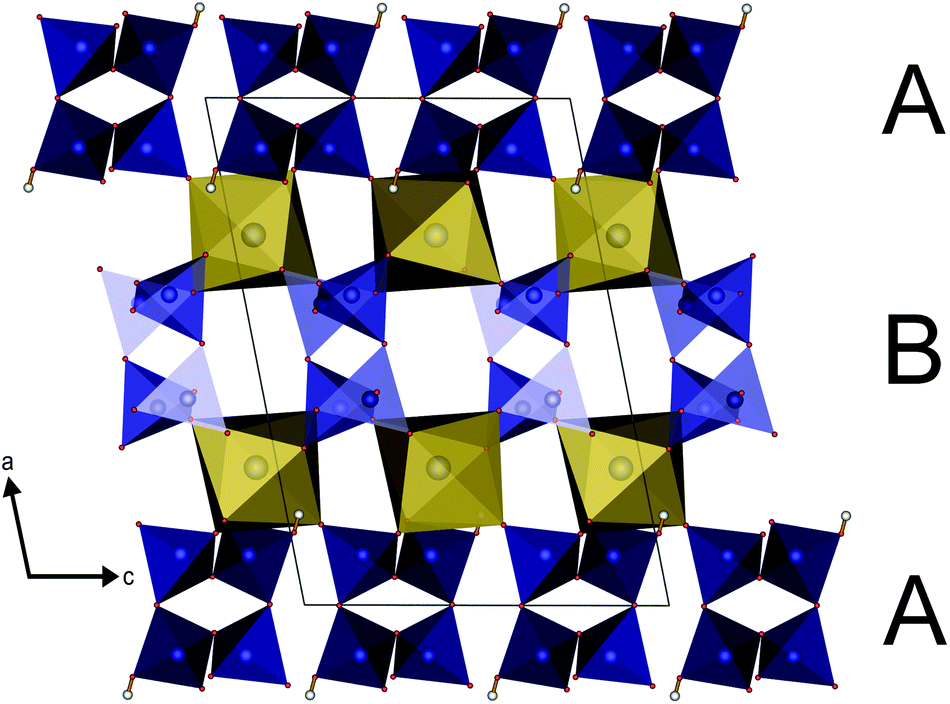
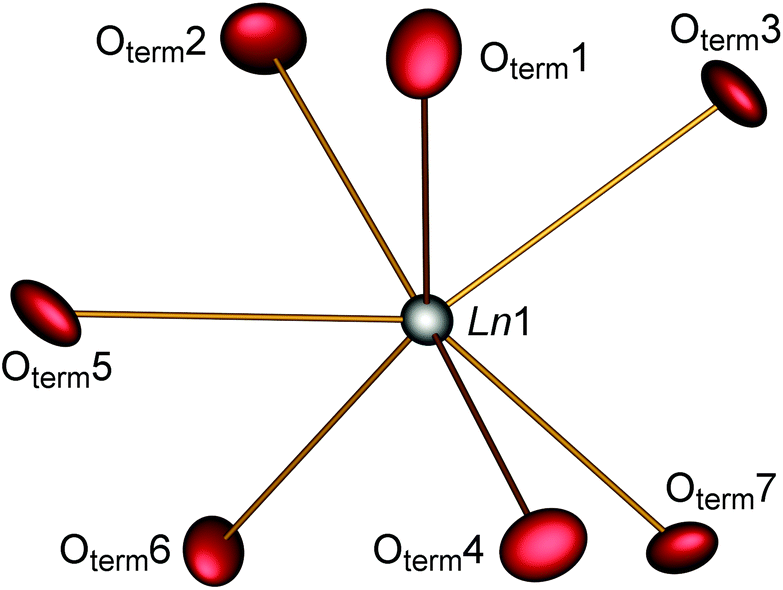
The parallel poly-hydrogenphosphate and polyphosphate chains of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) are interconnected via common O corners of LnO7 polyhedra (Fig. 4). The single-crystal structure reveals a single site of Ln3+, which is coordinated capped trigonal prismatic by seven terminal oxygen atoms of the polyphosphate chains (Fig. 5 and 6). The Ln–O bond lengths range between 225 and 245 pm (Table 10). These values are in the same range as the Er–O bond lengths in Er[H(PO3)4] determined by Palkina et al.17
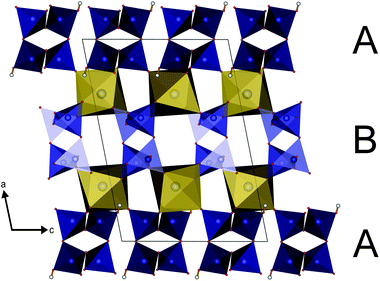 |
| | Fig. 4 Infinite helical chains of poly-hydrogenphosphate (A) and polyphosphate (B) tetrahedra in Ln[H(PO3)4] (Ln = Tb, Dy, Ho) in the [010] direction interconnected via LnO7 polyhedra (yellow); phosphate tetrahedra blue, oxygen red, hydrogen white. | |
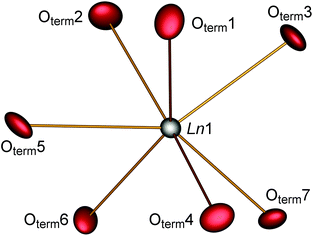 |
| | Fig. 5 Coordination sphere of the lanthanide in Ln[H(PO3)4] (Ln = Tb, Dy, Ho). The displacement ellipsoids are drawn on a probability level of 70%. | |
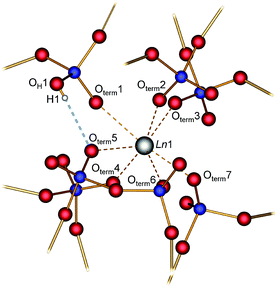 |
| | Fig. 6 Moderate hydrogen bond (dotted blue line) and coordination sphere of the Ln atom in Ln[H(PO3)4] (Ln = Tb, Dy, Ho); phosphorus blue, oxygen red, hydrogen white. | |
In Ln[H(PO3)4] (Ln = Tb, Ho) a single hydrogen bond was found which can be considered as moderate according to Steiner.30 Tb[H(PO3)4] exhibits a hydrogen to acceptor distance d(H1–Oterm5) of 176 pm and a bond angle ∢(DHA) of 165° (the donor to hydrogen distance d(OH1–H1) = 93 pm, and the donor to acceptor distance d(OH1–Oterm5) = 267 pm). In Ho[H(PO3)4] the hydrogen to acceptor distance d(H1–Oterm5) is 185 pm and the bond angle ∢(DHA) is 145° (the donor to hydrogen distance d(OH1–H1) = 94 pm, and the donor to acceptor distance d(OH1–Oterm5) = 266 pm) (Fig. 6). In contrast to moderate hydrogen bonds with a hydrogen to acceptor distance between 150 and 220 pm and bond angles ∢(DHA) larger than 130°, strong hydrogen bonds exhibit a distance of 120–150 pm and an angle of 170–180°.
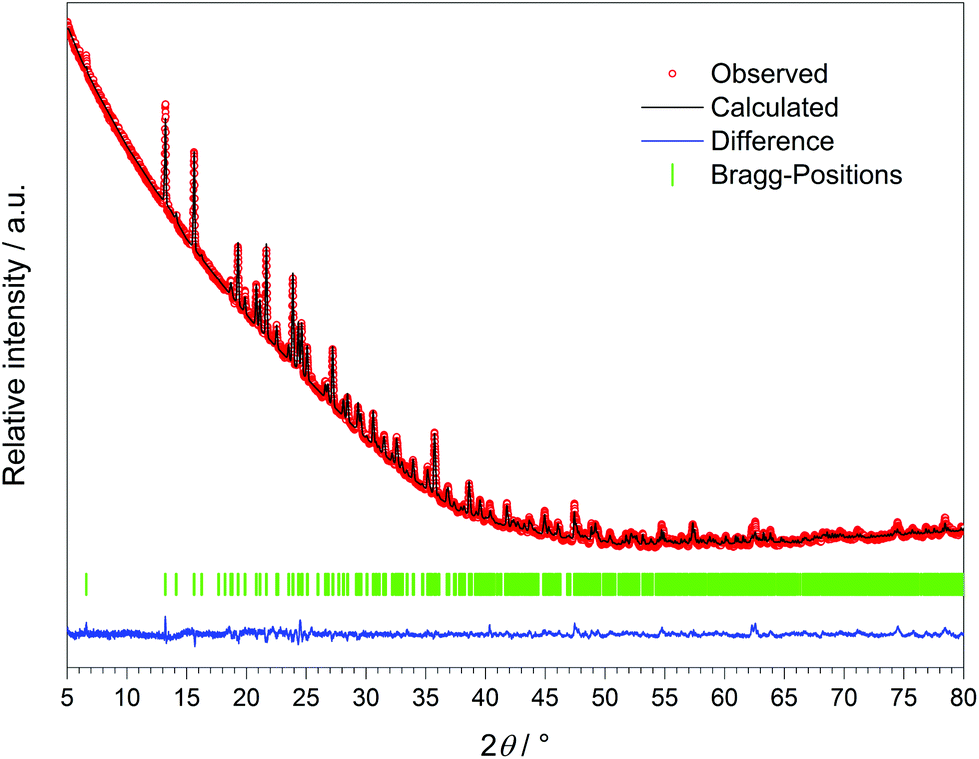
The crystal structure of Dy[H(PO3)4] was refined via the Rietveld method (Fig. 8 and Table 11) based on the structure model of Tb[H(PO3)4].
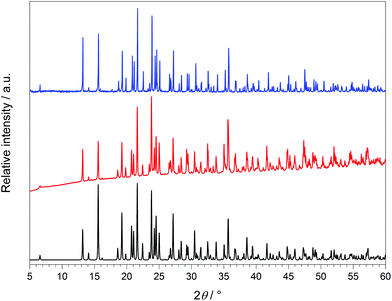 |
| | Fig. 7 X-ray powder diffraction patterns of Tb[H(PO3)4] (red) and Ho[H(PO3)4] (blue) and the calculated X-ray powder diffraction pattern from single-crystal data of Tb[H(PO3)4] (black) (λCu-Kα = 1.54178 Å). | |
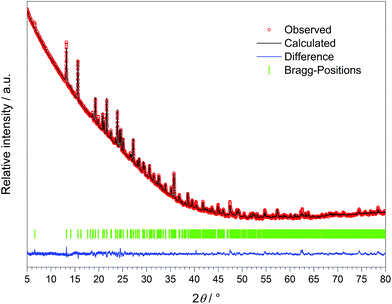 |
| | Fig. 8 X-ray powder diffraction pattern and the result of the Rietveld refinement of Dy[H(PO3)4] (λCu-Kα = 1.54178 Å). | |
3 Electrostatic calculations
The coordination numbers of the lanthanide ions and the electrostatic consistency of the structure model were confirmed by calculations based on the MAPLE concept (Madelung Part of Lattice Energy).31–33 The presence of one proton could also be proven. A structure model is electrostatically consistent if the sum of MAPLE values of chemically similar compounds deviates from the MAPLE value of the compound of interest by less than approximately 1%. Thus the structure models of Tb[H(PO3)4] and Ho[H(PO3)4] show electrostatic consistency (Table 1).
4 Infrared spectroscopy
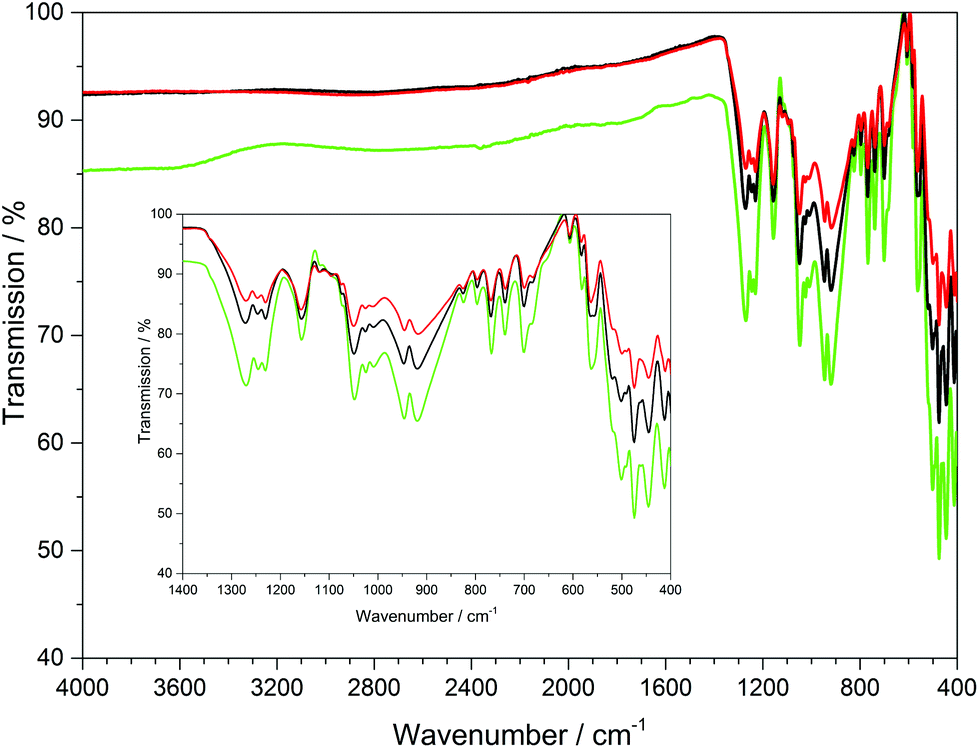
The infrared spectra of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) were recorded between 4000 and 400 cm−1 and are shown in Fig. 9.
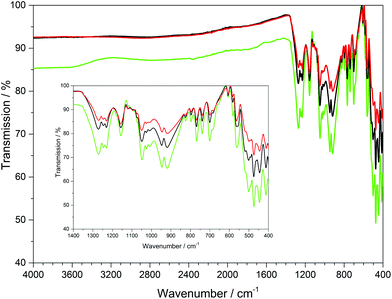 |
| | Fig. 9 Infrared spectra of Ln[H(PO3)4] (Ln = Tb, Dy, Ho); terbium green, dysprosium black, holmium red. | |
The bands between 1350 and 1200 cm−1 can be assigned to the asymmetric stretching vibrations of PO2, while vibrations between 1200 and 990 cm−1 can be assigned to the stretching vibrations of POterm. The νas(PObr) and νas(POP) vibrational bands range between 985 and 845 cm−1.11,36–38 The four symmetric stretching modes of the PObr vibrations are detected between 780 and 650 cm−1 peaking at 681, 700, 741 and 770 cm−1, which had been empirically correlated with the periodicity of polyphosphate chains in catena-polyphosphates.11,36,39 This also holds for Ln[H(PO3)4] (Ln = Tb, Dy, Ho), due to the periodicity P = 4 of its poly-hydrogenphosphate chains. Vibrations observed below 620 cm−1 are assigned to the bending vibrations of the PO4 tetrahedra.11,36–38 Surprisingly, no vibrations of the hydroxyl groups were detected, which leads to the assumption that presumably the ratio of O–H/P–O vibrations of 1:16 is too small to be detected via ATR.
5 UV/Vis spectroscopy
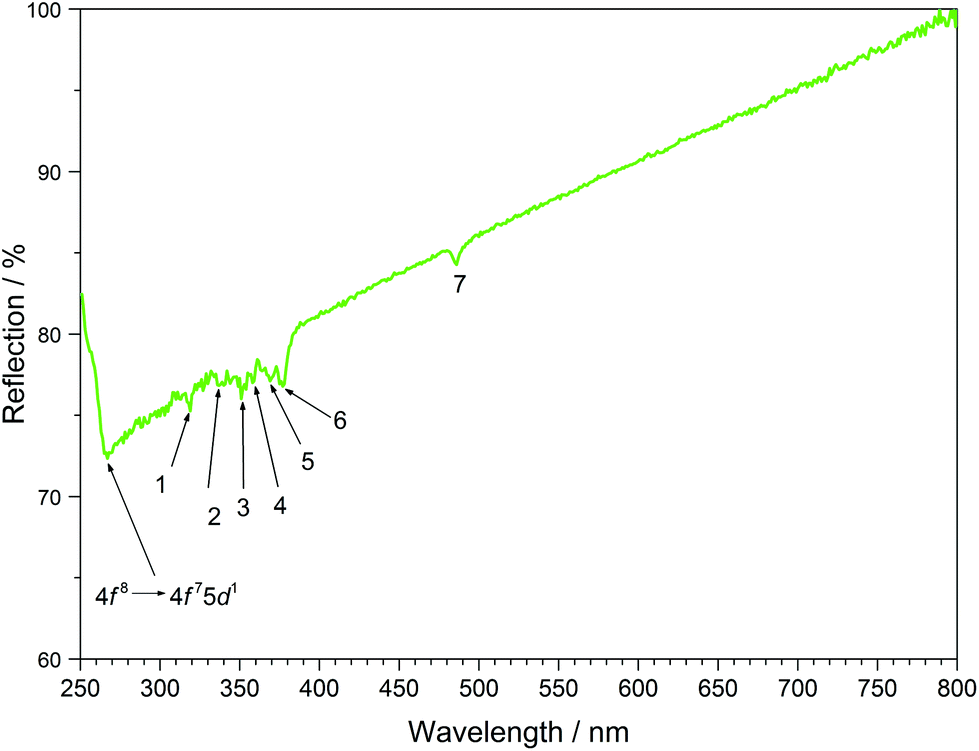
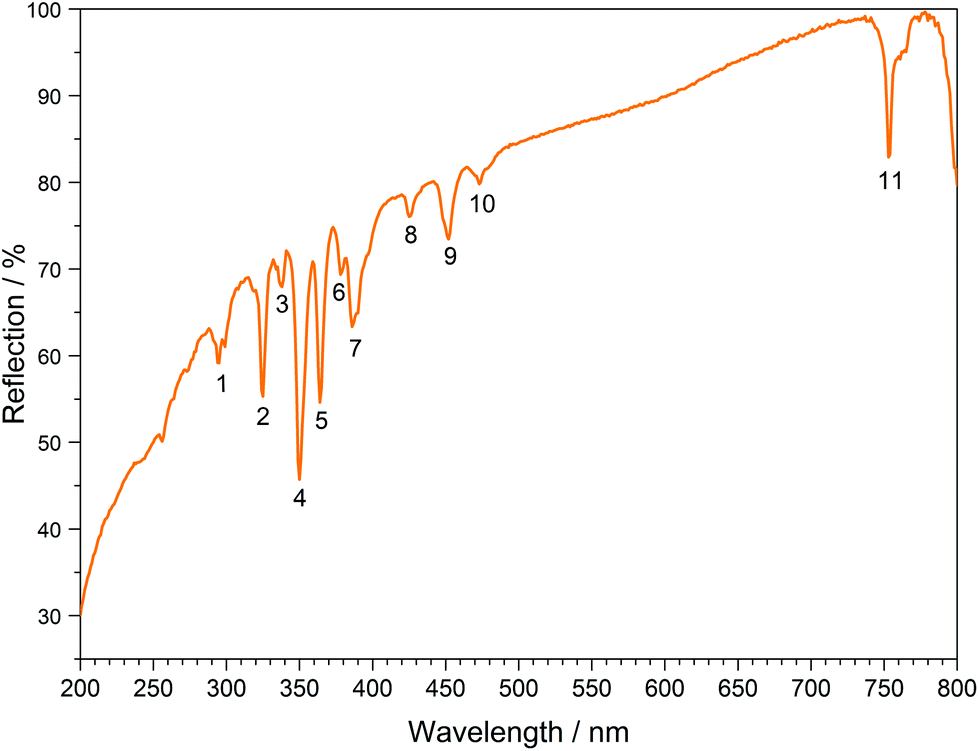
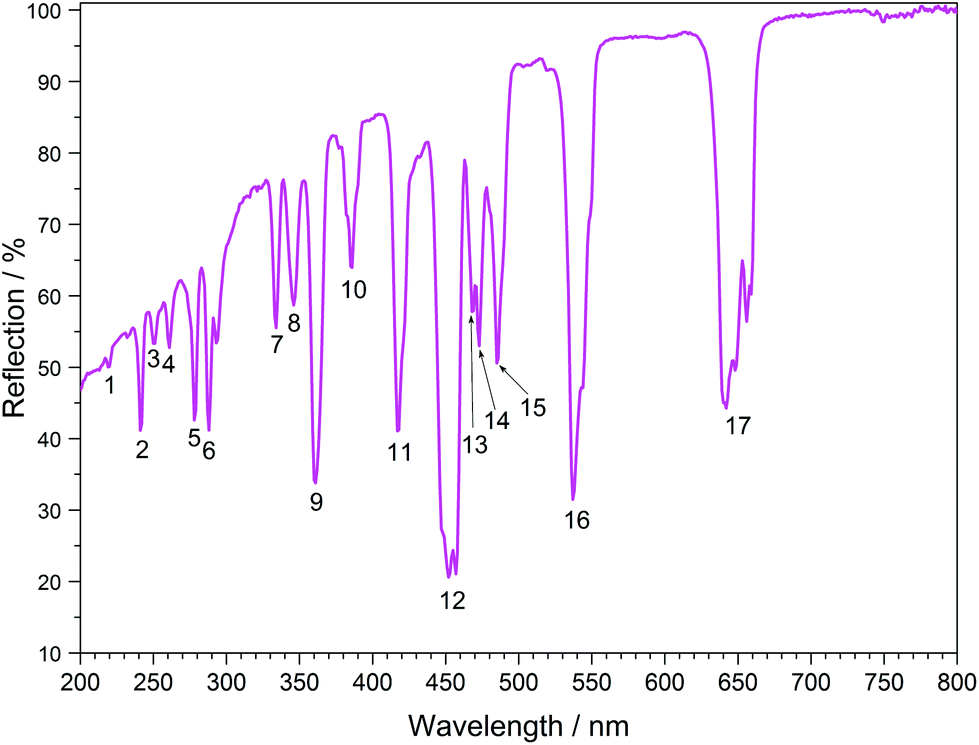
The UV/Vis reflection spectra of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) (Fig. 10–12) reveal the typical absorption bands of the f–f transitions of Tb3+, Dy3+ and Ho3+ ions, according to the well-known energy level schemes.40,41 All transitions start from the respective ground states 7F6, 6H15/2 and 5I8 with the electronic configuration [Xe] 4f8, [Xe] 4f9 and [Xe] 4f10, respectively. The corresponding assignments are listed in Tables 2–4.
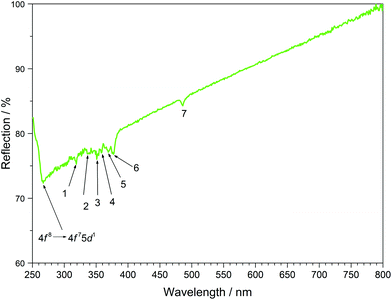 |
| | Fig. 10 UV/Vis spectrum of Tb[H(PO3)4]. | |
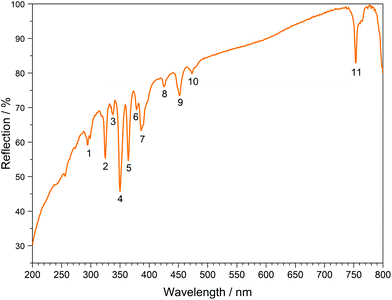 |
| | Fig. 11 UV/Vis spectrum of Dy[H(PO3)4]. | |
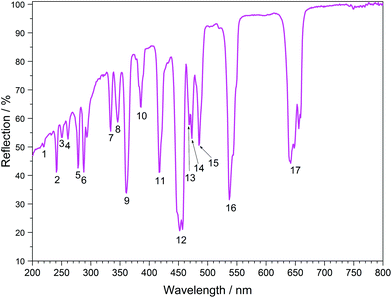 |
| | Fig. 12 UV/Vis spectrum of Ho[H(PO3)4]. | |
No significant deviations from the spectra of other inorganic salts of terbium, dysprosium and holmium were detected and the spectra are in accordance with the powder colours white (Tb[H(PO3)4] and Dy[H(PO3)4]) and pink/yellow (Ho[H(PO3)4]). Like other holmium compounds Ho[H(PO3)4] shows the alexandrite effect.43,44 Due to an absorption gap in the yellow region, it exhibits a yellow body colour in ambient day light. In artificial light, e.g. fluorescent lamps, exhibiting distinct emissions in the blue (∼450 nm), green (∼540 nm) and red (∼610 nm) range, it reveals a pink body colour, due to the reflection of the wavelengths between 550 and 620 nm.4
6 Fluorescence spectroscopy
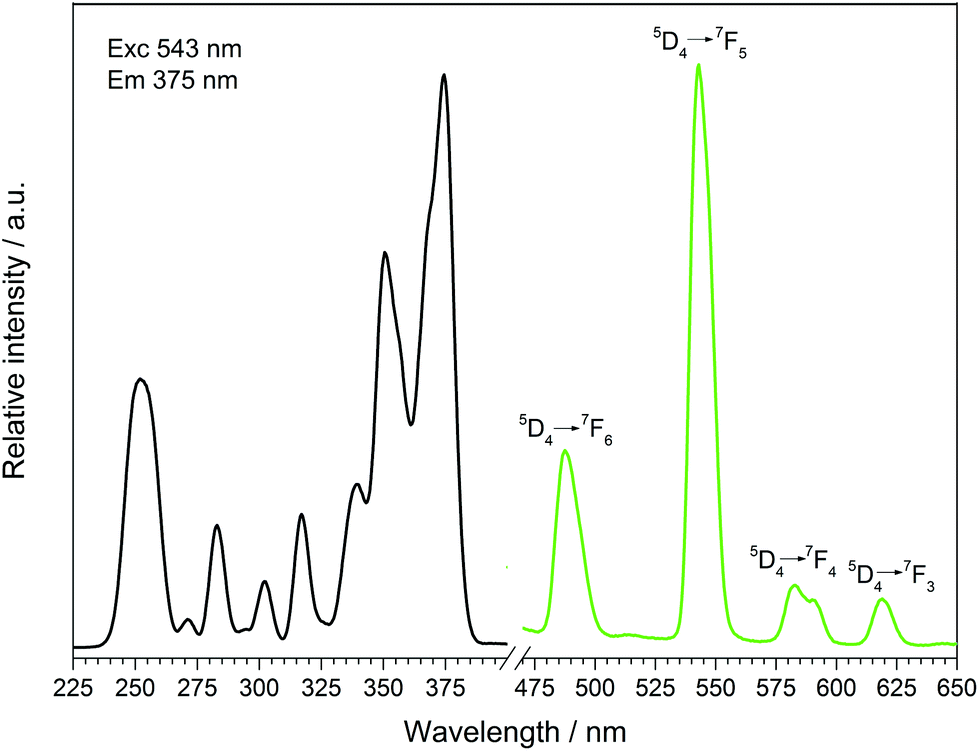
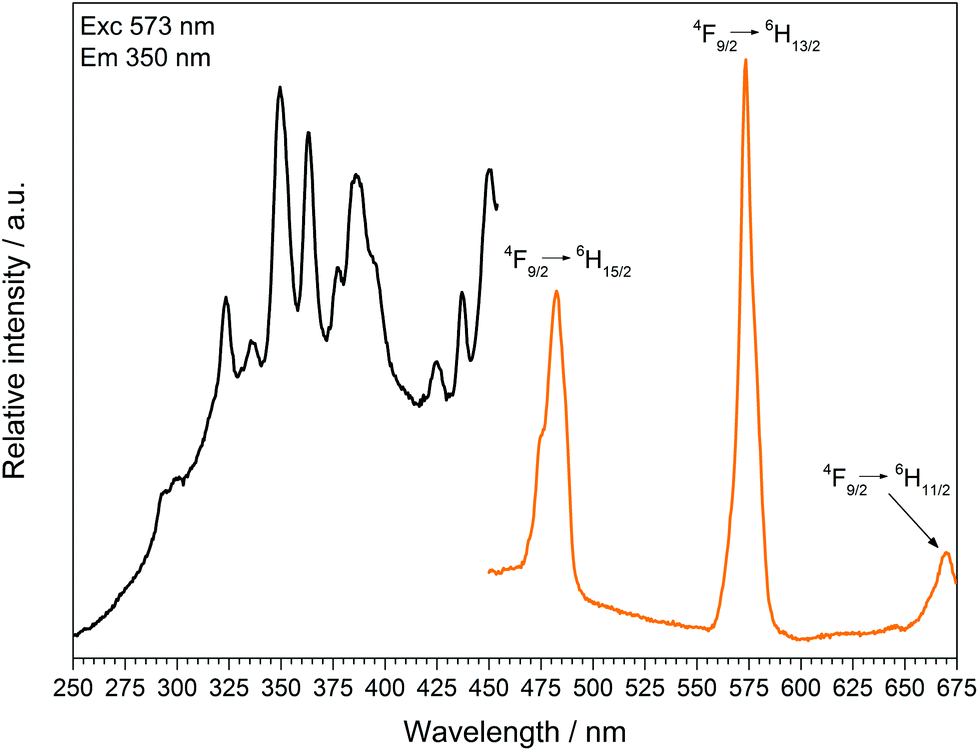
The fluorescence spectra of Tb[H(PO3)4] (Fig. 13) and Dy[H(PO3)4] (Fig. 14) reveal the characteristic f–f emissions of Tb3+ and Dy3+, respectively.40–42
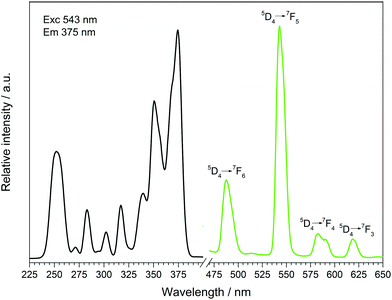 |
| | Fig. 13 Excitation (black), recorded at λEm = 543 nm, and emission (green) spectrum, recorded at λExc = 375 nm, of Tb[H(PO3)4]. | |
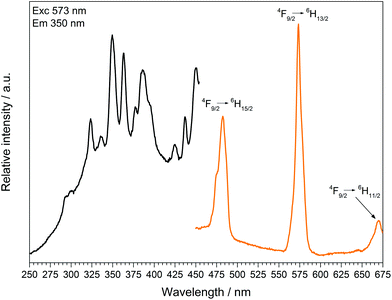 |
| | Fig. 14 Excitation (black), recorded at λEm = 573 nm, and emission (orange) spectrum, recorded at λExc = 350 nm, of Dy[H(PO3)4]. | |
Between 230 and 267 nm a broader band in the excitation spectrum of Tb[H(PO3)4] reveals the parity allowed 4f8 → 4f75d1 transition. Under excitation at 375 nm Tb[H(PO3)4] exhibits sharp emission lines between 475 and 630 nm, which can be assigned to the 5D4 → 7F6 (488 nm), 5D4 → 7F5 (543 nm) showing the highest intensity, 5D4 row 7F4 (583 nm) and 5D4 → 7F3 (619 nm) transitions.42
The excitation spectrum of Dy[H(PO3)4] exhibits a broad band around 290 nm, which reveals the 4f9 → 4f85d1 transition. Under excitation at 349 nm Dy[H(PO3)4] exhibits sharp emission lines between 460 and 675 nm, which correspond to the f–f electronic transitions 4F9/2 → 6H15/2 (482 nm), 4F9/2 → 6H13/2 (573 nm) and 4F9/2 → 6H11/2 (671 nm).41 The most intense transition 4F9/2 → 6H13/2 is sensitive to the surrounding of Dy3+, and is thus called hypersensitive. The surrounding of Dy3+ in Dy[H(PO3)4] is of relatively low symmetry (single capped trigonal prismatic), therefore the intensity of the hypersensitive transition is increased with respect to the 4F9/2 → 6H15/2 transition.45,46
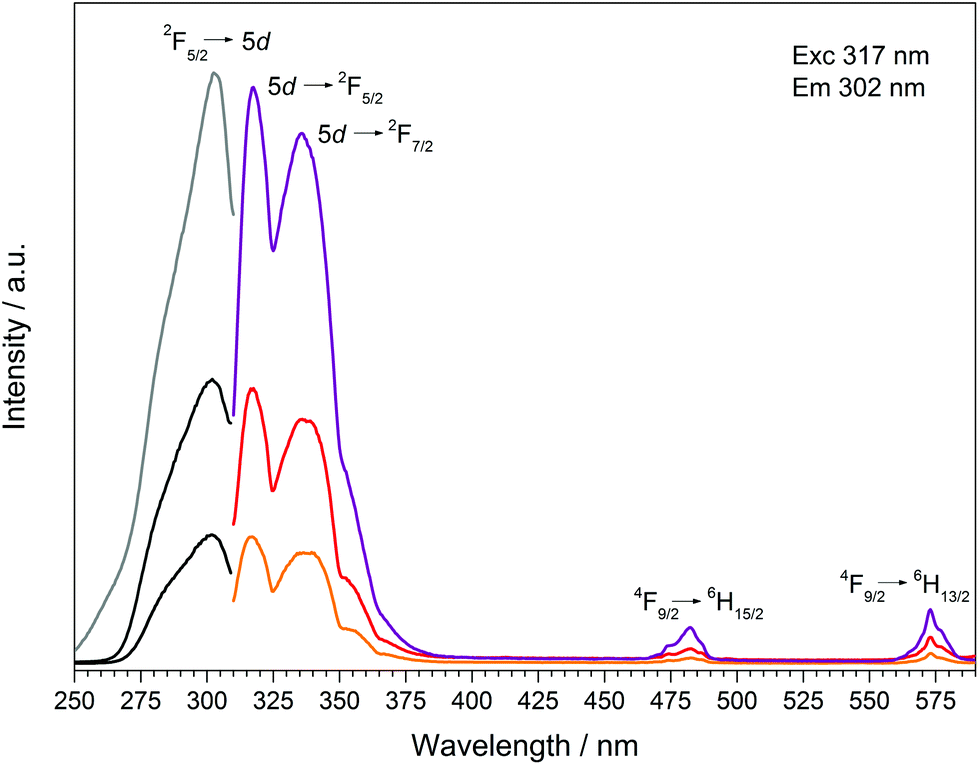
Doping of Dy[H(PO3)4] with increasing concentrations of Ce3+ (5, 10, 30 mol%) was carried out to investigate the effect of Ce3+ as a sensitizer. The presence of cerium and the corresponding ratios of Dy:Ce were investigated via EDX spectroscopy. The resulting ratios are 1:0.04 (expected: 1:0.05), 1:0.07 (1:0.10) and 1:0.26 (1:0.30), which are within the limits of accuracy of the measurements very close to the expected values. Due to an overlap in the wavelength range of the emission bands of Ce3+ and the excitation spectrum of Dy3+ energy transfer could be expected. Transitions in Ce3+ are f–d transitions, which are allowed due to the selection rules, and thus lead to broad and intense bands in the fluorescence spectrum.13 The fluorescence spectra of the doped samples Dy[H(PO3)4]:Ce3+ are shown in Fig. 15. The excitation spectra exhibit strongly increasing broad band intensities of Ce3+ at 302 nm (2F5/2 → 5d). In the emission spectra, next to the typical broad band emissions of Ce3+ peaking at 317 nm (5d → 2F5/2) and 336 nm (5d → 2F7/2)47,48 two comparably weak emission bands of Dy3+, corresponding to the f–f electronic transitions 4F9/2 → 6H15/2 and 4F9/2 → 6H13/2, can be observed. Despite the low Dy3+ emission intensities the hypersensitive transition 4F9/2 → 6H13/2 can still be observed with higher intensity compared to the 4F9/2 → 6H15/2 transition. Doubling the content of Ce3+ from 5 to 10% the emission intensity of Dy3+ also increases equally (4F9/2 → 6H15/2), whereas the hypersensitive transition 4F9/2 → 6H13/2 even triples. A further increase of the Ce3+ content to 30% leads to a raised intensity of a factor of 2.5 for the 4F9/2 → 6H15/2 transition and a factor of 1.7 for the 4F9/2 → 6H13/2 transition. Thus the emission intensities of Dy3+ increase slightly with the increasing Ce3+ content but do not prove efficient Ce3+ → Dy3+ energy transfer. The intensity of the f–d transition in the excitation spectrum increases corresponding to the increasing Ce3+ content. According to Luo and Yang satisfying energy transfer rates in the system Dy3+/Ce3+ could be reached, if the ratio of Dy3+ to Ce3+ ions is decreased to about 1:3, before the luminescence quenching of Ce3+ occurs.49–51 By reversing the Dy3+/Ce3+ ratio and increasing the content of Ce3+ to Ce0.75Dy0.25[H(PO3)4] efficient energy transfer might be reached, which represents a substitution process rather than a doping process.
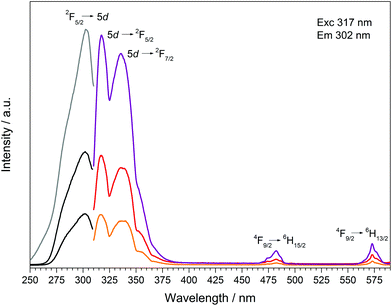 |
| | Fig. 15 Excitation, recorded at λEm = 317 nm, and emission spectra, recorded at λExc = 302 nm, of Dy[H(PO3)4]:Ce3+(5% black/orange, 10% dark grey/red, 30% grey/violet). | |
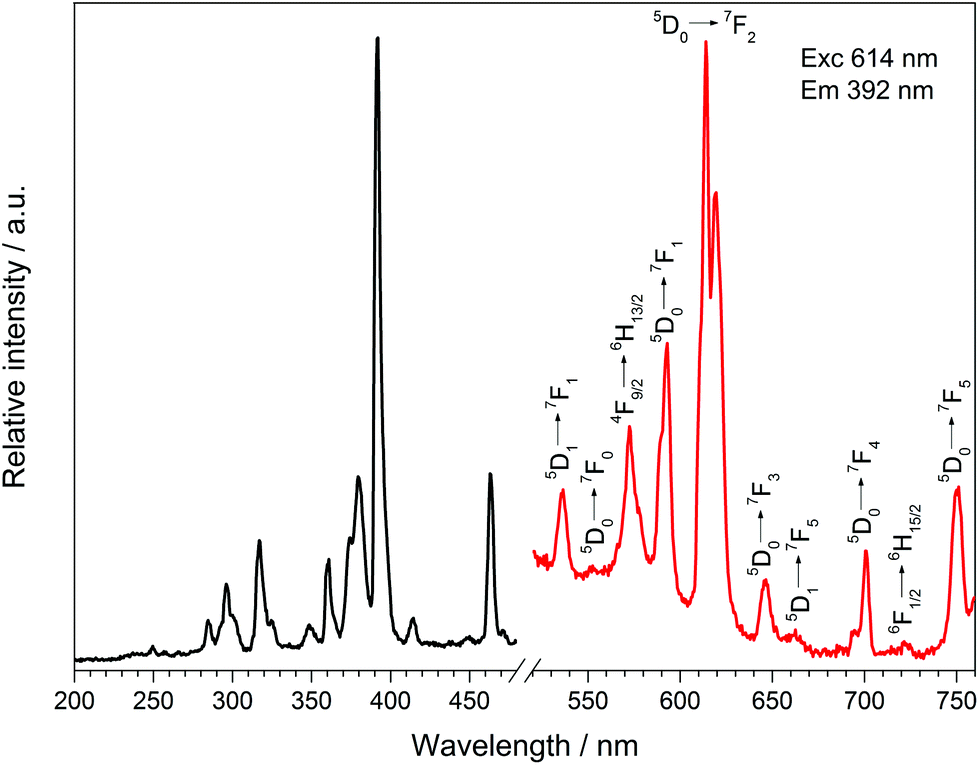
Doping Dy[H(PO3)4] with 5 mol% Eu3+ leads to the typical sharp excitation and emission bands of Dy3+ and Eu3+ (Fig. 16). The ratio of Dy:Eu was investigated via EDX spectroscopy exhibiting a ratio of 1:0.04, which is very close to the expected ratio (1:0.05). Under excitation at 392 nm Dy[H(PO3)4]:Eu3+ exhibits sharp emission lines between 530 and 757 nm, which can be assigned to the 5D1 → 7F1 (536 nm), 5D0 → 7F0 (553 nm), 5D0 → 7F1 (593 nm), 5D0 → 7F2 (614 nm), 5D0 → 7F3 (646 nm), 5D1 → 7F5 (663 nm), 5D0 → 7F4 (700 nm) and 5D0 → 7F5 (750 nm) transitions of Eu3+.52 The most intense transition 5D0 → 7F2 at 614 nm is called hypersensitive due to its sensitivity to the surrounding of Eu3+ and is responsible for the weak red emission. It is dominant over the 5D0 → 7F1 transition because of the asymmetric surrounding of Eu3+ in Dy[H(PO3)4]:Eu3+, which proves its partial substitution of Dy3+.53,54 Besides the emission bands of Eu3+ two emission bands of Dy3+ can be observed at 573 and 722 nm, which can be assigned to the 4F9/2 → 6H13/2 and 6F1/2 → 6H15/2 transitions of Dy3+, respectively.41
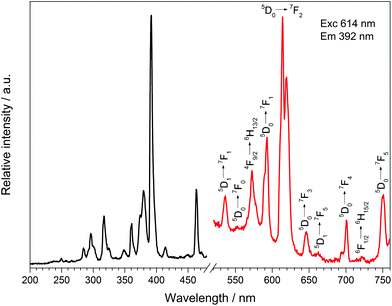 |
| | Fig. 16 Excitation (black), recorded at λEm = 614 nm, and emission (red) spectrum, recorded at λExc = 392 nm, of Dy[H(PO3)4]:Eu3+. | |
7 Magnetic properties
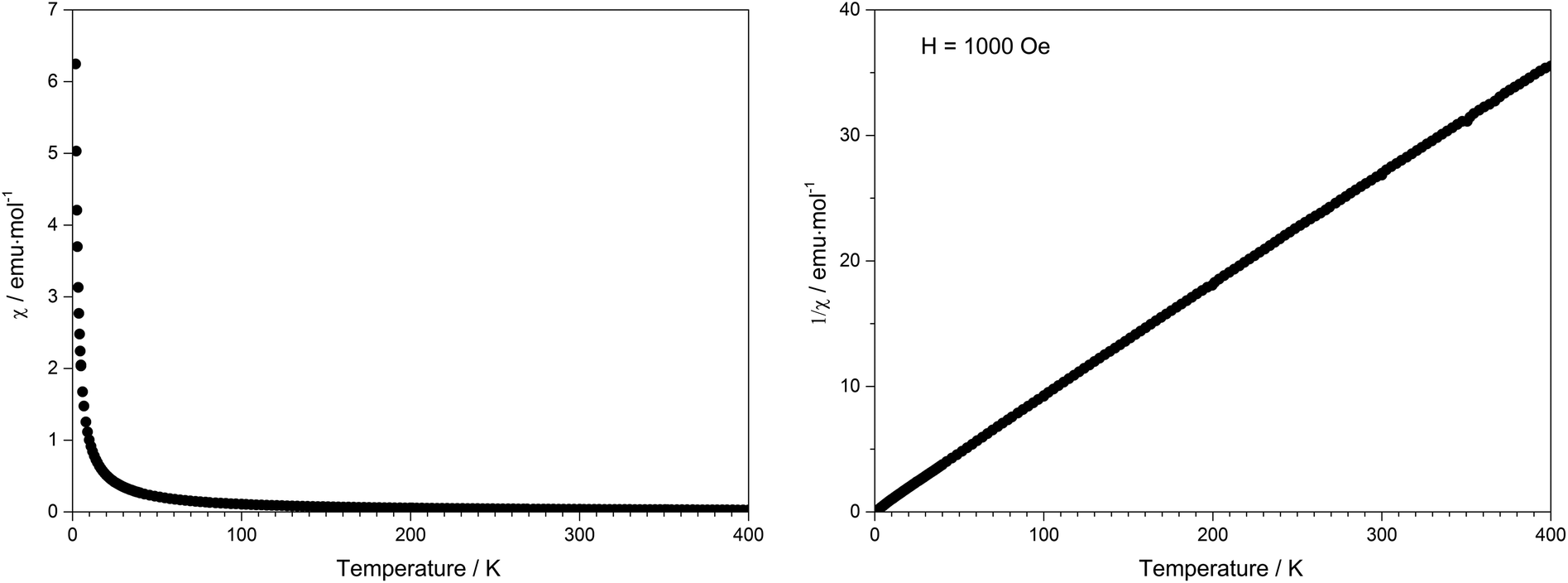
Due to the presence of one proton and the thus postulated sum formula of Tb[H(PO3)4], the valence state of terbium was confirmed by the magnetic susceptibility measurement, which was recorded in a field of 1000 Oe over the temperature range of 1.8 K < T < 400 K (Fig. 17). In the whole temperature range the molar susceptibility obeys Curie's law (χm = C/T) very well with a Curie constant of C = 11.3792 emu mol−1 K−1. The Curie constant corresponds to an effective magnetic moment per Tb3+ ion of μeff = 9.54μB, which is close to the theoretical value (μeff = 9.72μB) and to experimental effective magnetic moments of Tb3+ ions (9.7–9.8μB).55
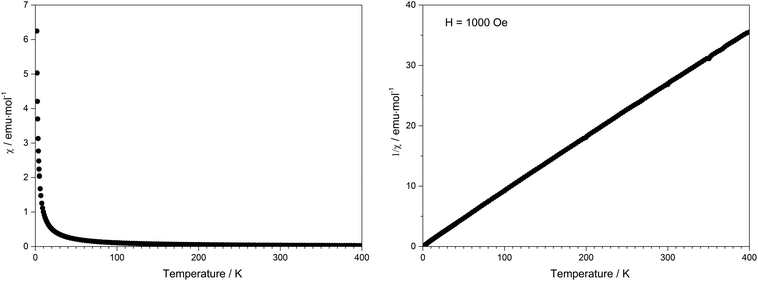 |
| | Fig. 17 Temperature dependence of the magnetic susceptibility χ of Tb[H(PO3)4]. | |
8 Thermal analysis
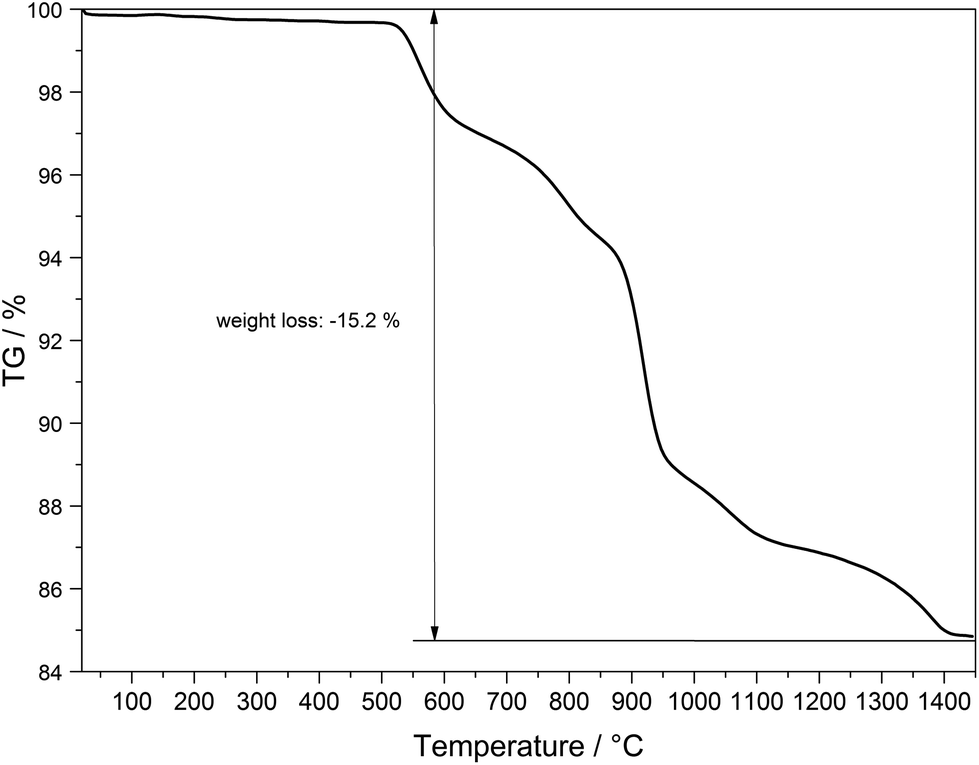
The thermal behaviour of Tb[H(PO3)4] was investigated between room temperature and 1450 °C (Fig. 18). The thermogravimetric curve revealed several undefined steps with a total mass loss of 15.2 wt% in the temperature range of 500–1450 °C. Assuming that besides 0.5 moles of H2O a further 0.5 moles of P2O5 evaporate (theor. mass loss: 16.8 wt%) a final composition of TbP3O9 may be assumed. After the thermal treatment the product was glazed and the expected composition could not be confirmed via X-ray powder diffraction.
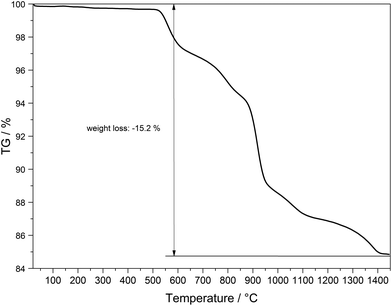 |
| | Fig. 18 Thermogravimetric analysis of Tb[H(PO3)4]. | |
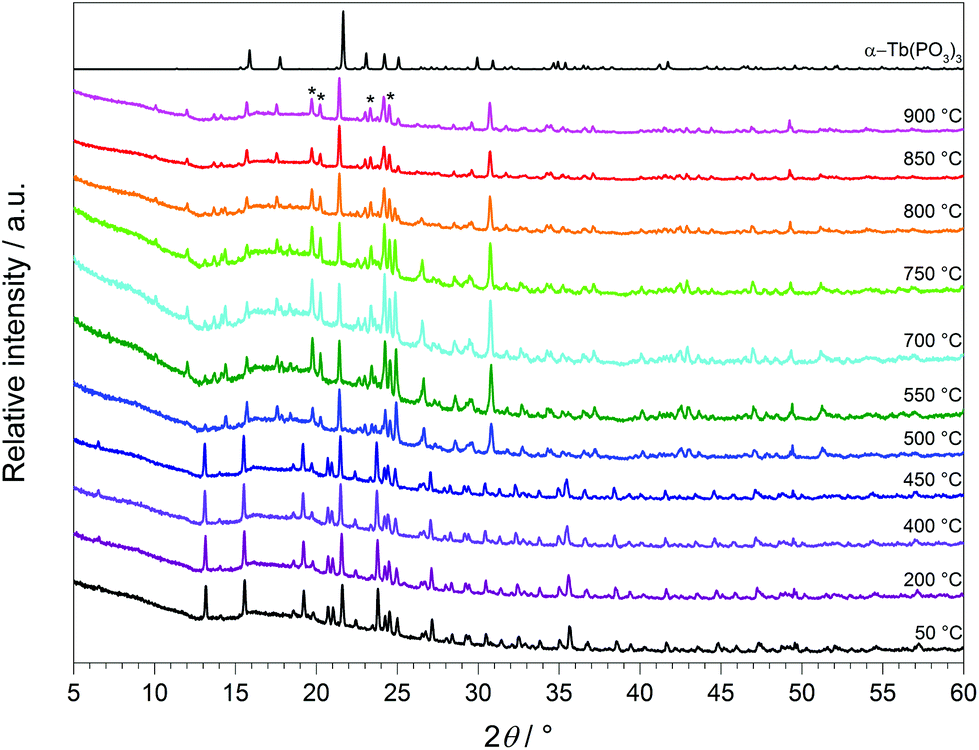
Temperature-dependent X-ray powder diffraction confirms that Tb[H(PO3)4] is stable up to 500 °C (Fig. 19). At 900 °C α-TbP3O9 (grey) represents the main phase next to a small part of TbP5O14 (marked with *).56 This leads to the conclusion that the assumption of losing half a mole of each, H2O and P2O5, in the thermal analysis is correct. Tb[H(PO3)4] exhibits infinite chains of [H(PO3)4]3− and [P3O9]3− anions. Hence it is not surprising that thermal decomposition leads to the elimination of the hydroxyl groups by evaporation of water. Astonishingly, under nitrogen flow this process does not occur below 500 °C.
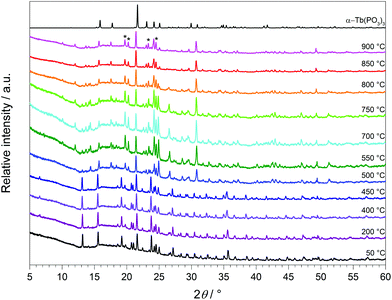 |
| | Fig. 19 Temperature-dependent X-ray powder diffraction patterns of Tb[H(PO3)4] recorded between 50 and 900 °C and the calculated powder diffraction pattern from the single-crystal data of α-TbP3O9 (grey) (λCu-Kα = 1.54178 Å). | |
The thermal decomposition of Ln[H(PO3)4] (Ln = Y, Gd–Er) was determined between 350 and 600 °C by Selevich et al.57 According to the authors all investigations were carried out in atmospheric humidity. Thus thermal analysis was carried out in air and naturally proceeds in a shorter and lower temperature range (350–600 °C) rather than under an inert atmosphere (500–1440 °C) as performed in this work. Since an increasing stability is to be expected with an increasing connectivity, the thermal stability of Tb[H(PO3)4], exhibiting infinite polyphosphate chains, is estimated to be higher than for non-condensed cyclotetraphosphates, e.g. Ba2(P4O12)·3.5H2O,27 which decomposes around 380 °C. In contrast, thermal decomposition of the ultraphosphate YP5O14, which reveals a higher degree of condensation, does not occur until 760 °C.58
9 Conclusions
In this contribution we demonstrated the crystal structures of Ln[H(PO3)4] (Ln = Tb, Dy, Ho), which were solved and refined based on single-crystal X-ray diffraction (Tb and Ho) and refined by a Rietveld refinement (Dy), respectively. Via MAPLE calculations the presence of a single proton could be proved on the one hand, and on the other hand the electrostatic consistency of the structure model of Ln[H(PO3)4] (Ln = Tb, Ho) was confirmed. All the observed bands in the IR spectrum are in agreement with our structure model. The optical properties reveal the typical UV/Vis reflection and fluorescence spectra of Tb3+, Dy3+ and Ho3+ ions exhibiting their characteristic absorption and emission bands. Doping of Dy[H(PO3)4] with increasing amounts of Ce3+ revealed only slightly more intense emission bands of Dy3+ in the blue and yellow regions. Doping of Dy[H(PO3)4] with Eu3+ leads to a partial occupancy of Dy3+ sites, which is in accordance with the relatively high intensity of the hypersensitive 5D0 → 7F2 transition of Eu3+ in a non-centrosymmetric surrounding. The magnetic measurements reveal no interaction between the Tb3+ centres and confirmed the oxidation state of Tb3+ and thus the composition of Tb[H(PO3)4]. Though Tb[H(PO3)4] only reveals a condensation in one dimension, it exhibits a remarkable thermal stability up to 500 °C before presumably H2O and P2O5 are released.
10 Experimental section
10.1 Syntheses of Ln[H(PO3)4] (Ln = Tb, Dy)
Ln[H(PO3)4] (Ln = Tb, Dy) were synthesised via solid-state reactions starting from the respective lanthanide oxides and phosphorous acid. Tb4O7 (94.70 mg, 0.14 mmol, Chempur, 99.9%) and Dy2O3 (94.00 mg, 0.25 mmol, Kristallhandel Kelpin, 99.99%), respectively, and H3PO3 (264.10 mg, 3.22 mmol, Sigma Aldrich, 99%) were ground in an agate mortar and transferred into a silica glass crucible. The reaction mixtures were covered with H2O2 (20 ml, Merck, 30%) to suppress the formation of LnP3O9 (Ln = Tb, Dy) as the side phase and heated up to 380 °C with a heating rate of 100 °C/h using a muffle furnace. The temperature was maintained for three hours before cooling down to room temperature by switching off the furnace. The products were washed with hot water and dried in air overnight. Different experiments showed that addition of H2O2 to the reaction mixtures leads to phase pure samples of colourless Tb[H(PO3)4] and Dy[H(PO3)4].
10.2 Synthesis of Ho[H(PO3)4]
Ho[H(PO3)4] was synthesised analogously to Ln[H(PO3)4] (Ln = Tb, Dy) but could be obtained as phase pure yellow/pink powder without addition of H2O2 to the reaction mixture.
10.3 X-Ray powder diffraction
The crystalline sample was finely ground, enclosed in a Hilgenberg glass capillary of 0.3 mm outer diameter and investigated at room temperature with a Bruker D8 Advance diffractometer using Cu-Kα radiation (LynxEye 1-D detector, steps of 0.2°, acquisition time 7 s per step, Soller slits 2.5°, fixed divergence slit 8 mm, transmission geometry). The obtained products were phase pure according to X-ray powder diffractometry (Fig. 7). The structure models of Tb[H(PO3)4] and Ho[H(PO3)4] were confirmed and the structure model of Dy[H(PO3)4] was refined by Rietveld analysis59,60 (Fig. 8) using the FullProf program suite and the WinPlotR graphical user interface.61 The single-crystal data of Tb[H(PO3)4] served as the starting structure model. The structure of Dy[H(PO3)4] was refined to excellent residuals of Rp = 0.009, Rwp = 0.012 and χ2 = 4.04 (Table 11). The phase purity of Dy[H(PO3)4] could thus be confirmed. In Table 11 the data of the Rietveld refinement of Dy[H(PO3)4] are summarised.
The composition of Ln[H(PO3)4] (Ln = Tb, Dy, Ho) was checked via EDX (Energy-dispersive X-ray) spectroscopy. For the doped samples Dy[H(PO3)4]:Ce and Dy[H(PO3)4]:Eu the ratios of Dy:Ce and Dy:Eu were also confirmed. Within the accuracy of the measurement limits no other elements were found.
10.4 Crystal structure determination of Ln[H(PO3)4](Ln = Tb, Ho)
Single-crystals suitable for single-crystal X-ray diffraction analysis were selected under a polarizing microscope. Diffraction data were collected with a Bruker D8 Venture diffractometer using Mo-Kα radiation (λ = 0.7093 Å) at a temperature of 293 ± 2 (Ln = Tb) and 296 ± 2 K (Ln = Ho), respectively.
The structure of the non-merohedrally twinned crystal of Tb[H(PO3)4] was solved by defining the twinned components using TWINABS (twin matrix: 0 0 1 0 −1 0 1 0 0; BASF = 0.430).62 Both crystal structures were solved by direct methods and refined by the full-matrix least-squares technique with the SHELXTL crystallographic software package.63,64 The Tb/Ho, P and O atoms could be clearly located. The hydrogen atom was added geometrically and was confirmed by MAPLE calculations.31–33 Relevant crystallographic data and further details of the structure determination are summarised in Table 5. Tables 6–9 show the atomic coordinates and displacement parameters.
Further details of the crystal structure investigations presented in this work may be obtained from the Fachinformationszentrum Karlsruhe, D-76344 Eggenstein-Leopoldshafen, Germany (Fax: +49-7247-808-666; E-mail: crysdata@fiz-karlsruhe.de, http://www.fiz-karlsruhe.de/request for depositeddata.html) on quoting the depository numbers CSD 429861 (Tb[H(PO3)4]) and CSD 429860 (Ho[H(PO3)4]).
10.5 Infrared spectroscopy
Infrared spectra were recorded at room temperature with a Bruker Equinox 55 FT-IR spectrometer using a Platinum ATR device with a scanning range from 4000 to 400 cm−1.
10.6 UV/Vis spectroscopy
The optical reflection spectra were recorded with a Varian Cary 300 Scan UV/Vis spectrophotometer in the range of 200–800 nm.
10.7 Fluorescence spectroscopy
Excitation and emission spectra were recorded at room temperature with a Fluoromax-4 spectrofluorometer with a Xe plasma lamp (Horiba Scientific, Unterhaching).
10.8 Magnetic investigations
The temperature-dependent magnetic susceptibility data were recorded with a Quantum Design MPMS-XL super-conducting quantum-interference device (SQUID) magnetometer in the field of 1000 Oe between 1.8 K < T < 400 K.
10.9 Thermal analysis
The thermal analysis was carried out with a Netzsch STA-409 PC thermal analyzer in the temperature range of 25–1450 °C in air with a heating rate of 5 K min−1.
Acknowledgements
The authors thank Dana Vieweg, Lehrstuhl für Experimentalphysik V, Universität Augsburg, for recording the magnetic susceptibility measurements and Dr Stefan Riegg, Lehrstuhl für Experimentalphysik V, Universität Augsburg, for valuable discussions about the magnetic measurement and the Rietveld refinement.
References
-
A. Holleman and E. Wiberg, Lehrbuch der Anorganischen Chemie, deGruyter, Berlin, New York, 102nd edn, 2007 Search PubMed.
-
A. Durif, Crystal Chemistry of Condensed Phosphates, Plenum Press, New York, 1995 Search PubMed.
- C. Feldmann, T. Jüstel, C. Ronda and P. Schmidt, Adv. Funct. Mater., 2003, 13, 511–516 CrossRef CAS PubMed.
- T. Jüstel, H. Nikol and C. Ronda, Angew. Chem., Int. Ed., 1998, 37, 3084–3103 CrossRef.
- T. Jüstel, C. Feldmann and C. R. Ronda, Phys. Biol., 2000, 56, 55–58 CrossRef PubMed.
- T. Jüstel, J.-C. Krupa and D. U. Wiechert, J. Lumin., 2001, 93, 179–189 CrossRef.
- C. R. Ronda, T. Jüstel and H. Nikol, J. Alloys Compd., 1998, 275–277, 669–676 CrossRef CAS.
- H. A. Höppe, K. Kazmierczak, S. Kacprzak, I. Schellenberg and R. Pöttgen, Dalton Trans., 2011, 40, 9971–9976 RSC.
- H. A. Höppe, M. Daub and M. C. Bröhmer, Chem. Mater., 2007, 19, 6358–6362 CrossRef.
- H. A. Höppe, Angew. Chem., Int. Ed., 2009, 48, 3572–3582 CrossRef PubMed.
- H. A. Höppe and J. M. U. Panzer, Eur. J. Inorg. Chem., 2009, 3127–3130 CrossRef PubMed.
- G. H. Dieke and H. M. Crosswhite, Appl. Opt., 1963, 2, 675–686 CrossRef CAS.
-
K. Ogasawara, S. Watanabe, H. Toyoshima and M. G. Brik, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 2007, vol. 37 Search PubMed.
- L. Ozawa, Chem. Rev., 2003, 103, 3835–3855 CrossRef CAS PubMed.
- K. Palkina and K. H. Jost, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2285–2290 CrossRef.
- K. Palkina, N. Kuzmina, A. Selevich and A. Lesnikovich, Russ. J. Inorg. Chem., 2003, 48, 161–164 Search PubMed.
- K. Palkina, N. Chudinova and G. Balagina, Izves. Akad. Nauk SSSR, 1982, 18, 1337–1341 Search PubMed.
- K. H. Jost, Z. Kristallogr., 1963, 16, 623–626 CAS.
- M. Rzaigui and K. Arigiub, J. Solid State Chem., 1983, 50, 240–246 CrossRef CAS.
- M. Graia, A. Driss and T. Jouini, Solid State Sci., 2003, 5, 393–402 CrossRef CAS.
- H. A. Höppe, Solid State Sci., 2005, 7, 1209–1215 CrossRef PubMed.
- H. A. Höppe, J. Solid State Chem., 2009, 182, 1786–1791 CrossRef PubMed.
- S. J. Sedlmaier and W. Schnick, Z. Anorg. Allg. Chem., 2008, 634, 1501–1505 CrossRef CAS PubMed.
- T. Balić-Žunić and E. Makovicky, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 78–81 CrossRef.
- E. Makovicky and T. Balić-Žunić, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 766–773 CrossRef.
- H. A. Höppe, M. Daub and O. Oeckler, Solid State Sci., 2009, 11, 1484–1488 CrossRef PubMed.
- H. A. Höppe, K. Kazmierczak and M. Daub, Z. Anorg. Allg. Chem., 2010, 636, 1106–1110 CrossRef PubMed.
- H. A. Höppe and M. Daub, Z. Kristallogr., 2012, 227, 535–539 Search PubMed.
- M. Daub, A. J. Lehner and H. A. Höppe, Dalton Trans., 2012, 41, 12121–12128 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- R. Hoppe, Angew. Chem., Int. Ed. Engl., 1966, 5, 95–106 CrossRef CAS PubMed.
- R. Hoppe, Angew. Chem., Int. Ed. Engl., 1970, 9, 25–34 CrossRef CAS PubMed.
-
R. Hübenthal, MAPLE, MAPLE, Program for the Calculation of the Madelung Part of Lattice Energy, University of Gießen, Germany, 1993 Search PubMed.
- A. Goto, T. Hondoh and S. Mae, J. Chem. Phys., 1990, 93, 1412–1417 CrossRef CAS PubMed.
- C. H. MacGillavry, H. C. J. de Decker and L. M. Nijland, Nature, 1949, 164, 448–449 CrossRef CAS PubMed.
- M. Weil, M. Puchberger, J. Schmedt auf der Günne and J. Weber, Chem. Mater., 2007, 19, 5067–5073 CrossRef CAS.
-
J. Weidlein, U. Müller and K. Dehnicke, Schwingungsfrequenzen I Hauptgruppenelemente, Georg Thieme Verlag Stuttgart, New York, 1981 Search PubMed.
- A. Rulmont, R. Cahay, M. Liegeois-Duyckaerts and P. Tarte, Eur. J. Solid State Inorg. Chem., 1991, 28, 207–219 CAS.
- O. Y. Miroshnichenko and V. V. Mombelli, Russ. J. Inorg. Chem. Engl. Transl., 1979, 24, 1631–1634 Search PubMed.
- W. T. Carnall, J. Phys. Chem., 1963, 67, 1206–1211 CrossRef CAS.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4424–4442 CrossRef CAS PubMed.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4447–4449 CrossRef CAS PubMed.
- H. A. Höppe, G. Kotzyba, R. Pöttgen and W. Schnick, J. Mater. Chem., 2001, 11, 3300–3306 RSC.
- K. Kazmierczak and H. A. Höppe, J. Solid State Chem., 2011, 184, 1221–1226 CrossRef CAS PubMed.
- Y.-C. Li, Y.-H. Changa, Y.-F. Lin, Y.-S. Changb and Y.-J. Lin, J. Alloys Compd., 2007, 439, 367–375 CrossRef CAS PubMed.
- Z.-J. Zhang, J.-L. Yuan, H.-H. Chen, X.-X. Yang and J.-T. Zhao, Solid State Sci., 2009, 11, 549–555 CrossRef CAS PubMed.
- D. Bimberg, D. J. Robbins, D. R. Wight and J. P. Jeser, Appl. Phys. Lett., 1975, 27, 67–68 CrossRef CAS PubMed.
- M. Guan, J. Sun, T. Shang, Q. Zhou, J. Han and A. Ji, J. Mater. Sci. Technol., 2010, 26, 45–48 CAS.
- C. Yang, Y. Pan and Q. Zhang, Mater. Sci. Eng., B, 2007, 137, 195–199 CrossRef CAS PubMed.
- Q. Luo, X. Qiao, X. Fan, H. Yang, X. Zhang, S. Cui, L. Wang and G. Wang, J. Appl. Phys., 2009, 105, 043506 CrossRef PubMed.
- H. Yu, W. Zi, S. Lan, S. Gan, H. Zou, X. Xu and G. Hong, Luminescence, 2013, 28, 679–684 CrossRef CAS PubMed.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4450–4455 CrossRef CAS PubMed.
- G. Blasse, A. Bril and W. Nieuwpoort, J. Phys. Chem. Solids, 1966, 27, 1587–1592 CrossRef CAS.
- G. Blasse and A. Bril, J. Chem. Phys., 1967, 47, 5442–5443 CrossRef CAS PubMed.
-
H. Lueken, Magnetochemie, B. G. Teubner Stuttgart, Leipzig, 1999 Search PubMed.
- A. Durif, Bull. Soc. Fr. Miner. Crystallogr., 1971, 94, 314–318 CAS.
- A. Selevich, A. Lyakhov and A. I. Lesnikovich, Phosphorus Res. Bull., 1999, 10, 171–176 CrossRef CAS.
- A. Mbarek, M. Graia, G. Chadeyron, D. Zambon, J. Bouaziz and M. Fourati, J. Solid State Chem., 2009, 182, 509–516 CrossRef CAS PubMed.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- H. M. Rietveld, Z. Kristallogr., 2010, 225, 545–547 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. Rev. B: Condens. Matter, 1993, 192, 55–69 CrossRef.
-
G. M. Sheldrick, TWINABS, Universität Göttingen, 1996 Search PubMed.
-
G. Sheldrick, SHELXTL, Version 6.14, Bruker AXS, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt02648b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a