
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-soluble Ir(III) complexes of deprotonated N-methylbipyridinium ligands: fluorine-free blue emitters†
Benjamin J.
Coe
*a,
Madeleine
Helliwell
a,
Sergio
Sánchez
a,
Martyn K.
Peers
b and
Nigel S.
Scrutton
b
aSchool of Chemistry, The University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: b.coe@manchester.ac.uk
bManchester Institute of Biotechnology, Faculty of Life Sciences, The University of Manchester, 131 Princess Street, Manchester M1 7DN, UK
First published on 6th August 2015
Abstract
New blue or blue-green emitting iridium complexes have been synthesised with cyclometalating ligands derived from the 1-methyl-3-(2′-pyridyl)pyridinium cation. Efficient luminescence is observed in MeCN or aqueous solutions, with a large range of lifetimes in the μs region and relatively high quantum yields.
The creation of bright and stable blue emitting compounds is a major challenge in the development of light-emitting electrochemical cells and organic light-emitting diodes (OLEDs).1,2 Iridium complexes have been investigated extensively due to their widely tunable and efficient luminescence.2–12 A common structural type is [IrIII(C^N)2(N^N)]+, where C^N is a cyclometalating ligand like deprotonated 2-phenylpyridine, and N^N is an α-diimine. A popular strategy to blue-shift the emission is to derivatise C^N with electron-withdrawing groups (often –F or –CF3) and/or place electron donors on N^N. Avoiding the use of fluorine is desirable to maximise the stability of the complexes in devices, and from an environmental perspective. Hence, we present an alternative approach, creating new fluorine-free IrIII luminophores by using 1-methyl-3-(2′-pyridyl)pyridinium to generate C^N. The quaternised nitrogen opposite the cyclometalating carbon is key to blue-shifting the emission.
IrIII complexes of C^N ligands derived from pyridinium species are extremely scarce.13 Notably, these known complexes are not suitable for luminescence, but were prepared in the context of catalytic studies. Complexes with quaternary N units as part of N^N have been reported, but these groups are generally not strongly coupled electronically to the IrIII centre.14–17 Remote ammonium groups have been attached to C^N18 or acetylacetonate19 ancillary ligands. Using IrIII complexes in bio-sensing/imaging20 is often restricted by poor water solubility,21 so increased positive charge is beneficial. Given this context and our general interest in photoactive complexes with quaternised pyridinium moieties,22 we targeted unusual IrIII species combining attractive emission and solubility properties.
The new complexes 1–3 were synthesised by a standard approach, i.e. cleaving a cyclometalated chloride-bridged dimer with a N^N ligand (Scheme 1). The PF6− and Cl− salts were characterised by 1H NMR spectroscopy, electrospray mass spectrometry and elemental analyses (see ESI†). In addition, single-crystal X-ray structures have been solved for 1P·2MeCN and 3P·3Me2CO (Tables S1 and S3, Fig. S1 and S2, ESI†). As expected, both complexes exhibit pseudooctahedral coordination at Ir, with the pyridyl rings of the C^N ligands in a trans geometry (Fig. 1). Their chemical structures bear some resemblance to the widely studied complexes of N-heterocyclic carbenes derived from imidazolium species, although such complexes are typically neutral or only +1 charged.23–26
 | ||
| Scheme 1 Synthesis of the complex salts 1P–3P; their chloride counterparts 1C–3C were prepared by treating purified 1P–3P with [nBu4N]Cl in acetone. | ||
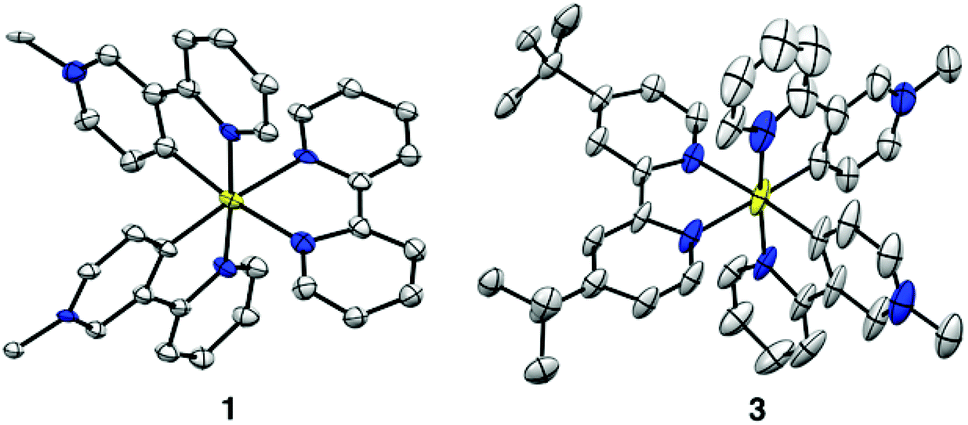 | ||
| Fig. 1 Representations of the molecular structures of the complex cations in 1P·2MeCN and 3P·3Me2CO, with the PF6− anions, solvent molecules and H atoms removed for clarity (50% probability ellipsoids). | ||
UV–vis absorption spectroscopic data are shown in Table 1. These spectra are almost unaffected by changing the counter-anions and solvent. They are dominated by intense bands at λ < 320 nm (Fig. S3 and S4, ESI†), assigned to π → π* and high energy metal-to-ligand charge-transfer (MLCT) transitions, involving both the C^N and N^N ligands. Weaker bands (λmax ≈ 350–356 nm) are observed also. Cyclic voltammograms of 1P–3P in MeCN (Fig. S5, Table S2, ESI†) show an irreversible oxidation, formally assigned to a IrIV/III couple. The reductive region includes multiple irreversible processes, and a sharp return peak is observed for 1P and 3P, indicating adsorption onto the electrode surface.
| Complex salt | Absorption, λmax/nm (ε/103 M−1 cm−1) | Emission, λ/nm |
τ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c/μs c/μs |
ϕ (%) | ||
|---|---|---|---|---|---|---|
| deox | ox | deox | ox | |||
| a In MeCN. b In water. c Estimated experimental errors ±10%. | ||||||
| 1P | 237 (54.3), 255sh (48.5), 302 (25.9), 313 (24.5), 352sh (5.3) | 444, 474max, 504, 548 | 3.5 | 1.2 | 24 | 4.7 |
| 1C | 237 (48.3), 255sh (42.4), 302 (22.8), 312 (21.8), 353sh (4.8) | 442, 470max, 504, 547 | 12.1 | 3.9 | 27 | 9.7 |
| 2P | 236 (47.4), 257 (43.2), 308 (21.6), 316 (20.9), 350sh (5.4) | 466, 494max, 525, 574 | 3.8 | 1.5 | 43 | 16 |
| 2C | 237 (52.7), 259 (52.1), 308 (25.2), 317 (24.3), 350sh (5.8) | 462, 494max, 529, 575 | 4.3 | 2.6 | 42 | 24 |
| 3P | 236 (51.8), 259sh (44.9), 299 (23.9), 311 (23.2), 356sh (4.5) | 440, 470max, 502, 546 | 3.8 | 1.2 | 43 | 11 |
| 3C | 237 (58.0), 260 (49.9), 300 (25.4), 312 (25.9), 355sh (5.5) | 440, 468max, 500, 547 | 9.5 | 2.9 | 45 | 14 |
Excitation at 315–400 nm in deoxygenated and oxygenated MeCN or aqueous solutions leads to bright blue (1 and 3) or blue-green (2) luminescence (Table 1). The spectra show significant fine structure, indicating primarily ligand-centred emission. As for the absorption spectra, changing the counter-anion and solvent has only slight effects, and the excitation profiles remain constant in all cases while monitoring at all the emission maxima. The spectra are very similar for R = H or tBu (λem = 468–474 nm), but shifted significantly to lower energy when R = CF3 (λem = 494). The fact that replacing H with tBu has little effect while –CF3 groups give a red-shift suggests that the character of the emitting state varies. The almost identical spectra of 1 and 3 (Fig. 2) indicate mainly 3LC emission involving C^N with little 3MLCT contribution. However, the red-shift for 2 suggests that the emission is associated with N^N. The quantum yields ϕ are not affected significantly by the counter-anions under deoxygenated conditions, but are substantially enhanced when R = CF3 or tBu (ϕ ≈ 42–45%) as opposed to H (ϕ ≈ 24–27%). In oxygenated conditions, 2 shows the largest ϕ values. All complexes have emission lifetimes τ in the μs region, covering a large range of values (ca. 1–12 μs), with monoexponential decay kinetics. 1C and 3C show relatively long τ values in water, considerably longer than for 2C (Fig. 3).
 | ||
| Fig. 2 Emission spectra of 1P (blue), 2P (green) and 3P (red) with excitation at 350 nm in MeCN. | ||
 | ||
| Fig. 3 Emission decay traces in water of 1C (blue), 2C (green) and 3C (red) following 375 nm excitation with a ps pulsed diode laser. | ||
The observed blue emissions from the fluorine-free complexes 1 and 3 are remarkable since, as mentioned above, decorating the C^N ligands with F or fluorinated groups is a common strategy to blue-shift the emission of this type of complex. The influence of the pyridinium fragment located para to the cyclometalating carbon is clearly shown by comparing the emission properties of 3P (λmax = 470 nm, ϕdeox = 43%, τdeox = 3.8 ms) with other reported complexes [IrIII(C^N)2{4,4′-(tBu)2bpy}]+. When using the heavily fluorinated cyclometalating ligand derived from 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine, the emission in MeCN (λmax = 470 nm, ϕdeox = 68%, τdeox = 2.3 ms) is similar to that of 3P.27 On the other hand, when C^N is deprotonated 2-phenylpyridine, the emission in MeCN is red-shifted strongly (λmax = 581 nm) with a lower ϕdeox (24%) and shorter τdeox (0.56 ms).28
The singlet ground (S0) and lowest triplet excited (T1) states of 1–3 were optimised by using density functional theory (DFT) (Fig. S6–S9, Tables S3–S8, ESI†). The calculated ground-state structures for 1 and 3 reproduce well the X-ray crystallographic ones. The LUMO is located on the C^N (69–90%) and N^N (6–27%) ligands. The HOMO is located at the Ir atom (50–55%) and the C^N ligands (40–46%), and is essentially invariant. Such relatively high C^N contributions are consistent with the irreversible oxidations observed by cyclic voltammetry (see above). The spectra simulated by time-dependent DFT agree relatively well with those measured. The weak low energy bands are due to a single transition for 1 and 3, a mixture of HOMO → LUMO and HOMO → LUMO+1 (347 nm, 1) or HOMO → LUMO (348 nm, 3). For 2, transitions occur at 342 nm (HOMO → LUMO+1) and 336 nm (HOMO−2 → LUMO), with the latter being ca. 5-fold more intense. These results indicate 1MLCT character with 1ML′CT and also 1LL′CT for 1 and 2 (L = C^N; L′ = N^N).
The T1 geometries resemble the S0 ones, except that 1 and 3 now have unequal Ir–C and (chemically equivalent) Ir–N distances, while in 2, these pairs of distances are equal. The calculated emission energies (Table 2) follow the experimental trend (1 ≈ 3 > 2). The spin densities for the T1 state (Fig. 4) show mainly 3LC involving one C^N ligand with some 3MLCT contribution for 1 and 3. In contrast, for 2, the spin density is located on N^N largely, indicating that the emission has 3L′C character with some 3ML′CT. Therefore, on excitation from the C^N/Ir-centred HOMO−2 to the C^N-centred LUMO (336 nm transition) in 2, there is efficient inter-ligand energy transfer to the emitting 3L′C excited state of N^N. Such energy transfer is expected if the 3L′C state lies below the 3MLCT.29 In 2, the presence of the electron-withdrawing –CF3 groups stabilises the π* orbitals of the N^N ligand, lowering the energy of the 3L′C state.
 | ||
| Fig. 4 M06/Def2-QZVP/SVP-calculated spin density plots for the T1 state of complexes 1–3. | ||
| Complex | E 0,0/eV (λ/nm) | E AE/eV (λ/nm) | λ max/nm (exp) |
|---|---|---|---|
| E 0,0 calculated by using the DFT-optimised geometries for T1 and S0. EAE calculated by using the DFT-optimised T1 geometry for both states (adiabatic electronic emission). | |||
| 1 | 2.91 (426) | 2.46 (504) | 474 |
| 2 | 2.76 (450) | 2.28 (544) | 494 |
| 3 | 2.91 (426) | 2.47 (503) | 470 |
To conclude, using 1-methyl-3-(2′-pyridyl)pyridinium to generate C^N affords new water-soluble IrIII complexes. Their excited-state and emissive behaviour can be switched between two types by modifying N^N. The bright blue emission in MeCN and water of the fluorine-free complexes 1 and 3 suggests potential uses in highly efficient OLEDs or bioimaging. The tunability of the emission properties is shown, not only by the emission maxima, but also in the range of quantum yields (ca. 5–45%) and lifetimes, from quite short (ca. 1 μs) to relatively long (ca. 12 μs). Much further scope exists for modifying properties, for example by using groups other than methyl on the quaternised N atom.
We thank the EPSRC for support (grant EP/J018635/1), and the BBSRC for a PhD studentship (M.K.P.). N.S.S. is supported by an EPSRC Established Career Fellowship (grant EP/J020192/1) and was funded by a Royal Society Wolfson Merit Award. We thank Dr Louise S. Natrajan and Michael B. Andrews for assistance with the luminescence measurements.
Notes and references
- X.-L. Yang, X.-B. Xu and G.-J. Zhou, J. Mater. Chem. C, 2015, 3, 913–944 RSC.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- J. A. G. Williams, A. J. Wilkinson and V. L. Whittle, Dalton Trans., 2008, 2081–2099 RSC.
- M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. De Cola, Adv. Funct. Mater., 2010, 20, 1812–1820 CrossRef CAS.
- C.-H. Yang, M. Mauro, F. Polo, S. Watanabe, I. Muenster, R. Fröhlich and L. De Cola, Chem. Mater., 2012, 24, 3684–3695 CrossRef CAS.
- P. Brulatti, R. J. Gildea, J. A. K. Howard, V. Fattori, M. Cocchi and J. A. G. Williams, Inorg. Chem., 2012, 51, 3813–3826 CrossRef CAS PubMed.
- N. Mohd Ali, V. L. MacLeod, P. Jennison, I. V. Sazanovich, C. A. Hunter, J. A. Weinstein and M. D. Ward, Dalton Trans., 2012, 41, 2408–2419 RSC.
- N. M. Shavaleev, F. Monti, R. D. Costa, R. Scopelliti, H. J. Bolink, E. Ortí, G. Accorsi, N. Armaroli, E. Baranoff, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2012, 51, 2263–2271 CrossRef CAS PubMed.
- E. Baggaley, D.-K. Cao, D. Sykes, S. W. Botchway, J. A. Weinstein and M. D. Ward, Chem. – Eur. J., 2014, 20, 8898–8903 CrossRef CAS.
- N. Darmawan, C.-H. Yang, M. Mauro, R. Fröhlich, L. De Cola, C.-H. Chang, Z.-J. Wu and C.-W. Tai, J. Mater. Chem. C, 2014, 2, 2569–2582 RSC.
- J.-B. Kim, S.-H. Han, K. Yang, S.-K. Kwon, J.-J. Kim and Y.-H. Kim, Chem. Commun., 2015, 51, 58–61 RSC.
- G. Song, Y. Zhang, Y. Su, W. Deng, K. Han and X. Li, Organometallics, 2008, 27, 6193–6201 CrossRef CAS.
- E. Zysman-Colman, J. D. Slinker, J. B. Parker, G. G. Malliaras and S. Bernhard, Chem. Mater., 2008, 20, 388–396 CrossRef CAS.
- Y. Ma, S.-J. Liu, H.-R. Yang, Y.-Q. Wu, C.-J. Yang, X.-M. Liu, Q. Zhao, H.-Z. Wu, J.-C. Liang, F.-Y. Li and W. Huang, J. Mater. Chem., 2011, 21, 18974–18982 RSC.
- F. Cucinotta, A. Guenet, C. Bizzarri, W. Mróz, C. Botta, B. Milián-Medina, J. Gierschner and L. De Cola, ChemPlusChem, 2014, 79, 45–57 CrossRef CAS.
- H. Ahmad, A. Wragg, W. Cullen, C. Wombwell, A. J. H. M. Meijer and J. A. Thomas, Chem. – Eur. J., 2014, 20, 3089–3096 CrossRef CAS PubMed.
- P. Sun, X. Lu, Q. Fan, Z. Zhang, W. Song, B. Li, L. Huang, J. Peng and W. Huang, Macromolecules, 2011, 44, 8763–8770 CrossRef CAS.
- H.-F. Shi, H.-B. Sun, H.-R. Yang, S.-J. Liu, G. Jenkins, W. Feng, F.-Y. Li, Q. Zhao, B. Liu and W. Huang, Adv. Funct. Mater., 2013, 23, 3268–3276 CrossRef CAS.
- S.-J. Liu, H. Liang, K. Y. Zhang, Q. Zhao, X.-B. Zhou, W.-J. Xu and W. Huang, Chem. Commun., 2015, 51, 7943–7946 RSC.
- S. Stimpson, D. R. Jenkinson, A. Sadler, M. Latham, A. Wragg, A. J. H. M. Meijer and J. A. Thomas, Angew. Chem., Int. Ed., 2015, 54, 3000–3003 CrossRef CAS PubMed.
- B. J. Coe, Coord. Chem. Rev., 2013, 257, 1438–1458 CrossRef CAS and references therein.
- C.-F. Chang, Y.-M. Cheng, Y. Chi, Y.-C. Chiu, C.-C. Lin, G.-H. Lee, P.-T. Chou, C.-C. Chen, C.-H. Chang and C.-C. Wu, Angew. Chem., Int. Ed., 2008, 47, 4542–4545 CrossRef CAS PubMed.
- H. Sasabe, J.-i. Takamatsu, T. Motoyama, S. Watanabe, G. Wagenblast, N. Langer, O. Molt, E. Fuchs, C. Lennartz and J. Kido, Adv. Mater., 2010, 22, 5003–5007 CrossRef CAS PubMed.
- S. B. Meier, W. Sarfert, J. M. Junquera-Hernández, M. Delgado, D. Tordera, E. Ortí, H. J. Bolink, F. Kessler, R. Scopelliti, M. Grätzel, M. K. Nazeeruddin and E. Baranoff, J. Mater. Chem. C, 2013, 1, 58–68 RSC.
- F. Monti, M. G. I. La Placa, N. Armaroli, R. Scopelliti, M. Grätzel, M. K. Nazeeruddin and F. Kessler, Inorg. Chem., 2015, 54, 3031–3042 CrossRef CAS PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal Jr., G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J.-J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS PubMed.
- Y. You and S. Y. Park, J. Am. Chem. Soc., 2005, 127, 12438–12439 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthetic, characterisation and theoretical studies. CCDC 1407686 and 1407687. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02591e |
| This journal is © The Royal Society of Chemistry 2015 |