
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the formation of chiral second sphere coordination complexes with aromatic tris amines: combined single crystal X-ray crystallography and molecular modeling analyses†‡
Hong-Cui
Yu
a,
Lei
Li
a,
Ji
Gao
a,
Jian
Tong
a,
Wenxu
Zheng
b,
Massimo
Cametti
c,
Antonino
Famulari
c,
Stefano Valdo
Meille
c,
Fang
Guo
*a and
Javier
Martí-Rujas
*d
aCollege of Chemistry, Liaoning University, Shenyang 110036, China. E-mail: fguo@lnu.edu.cn; Fax: +86 24 62202380; Tel: +86 24 62207831
bFujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, State Key Laboratory of Structural Chemistry, Fuzhou, China
cDipartimento di Chimica Materiali e Ingegneria Chimica, “Giulio Natta”, Politecnico di Milano, Via L. Mancinelli 7, 20131 Milan, Italy
dCenter for Nano Science and Technology@Polimi, Istituto Italiano di Tecnologia, Via Pascoli 70/3, 20133 Milano, Italy. E-mail: javier.rujas@iit.it; Fax: +392399 9866; Tel: +392399 9890
First published on 3rd August 2015
Abstract
A family of isostructural, chiral supramolecular networks have been obtained in the solid state by exploiting second sphere coordination interactions in the self-assembly of achiral tris amines L1 and L2 with tetrahalometallate and halide ions. Quantum-Mechanical (QM) calculations specific for solid phases provided additional insights into the intramolecular and packing interactions which determine chirality, pointing to a direct effect of the methyl groups of the central benzene ring.
Control over the formation of non-centrosymmetric chiral materials is highly desirable due to their potential applications in areas such as ferroelectricity, piezo- and pyroelectricity, and second harmonic generation.1 Unfortunately, the development of a reliable approach to induce the formation of a chiral material from achiral molecules remains a great challenge in the field of molecular chemistry.2 Furthermore, the understanding of the driving forces behind those aggregations is a prerequisite for the design and construction of chiral molecular arrays. C3-symmetrical tripodal molecules have emerged as attractive organic frameworks for the construction of chiral coordination compounds.3
Among the various intermolecular interactions which direct crystallization processes, second sphere coordination (SSC),4 namely any set of interactions established by ligands already involved in primary coordination to a metal ion, has attracted interest in various areas. For instance SSC has been applied in supramolecular chemistry,5 biological systems,6 in the selective extraction and transport of metal anions,7 proton conductivity,8 solid-state single-crystal-to-polycrystalline transformations9 and in the synthesis of dynamic host–guest adducts.10
Here, we report on the synthesis and structural analysis of chiral complexes of the formula [(L1–2·(H+)3·(MX42−)·(X−)] (M = ZnII, CdII, MnII, CoII, HgII; X = Cl, Br) made of tripodal compounds L1 and L2 which, upon protonation, in the presence of MX2 metal halide salts, afford highly symmetric (cubic, (P213)) and isostructural complexes 1–5 (L1) and 6–9 (L2) (Scheme 1). In all cases, SSC interactions are on the basis of the observed lattice organization.
 | ||
| Scheme 1 Synthesis of chiral or achiral SSC adducts using second sphere coordination. | ||
In order to offer a rationale on the emergence of chirality, a study of the second sphere coordination adduct using the derivative L3,11 which shows a different behaviour, under the same experimental conditions, is presented. Indeed, it crystallizes in the achiral (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) space group (Scheme 1). QM calculations provide insights into this unanticipated difference pointing to the effect of the methyl groups, which could otherwise be erroneously considered a minor structural difference. Finally, using a different procedure, an achiral L1 adduct in which a direct anion–water interaction disrupts the supramolecular architecture observed in the other (chiral) cases is also described.
) space group (Scheme 1). QM calculations provide insights into this unanticipated difference pointing to the effect of the methyl groups, which could otherwise be erroneously considered a minor structural difference. Finally, using a different procedure, an achiral L1 adduct in which a direct anion–water interaction disrupts the supramolecular architecture observed in the other (chiral) cases is also described.
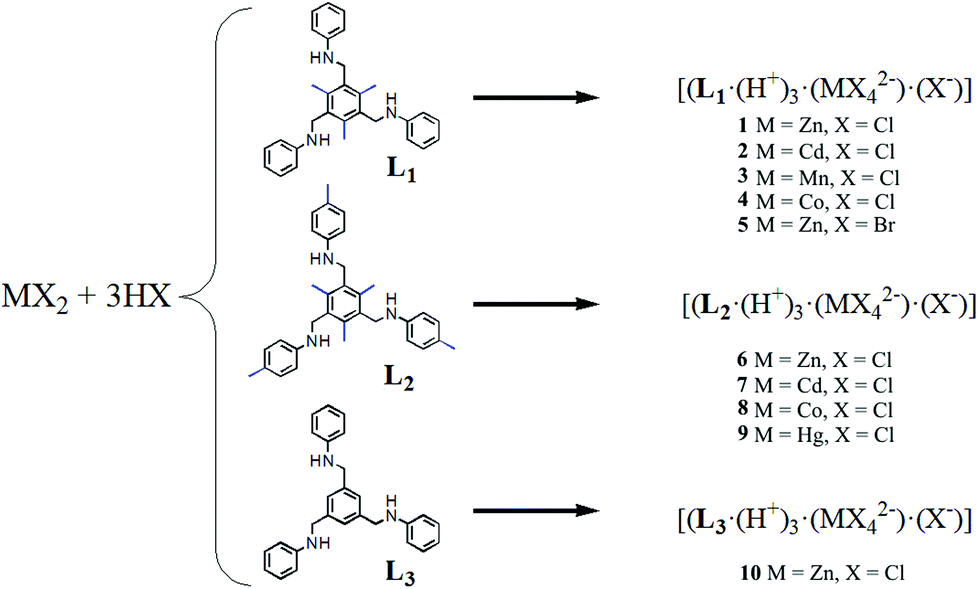
Single crystals of isostructural [(L1·(H+)3·(MX42−)·(X−)] complexes 1–5 (where M = ZnII, CdII, MnII, CoII; X = Cl, Br) were obtained by slow evaporation of a CH2Cl2/EtOH solution containing the organic tris amine, the metal salt and the acid.12L1 crystallizes in the C2 space group (Fig. S4‡) and upon protonation by HX (as in 1–5) its conformational freedom changes considerably (vide ante). Indeed, in 1–5, L1 shows a propeller-like 3-fold C3 symmetry, with only one tripodal arm present in the asymmetric unit.12 Under the experimental conditions employed, metal salts MX2 and acids HX react to form tetrahalometallates MX42−. 1–5 being isostructural, a detailed description of complex 1 (M = Zn) will suffice for all. Crystallographic analysis of 1 shows that the organic frame is fixed in a cis,cis,cis-conformation due to the interaction with the dianion [ZnCl4]2− (Fig. 1a and b). The tetrahedral [ZnCl4]2− anion is placed over the tripodal cation, matching its threefold symmetry. This allows three metal bound halides to establish significant charge assisted Zn–Cl−⋯+H–N (i) (Fig. 1a) hydrogen bond interactions with the ammonium groups (3.288 Å), all oriented in the same direction. This makes one Zn–Cl bond, which corresponds to the Zn–Cl bond that does not show any significant hydrogen bonds with the surrounding, shorter (2.249 Å) than the other three (2.271 Å).
 | ||
| Fig. 1 Single crystal structure of 1. (a) Interactions between the H3L2 trication, [ZnCl4]2− and Cl− anions; (b) view of the cis,cis,cis-conformation in 1 showing the charge assisted hydrogen bonds among trications and dianions; (c) crystal packing formed by neighbouring propellers. The inset shows the hydrogen bonds of Cl− with the NH+ groups and the distances with the centroids of the aromatic arms. Dashed black lines show the intermolecular interactions. Colour code: carbon (grey); nitrogen (blue); zinc (dark grey); chloride (green); hydrogen (white). | ||
The cis,cis,cis-conformation of L1 also allows the ammonium groups to interact with the free Cl− ions through strong N–H+⋯Cl− hydrogen bond interactions (3.095 Å) (ii) (Fig. 1a), constructing a trigonal block unit in a left-screw propeller-like trigonal fashion. The Cl− ion, surrounded by six benzene rings, where no anion–π interactions can be detected, can be seen as a templating unit (distances between the Cl− and the centroid of benzene rings are between 4.5 Å and 7 Å), Fig. 1c. The C3 symmetric feature of L1 transfers to the supramolecular complexes through the interaction with [ZnCl4]2− in the presence of chloride ions, thereby the 3-fold symmetry is retained.
It is well known that the hydrogen bonding strength involving halides as acceptors decreases along the halogen group. As a test, aimed at studying the effect of the anion in the formation of isostructural chiral materials, L1 was crystallized with ZnBr42− (i.e., using HBr for the triple protonation of L1) using the same protocol employed to synthesize chiral crystals 1–4. The results obtained from single crystal X-ray diffraction on 5 clearly showed that the crystal structure is isostructural to 1–4 and that the change in the anion interaction for this particular case is not affecting the crystal packing (Fig. S14‡). However, one interesting feature in 5 is constituted by the M–Br−⋯+H–N interactions which are weaker compared to those observed in 1–4, as evidenced by the larger distance observed in 5 (3.424 Å). Moreover, crystallization using ZnI2 did not form the chiral isostructural compounds, suggesting that the strength in such hydrogen bonding interactions is important directing the formation of the chiral crystals.
To further analyse the factors governing the generation of chirality, an investigation on the effect of small structural changes (i.e., introduction/removal of methyl substituents) at different positions in the tris-amine molecules was pursued. Compound L2 (see Scheme 1) bearing additional CH3 groups in the aromatic arms was synthesized. L2 also shows chirality with [MCl4]2− under the same experimental conditions used for the synthesis of complexes 1–5. The new second sphere complexes 6–9 crystallized in the same space group (P213), thus forming an identical structural arrangement to SSC adducts 1–5 (Fig. S16–22‡).
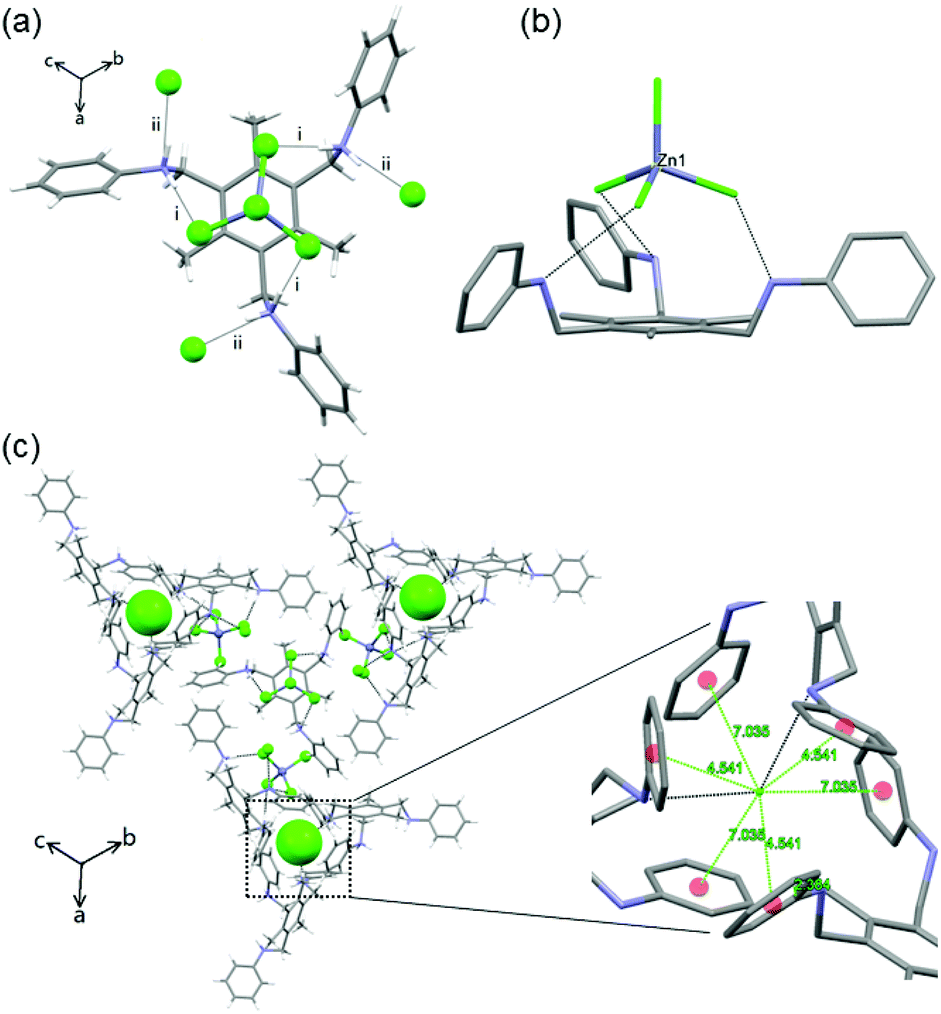
More significant is the role of the CH3 group located in the central benzene core. A tris amine derivative without the CH3 groups on the central ring (L3)11 shows that the absence of these CH3 groups as in L1 and L2 yields achiral crystals of the space group P and formula [(L3·(H+)3·(MX42−)·(X−)] (where M = Zn) (10) under the same crystallization conditions employed to form 1–5. In the structure, the C3 symmetric feature of the supramolecular interactions between L3 and tetrahalometallate anions was absent (Fig. 2). The N atoms in the three arms are hydrogen bonded with three free Cl− ions through N–H+⋯Cl− interactions (i–iii) (Fig. 2a), but the [ZnCl4]2− interacts only with two protonated N atoms (iv and v). Such difference might possibly arise due to the absence of CH3 groups in L3, because the three symmetric C–H⋯Cl weak interactions from the three CH3 groups in compound L1 contribute in a non-negligible way to the packing of the tetrahalometallate anion. As a result, the loss of helicity of L3 complexes leads to a layered structure along with the loss of chirality.
 | ||
| Fig. 2 Crystal structure of 10: (a) the hydrogen bonding between [H3L3]3+ and [ZnCl4]2−. (b) Crystal packing viewed along the a-axis. Colour code as in Fig. 1. Dashed black lines show the intermolecular interactions. | ||
Complementary insights into the field of solid-state reactions can be obtained by theoretical methods, in particular, by making use of QM calculations specific for solid phases.13,14 A similar computational approach was proved to be adequate in a number of cases, such as large supramolecular complexes,15 systems containing charged particles16 and crystalline phases of thiophene based oligomers and polymers.17 The effects of intra- and intermolecular interactions not included in standard DFT functionals have been accounted for by using the well-known Grimme corrections scheme in the framework of a DFT-D approach.18
First of all, we compared the conformational stability (gas phase calculations) of tri-protonated [H3L1]3+ and [H3L3]3+ species as found in molecular salt crystals 1 (chiral) and 10 (achiral). This implies the comparison of the energy of isolated [H3L3]3+ cations with that of [H3L1]3+ cations where the methyl groups are substituted by hydrogen atoms at optimal bond lengths (hereafter [H3L*3]3+, see Scheme 2).
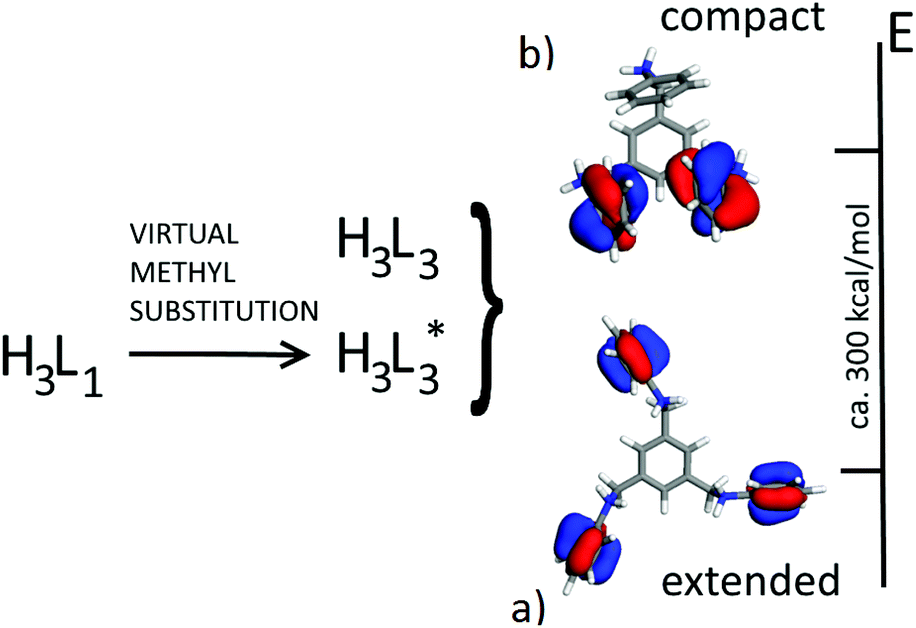 | ||
| Scheme 2 Cartoon showing the new notation after virtually substituting the methyl groups in the central ring of [H3L1]3+ for hydrogen atoms to form [H3L*3]3+. View of the HOMO orbitals in [H3L*3]3+(a) and [H3L3]3+ (b). In this case ([H3L3]3+) the asymmetry of the molecule brings the non-symmetrical HOMO, while in [H3L*3]3+ and [H3L1]3+ there is a perfect equivalence in the phenyl tails. | ||
At this stage, we employed a series of DFT and DFT-D functionals to assess a reliable energy gap between the conformation of the two species [H3L*3]3+ and [H3L3]3+. A more stable molecular conformation is expected to be the driving force for the preferential formation of a specific second sphere complex. At the PBE/DND and B3LYP/6-311G** levels, [H3L*3]3+ is more stable than [H3L3]3+ by about 300 kcal mol−1, an apparently large quantity that is, however, less than the energy pertaining to the corresponding packing processes into molecular salts. The stability order in the gas phase is easily rationalised by observing the conformations of [H3L3]3+ and [H3L*3]3+: considering that both species are tri-protonated cations and that [H3L3]3+ has a more “compact” structure (i.e., reduced molecular volume), the large positive charge in [H3L*3]3+ is more delocalized, which results in a lower conformational energy (Scheme 2).
The effect of the protonation can be also observed in the drastic changes of the HOMO (Highest Occupied Molecular Orbitals) as seen in Fig. 3 where the structures and isoenergetic HOMO in L1 and [H3L1]3+ are presented.
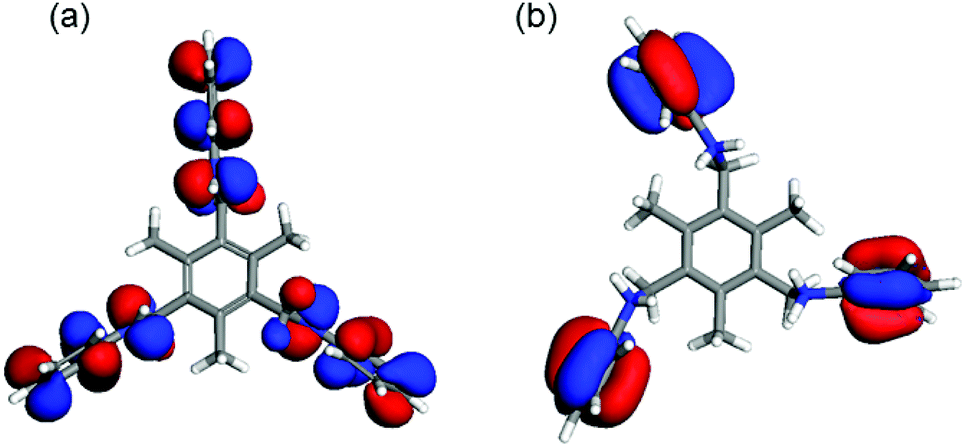 | ||
| Fig. 3 (a) Isoenergetic HOMO orbitals in L1 showing the conjugation of N unpaired electrons with phenyl terminal moieties. The involved groups have a strong tendency to be planar. These HOMOs are highly energetic (i.e., neutral molecule) and are the more reactive electrons of the system. (b) Isoenergetic HOMO orbitals in [H3L1]3+ with lower energy (i.e., compared to the neutral L1) as expected by the triple protonated system. The HOMO orbitals are strictly confined into phenyl terminal moieties. There are no N unpaired electrons. | ||
Concerning the calculations in the solid-state, here we present the data based on the analysis of SSC adduct 1 containing [ZnCl4]2− and Cl− ions. Calculations on the other crystalline systems presented similar outcomes.
In order to obtain the energies of the SSC in the crystalline state, we calculated the energy required to remove [H3L3]3+ or [H3L1]3+, in their tri-protonated form and assuming experimental frozen geometries, from the corresponding chiral and achiral crystals, respectively. Since these calculations are performed in the crystalline phase (i.e., within periodical boundary conditions), the interaction forces with the surrounding anions and cations are implicitly accounted for. DFT and DFT-D outcomes (with different schemes and functionals) showed that this energy is always larger in the case of [H3L3]3+ in the achiral structure compared to [H3L1]3+ in chiral crystals (i.e., 300 kcal mol−1). In particular, we can assess that these quantities for [H3L3]3+ and [H3L1]3+ are of the order of magnitude of 500 kcal mol−1 at the PBE/DND and PBE/DND plus Grimme (i.e., PBE/DND plus van der Waals contribution by Grimme scheme) levels. These values are larger with respect to the above mentioned differences in conformational energies. In particular, the packing efficiency is higher in the case of [H3L3]3+ into achiral crystals compared to [H3L1]3+ into chiral structures, i.e., the energy required to remove [H3L3]3+ from its crystals is higher with respect to that required to remove [H3L1]3+ by 20.5 kcal mol−1 and 5.8 kcal mol−1 calculated at PBE/DND and PBE/DND plus Grimme levels, respectively. These values increase to 24.2 kcal mol−1 and 9.6 kcal mol−1 at PBE/DNP and PBE/DNP plus Grimme levels, respectively. Such differences are even more important considering that L1 has a higher molecular mass than L3.
The different stabilities of [H3L3]3+ and [H3L1]3+ in the solid phase can be ascribed to the different molecular volumes of the two species. [H3L3]3+ adopts a conformation which is very compact when packed into the achiral crystalline structure, so that packing interactions are more efficient. In addition, the presence of methyl groups in [H3L1]3+ prevents the compact conformation observed in [H3L3]3+. So [H3L1]3+ is forced to retain the extended conformation and thus the chiral structure by the presence of methyl groups on the central ring. This extended conformation can be adopted also by the L3 trication but, in the crystalline state, the packing efficiency is higher with the more compact structure experimentally found in the [H3L3]3+ cations.
From the computational point of view, we can conclude that: (i) [H3L1]3+ cations show a good conformational stability due to charge delocalization on a more extended structure; (ii) [H3L3]3+ cations can adopt the same conformation as [H3L1]3+, or a less stable but more compact conformation; (iii) these compact [H3L3]3+ cations assemble better into achiral crystals (i.e., more effective packing interactions) so they are more stable than [H3L1]3+ in chiral crystals; (iv) due to the presence of methyl groups, [H3L1]3+ cannot form a compact conformation as [H3L3]3+ does. So [H3L1]3+ is forced to retain the C3 conformation (conformational restrains) and thus it forms the chiral structure.
Finally, a solvent-assisted mechanochemical synthesis forming an interesting hydrated form of complex 1 showing no chirality is described. Grinding L1 with CdCl2 in the presence of a few drops of concentrated HCl produced a fine powder. Crystallization of this product in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 CH2Cl2/ethanol mixture produced good quality single crystals of 11 (Scheme 3).19
:5 CH2Cl2/ethanol mixture produced good quality single crystals of 11 (Scheme 3).19
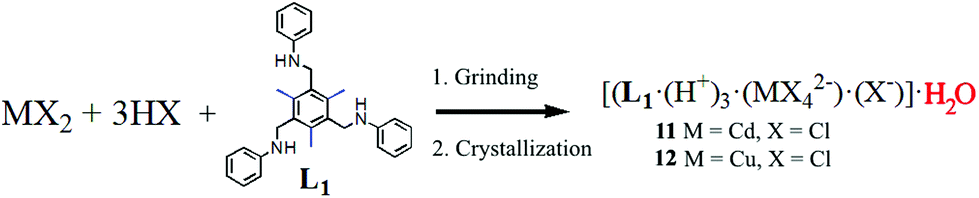 | ||
| Scheme 3 Synthesis of achiral SSC adducts 11 and 12 using second sphere coordination upon mechanochemical grinding followed by recrystallization. | ||
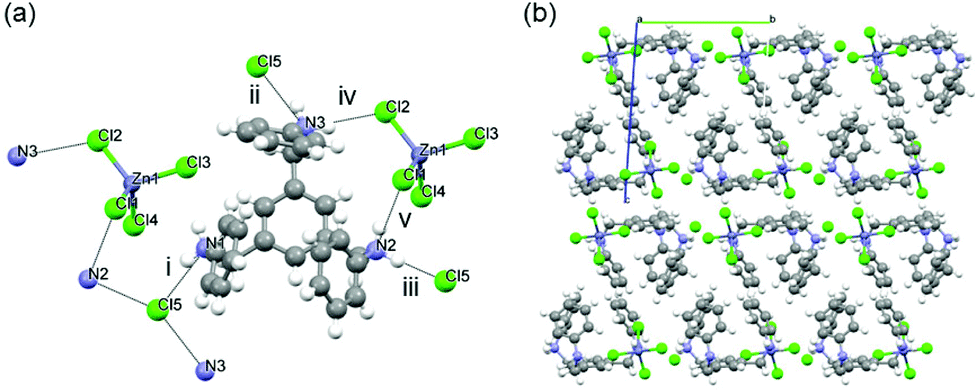
Crystallographic analysis shows that two of the three protonated N atoms are interacting with one [CdCl4]2− anion, while the third one interacts with the Cl− which is hydrogen bonded to the included water molecule (Fig. 4). The water molecule is also interacting with the protonated N in the ammonium group. The presence of a water molecule is clearly preventing the formation of the chiral adduct by such hydrogen bond interactions. We note that the conformation of the organic fragment in 11 is not compact, like in 10, but more similar to that of chiral complexes 1–5.
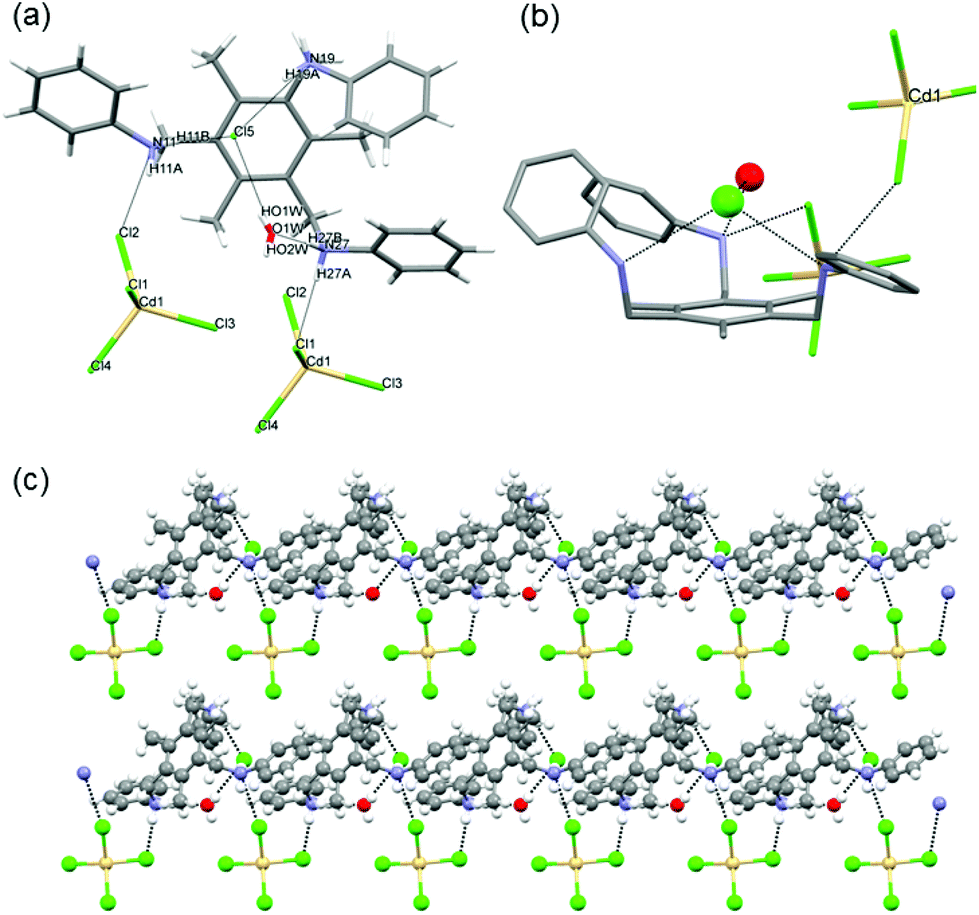 | ||
| Fig. 4 (a) View of the hydrogen bonding interactions between [H3L1]3+ [CdCl4]2−, Cl− anions and H2O. (b) Side view showing the tripodal tris amine with the arms in a similar conformation to that of adducts 1–5 as a result of the hydrogen bond interactions. (c) View of the layered structure of 11. Dashed black lines show the intermolecular interactions. Colour code: carbon (grey); nitrogen (blue); zinc (dark grey); chloride (green); oxygen (red); hydrogen (white). | ||
In conclusion, a novel synthetic strategy toward 3D chiral coordination frameworks with helical chains driven by SSC interactions is reported. Such design relies on the interaction between tetrahalometallates [MCl4]2−/[MBr4]2− (M = ZnII, CdII, MnII, CoII, HgII), a C3-symmetrical tripodal tris amine (L1/L2), in its fully protonated state, and chloride or bromine ions, which can be seen as templating species. The [H3L1]3+ cations show a helical arrangement of the three aromatic arms, resulting in conformational chirality. Such a C3 symmetric conformation induces a tripodal second sphere chiral organization of the counter anions.
While enantiomers can in principle pair, giving rise to a racemic packing and indeed often form centrosymmetric crystals, in the adducts formed with L1 and L2 this does not happen. We can speculate that in this instance, the chiral organization of charged species establishes a chiral environment propagating substantially beyond the immediate periphery of cations due to the long range nature of coulombic interactions. Hence the growth of individual non-centrosymmetric crystals adopting the P213 space group, in which only one of the enantiomers packs, is favoured. Clearly, none of the above applies to the non-chiral conformations of [H3L3]3+ cations, where the absence of the central methyl groups allows for a more compact conformation which better assembles in a achiral form, as shown by DFT calculations specific for solid phases. Interestingly, L1 under liquid assisted grinding conditions produces achiral supramolecular complexes, where direct water–halide interactions disrupt the lattice architecture observed in the chiral materials.
This research was supported by NSFC (no. 20903052), the Program for Liaoning Excellent Talents in University (LJQ 2011003) and the support program of Liaohe Scholar of Liaoning University. M. C. acknowledges Progetto per Giovani Ricercatori “Rita Levi Montalcini” 2009. A. F. acknowledges CINECA for the computational resources (IsC26_MODINT) and MIUR (PRIN 2010XLLNM3_007) for financial support.
Notes and references
- (a) P. A. Maggard, C. L. Stern and K. R. Poeppelmeier, J. Am. Chem. Soc., 2001, 123, 7742–7743 CrossRef CAS; (b) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115(15), 7304–7397 CrossRef CAS PubMed.
- P. S. Halasyamani and K. R. Poeppelmeyer, Chem. Mater., 1998, 10, 2753–2769 CrossRef CAS.
- Z. Dai and J. W. Canary, New J. Chem., 2007, 31, 1708–1718 RSC.
- D. A. Beauchamp and S. J. Loeb, Chem. – Eur. J., 2002, 8, 5084–5088 CrossRef CAS.
- (a) D. Mercer and S. J. Loeb, Chem. Soc. Rev., 2010, 39, 3612–3620 RSC; (b) S. A. Dalrymple and G. K. H. Shimizu, Supramol. Chem., 2003, 15, 591–606 CrossRef CAS PubMed; (c) S. A. Dalrymple, M. Parvez and G. K. H. Shimizu, Inorg. Chem., 2002, 41, 6986–6996 CrossRef CAS PubMed.
- (a) D. R. Williams, The Metals of Life: the Solution Chemistry of Metal Ions in Biological Systems, Van Nostrand Reinhold, London, 1971 Search PubMed; (b) J. Shaw and G. W. Everett, Inorg. Chem., 1985, 24, 1917–1920 CrossRef CAS; (c) S. E. Sherman and S. J. Lippard, Chem. Rev., 1987, 87, 1153–1181 CrossRef CAS; (d) M. Botta, Eur. J. Inorg. Chem., 2000, 3, 399–407 CrossRef.
- R. J. Warr, A. N. Westra, K. J. Bell, J. Chartres, R. Ellis, C. Tong, T. G. Simmance, A. Gadzhieva, A. J. Blake, P. A. Tasker and M. Schröder, Chem. – Eur. J., 2009, 15, 4836–4850 CrossRef CAS PubMed.
- L. Li, F. Maddalena, M. Oliveros, M. Caironi, F. Guo and J. Martí-Rujas, CrystEngComm, 2014, 16, 10888–10892 RSC.
- J. Martí-Rujas and M. Cametti, New J. Chem., 2014, 38, 1385–1388 RSC.
- F. Guo, X. Wang, H.-Y. Guan, H.-B. Yu, L. Li, S.-S. Chen, A. Famulari and J. Martí-Rujas, Cryst. Growth Des., 2015, 15, 2842–2852 CAS.
- D. T. Glatzhofer and M. A. Khan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 2128–2133 CrossRef.
- See the ESI‡ for further details.
- Density functional theory (DFT) approaches have been employed herein. The PBE (Perdew–Burke–Ernzerhof) exchange–correlation functions have been used for both gas and solid phases (i.e., under periodical conditions): (a) J. P. Perdew, K. Burke and M. E. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS; (b) J. P. Perdew, K. Burke and M. E. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS. For the sake of consistency, all the calculations were accomplished by using the DMol3 software: B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS PubMed.
- A combination of numerical double-ζ quality basis set, either including polarization functions on all atoms (DNP) and not (DND), and an effective core potential for the metal atoms were adopted. In all the calculations, we used the experimental X-ray determined unit cell and geometries for heavy atoms while X–H bond lengths were optimised.
- (a) Y. Kolokoltsev, O. Amelines-Sarria, T. Y. Gromovoy and V. A. Basiuk, J. Comput. Theor. Nanosci., 2010, 7, 1095–1103 CrossRef CAS PubMed; (b) O. Amelines-Sarria, Y. Kolokoltsev and V. A. Basiuk, J. Comput. Theor. Nanosci., 2010, 7, 1996–2003 CrossRef CAS PubMed; (c) V. A. Basiuk, O. Amelines-Sarria and Y. Kolokoltsev, J. Comput. Theor. Nanosci., 2010, 7, 2322–2330 CrossRef CAS PubMed; (d) V. A. Basiuk, Int. J. Quantum Chem., 2011, 15, 4197–4205 CrossRef PubMed.
- (a) G. Yu, S. Yin, Y. Liu, Z. Shuai and D. Zhu, J. Am. Chem. Soc., 2003, 125, 14816–14824 CrossRef CAS PubMed; (b) F. Guo, M.-Q. Zhang, A. Famulari and J. Martí-Rujas, CrystEngComm, 2013, 15, 6237–6243 RSC; (c) E. Maccaroni, L. Malpezzi, A. Famulari and N. Masciocchi, J. Pharm. Biomed. Anal., 2012, 60, 65–70 CrossRef CAS PubMed; (d) H. Y. Guan, Z. Wang, A. Famulari, X. Wang, F. Guo and J. Martí-Rujas, Inorg. Chem., 2014, 53, 7438–7445 CrossRef CAS PubMed; (e) F. Guo, H. D. Shao, Q. Yang, A. Famulari and J. Martí-Rujas, CrystEngComm, 2014, 16, 969–973 RSC; (f) H.-L. Liu, Y.-F. Xie, Z. G. Pan, A. Famulari, F. Guo, Z. Zhou and J. Martí-Rujas, Cryst. Growth Des., 2014, 14, 6528–6536 CrossRef CAS.
- (a) A. Famulari, G. Raos, A. Baggioli, M. Casalegno, R. Po and S. V. Meille, J. Phys. Chem. B, 2012, 116, 14504–14509 CrossRef CAS PubMed; (b) M. Casalegno, A. Baggioli, A. Famulari, S. V. Meille, T. Nicolini, R. Po and G. Raos, 02002-1 02002-8, Materials for organic photovoltaics: insights from detailed structural models and molecular simulations, EPJ WEB OF CONFERENCES, ISSN 2100-014X, 2012, DOI: DOI:10.1051/epjconf/20123302002; (c) T. Nicolini, A. Famulari, T. Gatti, J. Martí-Rujas, F. Villafiorita Monteleone, E. V. Canesi, C. Botta, E. Parisini, S. V. Meille and C. Bertarelli, J. Phys. Chem. Lett., 2014, 5, 2171–2176 CrossRef CAS.
- (a) S. Grimme, J. Chem. Phys., 2006, 124, 34108–34134 CrossRef PubMed; (b) A. Baggioli, S. V. Meille, G. Raos, R. Po, M. Brinkmann and A. Famulari, Int. J. Quantum Chem., 2013, 113, 2154–2162 CrossRef CAS PubMed; (c) A. Baggioli and A. Famulari, Phys. Chem. Chem. Phys., 2014, 16, 3983–3994 RSC.
- An isostructural SSC complex is also obtained by grinding L1 with CuCl2·2H2O. See the ESI‡ for further information.
Footnotes |
| † Single crystal X-ray diffraction. Single crystal data collection of 1–12 was performed with Mo-Kα radiation (λ = 0.71073 Å) on a Bruker SMART Apex CCD diffractometer. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, crystal structure description, and additional figures. CCDC 1406455–1406466 (1–12). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02387d |
| This journal is © The Royal Society of Chemistry 2015 |