
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkynyl-functionalized gold NHC complexes and their coinage metal clusters†‡
Claude Kiefera,
Sebastian Bestgena,
Michael T. Gamera,
Sergei Lebedkinbc,
Manfred M. Kappes*bc and
Peter W. Roesky*a
aInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstraße 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu; Fax: (+)49 721 6084 4854
bInstitute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber Weg 2, 76131 Karlsruhe, Germany
cInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 29th June 2015
Abstract
Phenylpropynyl-functionalized imidazolium salts, as well as their gold complexes, were prepared in excellent yields affording suitable starting materials for metal cluster synthesis. The reactions of these gold complexes with coinage metal phenylacetylides [M(CCPh)]x (M = Cu, Ag) resulted in the formation of novel heterometallic hexanuclear clusters which exhibit mixed metallophillic interactions and intense white photoluminescence at low temperature.
Introduction
N-Heterocyclic carbenes (NHCs) have gained increasing and remarkable relevance in transition metal and main group chemistry since their discovery by Wanzlick1,2 and Öfele3 in the 1960s and the isolation of a free NHC by Arduengo4 in 1991. Nowadays, NHCs are widely used in organometallic synthesis to establish novel complexes, which can be investigated in terms of their catalytic5,6 or photophysical7,8 properties. One particular advantage of NHCs is their broad variety as many different substituents can be attached to the nitrogen atoms.9–12 This allows for modification of the ligand with respect to its steric and electronic properties, as well as its denticity, and thus affects the range of applications. At the same time, research activity into the chemistry of gold has experienced a steep rise over the last decades, which has led to an increasing number of gold NHC complexes being investigated as catalysts13,14 for cyclization reactions, hydroamination or for their photophysical15,16 properties. Furthermore, gold complexes feature an unique phenomenon that is commonly known as aurophilicity.17–19 This attractive d10–d10-dispersion interaction, in which the distance between two gold(I) ions is well below the sum of their van der Waals radii, is a special case of metal–metal bonds (metallophilicity), which is also observed for other metals like silver and mercury.20–22 Therefore, multi- and heterometallic gold complexes are being thoroughly investigated for the influence of metallophilic interactions on luminescence,23 and cooperative effects in certain catalytic steps.24–26 Given our7 and others’27,28 interest in polynuclear coinage metal complexes, we intended to extend our studies from phosphine-functionalized NHCs to alkynyl-functionalized carbene complexes aiming for the synthesis of heteronuclear clusters. A combination of gold–metal bonds, enabled by the relativistic effects of gold, and alkynyl groups attached to a carbene complex, should facilitate the synthesis of oligomeric or supramolecular coinage metal assemblies.29 Although terminal alkynes can act as bidentate ligands through end-on and side-on coordination, we initially focused on phenyl-protected alkyne substituents, which should allow for side-on coordination of different metals through π-bonding.Results and discussion
The reaction of 1H-mesityl-1H-imidazole with propargyl chloride/bromide in acetonitrile afforded the phenylacetylene-substituted imidazolium salts 1 and 2 in almost quantitative yields (Scheme 1, Fig. S1‡). | ||
| Scheme 1 Synthesis of phenylacetylene-functionalized imidazolium salts. | ||
Subsequent transformation of 1 and 2 to the corresponding gold complexes was achieved in two different ways, namely by applying the well-known silver oxide30 route and the mild-base route as described by Nolan et al.31 and Gimeno et al.32 The silver carbene complex was obtained by stirring of the imidazolium salts with Ag2O for four days. By reaction of this solution with one equiv. [AuCl(tht)], transmetallation took place and afforded the gold complex [(NHC(1))(AuCl)] (3) in good yields (Scheme 2). As this reaction was rather time-consuming and only allowed for the synthesis of the gold chloride derivative, the mild-base route was used as an improved synthetic procedure for the desired complexes. Since the anion of the deployed imidazolium salt finally occurs in the product, the [(NHC(1))(AuBr)] complex (4) could also be obtained from 2 in excellent yields. In the 1H NMR spectra, the absence of any resonances above 8 ppm clearly indicates the successful deprotonation of the imidazolium salt and thus the generation of carbene complexes. This is supported further by the presence of singlets at 172.5 (3) and 175.7 (4) ppm in the 13C{1H} NMR spectra, which are in the typical range for NHC gold compounds.31
 | ||
| Scheme 2 Synthesis of the gold complexes 3, 4 and 5. | ||
The proposed structures were further confirmed by FAB-MS and elemental analysis. Single crystal X-ray crystallography confirmed the structures. For 3, gold–carbene and gold–chloride bond lengths of 1.995(9) Å and 2.317(2) Å are observed, which is in the expected range for such complexes (Fig. 1).33 Furthermore, the gold atom is nearly linearly coordinated with a C1–Au1–Cl1 angle of 175.3(3)°. In compound 4 (Fig. 2), the bond distances between the gold and the carbon atom (1.983(4) Å) are similar to 3. With a C1–Au1–Br1 angle of 175.33(11)°, almost linear coordination geometry is also achieved in 4. Both compounds feature a weak intermolecular aurophilic interaction in the solid state, which is known as unsupported aurophilicity.19 For 3 and 4, the Au1–Au2 distances are very similar at 3.5387(1) Å and 3.5636(1) Å respectively, which is in the upper range for aurophilic interactions and hence implies a weak bonding situation.
 | ||
| Fig. 1 Solid-state structure of 3 showing two molecules weakly linked by aurophilic interactions. | ||
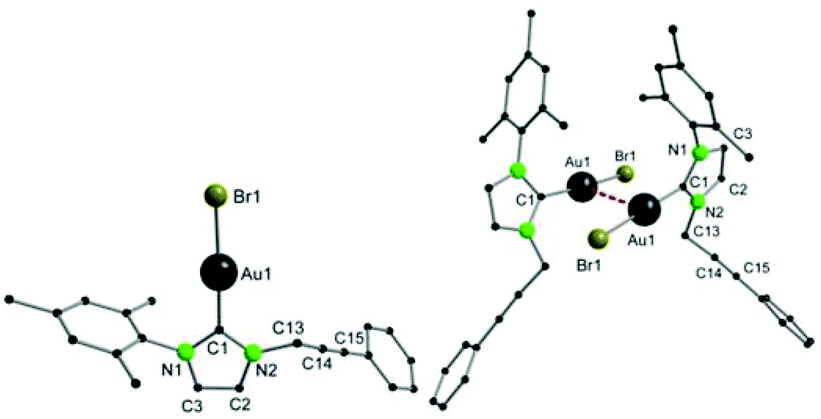 | ||
| Fig. 2 Solid-state structure of 4 (left), and an extended view illustrating both molecules weakly linked by Au–Au contacts (d(Au1–Au1′) = 3.5636(1) Å) (right). | ||
In order to extend our studies on the reactivity of the ligand, two more complexes were synthesized. First, the same reaction as for 3 and 4 was conducted, but with only half an equiv. of [AuCl(tht)] leading to the charged bis-NHC-complex [(NHC(1))2Au][Br] 5 in good yield (Scheme 2). This complex was fully characterized by standard analytic and spectroscopic methods. Again, the absence of the resonance for the imidazolium proton NCHN indicates the formation of a carbene complex, which is in accordance with the 13C{1H} spectrum exhibiting a signal at 185.1 ppm that can be attributed to a carbene carbon atom coordinated to a gold ion. ESI-MS was conducted to confirm the conformation of the complex showing the molecular ion peak at m/z = 797.29. Secondly, the gold NHC complex 3 was ground together with phenylacetylene and excess KOH according to a procedure developed by Nolan et al.34 to accomplish ligand exchange and to introduce a second alkyne group onto the metal complex by postfunctionalization. After extraction of the mixture with dichloromethane, [(NHC(1))(AuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)] (6) was obtained in very good yield as a pure solid (Scheme 3). In the 13C{1H} NMR spectrum, the resonance for the carbene atom is observed at 188.6 ppm, which is shifted far downfield compared to the halogenide complexes 3 (172.5 ppm, +14.1 ppm), 4 (175.8 ppm, +12.9 ppm) and to the bis-NHC complex 5 (185.1 ppm, +3.5 ppm). Consequently, the nature of different complexes in solution can be simply observed by 13C NMR spectroscopy, which proved to be important for the reactions described below. The gold carbene distances are slightly increased to 2.014(3) Å compared to 3, whereas the bond length to C22 amounts to 1.985(4) Å. The C1–Au1–C22 bond angle of 175.63(15)° only differs marginally from the ideal 180° (Fig. 3).
CPh)] (6) was obtained in very good yield as a pure solid (Scheme 3). In the 13C{1H} NMR spectrum, the resonance for the carbene atom is observed at 188.6 ppm, which is shifted far downfield compared to the halogenide complexes 3 (172.5 ppm, +14.1 ppm), 4 (175.8 ppm, +12.9 ppm) and to the bis-NHC complex 5 (185.1 ppm, +3.5 ppm). Consequently, the nature of different complexes in solution can be simply observed by 13C NMR spectroscopy, which proved to be important for the reactions described below. The gold carbene distances are slightly increased to 2.014(3) Å compared to 3, whereas the bond length to C22 amounts to 1.985(4) Å. The C1–Au1–C22 bond angle of 175.63(15)° only differs marginally from the ideal 180° (Fig. 3).
 | ||
| Scheme 3 Synthesis of complex 6. | ||
 | ||
| Fig. 3 Solid-state structure of 6. | ||
As it is well-known that copper, silver, and gold ions preferentially coordinate to alkynes via end-on σ- or side-on π-bonding,27,29,35–40 we intended to synthesize heteronuclear clusters by the reaction of carbene complexes 3 and 4 with coinage metal phenylacetylenes. Thus, by stirring these compounds with copper or silver phenylacetylide compounds [M(CCPh)]x (M = Cu, Ag) for three days and subsequent fractional crystallization of the crude reaction mixture, we could isolate two isostructural heterohexanuclear Au4M2 clusters 7 and 8 with mixed metallophilic interactions (Scheme 4).
 | ||
| Scheme 4 Synthesis of the heteronuclear clusters 7 and 8. | ||
In both cases, the reaction was monitored by 13C{1H} NMR spectroscopy and ESI-MS. For the reaction with [Cu(CCPh)]x, the spectrum of the crude product exhibited three resonances between 150 and 200 ppm indicating the formation of three different carbene species. Based on our previous work, the peaks at 172.5 ppm, 185.0 ppm and 188.3 ppm could be most certainly assigned to the starting material 3, the bis-NHC complex 5 and the doubly alkyne-functionalized product 6. Furthermore, ESI-MS confirmed the presence of species 5 (m/z = 797.31) and a second species that was identified as adduct of 5 and 6 at m/z = 1395.43. Nevertheless, by crystallization of the yellow solid from DCM/pentane, yellow prisms appeared which were identified by X-ray crystallography as the desired heteronuclear complex [{((NHC(1))Au(CCPh))2}{((PhCC)2Au)2Cu2}] 7 (Fig. 4).
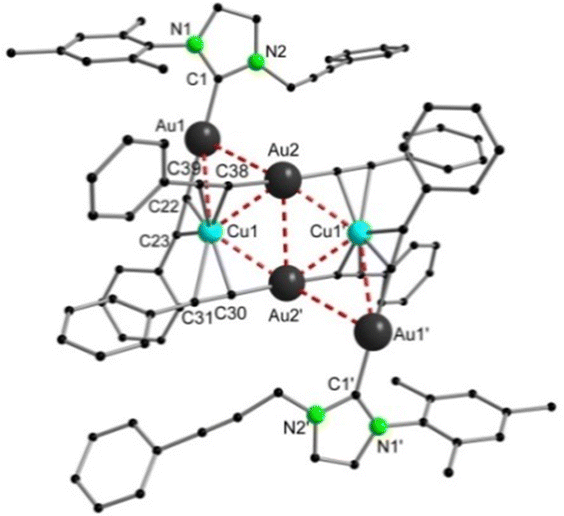 | ||
| Fig. 4 Solid-state structure of the heterohexanuclear complex 7. | ||
In the solid-state structure of compound 7 an inversion centre is located in the middle of the rhomboid spanned by Cu1–Au2–Cu1′–Au2′, whose angles were found to be Cu1–Au2–Cu1′ 109.21(2)° and Au2–Cu1–Au2′ 70.79(2)°. Hence, the Au2Cu2-unit is in a perfectly planar arrangement as the interior angles sum up to 360°. As can be seen in the structure, transfer of the alkynyl groups from copper to the gold atoms has taken place since every gold atom is attached end-on to the phenylacetylides with Au–C bond lengths of 2.004(5) Å (Au2–C30) and 2.028(5) Å (Au2–C38). On the other hand, the copper ions are now coordinated side-on by three alkynyl groups in an almost trigonal fashion, whereas the Cu–C distances vary between 2.071(5) Å and 2.815(2) Å. Consequently, every phenylacetylide unit acts as a bidentate bridging ligand between copper and gold and therefore, metal–metal interactions can be classified as semisupported interactions.19 The structure can also be described as two molecules of 6 which enclose a tetranuclear [{(PhCC)2Au}2Cu2] cluster. This is in good accordance with the resonance at 188.3 ppm in the 13C{1H} NMR spectrum. To the best of our knowledge, this is the first heteronuclear alkynyl cluster, which is surrounded by NHC complexes. The intermetallic distances between Au1–Au2 (3.4384(5) Å) and Au2–Au2′ (3.3118(7) Å) clearly indicate metallophilic interactions, whose strength can be estimated to around 7.5–11.8 kJ mol−1.41 Also, mixed metal–metal contacts are present as shown by the moderately short Au2–Cu1 distances of 2.7920(9) Å and 2.9230(8) Å, which are much shorter than the sum of their van der Waals radii (3.06 Å) and in a similar region of comparable Au–Cu–Alkyne complexes.20,42 Surprisingly, no coordination of any metal to the NHC–alkynyl side chains could be observed.
For the reaction with the silver derivative [Ag(CCPh)]x, after three days only a single resonance in the carbene region in the 13C{1H} NMR spectrum was observed at 188.7 ppm, which can again be ascribed to a complex similar to 6. By fractional crystallization, two products were identified by X-ray crystallography, namely 6 and the analogous heterometallic cluster [{((NHC(1))Au(CCPh))2}{((PhCC)2Au)2Cu2}] 8 (Scheme 4 and Fig. 5). Although numerous attempts have been made to separate these complexes from each other, complex 8 was always contaminated with a minor amount of 6. The Au4Ag2 cluster is isostructural with the copper analogue. As the covalent and van der Waals radii of silver(I) (2 × radius: 2.90(5) Å, 3.44 Å) are considerably longer than for copper(I) (2 × radius: 2.64(4) Å, 2.80 Å), the stretched parallelogram is markedly widened as evidenced by the Au2–Au2′ distance of 3.8900(2) Å. The internal angles Ag1–Au2–Ag1′ 98.06(2)° and Au2–Ag1–Au2′ 81.94(2)° add up to 360° demonstrating again a perfectly planar arrangement of the metals. Thus, significant aurophilic interactions cannot be observed in 8 as the Au1–Au2 distance (3.8365(2) Å) is beyond an energetically relevant magnitude. However, the gold-silver bond lengths (Ag1–Au2′ 2.9814(9) Å, Ag1–Au2 2.9513(10) Å, Ag1–Au1 3.1805(9) Å) are well below the sum of their van der Waals radii,20,29 which suggests an attractive affinity between both metals and a certain degree of metallophilicity-driven structural architecture.43
 | ||
| Fig. 5 Solid-state structure of the hexanuclear Au4Ag2-cluster 8. | ||
The photoluminescent (PL) properties of compounds 6 and 7 were investigated in order to ascertain the effect of metallophilic interactions in 7. Both compounds show very weak PL at ambient temperature, but emit bright blue and white light, respectively, at cryogenic temperatures below ∼100 K. In this regime, the PL of 6 and 7 is rather complicated as illustrated by the multi-band PL excitation (PLE) and emission spectra (Fig. 6 and 7). The emission of 6 is dominated by a vibronically structured band at ∼450 nm, which is effectively excited below ∼300 nm. The broad red emission at ∼700 nm is very weak by comparison. Their relative intensities are well illustrated by the PL excitation-emission map shown in Fig. S2.‡ In contrast, 7 manifests two major PL bands at ∼490 and ∼700 nm, which sum up to produce white luminescence. The PL of both compounds decays on a time scale of a few to tens of microseconds, depending on the emission wavelength (Fig. S3‡).
 | ||
| Fig. 6 PL excitation (PLE) and emission spectra of solid complex 6 at different emission and excitation wavelengths, respectively, and T = 20 K. The spectra are vertically shifted for clarity. | ||
 | ||
| Fig. 7 PL excitation (PLE) and emission spectra of solid complex 7 at 20 K. The emission spectra are vertically shifted for clarity. | ||
The red phosphorescence in 7 – nearly absent in the structurally related complex 6 – may be attributed to low-energy excited states involving gold and copper atoms (of a charge-transfer or metal-localized character), i.e. to states associated with (intermolecular) metallophilic interactions.21 Such interactions are absent in 6. Our inference that these interactions give rise to the red emission of 7 is supported by the following observation. This emission was found to increase in relative intensity by a factor of ∼10 after several months storage of 7 under ambient conditions in the dark (cf. a PL map in Fig. S4‡). By contrast, no change of the PL properties was observed for co-stored complex 6. Some loss of co-crystallized dichloromethane molecules (see Crystallographic Table S1‡) is expected after such a long-term storage. This may lead to slight changes of the Au–Cu and Cu–alkynyl distances in a correspondingly ‘dried’ sample of 7 (while the same orange crystallite appearance is otherwise retained), accompanied by changes in the metallophilic interactions. In 7, this apparently results in more efficient energy transfer into/radiative relaxation from the low-energy excited states. Note that the above effect of solvent molecules is expected to be especially pronounced for intermolecular metallophilic interactions – as in complex 7. We also remark close analogy to the gold complexes demonstrating aggregation-induced bright emission in solid state (also attributed to intermolecular metallophilic interactions) – in contrast to their weak emission in a dilute solution.37,44
Conclusions
We prepared two alkynyl-functionalized imidazolium salts in excellent yields, which could be successfully converted to their corresponding homo- and heteroleptic gold complexes via a mild base route in very good yields. Post-functionalization of the complexes was achieved by the reaction with KOH and phenylacetylene to introduce a second alkyne group onto the gold atom. All compounds have been fully characterized by analytic, spectroscopic and X-ray techniques. By reaction with the copper and silver phenylacetylides, heterometallic hexanuclear coinage metal cluster were synthesized in which tetranuclear [((PhCC)2Au)2M2] (M = Cu, Ag) units are embedded by NHC gold complexes for the first time. Closely related compounds 6 and 7 show, respectively, bright blue and white phosphorescence in the solid state at low temperatures. This spectral difference is due to a significant red emission contribution in 7. The origin of that emission can be attributed to intermolecular metallophilic interactions in 7 (absent in 6).Experimental
General procedures
All manipulations were performed under rigorous exclusion of moisture and oxygen in flame-dried Schlenk-type glassware or in an argon-filled MBraun glovebox. Furthermore, for the most part the silver and gold compounds were handled with the exclusion of light by wrapping the compound-containing flasks in aluminium foil. Prior to use, CH2Cl2 and MeCN were distilled under nitrogen from CaH2. Hydrocarbon solvents (THF, diethyl ether, n-pentane) were dried using an MBraun solvent purification system (SPS-800). Deuterated solvents were obtained from Carl Roth GmbH (99.5 atom% D). NMR spectra were recorded on a BrukerAvance II 300 MHz or Avance 400 MHz. Chemical shifts are referenced to the residual 1H and 13C resonances of the deuterated solvents and are reported relative to tetramethylsilane. IR spectra were obtained on a Bruker Tensor 37. EI-Mass spectra were recorded at 70 eV on a Thermo Scientific DFS. ESI mass spectra were obtained using a FT-ICR (Fourier transform ion cyclotron resonance) IonSpecUltima mass spectrometer equipped with a 7 T magnet (Cryomagnetics). FAB-Mass spectra were recorded with a Finnigan MAT90 spectrometer. Elemental analyses were carried out with an ElementarVario EL or Micro Cube. [AuCl(tht)],45 mesitylimidazole,46 phenylpropargyl bromide,47 [Cu(CCPh)]x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48 and [Ag(CCPh)]x49 were prepared according to literature procedures. Ag2O was purchased from Alfa-Aesar (99.99%) and used as received.
48 and [Ag(CCPh)]x49 were prepared according to literature procedures. Ag2O was purchased from Alfa-Aesar (99.99%) and used as received.
The PL measurements were performed with a Horiba JobinYvon Fluorolog-322 spectrometer equipped with a closed-cycle optical cryostat (Leybold) operating at ∼20–300 K. A Hamamatsu R9910 photomultiplier was used as the detector for the emission spectral range of ∼300–850 nm. Solid samples (crystalline powders) were measured as dispersions in a thin layer of polyfluoroester oil (ABCR GmbH) placed between two 1 mm quartz plates. All emission spectra were corrected for the wavelength-dependent response of the spectrometer and detector (in relative photon flux units). Emission decay traces were recorded by connecting the photomultiplier to an oscilloscope (typically via a 500 Ohm load) and using a N2-laser for pulsed excitation at 337 nm (∼2 ns, ∼5 μJ per pulse).
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 3139 (w), 3100 (w), 3031 (w), 2975 (w), 2925 (w), 1772 (vw), 1734 (vw), 1717 (vw), 1699 (vw), 1684 (vw), 1653 (vw), 1636 (w), 1598 (w), 1550 (vw), 1507 (vw), 1487 (w), 1440 (w), 1375 (w), 1358 (w), 1326 (vw), 1230 (vw), 1227 (vw), 1200 (m), 1161 (w), 1117 (vw), 1068 (w), 1033 (w), 981 (vw), 923 (vw), 891 (vw), 864 (m), 758 (m), 744 (w), 694 (m), 672 (w), 628 (w), 598 (w), 584 (vw), 552 (w), 538 (w), 520 (w). ESI-MS (DMSO): m/z = [C21H21N2+]: calcd 301.1699; found 301.1940. EA calcd (%) for [C21H21ClN2](336.86): C 74.88, H 6.28, N 8.32; found C 74.85, H 6.38, N 8.28.
(cm−1) = 3139 (w), 3100 (w), 3031 (w), 2975 (w), 2925 (w), 1772 (vw), 1734 (vw), 1717 (vw), 1699 (vw), 1684 (vw), 1653 (vw), 1636 (w), 1598 (w), 1550 (vw), 1507 (vw), 1487 (w), 1440 (w), 1375 (w), 1358 (w), 1326 (vw), 1230 (vw), 1227 (vw), 1200 (m), 1161 (w), 1117 (vw), 1068 (w), 1033 (w), 981 (vw), 923 (vw), 891 (vw), 864 (m), 758 (m), 744 (w), 694 (m), 672 (w), 628 (w), 598 (w), 584 (vw), 552 (w), 538 (w), 520 (w). ESI-MS (DMSO): m/z = [C21H21N2+]: calcd 301.1699; found 301.1940. EA calcd (%) for [C21H21ClN2](336.86): C 74.88, H 6.28, N 8.32; found C 74.85, H 6.38, N 8.28.Single crystals suitable for X-ray crystallography were grown by slow diffusion of diethyl ether into a saturated solution of 2 in dichloromethane.
1H NMR (300.13 MHz, DMSO-d6): δ (ppm) = 9.70 (t, 4JHN = 1.6 Hz, 1H, N–CH–N), 8.24 (m, 1H, CH), 8.02 (m, 1H, CH), 7.58–7.55 (m, 2H, o-Ph–H), 7.47–7.42 (m, 3H, m-, p-Ph–H), 7.16 (s, 2H, m-Mes–H), 5.60 (s, 2H, CH2), 2.33 (s, 3H, p-Mes–CH3), 2.05 (s, 6H, o-Mes–CH3). 13C{1H} NMR (75.48 MHz, DMSO-d6): δ (ppm) = 140.3 (N–Mes–CH3), 137.5 (Mes), 134.3 (Mes), 131.7 (Mes), 131.2 (Ph), 129.5 (Ph), 129.2 (Mes), 128.8 (Ph), 124.2 (Ph), 123.1 (CH), 120.9 (CH), 87.1 (Cq), 81.2 (Cq), 39.9 (CH2), 20.6 (p-Mes–CH3), 16.9 (o-Mes–CH3). IR (ATR): (cm−1) = 3000 (w), 2893 (w), 1553 (m), 1488 (m), 1439 (w), 1416 (m), 1368 (w), 1339 (w), 1312 (w), 1267 (w), 1236 (w), 1199 (s), 1155 (m), 1090 (m), 1067 (s), 1030 (m), 984 (vw), 965 (w), 933 (w), 878 (w), 864 (s), 765 (s), 753 (vs), 727 (w), 715 (w), 691 (w), 668 (m), 619 (m), 606 (m), 581 (w), 552 (m), 534 (w), 526 (w), 507 (m). ESI-MS (DMSO): m/z = [C21H21N2+]: calcd 301.1699; found 301.1673. EA calcd (%) for [C21H21BrN2](381.32): C 66.15, H 5.55, N 7.35; found C 65.33, H 5.46, N 7.14.
Route B: Imidazolium salt 1 (200 mg, 0.59 mmol, 1.00 eq.) and [AuCl(tht)] (190 mg, 0.59 mmol, 1.00 eq.) were dissolved in dichloromethane and stirred for 15 minutes. Then finely ground K2CO3was added and the suspension was stirred for 3 hours. Filtration through Celite afforded a clear solution, which was concentrated to ∼2 mL. Addition of n-pentane or diethyl ether (25 mL) gave a colourless precipitate, which was further washed with n-pentane or diethyl ether (3 × 25 mL) and finally dried under vacuum. Yield: 293 mg (93%).
Single crystals suitable for X-ray crystallography were obtained by slow diffusion of n-pentane into a saturated solution of 3 in dichloromethane.
1H NMR (300.13 MHz, CD2Cl2): δ (ppm) = 7.55–7.52 (m, 3H, m-, p-Ph–H), 7.41–7.34 (m, 3H, o-Ph–H, CH), 7.04 (s, 2H, m-Mes–H), 7.00 (d, 4JHH = 2.0 Hz, 1H, CH), 5.36 (s, 2H, CH2), 2.37 (s, 3H, p-Mes–CH3), 2.06 (s, 6H, o-Mes–CH3). 13C{1H} NMR (75.48 MHz, CD2Cl2): δ (ppm) = 172.5 (N–C–N), 140.4 (Mes), 135.5 (Mes), 135.4 (Mes), 132.4 (Mes), 129.8 (Ph), 129.7 (Ph), 129.0 (Ph), 123.1 (Ph), 122.1 (CH), 121.0 (CH), 87.5 (Cq), 81.8 (Cq), 42.3 (CH2), 21.4 (p-Mes–CH3), 18.2 (o-Mes–CH3). IR (ATR): (cm−1) = 3158 (vw), 3131 (vw), 2920 (vw), 1559 (vw), 1489 (m), 1448 (w), 1439 (w), 1417 (m), 1406 (m), 1376 (vw), 1357 (vw), 1329 (w), 1264 (vw), 1231 (s), 1201 (w), 1165 (vw), 1117 (vw), 1087 (vw), 1071 (vw), 1027 (w), 981 (vw), 966 (vw), 919 (vw), 847 (m), 764 (vs), 741 (vs), 691 (vs), 636 (vs), 586 (w), 541 (w), 524 (w). FAB-MS: m/z (%) = 532.1 ([M]+, 13), 497.2 ([M − Cl]+, 61). EA calcd (%) for [C21H20AuClN2](532.82): C 47.34, H 3.78, N 5.26; found C 46.97, H 3.92, N 5.08.
Single crystals suitable for X-ray crystallography were obtained by slow diffusion of n-pentane into a saturated solution of 4 in dichloromethane.
1H NMR (300.13 MHz, CD2Cl2): δ (ppm) = 7.57–7.53 (m, 3H, m-Ph–H, p-Ph–H), 7.42–7.35 (m, 3H, CH, o-Ph–H), 7.05 (s, 2H, m-Mes–H), 7.00 (d, 4JHH = 2.0 Hz, 1H, CH), 5.37 (s, 2H, CH2), 2.37 (s, 3H, p-Mes–CH3), 2.07 (s, 6H, o-Mes–CH3).13C{1H} NMR (75.48 MHz, CD2Cl2): δ (ppm) = 175.8 (N–C–N), 140.3 (Mes), 135.4 (Mes), 135.3 (Mes), 132.3 (Mes), 129.7 (Ph), 129.7 (Ph), 128.9 (Ph), 123.0 (Ph), 122.1 (CH), 120.9 (CH), 87.4 (Cq), 81.7 (Cq), 42.1 (CH2), 21.4 (p-Mes–CH3), 18.1 (o-Mes–CH3). IR (ATR): (cm−1) = 3156 (vw), 3129 (vw), 2963 (w), 2918 (w), 2853 (vw), 1734 (vw), 1699 (vw), 1684 (vw), 1653 (vw), 1607 (vw), 1578 (vw), 1558 (w), 1540 (vw), 1522 (vw), 1507 (vw), 1488 (m), 1449 (m), 1438 (m), 1417 (m), 1405 (m), 1375 (w), 1356 (vw), 1328 (m), 1260 (s), 1230 (s), 1201 (m), 1164 (m), 1087 (s), 1070 (s), 1025 (vs), 981 (m), 966 (m), 935 (w), 918 (w), 846 (m), 798 (vs), 762 (vs), 737 (vs), 689 (vs), 634 (m), 611 (w), 585 (m), 540 (m), 522 (m), 510 (m). FAB-MS: m/z (%) = 576.0 ([M]+, 14), 497.1 ([M − Br]+, 82). EA calcd (%) for [C21H20AuBrN2](577.28): C 43.69, H 3.49, N 4.85; found C 42.65, H 3.43, N 4.38.
1H-NMR (300.13 MHz, CD2Cl2): δ (ppm) = 7.72 (d, 4JHH = 1.9 Hz, 2H, CH), 7.77–7.44 (m, 4H, o-Ph–H), 7.39–7.35 (m, 6H, CH, m-, p-Ph–H), 6.98 (d, 4JHH = 1.9 Hz, 2H, CH), 6.92 (s, 4H, m-Mes–H), 5.20 (s, 4H, CH2), 2.39 (s, 6H, p-Mes–CH3), 1.79 (s, 12H, o-Mes–CH3). 13C{1H} NMR (75.48 MHz, CD2Cl2): δ (ppm) = 185.1 (N–C–N), 140.1 (Mes), 135.3 (Mes), 135.1 (Mes), 132.2 (Mes), 129.7 (Ph), 129.6 (Ph), 129.0 (Ph), 123.4 (Ph), 122.4 (CH), 121.1 (CH), 87.3 (Cq), 82.3 (Cq), 41.9 (CH2), 21.5 (p-Mes–CH3), 17.8 (o-Mes–CH3). IR (ATR): (cm−1) = 3038 (w), 2972 (w), 2926 (w), 2893 (w), 2819 (w), 1734 (vw), 1699 (vw), 1685 (vw), 1653 (vw), 1597 (w), 1554 (m), 1507 (w), 1488 (m), 1439 (m), 1417 (m), 1368 (w), 1339 (w), 1313 (w), 1264 (m), 1233 (w), 1199 (s), 1155 (m), 1090 (m), 1067 (s), 1029 (m), 983 (w), 966 (w), 934 (w), 878 (w), 864 (m), 801 (w), 765 (vs), 753 (vs), 715 w), 691 (s), 668 (s), 620 (m), 606 (m), 581 (w), 552 (m), 537 (w), 526 (m), 507 (w). ESI-MS (DCM): m/z = [C42H40AuN4+]: Calcd 797.2918; found 797.3034. EA calcd (%) for [C42H40AuBrN4] (877.68): C 57.48, H 4.59, N 6.38; found C 57.43, H 4.68, N 6.28.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CPh)] (6). The NHC–AuCl complex 3 (200 mg, 0.375 mmol, 1.00 eq.) and phenylacetylene (0.12 mL, 1.12 mmol, 3.00 eq.) were ground together with KOH pellets (excess) for ten minutes. The greyish residue was extracted with dichloromethane (3 × 15 mL) and filtered. After concentration of the solution to about 2 mL, n-pentane was added to precipitate the product as a colourless solid, which was dried under vacuum. Yield: 199 mg (89%).
CPh)] (6). The NHC–AuCl complex 3 (200 mg, 0.375 mmol, 1.00 eq.) and phenylacetylene (0.12 mL, 1.12 mmol, 3.00 eq.) were ground together with KOH pellets (excess) for ten minutes. The greyish residue was extracted with dichloromethane (3 × 15 mL) and filtered. After concentration of the solution to about 2 mL, n-pentane was added to precipitate the product as a colourless solid, which was dried under vacuum. Yield: 199 mg (89%).Single crystals suitable for X-ray crystallography were obtained by slow diffusion of n-pentane into a saturated solution of 6 in dichloromethane.
1H NMR (300.13 MHz, CD2Cl2): δ (ppm) = 7.56–7.51 (m, 3H, m-, p-PhNHC–H), 7.41–7.37 (m, 3H, CH, o-PhNHC–H), 7.33–7.29 (m, 2H, o-PhAlkin–H), 7.23–7.12 (m, 3H, m-, p-PhAlkin–H), 7.06 (s, 2H, m-Mes–H), 6.97 (d, 4JHH = 1.9 Hz, 1H, CH), 5.41 (s, 2H, CH2), 2.39 (s, 3H, p-Mes–CH3), 2.08 (s, 6H, o-Mes–CH3). 13C{1H} NMR (75.48 MHz, CD2Cl2): δ (ppm) = 188.6 (N–C–N), 140.2 (Mes), 135.6 (Mes), 135.5 (Mes), 132.4 (Mes), 132.3 (Ph), 130.6 (Ph), 129.7 (Ph), 129.6 (Ph), 129.0 (Ph), 128.4 (Ph), 126.7 (Ph), 126.5 (Ph), 123.1 (CH), 122.3 (CAlkinq), 120.9 (CH), 104.3 (CAlkinq), 87.3 (CNHCq), 82.1 (CNHCq), 41.9 (CH2), 21.5 (p-Mes–CH3), 18.2 (o-Mes–CH3). IR (ATR): (cm−1) = 2963 (w), 2915 (w), 2113 (vw), 1595 (w), 1559 (w), 1485 (m), 1441 (m), 1399 (m), 1380 (w), 1355 (w), 1317 (w), 1260 (m), 1229 (m), 1211 (m), 1165 (w), 1089 (m), 1068 (m), 1024 (s), 966 (m), 935 (w), 912 (w), 854 (m), 799 (s), 755 (vs), 690 (vs), 632 (m), 587 (m), 527 (m), 510 (m). FAB-MS: m/z (%) = 598.2 ([M]+, 12), 497.1 ([M − C8H5]+, 31). EI-MS: m/z (%) = 598.46 ([M+], 36). EA calcd (%) for [C29H25AuN2] (598.50): C 58.20, H 4.21, N 4.68; found C 57.63, H 4.21, N 4.65.
CPh))2}{((PhCC)2Au)2Cu2}] (7). The NHC–AuCl complex 6 (100 mg, 0.18 mmol, 1.00 eq.) and [(CuCCPh)x] (62 mg, 0.37 mmol, 2.00 eq.) were suspended in dichloromethane and stirred for 72 hours at ambient temperature. After filtration through Celite, the volume of the solution was reduced to 3 mL and n-pentane was added to precipitate the product as a yellow solid. The solid was a mixture of the gold complex 6 and the desired product 7. The compounds can be obtained pure by fractional crystallization, which was realized by layering a solution of the solid in dichloromethane with n-pentane. Yield: 48 mg (48%, single crystalline material). 1H NMR (300.13 MHz, CD2Cl2): δ (ppm) = 7.52–7.49 (m, 6H, m-, p-PhNHC–H), 7.41–7.37 (m, 4H, o-PhNHC–H), 7.35–7.31 (m, 6H, m-, p-PhAlkin–H), 7.20–7.19 (m, 4H, o-PhAlkin–H), 7.17 (d, 4JHH = 1.7 Hz, 2H, CH) 7.08–7.06 (m, 12H, m-, p-PhDi-Alkin–H), 7.01 (s, 4H, m-Mes–H), 6.97 (d, 4JHH = 1.9 Hz, 2H, CH), 6.90–6.84 (m, 8H, o-PhDi-Alkin–H), 5.45 (s, 4H, CH2), 2.34 (s, 6H, p-Mes–CH3), 2.08 (s, 12H, o-Mes–CH3). 13C{1H}NMR (75.48 MHz, CD2Cl2): δ (ppm) = 188.3 (N–C–N), 140.4 (Mes), 135.5 (Mes), 135.4 (Mes), 135.3 (Mes), 132.9 (Ph), 132.4 (Ph), 129.9 (Ph), 129.8 (Ph), 129.7 (Ph), 129.6 (Ph), 129.0 (Ph), 129.0 (Ph), 128.4 (Ph), 126.6 (Ph), 123.5 (Ph), 123.1 (Ph), 122.2 (CH), 122.0 (CH), 121.0 (CAlkinq), 104.6 (CAlkinq), 87.5 (CLigandq), 82.2 (CLigandq), 81.8 (CDi-Alkinq), 74.2 (CDi-Alkinq) 42.3 (CH2), 21.4 (p-Mes–CH3), 18.2 (o-Mes–CH3). IR (ATR): (cm−1) = 2963 (w), 1595 (w), 1559 (w), 1485 (m), 1441 (m), 1400 (m), 1355 w), 1260 (m), 1231 (m), 1197 (m), 1088 (s), 1068 (s), 1025 (s), 935 (m), 913 (m), 852 (m), 799 (s), 753 (vs), 689 (vs), 586 (m), 526 (s). ESI-MS (DCM): m/z = [(C29H21AuN2·C42H32AuN4)+]: Calcd 1395.460, found 1395.431, [C16H10Au−]: calcd 399.0453, found 399.0748. EA calcd (%) for [C90H70Au4Cu2N4](2124.55): C 50.93, H 3.32, N 2.64; found C 51.98, H 3.87, N 3.51.
CPh))2}{((PhCC)2Au)2Ag2}] (8). The NHC–AuCl complex 6 (100 mg, 0.18 mmol) and [(AgCCPh)x] (excess) were suspended in dichloromethane and stirred for 72 hours at ambient temperature. After filtration through Celite, the volume of the solution was reduced to 3 mL and n-pentane was added to precipitate the product as a yellow solid. The solid was a mixture of the gold complex 6 and the desired product 8. Compound 8 could not be separated quantitatively from the side product 6. In solution and based on NMR spectroscopy, mainly compound 6 is observed whereas the heterometallic Au–Ag complex could be established by X-ray crystallography.Single crystals suitable for X-ray crystallography were obtained by layering a solution of the mixture in dichloromethane with n-pentane.
1H NMR (300.13 MHz, CD2Cl2): δ (ppm) = 7.56–7.52 (m, 6H, m-, p-PhNHC–H), 7.42–7.37 (m, 6H, m-, p-PhAlkin–H), 7.33–7.31 (m, 4H, o-PhNHC–H), 7.23–7.14 (m, 7H, sollte 26H sein, CH, o-PhAlkin–H, o-, m-, p-PhDi-Alkin–H), 7.06 (s, 4H, m-Mes–H), 6.97 (d, 4JHH = 1.9 Hz, 2H, CH), 5.41 (s, 4H, CH2), 2.39 (s, 6H, p-Mes–CH3), 2.08 (s, 12H, o-Mes–CH3). 13C{1H} NMR (75.48 MHz, CD2Cl2): δ (ppm) = 188.7 (N–C–N), 140.3 (Mes), 135.6 (Mes), 135.6 (Mes), 132.4 (Mes), 132.3 (Ph), 130.6 (Ph), 129.8 (Ph), 129.7 (Ph), 129.5 (Ph), 129.1 (Ph), 129.0 (Ph), 128.5 (Ph), 128.3 (Ph), 126.7 (Ph), 126.5 (Ph), 123.1 (Ph), 122.3 (CH), 122.0 (CH), 120.1 (CAlkinq), 104.4 (CAlkinq), 87.3 (CLigandq), 82.2 (CLigandq), 42.0 (CH2), 21.5 (p-Mes–CH3), 18.2 (o-Mes–CH3). IR (ATR): (cm−1) = 3128 (w), 3053 (w), 2968 (w), 2895 (m), 2855 (m), 2227 (vw), 1712 (vw), 1573 (m), 1490 (s), 1442 (m), 1407 (w), 1339 (m), 1321 (m), 1233 (vw), 1182 (w), 1153 (w), 1137 (vs), 1107 (w), 1069 (w), 1047 (w), 1030 (w), 1000 (w), 981 (w), 919 (w), 911 (w), 879 (w), 845 (w), 818 (s), 785 (m), 772 (s), 761 (vs), 723 (s), 695 (vs), 689 (vs), 624 (w), 608 (vs), 557 (w), 543 (w), 529 (s), 511.741. ESI-MS (DCM): m/z = [(C29H25AuN2)2Ag+]: Calcd 1303.2417; found 1303.353, [C16H10Au−]: calcd 399.0453; found 399.0517.
X-ray crystallographic studies
A suitable crystal was covered in mineral oil (Aldrich) and mounted on a glass fibre. The crystal was transferred directly to the cold stream of a STOE IPDS 2 or a STOE StadiVari diffractometer. All structures were solved by direct methods or by the Patterson method (SHELXS-2013).50 The remaining non-hydrogen atoms were located from difference in Fourier map calculations. The refinements were carried out by using full-matrix least-squares techniques on F, minimizing the function (Fo − Fc)2, where the weight is defined as 4Fo2/2(Fo2) and Fo and Fc are the observed and calculated structure factor amplitudes using the program SHELXL-2013.50 Carbon-bound hydrogen atom positions were calculated. The locations of the largest peaks in the final difference Fourier map calculation as well as the magnitude of the residual electron densities in each case were of no chemical significance. Data collection parameters are given in Table S1.‡Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as a supplementary publication no. CCDC 1059550–1059555.
Acknowledgements
Financial support by the DFG-funded transregional collaborative research centre SFB/TRR 88 “Cooperative Effects in Homo- and Heterometallic Complexes (3MET)” is gratefully acknowledged (projects C3 and C7). S.B. thanks the Fonds der Chemischen Industrie (FCI) for a generous fellowship. Dr Rory P. Kelly is gratefully acknowledged for help with preparing the manuscript.Notes and references
- H. W. Wanzlick, Angew. Chem., Int. Ed. Engl., 1962, 1, 75–80 CrossRef PubMed.
- H. W. Wanzlick and H. J. Schönherr, Angew. Chem., Int. Ed. Engl., 1968, 7, 141–142 CrossRef CAS PubMed.
- K. Öfele, J. Organomet. Chem., 1968, 12, P42–P43 CrossRef.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin, Heidelberg, New York, 2007 Search PubMed.
- N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Dordrecht, Heidelberg, London, New York, 2011 Search PubMed.
- S. Bestgen, M. T. Gamer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2015, 21, 601–614 CrossRef CAS PubMed.
- R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- O. Kühl, Chem. Soc. Rev., 2007, 36, 592–607 RSC.
- D. Yuan, H. Tang, L. Xiao and H. V. Huynh, Dalton Trans., 2011, 40, 8788–8795 RSC.
- S. Gaillard and J.-L. Renaud, Dalton Trans., 2013, 42, 7255–7270 RSC.
- A. John and P. Ghosh, Dalton Trans., 2010, 39, 7183–7206 RSC.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766–1775 RSC.
- V. W.-W. Yam and E. C.-C. Cheng, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer, Berlin, Heidelberg, 2007, vol. 281, pp. 269–309 Search PubMed.
- V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC.
- F. Scherbaum, A. Grohmann, B. Huber, C. Krüger and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1988, 27, 1544–1546 CrossRef PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC.
- H. Schmidbaur and A. Schier, Organometallics, 2015, 34, 2048–2066 CrossRef CAS.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- C. Sarcher, A. Lühl, F. C. Falk, S. Lebedkin, M. Kühn, C. Wang, J. Paradies, M. M. Kappes, W. Klopper and P. W. Roesky, Eur. J. Inorg. Chem., 2012, 2012, 5033–5042 CrossRef CAS PubMed.
- A. Gómez-Suárez and S. P. Nolan, Angew. Chem., Int. Ed., 2012, 51, 8156–8159 CrossRef PubMed.
- D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- I. O. Koshevoy, L. Koskinen, M. Haukka, S. P. Tunik, P. Y. Serdobintsev, A. S. Melnikov and T. A. Pakkanen, Angew. Chem., Int. Ed., 2008, 47, 3942–3945 CrossRef CAS PubMed.
- P. Ai, A. A. Danopoulos, P. Braunstein and K. Y. Monakhov, Chem. Commun., 2014, 50, 103–105 RSC.
- I. O. Koshevoy, P. V. Ostrova, A. J. Karttunen, A. S. Melnikov, M. A. Khodorkovskiy, M. Haukka, J. Janis, S. P. Tunik and T. A. Pakkanen, Dalton Trans., 2010, 39, 9022–9031 RSC.
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS.
- A. Collado, A. Gomez-Suarez, A. R. Martin, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 5541–5543 RSC.
- R. Visbal, A. Laguna and M. C. Gimeno, Chem. Commun., 2013, 49, 5642–5644 RSC.
- P. de Frémont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef.
- J. D. Egbert, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 2271–2274 CrossRef CAS.
- O. M. Abu-Salah, A.-R. A. Al-Ohaly and C. B. Knobler, J. Chem. Soc., Chem. Commun., 1985, 1502–1503 RSC.
- I. S. Krytchankou, D. V. Krupenya, A. J. Karttunen, S. P. Tunik, T. A. Pakkanen, P.-T. Chou and I. O. Koshevoy, Dalton Trans., 2014, 43, 3383–3394 RSC.
- I. O. Koshevoy, Y.-C. Chang, A. J. Karttunen, J. R. Shakirova, J. Jänis, M. Haukka, T. Pakkanen and P.-T. Chou, Chem. – Eur. J., 2013, 19, 5104–5112 CrossRef CAS PubMed.
- I. O. Koshevoy, Y.-C. Chang, A. J. Karttunen, M. Haukka, T. Pakkanen and P.-T. Chou, J. Am. Chem. Soc., 2012, 134, 6564–6567 CrossRef CAS PubMed.
- I. O. Koshevoy, A. J. Karttunen, Y.-C. Lin, C.-C. Lin, P.-T. Chou, S. P. Tunik, M. Haukka and T. A. Pakkanen, Dalton Trans., 2010, 39, 2395–2403 RSC.
- A. Gimeno, A. B. Cuenca, S. Suárez-Pantiga, C. R. de Arellano, M. Medio-Simón and G. Asensio, Chem. – Eur. J., 2014, 20, 683–688 CrossRef CAS PubMed.
- P. Schwerdtfeger, A. E. Bruce and M. R. M. Bruce, J. Am. Chem. Soc., 1998, 120, 6587–6597 CrossRef CAS.
- S.-K. Yip, C.-L. Chan, W. H. Lam, K.-K. Cheung and V. W.-W. Yam, Photochem. Photobiol. Sci., 2007, 6, 365–371 CAS.
- S. Sculfort, P. Croizat, A. Messaoudi, M. Bénard, M.-M. Rohmer, R. Welter and P. Braunstein, Angew. Chem., Int. Ed., 2009, 48, 9663–9667 CrossRef CAS PubMed.
- J. Liang, Z. Chen, J. Yin, G.-A. Yu and S. H. Liu, Chem. Commun., 2013, 49, 3567–3569 RSC.
- S. Ahrland, K. Dreisch, B. Norén and Å. Oskarsson, Mater. Chem. Phys., 1993, 35, 281–289 CrossRef CAS.
- J. Liu, J. Chen, J. Zhao, Y. Zhao, L. Li and H. Zhang, Synthesis, 2003, 2661–2666 CAS.
- D. R. Denomme, S. M. Dumbris, I. F. D. Hyatt, K. A. Abboud, I. Ghiviriga and L. McElwee-White, Organometallics, 2010, 29, 5252–5256 CrossRef CAS.
- W. Shi, Y. Luo, X. Luo, L. Chao, H. Zhang, J. Wang and A. Lei, J. Am. Chem. Soc., 2008, 130, 14713–14720 CrossRef CAS PubMed.
- O. M. Abu-Salah and A. R. A. Al-Ohaly, J. Chem. Soc., Dalton Trans., 1988, 2297–2300 RSC.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Dr Manfred Scheer on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1040816–1040823. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02228b |
| This journal is © The Royal Society of Chemistry 2015 |