
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompetition between vinylidene rearrangement and 1,2-insertion of carbon-disubstituted internal alkynes at a Cp*Ir(III) complex†
Yousuke
Ikeda
a,
Shintaro
Kodama
a,
Noriko
Tsuchida
b and
Youichi
Ishii
*a
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Chuo University, 1-13-27 Kasuga, Bunkyo-ku, Tokyo, Japan. E-mail: yo-ishii@kc.chuo-u.ac.jp; Fax: +81-3-3817-1895; Tel: +81-3-3817-1901
bDepartment of Liberal Arts, Faculty of Medicine, Saitama Medical University, 38 Morohongo, Moroyama-machi, Iruma-gun, Saitama 350-0495, Japan. E-mail: n_tsuchi@saitama-med.ac.jp; Fax: +81-49-276-1901; Tel: +81-49-276-1901
First published on 20th August 2015
Abstract
Competition between vinylidene rearrangement/1,1-insertion and 1,2-alkyne insertion into the Ir–Ar bond of [Cp*Ir(ppy-F4)]+ was observed on reaction with diarylacetylenes. The former process afforded the iridacycle 2via the subsequent 1,4-Ir migration, whereas the latter led to the pyridoisoquinolinium complex 4. Detailed analysis revealed that 4 isomerizes to 2 by heating at 50 °C.
Vinylidene rearrangement is now recognized as one of the most valuable methods for the transformation of terminal alkynes.1 Details of this tautomerization involving mechanistic studies1f,2 and applications to organic synthesis1 have been well discussed. The rich reactivities of vinylidenes as well as the accumulated mechanistic information concerning their formation brought about interest in the vinylidene rearrangement of more general alkynes.1l However, the tautomerization of internal alkynes is still not recognized as a common process,1b although a few examples of the internal alkyne-disubstituted vinylidene rearrangement have been studied by us and other groups during the last decade.3,4 This is partly because the vinylidene rearrangement of internal alkynes is a slower process than that of terminal alkynes, hence cannot compete favorably with other metal promoted reactions. For example, internal alkynes readily insert into the metal–carbon bond of an alkyl– or aryl–metal complex in a 1,2 fashion, and therefore the vinylidene rearrangement of the alkynes does not take place. A representative reaction is found in Cp*Rh(III)-mediated annulation of 2-phenylpyridine with internal alkynes, which is first developed by Jones in 2008,5 and the mechanism of this interesting reaction has been proposed to involve the first ortho-metallation of phenylpyridine to form a five-membered metallacycle [Cp*RhCl(ppy)] (ppy = 2-(2-pyridyl)phenyl) followed by the 1,2-insertion of the alkyne into the Rh–aryl bond and the reductive elimination of a C–N bond. This strategy has been applied to a variety of substrates other than 2-phenylpyridine and also expanded to catalytic versions.6 Needless to say, vinylidene rearrangement of internal alkynes and subsequent 1,1-insertion of the disubstituted vinylidene ligand has never been observed throughout these studies.
Obviously, the general kinetic trend that the 1,2-insertion of internal alkynes is faster than their vinylidene rearrangement/1,1-insertion limits the synthetic utility of the latter process. To overcome this drawback and broaden applicability of the vinylidene rearrangement, we have explored how we can control the preference between these processes. As a model system, we have adopted [Cp*IrCl(ppy-F4)] (1) (ppy-F4 = 2,3,4,5-tetrafluoro-6-(2-pyridyl)phenyl), in which the C6F4 group is expected to bind to the metal more strongly than the C6H4 group in ppy and hence to slow down the 1,2-insertion. To our surprise, the products derived from the vinylidene rearrangement of diarylacetylenes were obtained in preference to those from the 1,2-insertion as the thermodynamic product, but not necessarily the kinetic product. This also provides the first example of vinylidene rearrangement of internal alkynes at metal complexes other than group 8 metals.
The iridium precursor 1 was readily synthesized by the reaction of [Cp*IrCl2]2 with 2-(2,3,4,5-tetrafluorophenyl)pyridine in the presence of NaOAc·3H2O and fully characterized by spectroscopic as well as crystallographic analysis (see ESI†). When 1 was allowed to react with diphenylacetylene and NaBArF4 in C2H4Cl2 (1,2-dichloroethane) at 50 °C for 4 h, the colour of the reaction mixture turned from yellow to dark purple (Scheme 1). Recrystallization of this mixture afforded the nine-membered iridacycle complex 2a with an Ir–(vinyl CH) agostic interaction as dark purple crystals in 87% isolated yield, and 2a was fully characterized by means of NMR analysis as well as a single-crystal X-ray diffraction study (Fig. 1, left). In the 1H NMR spectrum, the vinyl CH signal of 2a appears in a significantly low-field region (δ 4.16) as an agostic CH, whereas its 13C{1H} NMR signal exhibited notable high-field shift (δ 49.3). It is interesting to note that these spectroscopy data are in marked difference to those of the related (o-vinyl)aryliridium complex [Cp*Ir{o-C6H4C(Ph)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh}(PMe3)][BArF4] which showed its vinyl CH signal at δ −0.30 in the 1H NMR and δ 86.4 in the 13C{1H} NMR spectra.7 The molecular structure of 2a shows that the Ir1–C1 distance at 2.214(5) Å is explicitly shorter than common agostic iridium–CH distances,7,8 suggesting that the iridium center of 2a interacts more strongly with the C1 atom than the common Ir–CH agostic interaction. In fact, complex 2a easily dissociates the agostic vinyl proton by stirring in MeOH at room temperature for 30 min to form the neutral iridium(III) complex 3 in 73% isolated yield (Scheme 1). Complex 3 was fully characterized by spectroscopic as well as crystallographic analysis (Fig. 1, right). Importantly, the two Ph groups of 2a are bound to the same carbon atom (C2), indicating that one of the Ph groups of diphenylacetylene has migrated across the C
CHPh}(PMe3)][BArF4] which showed its vinyl CH signal at δ −0.30 in the 1H NMR and δ 86.4 in the 13C{1H} NMR spectra.7 The molecular structure of 2a shows that the Ir1–C1 distance at 2.214(5) Å is explicitly shorter than common agostic iridium–CH distances,7,8 suggesting that the iridium center of 2a interacts more strongly with the C1 atom than the common Ir–CH agostic interaction. In fact, complex 2a easily dissociates the agostic vinyl proton by stirring in MeOH at room temperature for 30 min to form the neutral iridium(III) complex 3 in 73% isolated yield (Scheme 1). Complex 3 was fully characterized by spectroscopic as well as crystallographic analysis (Fig. 1, right). Importantly, the two Ph groups of 2a are bound to the same carbon atom (C2), indicating that one of the Ph groups of diphenylacetylene has migrated across the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond during the reaction. In addition, one Ph group is ortho-metallated by the iridium center, and the H atom is transferred to the vinyl carbon (C1). These results clearly demonstrate that the formation of 2a occurred through the initial vinylidene rearrangement to generate the diphenylvinylidene intermediate followed by the 1,1-insertion of the vinylidene ligand into the Ir–C6F4 bond and 1,4-Ir(III) migration7,9 to the ortho position of the Ph group originated from the alkyne (vide infra). Similarly, para-substituted diphenylacetylene derivatives p-XC6H4CCC6H4X-p (X = Me, Cl) were applied to this transformation, and the corresponding cyclometallated complexes 2b and 2c were formed in good yield under appropriate conditions (Scheme 1). Apparently, in this system, vinylidene rearrangement of internal alkynes was more favored than 1,2-insertion into the Ir–Caryl bond. It should also be mentioned that the present reaction provides the first example of the vinylidene rearrangement of carbon-disubstituted internal alkynes at an Ir complex;10 related reactions have so far been observed only at group 8 metal complexes.3,4
C bond during the reaction. In addition, one Ph group is ortho-metallated by the iridium center, and the H atom is transferred to the vinyl carbon (C1). These results clearly demonstrate that the formation of 2a occurred through the initial vinylidene rearrangement to generate the diphenylvinylidene intermediate followed by the 1,1-insertion of the vinylidene ligand into the Ir–C6F4 bond and 1,4-Ir(III) migration7,9 to the ortho position of the Ph group originated from the alkyne (vide infra). Similarly, para-substituted diphenylacetylene derivatives p-XC6H4CCC6H4X-p (X = Me, Cl) were applied to this transformation, and the corresponding cyclometallated complexes 2b and 2c were formed in good yield under appropriate conditions (Scheme 1). Apparently, in this system, vinylidene rearrangement of internal alkynes was more favored than 1,2-insertion into the Ir–Caryl bond. It should also be mentioned that the present reaction provides the first example of the vinylidene rearrangement of carbon-disubstituted internal alkynes at an Ir complex;10 related reactions have so far been observed only at group 8 metal complexes.3,4
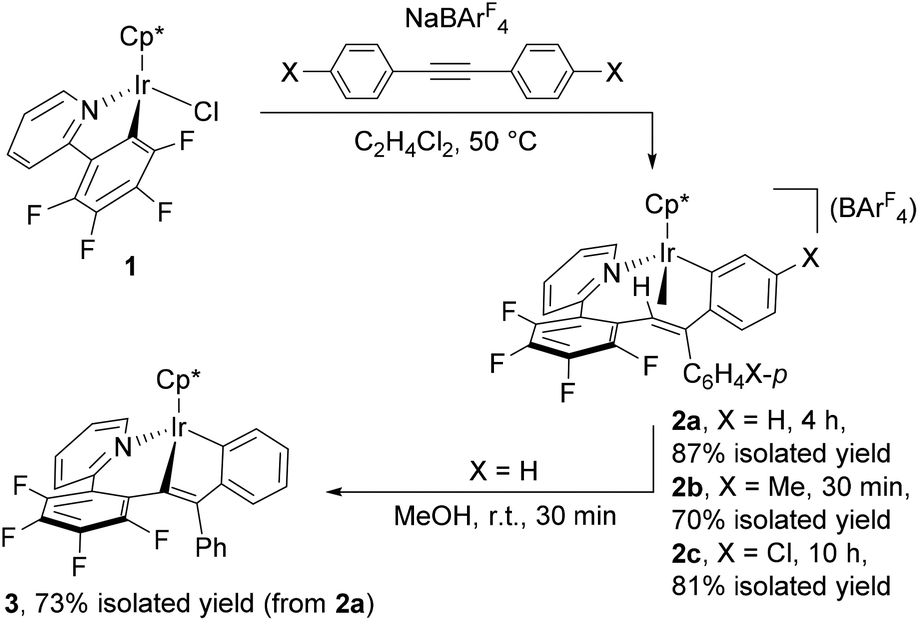 | ||
| Scheme 1 Reaction of 1 with diphenylacetylene derivatives and NaBArF4, and deprotonation of 2a to form 3. | ||
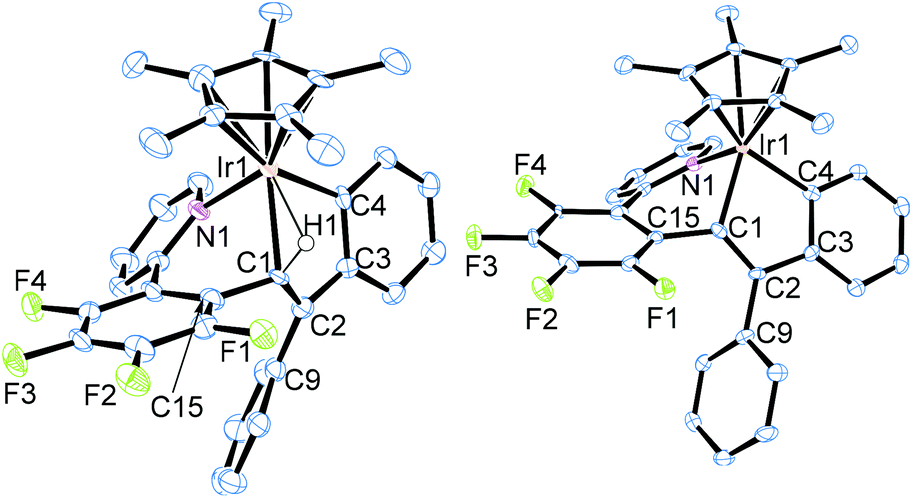 | ||
| Fig. 1 ORTEP drawings of 2a (left) and 3 (right). Anionic part and hydrogen atoms except for H1 of 2a are omitted for clarity. Selected bond lengths (Å) and angles (°): 2a, Ir1–N1, 2.121(5); Ir1–C1, 2.213(5); Ir1–C4, 2.026(6); Ir1–H1, 1.893; C1–C2, 1.461(9); C1–C2–C3, 114.0(5); C1–C2–C9, 123.0(6); C3–C2–C9, 122.5(6); C2–C1–C15, 119.3(5). 3, Ir1–N1, 2.085(3); Ir1–C1, 2.033(5); Ir1–C4, 2.056(4); C1–C2, 1.346(6); Ir1–C1–C2, 120.3(3); Ir1–C1–C15, 112.8(3); C2–C1–C15, 126.0(4); C1–C2–C3, 112.5(4); C1–C2–C9, 125.4(4); C3–C2–C9, 121.7(4). | ||
To gain deeper insight into this reaction, we monitored the progress of the formation of 2c at 50 °C in CDCl3 by means of 1H NMR.11 After 30 min, 1 was consumed completely, and two Cp* signals were observed at δ 1.58 and 1.63 in the intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.1. The former signal is assigned to 2c, whereas the latter species was isolated in 50% yield as yellow crystals by column chromatography on silica gel and characterized unambiguously by a X-ray diffraction study (see ESI†) as the Ir(I) η4-pyridoisoquinolinium complex 4c (Scheme 2). A structurally related rhodium complex was already reported by Huang.6a Obviously complex 4c is formed from the 1,2-insertion of the alkyne into the Ir–C6F4 bond to form the seven-membered metallacycle instead of the vinylidene rearrangement, and this species was further isomerized to 4c through the reductive elimination (vide infra).
:2.1. The former signal is assigned to 2c, whereas the latter species was isolated in 50% yield as yellow crystals by column chromatography on silica gel and characterized unambiguously by a X-ray diffraction study (see ESI†) as the Ir(I) η4-pyridoisoquinolinium complex 4c (Scheme 2). A structurally related rhodium complex was already reported by Huang.6a Obviously complex 4c is formed from the 1,2-insertion of the alkyne into the Ir–C6F4 bond to form the seven-membered metallacycle instead of the vinylidene rearrangement, and this species was further isomerized to 4c through the reductive elimination (vide infra).
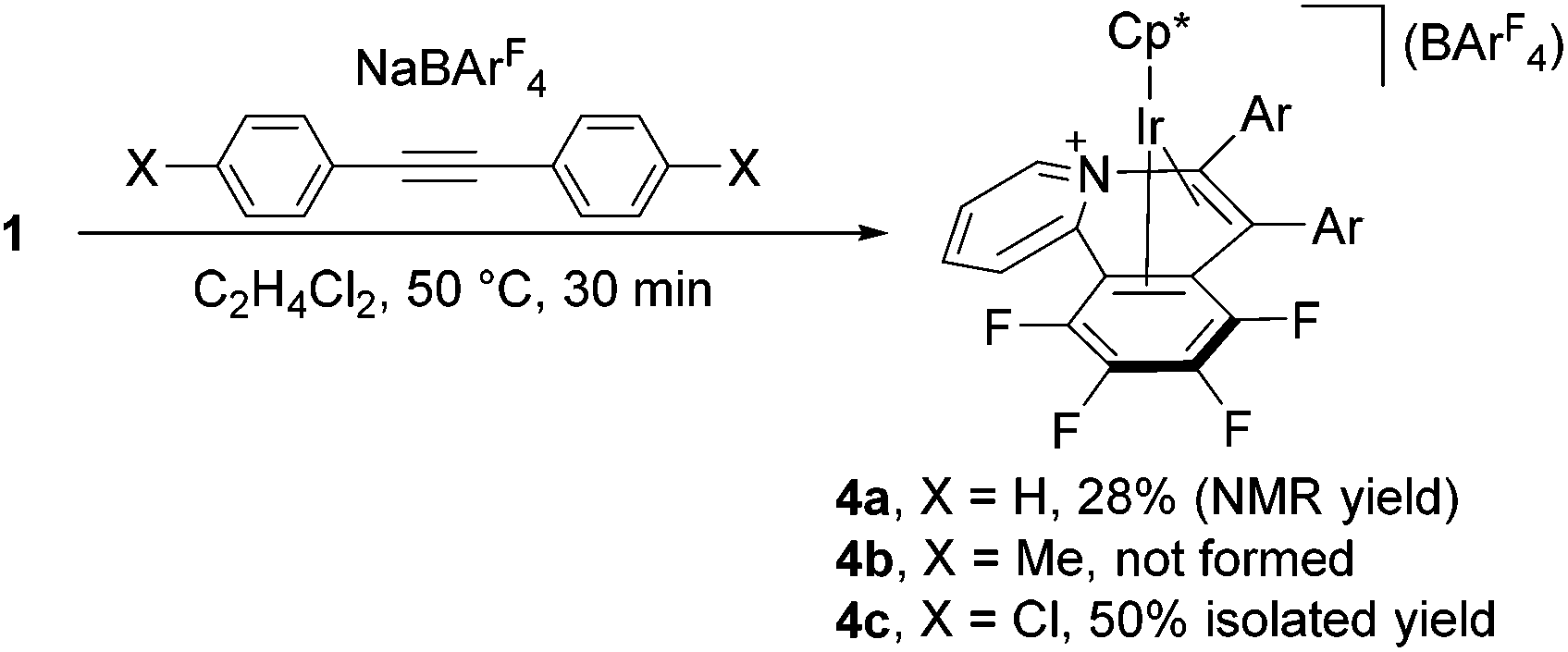 | ||
| Scheme 2 Formation of complexes 4. | ||
Surprisingly, as the reaction proceeded, isomerization of 4c to 2c was observed, and it was completed after 10 h (Scheme 3). Thus, complex 4c is kinetically formed at the early stage of this reaction, and on maintaining at 50 °C, 4c slowly isomerizes to 2c, the thermodynamic product of this reaction. Similarly, Cp*Ir(I) complex 4a was observed at the early stage of the reaction of 1 with PhCCPh as a minor species and isomerized to 2a over 4 h (Scheme 3), although it could not be isolated in a pure form (Scheme 2).12 On the other hand, the formation of 2b was so fast that the corresponding 4b could not be observed.
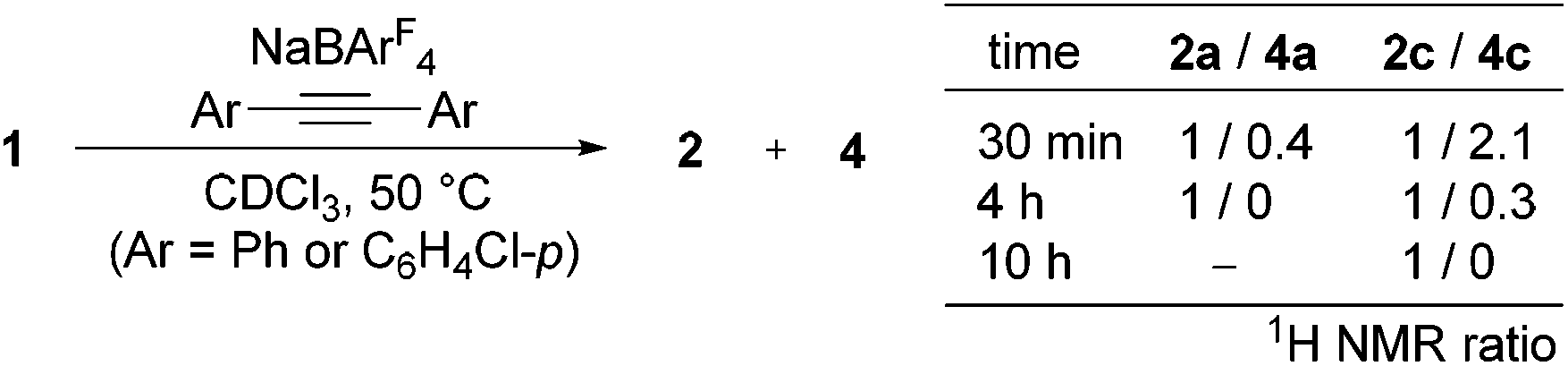 | ||
| Scheme 3 Isomerization of 4 to 2 at 50 °C. | ||
The above observation indicates that the rate of the formation of 2 is notably enhanced by introducing electron donating groups into the para positions of the diphenylacetylene. Recently we have shown both experimentally and theoretically that the internal alkyne-disubstituted vinylidene rearrangement at [CpRu(dppe)]+ is facilitated by an electron-donating substituent,4b,e and this tendency is in good agreement with the present observations, suggesting that the present vinylidene rearrangement at the Ir(III) center involves nucleophilic aryl migration in the rate determining step. In addition, the ppy-F4 ligand is essential for the formation of 2; the reaction of [Cp*IrCl(ppy)] (5) with PhCCPh in the presence of NaBArF4 at 50 °C for 30 min resulted in selective formation of 6 as the sole product in 67% yield, and the corresponding vinylaryliridium species was not formed any more (Scheme 4). For a better understanding of the above observations, preliminary density functional theory (DFT) calculations on the cationic part of 2a and 4a were performed with the B3PW91 functional. As expected, it was confirmed that 2a is more stable in energy than 4a by 8.52 kcal mol−1, which gives good explanation for the selective formation of 2 as the thermodynamic product (Scheme 5). Although we must await a more detailed theoretical study, several mechanisms are considered to be plausible for the conversion of 4 to 2. One is the C–N bond oxidative addition followed by the β-carbon elimination (back-reaction from 4) to regenerate the η2-alkyne complex,13 which then undergo vinylidene rearrangement. β-Carbon elimination from a vinyl complex to form the corresponding η2-alkyne complex is a rare process, but some examples are known in the literature.4f,14 Alternatively, direct isomerization of the seven-membered iridacycle A may be operative. In this case, concerted migration of the iridium center and an aryl group (Fig. 2(a)) or an aryl group migration to eliminate C5H4N-C6F4− anion followed by its nucleophilic attack at the vinylidene α-carbon (Fig. 2(b)) is assumed to be involved.15
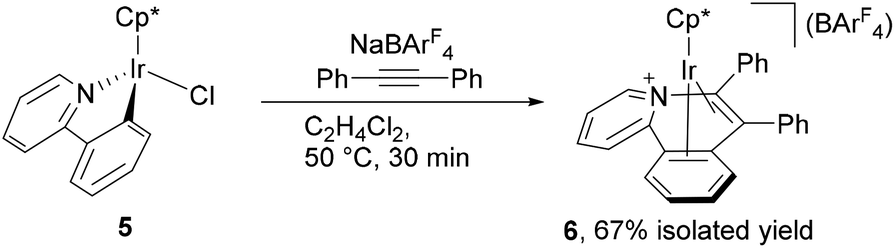 | ||
| Scheme 4 Reactions of 5 with PhCCPh. | ||
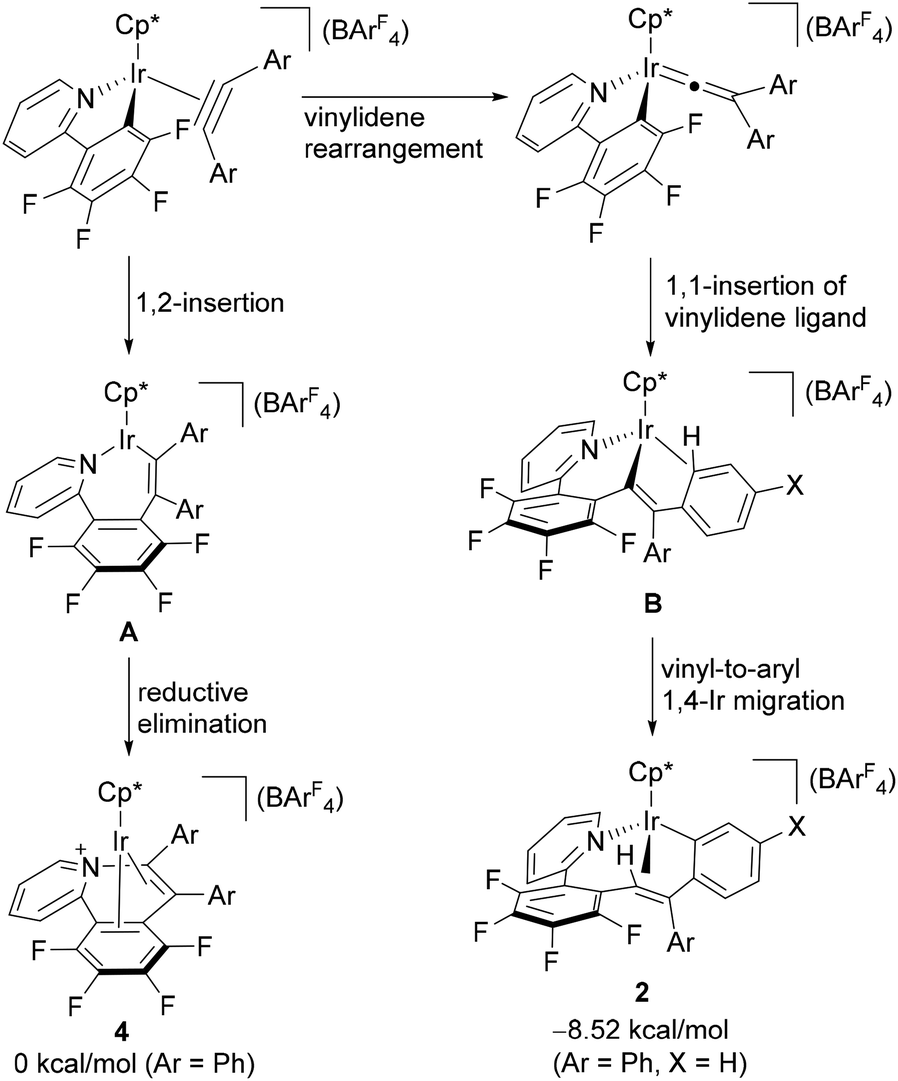 | ||
| Scheme 5 Plausible mechanism of formation of 2 and 4, and Gibbs energy differences between 2a and 4a. | ||
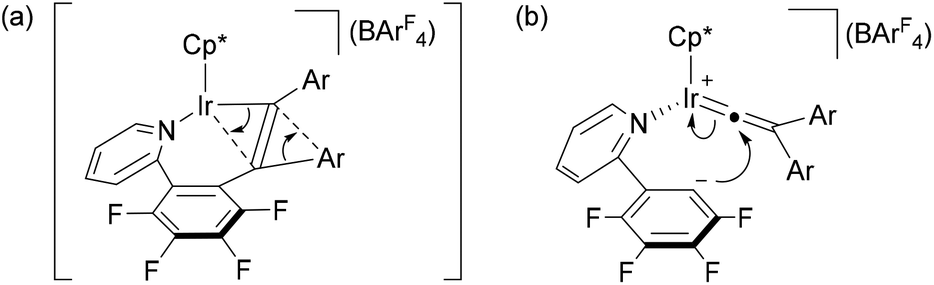 | ||
| Fig. 2 Possible structures for (a) the transition state in a concerted migration of A into B (Scheme 5) and (b) the intermediate derived from aryl migration of A to eliminate C5H4N-C6F4− anion. | ||
Finally, we have investigated the reactivity of 1 with acyl alkyne, which is known to be a reactive substrate towards a vinylidene rearrangement.3,4c When a mixture of 1 with PhCCCOPh and NaBArF4 in C2H4Cl2 was allowed to react at 50 °C for 30 min, a mixture of yellow and red crystals was obtained after recrystallization. X-ray diffraction studies disclosed that the yellow product is the ten-membered iridacycle complex 7, while the red one is attributed to the vinyliridium complex 8 (see ESI†).16 These complexes could be separated by column chromatography on silica gel and isolated in 36% and 38% yields, respectively.
Judging from these structures, the 1,2-insertion and vinylidene rearrangement of PhCCCOPh competitively occurred to generate the seven-membered vinyliridium species 9 and the iridium vinylidene species 10. Complex 7 was formed from 9 by the 1,4-Ir migration from the vinyl to the ortho position of the COPh group, whereas 8 was produced by the 1,1-insertion of the vinylidene ligand in 10 into the Ir–C6F4 bond and the final coordination of the oxygen atom of the carbonyl group (Scheme 6). Unlike the reaction with diarylalkynes, the ratio of 7 and 8 was not changed by further heating at 50 °C.17
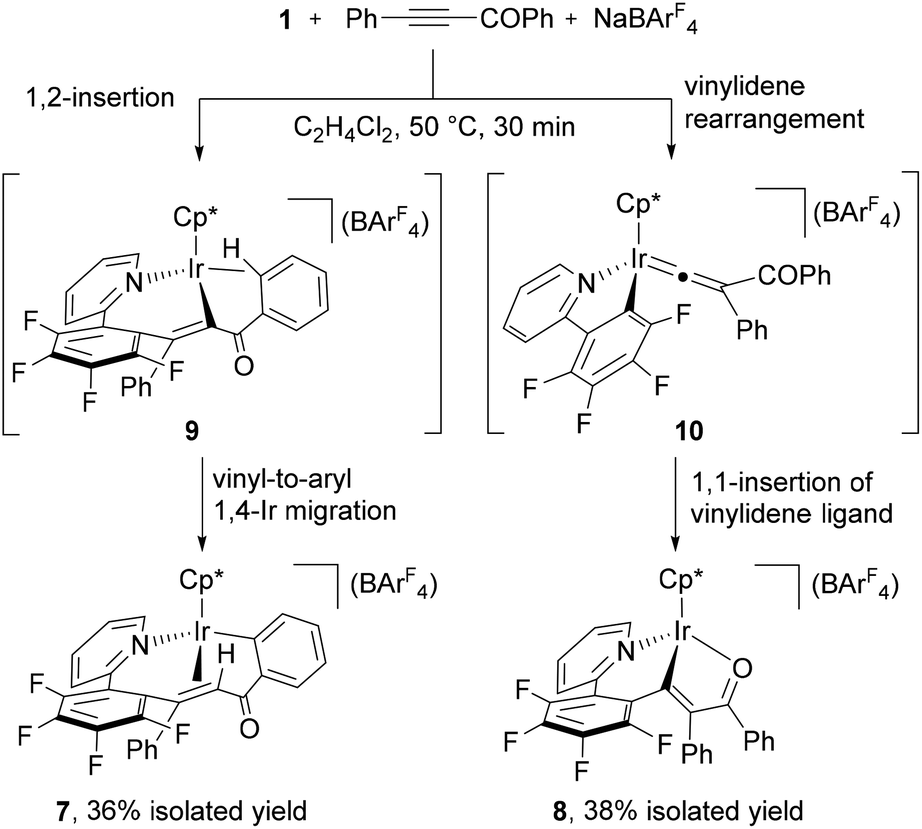 | ||
| Scheme 6 Reactions of 1 with PhCCCOPh and NaBArF4. | ||
In conclusion, we have revealed that the reaction of [Cp*Ir(ppy-F4)]+ with diphenylacetylene derivatives afforded the nine-membered metallacycle complex 2 by way of the sequential vinylidene rearrangement, 1,1-insertion of the vinylidene ligand, and the 1,4-Ir migration to the Ar group. Detailed analysis of this reaction disclosed that 2 and the Ir(I) pyridoisoquinolinium complex 4, the latter of which is a normal 1,2-insertion–reductive elimination product, are competitively generated at the early stage of the reaction, but 4 is gradually isomerized to 2, indicating that 2 is the thermodynamically favoured product. The present reaction provides not only the first example of the internal alkyne-disubstituted vinylidene rearrangement at an iridium complex but also a rare example of actual observation of the competition between vinylidene rearrangement and 1,2-insertion of internal alkynes.
We thank the Research Center for Computational Science in Okazaki, Japan, for the use of the computer facilities. This research was financially supported by JST ACT-C. Y. Ikeda thanks the Japan Society for the Promotion of Science (JSPS) Research Fellowships for Young Scientists.
Notes and references
- For reviews, see: (a) Metal Vinylidenes and Allenylidenes in Catalysis: From Reactivity to Applications in Synthesis, ed. C. Bruneau and P. H. Dixneuf, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed; (b) G. Marinelli, W. E. Streib, J. C. Huffma, K. G. Caulton, M. R. Gagné, J. Takats, M. Dartiguenave, C. Chardon, S. A. Jackson and O. Eisenstein, Polyhedron, 1990, 9, 1867–1881 CrossRef CAS; (c) M. I. Bruce, Chem. Rev., 1991, 91, 197–257 CrossRef CAS; (d) C. Bruneau and P. H. Dixneuf, Acc. Chem. Res., 1999, 32, 311–323 CrossRef CAS; (e) P. Valerga and M. C. Puerta, Coord. Chem. Rev., 1999, 193–195, 977–1025 Search PubMed; (f) Y. Wakatsuki, J. Organomet. Chem., 2004, 689, 4092–4109 CrossRef CAS; (g) V. Cadierno, M. P. Gamasa and J. Gimeno, Coord. Chem. Rev., 2004, 248, 1627–1657 CrossRef CAS; (h) B. M. Trost, M. U. Frederiksen and M. T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630–6666 CrossRef CAS; (i) C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2006, 45, 2176–2203 CrossRef CAS; (j) J. A. Varela and C. Saá, Chem. – Eur. J., 2006, 12, 6450–6456 CrossRef CAS; (k) B. M. Trost and A. McClory, Chem. – Asian J., 2008, 3, 164–194 CrossRef CAS; (l) J. M. Lynam, Chem. – Eur. J., 2010, 16, 8238–8247 CrossRef CAS.
- O. J. S. Pickup, I. Khazal, E. J. Smith, A. C. Whitwood and J. M. Lynam, Organometallics, 2014, 33, 1751–1761 CrossRef CAS.
- (a) P. J. King, S. A. R. Knox, M. S. Legge, A. G. Orpen, J. N. Wilkinson and E. A. Hill, J. Chem. Soc., Dalton Trans., 2000, 1547–1548 RSC; (b) M. J. Shaw, S. W. Bryant and N. Rath, Eur. J. Inorg. Chem., 2007, 3943–3946 CrossRef CAS; (c) E. Bustelo, I. de los Rios, M. C. Puerta and P. Valerga, Organometallics, 2010, 29, 1740–1749 CrossRef; (d) V. K. Singh, E. Bustelo, I. de los Ríos, I. Macías-Arce, M. C. Puerta, P. Valerga, M. A. Ortuño, G. Ujaque and A. Lledós, Organometallics, 2011, 30, 4014–4031 CrossRef CAS; (e) F. E. Fernández, M. C. Puerta and P. Valerga, Inorg. Chem., 2013, 52, 6502–6509 CrossRef.
- (a) Y. Ikeda, T. Yamaguchi, K. Kanao, K. Kimura, S. Kamimura, Y. Mutoh, Y. Tanabe and Y. Ishii, J. Am. Chem. Soc., 2008, 130, 16856–16857 CrossRef CAS; (b) Y. Mutoh, Y. Ikeda, Y. Kimura and Y. Ishii, Chem. Lett., 2009, 38, 534–535 CrossRef CAS; (c) Y. Mutoh, K. Imai, Y. Kimura, Y. Ikeda and Y. Ishii, Organometallics, 2011, 30, 204–207 CrossRef CAS; (d) Y. Mutoh, Y. Kimura, Y. Ikeda, N. Tsuchida, K. Takano and Y. Ishii, Organometallics, 2012, 31, 5150–5158 CrossRef CAS; (e) M. Otsuka, N. Tsuchida, Y. Ikeda, Y. Kimura, Y. Mutoh, Y. Ishii and K. Takano, J. Am. Chem. Soc., 2012, 134, 17746–17756 CrossRef CAS; (f) Y. Ikeda, Y. Mutoh, K. Imai, N. Tsuchida, K. Takano and Y. Ishii, Organometallics, 2013, 32, 4353–4358 CrossRef CAS; (g) M. Otsuka, N. Tsuchida, Y. Ikeda, N. Lambert, R. Nakamura, Y. Mutoh, Y. Ishii and K. Takano, Organometallics, 2015, 34, 3934–3943 CrossRef CAS.
- L. Li, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2008, 130, 12414–12419 CrossRef CAS.
- For the recent examples, see: (a) G. Zhang, L. Yang, Y. Wang, Y. Xie and H. Huang, J. Am. Chem. Soc., 2013, 135, 8850–8853 CrossRef CAS; (b) X.-C. Huang, X.-H. Yang, R.-J. Song and J.-H. Li, J. Org. Chem., 2014, 79, 1025–1031 CrossRef CAS; (c) W. Han, G. Zhang, G. Li and H. Huang, Org. Lett., 2014, 16, 3532–3535 CrossRef CAS; (d) C. Ma, C. Ai, Z. Li, B. Li, H. Song, S. Xu and B. Wang, Organometallics, 2014, 33, 5164–5172 CrossRef CAS; (e) D. Ghorai and J. Choudhury, Chem. Commun., 2014, 50, 15159–15162 RSC.
- Y. Ikeda, K. Takano, S. Kodama and Y. Ishii, Organometallics, 2014, 33, 3998–4004 CrossRef CAS.
- (a) A. C. Albéniz, G. Schulte and R. G. Crabtree, Organometallics, 1992, 11, 242–249 CrossRef; (b) C. S. Chin, M. Kim, H. Lee, S. Noh and K. M. Ok, Organometallics, 2002, 21, 4785–4793 CrossRef.
- Recent reviews of the 1,4-metal migration, see: (a) S. Ma and Z. Gu, Angew. Chem., Int. Ed., 2005, 44, 7512–7517 CrossRef CAS; (b) F. Shi and R. C. Larock, Top. Curr. Chem., 2010, 292, 123–164 CrossRef CAS.
- For recent examples concerning Ir-based vinylidene rearrangement of terminal alkynes, see: W. Iali, F. L. Paglia, X.-F. L. Goff, D. Sredojević, M. Pfeffer and J.-P. Djukic, Chem. Commun., 2012, 48, 10310–10312 RSC.
- Instead of CDCl3, C2H4Cl2 was used as the solvent for initial 30 min of this reaction because of the insolubility of NaBArF4 for CDCl3.
- The structure of 4a was determined by a preliminary single-crystal X-ray diffraction study.
- Preliminary DFT calculation indicated that the free energy barrier of activation for the β-carbon elimination is ca. 19 kcal mol−1 (see ESI†), and therefore we consider that the formation of the η2-alkyne complex from A is acceptable in the present reaction conditions, while we have not obtained satisfactory calculation results to explain the C–N bond oxidative addition yet.
- M. Etienne, R. Mathieu and B. Donnadieu, J. Am. Chem. Soc., 1997, 119, 3218–3228 CrossRef CAS.
- We appreciate one of the reviewers’ helpful suggestions.
- The 1H NMR analysis of the crude reaction mixture indicated that complexes 7 and 8 were formed in the ratio of 1.4:1.
- When isolated 7 and 8 are dissolved in C2H4Cl2 and kept at 50 °C overnight, no isomerization was observed for both complexes.
Footnote |
| † Electronic supplementary information (ESI) available: Text, figures and CIF files giving experimental procedures and crystallographic data. CCDC 1052465–1052470. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02207j |
| This journal is © The Royal Society of Chemistry 2015 |