
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal ion influences distortion of the ligand in the structure of [M{2-MeO(O)CC6H4NHC(S)NP(S)(OiPr)2}2] (M = ZnII, CdII) complexes: a driving force for intermolecular aggregation†‡
Damir A.
Safin
*a,
Maria G.
Babashkina
a,
Michael
Bolte
b,
Mariusz P.
Mitoraj
*c and
Axel
Klein
*a
aInstitut für Anorganische Chemie, Universität zu Köln, Greinstrasse 6, D-50939 Köln, Germany. E-mail: damir.a.safin@gmail.com; axel.klein@uni-koeln.de; Fax: +49 221 4705196; Tel: +49 221 4702913
bInstitut für Anorganische Chemie J.-W.-Goethe-Universität, Frankfurt/Main, Germany
cDepartment of Theoretical Chemistry, Faculty of Chemistry, Jagiellonian University, R. Ingardena 3, 30-060 Cracow, Poland. E-mail: mitoraj@chemia.uj.edu.pl
First published on 7th July 2015
Abstract
Reaction of the in situ deprotonated N-thiophosphorylated thiourea 2-MeO(O)CC6H4NHC(S)NHP(S)(OiPr)2 (HL) with MCl2 (M = ZnII, CdII) in aqueous ethanol leads to complexes of the formula [ML2]. Both compounds crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z = 2 and the metal cations are found in a tetrahedral S2S′2 coordination environment formed by the C–S and P–S sulfur atoms. The crystal structures reveal intramolecular N–H⋯O
with Z = 2 and the metal cations are found in a tetrahedral S2S′2 coordination environment formed by the C–S and P–S sulfur atoms. The crystal structures reveal intramolecular N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds formed within the 2-MeO(O)CC6H4NH fragments. Both structures are further stabilised by intermolecular π⋯π stacking interactions, which are more efficient in [CdL2]. Here, a pronounced dimeric intermolecular aggregate is observed which goes along with a pronounced distortion of the chelate [(S)CNP(S)]− backbone of the ligand upon coordination to CdII as well as a significantly distorted coordination tetrahedron CdS2S′2. The aggregation is also reflected in the positive electrospray ionisation (ESI) mass spectrum of the CdII complex, which exhibits peaks for the dimeric cations [Cd2L3]+, [Cd2L4 + H]+ and [Cd2L4 + Na]+, while for the ZnII analogue only monomeric species were observed. Quantum chemical ETS-NOCV (ADF) calculations confirm the higher stability of dimers in [CdL2] compared with [ZnL2]. The π⋯π stacking interactions are prodominantly due to dispersion contributions, though the electrostatic and orbital interaction components are also important. QTAIM (ADF) type calculations additionally quantify the covalent and non-covalent interactions in the momomers.
C hydrogen bonds formed within the 2-MeO(O)CC6H4NH fragments. Both structures are further stabilised by intermolecular π⋯π stacking interactions, which are more efficient in [CdL2]. Here, a pronounced dimeric intermolecular aggregate is observed which goes along with a pronounced distortion of the chelate [(S)CNP(S)]− backbone of the ligand upon coordination to CdII as well as a significantly distorted coordination tetrahedron CdS2S′2. The aggregation is also reflected in the positive electrospray ionisation (ESI) mass spectrum of the CdII complex, which exhibits peaks for the dimeric cations [Cd2L3]+, [Cd2L4 + H]+ and [Cd2L4 + Na]+, while for the ZnII analogue only monomeric species were observed. Quantum chemical ETS-NOCV (ADF) calculations confirm the higher stability of dimers in [CdL2] compared with [ZnL2]. The π⋯π stacking interactions are prodominantly due to dispersion contributions, though the electrostatic and orbital interaction components are also important. QTAIM (ADF) type calculations additionally quantify the covalent and non-covalent interactions in the momomers.
Introduction
The complexation properties of imidodiphosphinate [R2P(X)NP(Y)R′2]− (X, Y = O, S, Se, Te)1 and aroylthioureate [R2NC(X)NC(Y)R′]− (X, Y = O, S, Se)2 ligands towards ZnII and CdII have previously been reported. This is reflected in a total of 33 structures found in the Cambridge Structural Database.3 Recently, we have also studied the structures of ZnII and CdII complexes with N-(thio)phosphorylated thioamidate and thioureate [RC(S)NP(X)R′2]− (X = O, S) ligands, which are asymmetric derivatives of imidodiphosphinate and aroylthioureate anions. The overwhelming majority of these structures corresponds to the phosphorylated [RC(S)NP(O)R′2]− anions.4 In contrast to this, only three structures of thiophosphorylated [RC(S)NP(S)R′2]− anions are known for each ZnII and CdII.5 This is surprising since it has been postulated for a long time that complexes of the dithioderivatives are much more stable compared with those ligands containing oxygen and sulfur atoms simultaneously. Thus, every new structure of coordination compounds of [RC(S)NP(S)R′2]− with ZnII and CdII are of great importance and value. This becomes relevant when considering that ZnII and CdII complexes with imidodiphosphinate and aroylthioureate have been extensively used as single source precursors for nanomaterials.6 Furthermore, we have recently demonstrated that complexes of [RC(S)NP(X)R′2]− with AgI and NiII are efficient precursors for nanoparticles and nanofilms.7 In addition, the coordination chemistry of ZnIIvs. CdII towards thiolate ligands receives great interest from the binding of these metals in metallothioneins.8In this contribution we describe the synthesis of new ZnII and CdII complexes with the N-thiophosphorylated thiourea 2-MeO(O)CC6H4NHC(S)NHP(S)(OiPr)2 (HL).9 We also describe a complete structural investigation of the obtained complexes [ZnL2] and [CdL2] both in solution and solid state together with their thermal properties. The experimental results were supported by detailed quantum chemical calculations.
Results and discussion
The complexes [ZnL2] and [CdL2] were prepared by reacting the in situ deprotonated ligand, using KOH, with MCl2 (M = ZnII, CdII) (Scheme 1). The obtained colourless solid materials are soluble in most polar solvents. | ||
| Scheme 1 Synthesis of [ZnL2] and [CdL2]. | ||
The IR spectra of complexes are very similar and contain a band at about 560 cm−1 representing the P–S group of the anionic form L− with a delocalised 6 π electron S–C–N–P–S system (bond order ∼1.5).10 This band is shifted by ∼90 cm−1 to low frequencies compared with that in the spectrum of the parent HL.9 Further bands at 1530 and 1695 cm−1 correspond to the conjugated SCN fragment and CO group, respectively. In addition, there is a broad intense absorption arising from the POC group at about 970–990 cm−1. Also, the characteristic band for the arylNH group is found at about 3215 cm−1.
The 31P{1H} NMR spectra of [ZnL2] and [CdL2] in CDCl3 exhibit a unique signal at 55.7 and 55.9 ppm, respectively, which indicates the exclusive presence of 1,5-S,S′-coordinated ligands in the complexes.9–11 The 1H NMR spectra of complexes in CDCl3 each contain one set of signals. The signals of the isopropyl CH3 protons are observed at about 1.40 ppm, while the aryl CH3 protons are at 3.90 ppm. The isopropyl CH(O) protons appear as a doublet of septets at 4.81 ppm with the characteristic coupling constants 3JPOCH = 10.4 Hz and 3JH,H = 6.1 Hz. The phenylene protons are observed as four multiplet peaks at about 7.05, 7.45, 8.00 and 8.65 ppm with the typical coupling constants 3JH,H ranging from 7.9 to 8.3 Hz. The spectra also contain a doublet for the arylNH protons, observed at 11.25 ppm with a coupling constant 4JPNCNH of 7.5 Hz. The latter is exclusively observed when the structure of the H–N–C–N–P fragment meets the so-called “W-criterion” (marked by red in Scheme 1).12 The low-field shift of the signal for the arylNH protons is due to the formation of intramolecular hydrogen bonds of the type arylN–H⋯OC.
Crystals of the complexes were obtained by slow evaporation of the solvent from CH2Cl2–n-hexane solutions. Both structures represent spirocyclic chelates and were refined in the triclinic space group P, each containing one independent molecule in the unit cell. In the crystal, both Δ and Λ enantiomers of the complexes are present due to the inversion centre in the non-chiral space group P (Fig. S1 in the ESI†). Such pairs of enantiomers can be expected for nonplanar bis-chelate complexes.13 Both metal cations are found in a tetrahedral S2S′2 coordination environment formed by the C–S and P–S sulfur atoms (Fig. 1). The six-membered M–S–C–N–P–S metallocycles have an asymmetric boat form. The values of the endocyclic S–M–S angles are about 108.9° (Table 1) in the structure of [ZnL2] and very close to that of the ideal tetrahedron (109.5°). The same angles in the structure of [CdL2] deviate significantly with 101.0(1) and 104.3(1)°. The same trend is found for the exocyclic S(C)–M–S(C), S(P)–M–S(P) and S(C)–M–S(P) angles: the values fall in the range of 108.6–110.9° in the structure of the ZnII complex, while the corresponding values of about 116.3, 112.0, 107.5 and 115.6 were observed in the CdII analog. The two values for the N–P–S, Zn–S–C and Zn–S–P angles are very similar in [ZnL2] representing a quite symmetric surrounding. This is not the case for [CdL2] where the corresponding pairs show significant deviations (Table 1).
 | ||
| Fig. 1 Molecular structures of [ZnL2] (left) and [CdL2] (right). Ellipsoids are given with a 50% probability level. Hydrogen atoms not involved in H-bonding were omitted for clarity. Colour code: C = black, H = light grey, N = blue, O = red, P = brown, S = yellow, M = magenta. | ||
| [ZnL2] | [CdL2] | |
|---|---|---|
| CS |
1.743(3), 1.747(2) | 1.770(4), 1.778(4) |
| PS |
1.991(1), 1.991(1) | 1.972(1), 1.992(2) |
| P–N | 1.602(2), 1.603(2) | 1.583(3), 1.604(3) |
| C–N(C) | 1.356(3), 1.361(3) | 1.342(5), 1.351(4) |
| C–N(P) | 1.304(3), 1.305(3) | 1.291(5), 1.343(5) |
| M–S(C) | 2.3087(8), 2.3137(7) | 2.433(1), 2.513(1) |
| M–S(P) | 2.3365(7), 2.3505(7) | 2.531(1), 2.621(1) |
| S–C–N(C) | 110.5(2), 111.9(2) | 111.3(3), 111.9(3) |
| S–C–N(P) | 128.4(2), 129.2(2) | 128.4(2), 128.5(3) |
| N–C–N | 119.7(2), 120.3(2) | 119.7(3), 120.2(3) |
| N–P–S | 117.4(1), 117.5(1) | 112.4(1), 119.5(1) |
| C–N–P | 127.3(2), 128.4(2) | 127.2(2), 128.5(3) |
| M–S–C | 106.1(1), 108.9(1) | 97.1(1), 101.2(1) |
| M–S–P | 95.16(3), 97.34(3) | 93.63(5), 97.71(6) |
| S–M–Sendo | 108.89(2), 108.94(3) | 101.00(5), 104.32(4) |
| S(C)–M–S(C)exo | 110.77(3) | 116.32(4) |
| S(P)–M–S(P)exo | 108.71(3) | 112.04(5) |
| S(C)–M–S(P)exo | 108.62(3), 110.85(3) | 107.49(4), 115.62(4) |
The lengthening of the C–S and P–S and shortening of the C–N(P) and P–N bonds in both complexes, compared with the values for the parent ligand HL,9 are in agreement with the IR data. The relative higher symmetry of the coordination surrounding of the metal atoms in [ZnL2] compared with [CdL2] is also visible in the M–S(C) and M–S(P) bonds. While the each two values for the M–S(C) and M–S(P) bonds in [ZnL2] are very similar and range from 2.31 to 2.35 Å, the two M–S(C) and M–S(P) bonds in the structure of [CdL2] are rather dissimilar and range from 2.43 to 2.62 Å.
The arylNH protons are involved in intramolecular hydrogen bonds of the type arylN–H⋯OC (Fig. 1 and Table 2) in line with the structure concluded from NMR (Scheme 1). A closer inspection of the crystal structures revealed further H⋯X hydrogen bonds (Table S1 in the ESI†), however, based on established criteria14 we consider them not to be determining the crystal or molecular structures.
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | |
|---|---|---|---|---|---|
| [ZnL2] | N(2)–H(2)⋯O(3) | 0.79(3) | 2.02(3) | 2.677(3) | 142(3) |
| N(4)–H(4)⋯O(7) | 0.83(4) | 1.91(4) | 2.637(3) | 146(3) | |
| [CdL2] | N(2)–H(2)⋯O(3) | 0.89(4) | 1.94(4) | 2.680(4) | 140(4) |
| N(4)–H(4)⋯O(7) | 0.73(4) | 2.00(4) | 2.620(5) | 143(4) |
Both structures are further stabilised by an intermolecular π⋯π stacking interaction leading to dimers for both [ZnL2] and [CdL2] (Fig. 2, Table 3). The π⋯π stacking seems to be much more efficient in [CdL2] leading to an almost coplanar arrangement (interplanar angle α = 0.03°) and a quite short interplanar distance of 3.756(3) Å with a displacement angle β of 24.12°.15 For [ZnL2] a second, only slightly longer, π⋯π stacking interaction (Table 3) finally leads to a 1D polymeric chain (Fig. 3), which is not observed for the Cd derivative. This marked difference seems to be caused by the pronounced distortion of the chelate [(S)CNP(S)]− fragment of the ligands upon coordination in the structure of [CdL2] as well as the significantly distorted coordination tetrahedron CdS2S′2 (Fig. 4) discussed already above. Thus, the nature and especially the size of the metal cation (ZnII: 74 pm vs. CdII: 92 pm)16 drive the supramolecular aggregation of molecules in the structures of [ZnL2] and [CdL2]. A similar influence of ZnIIvs. CdII was found for the formation of supramolecular coordination complexes of the N-thiophosphorylated 2,5-dithiobiurea [NHC(S)NHP(S)(OiPr)2]2.5e The dinuclear mesocate structure was formed upon reacting with ZnII, while the tetranuclear nanoscaled aggregate was isolated in the reaction with CdII. Furthermore, the idea to use the different sizes of these two d10 configured so-called “spherical ions”17 to control the supramolecular aggregation has been worked out recently.17,18 However, in most cases the observed supramolecular aggregate is the product of several interactions and carefuly ligand design is necessary.
 | ||
| Fig. 2 Dimers formed via π⋯π stacking interactions in the structures of [ZnL2] (top) and [CdL2] (bottom). Hydrogen atoms except NH were omitted for clarity. Colour code: C = black, H = light grey, N = blue, O = red, P = brown, S = yellow, M = magenta. | ||
 | ||
| Fig. 3 1D polymeric chain formed via π⋯π stacking interactions of the π⋯π stacked dimers in the structure of [ZnL2]. Hydrogen atoms except NH were omitted for clarity. | ||
 | ||
| Fig. 4 Molecule overlay of [ZnL2] (red) and [CdL2] (black). Hydrogen atoms were omitted for clarity. | ||
| Cg(I) | Cg(J) | Cg–Cg (Å) | α (°) | β (°) | |
|---|---|---|---|---|---|
| a Symmetry codes: #1 1 − x, 1 − y, 1 − z; #2 −x, −y, −z. Cg(3): C(11)–C(12)–C(13)–C(14)–C(15)–C(16), Cg(4): C(31)–C(32)–C(33)–C(34)–C(35)–C(36). b Symmetry codes: #1 1 − x, 1 − y, 1 − z. Cg(4): C(31)–C(32)–C(33)–C(34)–C(35)–C(36). | |||||
| [ZnL2] | Cg(3) | Cg(3)#1 | 4.1083(15) | 0.02 | 34.75 |
| Cg(4) | Cg(4)#2 | 4.112(2) | 0.03 | 32.77 | |
| [CdL2] | Cg(4) | Cg(4)#1 | 3.756(3) | 0.03 | 24.12 |
Frequently, CdII exhibits higher coordination numbers than ZnII thus changing the crystal structure.19 It is also quite common that going from ZnII to CdII leads to a higher distortion of the same coordination polyhedron.5c,20 In contrast to this, the two here presented compounds represent a rare example in which the increasing size of the central metal ion well-nigh “switches on” a very strong π⋯π stacking interaction leading to a marked change in the supramolecular interactions and crystal structure; e.g., the cell volume of 2006.2(2) Å3 for [ZnL2] is reduced to 1949.9(7) Å3 for [CdL2].
The bulk samples of [ZnL2] and [CdL2] were studied by means of X-ray powder diffraction analysis (Fig. 5). The experimental X-ray powder patterns are in agreement with the calculated powder patterns obtained from a single crystal X-ray analysis, showing that the bulk materials of [ZnL2] and [CdL2] are free from phase impurities.
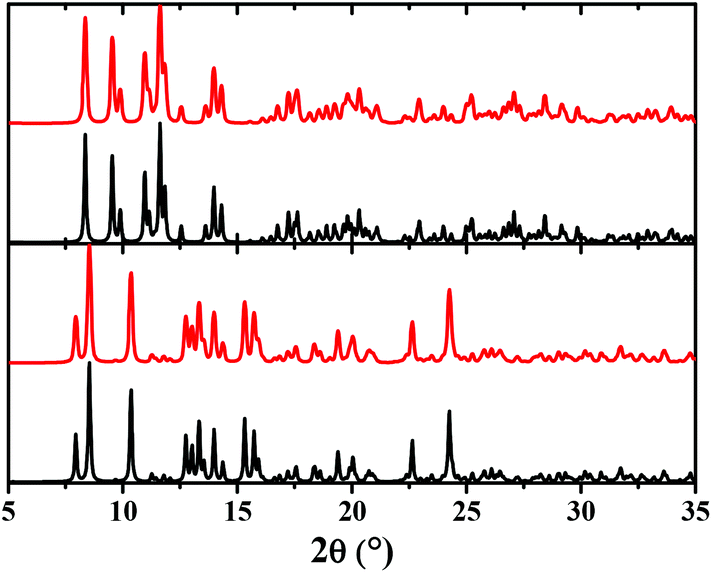 | ||
| Fig. 5 Calculated (black) and experimental (red) X-ray powder diffraction patterns of [ZnL2] (bottom) and [CdL2] (top). | ||
The formation of a rather stable dimeric aggregate via efficient intermolecular π⋯π stacking interactions is also reflected in the positive electrospray ionisation (ESI) mass spectrum of the CdII complex, which exhibits intense peaks for the dimeric cations [Cd2L3]+, [Cd2L4 + H]+ and [Cd2L4 + Na]+ (Fig. 6). For the ZnII analogue only monomeric species were found in the positive ion ESI mass spectrum.
 | ||
| Fig. 6 The positive ion ESI-mass spectrum of [CdL2] (top). Calculated (black) and experimental (red) patterns for the [Cd2L3]+ (middle) and [Cd2L4 + H]+ (bottom) cations. | ||
The thermal properties of [ZnL2] and [CdL2] in air atmosphere were studied by means of TG analyses in order to determine their respective stabilities (Fig. 7). The compound [ZnL2] is stable up to about 100 °C and decomposed in two steps. [CdL2] is stable up to 140 °C and decomposed in three steps with the third step to be poorly defined. The observed final residues of 11.6 and 16.4% are in excellent agreement with the calculated 11.54 and 16.21% for ZnS and CdS, respectively. The formation of these sulfides, each exhibiting a hexagonal form (wurtzite for ZnS and greenockite for CdS, respectively), was proved based on the powder X-ray diffraction analysis with full correspondence with data calculated from a corresponding single crystal analysis (Fig. 8).21
 | ||
| Fig. 7 TG analyses of [ZnL2] (bottom) and [CdL2] (top) performed in a dynamic air atmosphere. | ||
 | ||
| Fig. 8 Calculated (black) and experimental (red) X-ray powder diffraction patterns of ZnS (bottom) and CdS (top) obtained from annealing of [ZnL2] and [CdL2], respectively, at 750 °C. | ||
Thus, the complexes [ZnL2] and [CdL2], being easily obtained, and air/mosture stable might be very suitable single source precursors for the formation of ZnS and CdS, respectively.
In order to shed some light on the nature of bonding between the monomers in [ZnL2] and [CdL2], we have applied the charge and energy decomposition method (ETS-NOCV)22 as implemented in the ADF2012.01 program suite.23 We have used DFT/BLYP-D3 when performing the ETS-NOCV calculations as it was shown that such computational details provides satisfactory results for the noncovalent interactions as compared with the accurate CCSD(T) results.24 The coordinates of the dimers were extracted from the crystal structures.
The total interaction energy (ΔEtotal) in the dimer of [CdL2] is significantly more pronounced compared to that of [ZnL2] (Table 4). These results are in line with the ESI-mass spectra (Fig. 6), which also suggest a higher stability of the dimer of [CdL2]. Decomposition of the π⋯π stacking interaction energies shows that the dispersion contribution (ΔEdispersion) is the most important for the overall stabilisation and it contributes as much as −31.6 kcal mol−1 for [CdL2] and −40.3 kcal mol−1 for [ZnL2] (Table 4). Quantitatively less important is the electrostatic term (ΔEelstat), which appeared to be quite similar for both dimers and of about −10.0 kcal mol−1 (Table 4). Finally, the least contribution is the orbital interaction (ΔEorb), which is approximately twice more important for the cadmium-containing system. In order to shed further light on the ΔEorb term we have plotted the overall deformation density (Δρorb) upon formation of dimers (Fig. 9). The formation of both dimers leads rather to internal polarisations within the monomers, no typical charge transfer between the stacking rings is observed. It is noticeable that changes in the electron density are not only within the stacking rings but they also cover further regions including the OiPr groups as well as the sulphur atoms. The domination of the dispersion contribution in such stacking interactions is in line with the literature.25
 | ||
| Fig. 9 Contours (0.0004 a. u.) of the overall deformation density (Δρorb) together with the corresponding orbital interaction terms for the top (bottom) and side (top) views of dimers of [ZnL2] (left) and [CdL2] (right). Red colour of Δρorb shows the charge depletion, whereas blue colour indicates the electron density accumulation due to the formation of dimers. | ||
| [ZnL2] | [CdL2] | |
|---|---|---|
| ΔEtotal = ΔEelstat + ΔEPauli + ΔEorb + ΔEdispersion. | ||
| ΔEelstat | −9.7 | −10.6 |
| ΔEPauli | 20.8 | 26.0 |
| ΔEorb | −4.6 | −8.8 |
| ΔEdispersion | −31.6 | −40.3 |
| ΔEtotal | −25.2 | −33.7 |
We have further performed the geometry optimisation of the monomers of [ZnL2] and [CdL2] in the gas phase based on DFT/BLYP-D3/TZP (Fig. 10), and the calculated parameters are in qualitative agreement with the experimental values (Tables 1 and 2 and Table S2 in the ESI†).
 | ||
| Fig. 10 The optimised structures with the selected bond lengths (Å) for monomers of [ZnL2] (bottom) and [CdL2] (top) based on ADF/DFT/BLYP-D3/TZP. | ||
We have finally performed a preliminary study of bonding in the monomers of [ZnL2] and [CdL2] based on the Quantum Theory of Atoms in Molecules (QTAIM)26 method as implemented in the ADF program.23 It was found that in both cases not only the strongest coordinating bonds Zn–S and Cd–S are observed, but also less important secondary intramolecular non-covalent interactions of the types O⋯H–N, N⋯H–C and S⋯H–C. From density values at the bond critical points as well as distances one can further infer that the strength of these interactions decreases in the following order O⋯H–N > N⋯H–C > S⋯H–C. Finally, given the fact that the bond critical points implies that stabilisation exists from the electronic exchange channel between atoms, further studies are required to more deeply describe each contribution of intramolecular close contacts. Neverthless, we believe that these preliminary QTAIM based results indentify important factors that might influence the overall stability of the [ZnL2] and [CdL2] monomers (Fig. 11).
 | ||
| Fig. 11 The QTAIM molecular graphs together with the selected intramolecular interactions, characterising monomers of [ZnL2] (bottom) and [CdL2] (top). The density values (a. u.) at the bond critical points are shown for the selected contacts. | ||
Conclusions
We have synthesised the ZnII and CdII complexes [ZnL2] and [CdL2] of the deprotonated N-thiophosphorylated thiourea 2-MeO(O)CC6H4NHC(S)NHP(S)(OiPr)2 (HL). The molecular structures of the complexes were studied by IR and NMR spectroscopy revealing two deprotonated ligands with a delocalised 6 π electron S–C–N–P–S system. Intramolecular N–H⋯OC hydrogen bonds favour the all-trans arrangement of the ligands H–N–C–N–P fragment (the so-called “W-criterion”). In the solid, the structure was elucidated by single crystal X-ray diffraction analysis, revealing that both compounds crystallised isostructural in the triclinic space group P, each containing one independent molecule in the unit cell. The metal cations display a tetrahedral S2S′2 coordination environment formed by the C–S and P–S sulfur atoms and strong intramolecular N–H⋯OC hydrogen bonds were observed within the 2-MeO(O)CC6H4NH fragments. Both structures are further stabilised by intermolecular π⋯π stacking interactions, which are more efficient in [CdL2] leading to isolated stongly connected dimers. In contrast to this, a second, weaker π⋯π stacking is observed in the Zn analogue which leads to a 1D polymeric structure in the solid. This difference between the two complexes is also reflected in the positive electrospray ionisation (ESI) mass spectrum of the CdII complex, which exhibits peaks for the dimeric cations [Cd2L3]+, [Cd2L4 + H]+ and [Cd2L4 + Na]+. Only peaks for the monomeric species were found in the ESI mass spectrum of the ZnII analogue. Theoretical calculations confirm a higher stability of [CdL2] compared with [ZnL2]. Furthermore, the π⋯π stacking interactions are prodominantly due to dispersion contributions, though the electrostatic and orbital interaction components are also important. Thus, there is strong evidence from experiment and theory that formation of the dimeric intermolecular aggregate in the structure of [CdL2] is driven by the pronounced distortion of the chelate [(S)CNP(S)]− backbones of ligands upon coordination to CdII as well as by a significantly distorted coordination tetrahedron CdS2S′2. Thus, the nature and most of all the size of the metal cation (ZnIIvs. CdII) drives the supramolecular aggregation of molecules in the structures of [ZnL2] and [CdL2].
Experimental
General procedures
Infrared spectra (Nujol) were recorded with a Thermo Nicolet 380 FT-IR spectrometer in the range 400–3600 cm−1. NMR spectra in CDCl3 were obtained on a Bruker Avance 300 MHz spectrometer at 25 °C. 1H and 31P{1H} NMR spectra were recorded at 299.948, and 121.420 MHz, respectively. Chemical shifts are reported with reference to SiMe4 (1H) and 85% H3PO4 (31P{1H}). The electrospray ionisation (ESI) mass spectra were measured with a Finnigan-Mat TCQ 700 mass spectrometer. The speed of sample submission was 2 μL min−1. The ionisation energy was 4.5 kV. The capillary temperature was 200 °C. Thermogravimetric analysis (TGA) data were recorded using a Q5000 IR TGA instrument at a heating rate of 10 °C min−1 between room temperature and 800 °C under a constant flow of air (100 mL min−1). Elemental analyses were performed on a Thermoquest Flash EA 1112 Analyzer from CE Instruments.DFT calculations
We have used the ADF2012.01 program23 based on DFT/BLYP-D3/TZP. For topological description of electron density in the monomers the Quantum Theory of Atoms in Molecules (QTAIM)26 were applied.Synthesis of [ZnL2] and [CdL2]
A solution of HL (1 mmol, 0.390 g) in aqueous EtOH (10 mL) was mixed with KOH (1.1 mmol, 0.062 g). An aqueous (10 mL) solution of MCl2 (M = ZnII, CdII; 0.6 mmol, 0.082 and 0.110 g, respectively) was added dropwise under vigorous stirring to the resulting potassium salt. The mixture was stirred at room temperature for 1 h and left overnight. The resulting complex was extracted with CH2Cl2, washed with water and dried with anhydrous MgSO4. The solvent was then removed in vacuo. Colourless crystals were isolated by recrystallisation from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture of CH2Cl2 and n-hexane.
S), 970, 987 (POC), 1531 (SCN), 1694 (CO), 3211 (NH). 1H NMR δ (ppm): 1.37 (d, 3JH,H = 6.2 Hz, 24H, CH3, iPr), 3.92 (s, 6H, CH3, Me), 4.81 (d. sept, 3JPOCH = 10.4 Hz, 3JH,H = 6.1 Hz, 4H, OCH), 7.04 (d. t, 3JH,H = 8.2 Hz, 4JH,H = 1.1 Hz, 2H, p-H, C6H4), 7.44 (d. t, 3JH,H = 8.1 Hz, 4JH,H = 1.6 Hz, 2H, m-H, C6H4), 7.99 (d. d, 3JH,H = 8.0 Hz, 4JH,H = 1.5 Hz, 2H, o-H, C6H4), 8.65 (d, 3JH,H = 8.3 Hz, 2H, m-H, C6H4), 11.26 (d, 4JPNCNH = 7.4 Hz, 2H, arylNH). 31P{1H} NMR δ (ppm): 55.7. ES-MS positive ion, m/z (%): 454.2 (18.4) [ZnL]+, 866.4 (100) [ZnL2 + Na]+. ES-MS negative ion, m/z (%): 389.2 (100) [L]−, 1232.8 (37.2) [ZnL3]−. Anal. Calc. for C30H44N4O8P2S4Zn (844.27): C 42.68, H 5.25, N 6.64. Found: C 42.61, H 5.22, N 6.71%.
S), 970. 982 (POC), 1530 (SCN), 1695 (CO), 3215 (NH). 1H NMR δ (ppm): 1.38 (d, 3JH,H = 6.2 Hz, 24H, CH3, iPr), 3.93 (s, 6H, CH3, Me), 4.81 (d. sept, 3JPOCH = 10.3 Hz, 3JH,H = 6.2 Hz, 4H, OCH), 7.04 (br. t, 3JH,H = 7.9 Hz, 2H, p-H, C6H4), 7.43 (d. t, 3JH,H = 8.0 Hz, 4JH,H = 1.4 Hz, 2H, m-H, C6H4), 7.98 (d. d, 3JH,H = 8.1 Hz, 4JH,H = 1.5 Hz, 2H, o-H, C6H4), 8.62 (d, 3JH,H = 8.2 Hz, 2H, m-H, C6H4), 11.28 (d, 4JPNCNH = 7.8 Hz, 2H, arylNH). 31P{1H} NMR δ (ppm): 55.9. ES-MS positive ion, m/z (%): 892.7 (63.4) [CdL2 + H]+, 1394.1 (100) [Cd2L3]+, 1783.2 (27.1) [Cd2L4 + H]+, 1806.7 (7.1) [Cd2L4 + Na]+. ES-MS negative ion, m/z (%): 389.1 (100) [L]−. Anal. Calc. for C30H44CdN4O8P2S4 (891.30): C 40.43, H 4.98, N 6.29. Found: C 40.52, H 5.06, N 6.37%.
:4 mixture of CH2Cl2 and n-hexane.
S), 970, 987 (POC), 1531 (SCN), 1694 (CO), 3211 (NH). 1H NMR δ (ppm): 1.37 (d, 3JH,H = 6.2 Hz, 24H, CH3, iPr), 3.92 (s, 6H, CH3, Me), 4.81 (d. sept, 3JPOCH = 10.4 Hz, 3JH,H = 6.1 Hz, 4H, OCH), 7.04 (d. t, 3JH,H = 8.2 Hz, 4JH,H = 1.1 Hz, 2H, p-H, C6H4), 7.44 (d. t, 3JH,H = 8.1 Hz, 4JH,H = 1.6 Hz, 2H, m-H, C6H4), 7.99 (d. d, 3JH,H = 8.0 Hz, 4JH,H = 1.5 Hz, 2H, o-H, C6H4), 8.65 (d, 3JH,H = 8.3 Hz, 2H, m-H, C6H4), 11.26 (d, 4JPNCNH = 7.4 Hz, 2H, arylNH). 31P{1H} NMR δ (ppm): 55.7. ES-MS positive ion, m/z (%): 454.2 (18.4) [ZnL]+, 866.4 (100) [ZnL2 + Na]+. ES-MS negative ion, m/z (%): 389.2 (100) [L]−, 1232.8 (37.2) [ZnL3]−. Anal. Calc. for C30H44N4O8P2S4Zn (844.27): C 42.68, H 5.25, N 6.64. Found: C 42.61, H 5.22, N 6.71%.
S), 970. 982 (POC), 1530 (SCN), 1695 (CO), 3215 (NH). 1H NMR δ (ppm): 1.38 (d, 3JH,H = 6.2 Hz, 24H, CH3, iPr), 3.93 (s, 6H, CH3, Me), 4.81 (d. sept, 3JPOCH = 10.3 Hz, 3JH,H = 6.2 Hz, 4H, OCH), 7.04 (br. t, 3JH,H = 7.9 Hz, 2H, p-H, C6H4), 7.43 (d. t, 3JH,H = 8.0 Hz, 4JH,H = 1.4 Hz, 2H, m-H, C6H4), 7.98 (d. d, 3JH,H = 8.1 Hz, 4JH,H = 1.5 Hz, 2H, o-H, C6H4), 8.62 (d, 3JH,H = 8.2 Hz, 2H, m-H, C6H4), 11.28 (d, 4JPNCNH = 7.8 Hz, 2H, arylNH). 31P{1H} NMR δ (ppm): 55.9. ES-MS positive ion, m/z (%): 892.7 (63.4) [CdL2 + H]+, 1394.1 (100) [Cd2L3]+, 1783.2 (27.1) [Cd2L4 + H]+, 1806.7 (7.1) [Cd2L4 + Na]+. ES-MS negative ion, m/z (%): 389.1 (100) [L]−. Anal. Calc. for C30H44CdN4O8P2S4 (891.30): C 40.43, H 4.98, N 6.29. Found: C 40.52, H 5.06, N 6.37%.
X-Ray powder diffraction
X-Ray powder diffraction for bulk samples was carried out using a Rigaku Ultima IV X-ray powder diffractometer. The Parallel Beam mode was used to collect the data (λ = 1.541836 Å).Single crystal X-ray diffraction
The X-ray diffraction data for the crystals of [ZnL2] and [CdL2] were collected at 173(2) K on a STOE IPDS-II diffractometer with graphite-monochromatised Mo-Kα radiation generated by a fine-focus X-ray tube operated at 50 kV and 40 mA. The reflections of the images were indexed, integrated and scaled using the X-Area data reduction package.27 Data were corrected for absorption using the PLATON program.28 The structures were solved by direct methods using the SHELXS97 program29 and refined first isotropically and then anisotropically using SHELXL-97.29 Hydrogen atoms were revealed from Δρ maps and those bonded to carbon atoms were refined using appropriate riding models. Hydrogen atoms bonded to nitrogen atoms were freely refined. All figures were generated using the program Mercury.30, a = 10.4280(5), b = 11.4053(6), c = 17.3530(9) Å, α = 78.001(4), β = 84.932(4), γ = 85.131(4)°, V = 2006.19(18) Å3, Z = 2, ρ = 1.398 g cm−3, μ(Mo-Kα) = 0.949 mm−1, reflections: 32272 collected, 7511 unique, Rint = 0.0510, R1(all) = 0.0454, wR2(all) = 0.1055.
, a = 9.7368(19), b = 10.046(2), c = 21.882(4) Å, α = 97.42(3), β = 99.37(3), γ = 109.62(3)°, V = 1949.9(7) Å3, Z = 2, ρ = 1.518 g cm−3, μ(Mo-Kα) = 0.907 mm−1, reflections: 18402 collected, 7261 unique, Rint = 0.0853, R1(all) = 0.0539, wR2(all) = 0.1060.
Notes and references
- (a) D. Cupertino, R. Keyte, A. M. Z. S. Lawin, D. J. Williams and J. D. Woollins, Inorg. Chem., 1996, 35, 2695 CrossRef CAS; (b) V. Garcia-Montalvo, J. Novosad, P. Kilian, J. D. Woollins, A. M. Z. Slawin, P. Garcia y Garcia, M. Lopez-Cardoso, G. Espinosa-Perez and R. Cea-Olivares, J. Chem. Soc., Dalton Trans., 1997, 1025 RSC; (c) D. C. Cupertino, R. W. Keyte, A. M. Z. Slawin and J. D. Woollins, Polyhedron, 1998, 17, 4219 CrossRef CAS; (d) D. Cupertino, D. J. Birdsall, A. M. Z. Slawin and J. D. Woollins, Inorg. Chim. Acta, 1999, 290, 1 CrossRef CAS; (e) P. Sekar and J. A. Ibers, Inorg. Chim. Acta, 2001, 319, 117 CrossRef CAS; (f) A. Silvestru, D. Ban and J. E. Drake, Rev. Roum. Chim., 2002, 47, 1077 CAS; (g) M. Afzaal, D. Crouch, M. A. Malik, M. Motevalli, P. O'Brien and J.-H. Park, J. Mater. Chem., 2003, 13, 639 RSC; (h) M. Afzaal, D. Crouch, M. A. Malik, M. Motevalli, P. O'Brien, J.-H. Park and J. D. Woollins, Eur. J. Inorg. Chem., 2004, 171 CrossRef CAS PubMed; (i) T. Chivers, D. J. Eisler and J. S. Ritch, Dalton Trans., 2005, 2675 RSC; (j) M. Ghesner, A. Silvestru, C. Silvestru, J. E. Drake, M. B. Hursthouse and M. E. Light, Inorg. Chim. Acta, 2005, 358, 3724 CrossRef CAS PubMed; (k) E. Ferentinos, A. B. Tsoupras, M. Roulia, S. D. Chatziefthimiou, C. A. Demopoulos and P. Kyritsis, Inorg. Chim. Acta, 2011, 378, 102 CrossRef CAS PubMed.
- (a) W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1993, 619, 786 CrossRef CAS PubMed; (b) W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1993, 619, 791 CrossRef CAS PubMed; (c) X. Shen, X. Shi, B. Kang, Y. Liu, Y. Tong, H. Jiang and K. Chen, Polyhedron, 1998, 17, 4049 CrossRef; (d) X. Shen, X. Shi, B. Kang, Y. Liu, L. Gu and X. Huang, J. Coord. Chem., 1999, 47, 1 CrossRef CAS PubMed; (e) M. Reinel, R. Richter and R. Kirmse, Z. Anorg. Allg. Chem., 2002, 628, 41 CrossRef CAS; (f) M. Bolte and L. Fink, Private Communication, 2003; (g) M. Kampf, R. Richter, L. Hennig, A. Eidner, J. Baldamus and R. Kirmse, Z. Anorg. Allg. Chem., 2004, 630, 2677 CrossRef CAS PubMed; (h) K. E. Armstrong, J. D. Crane and M. Whittingham, Inorg. Chem. Commun., 2004, 7, 784 CrossRef CAS PubMed; (i) H. Arslan, U. Florke, N. Kulcu and M. F. Emen, J. Coord. Chem., 2006, 59, 223 CrossRef CAS PubMed; (j) K. Ramasamy, M. A. Malik, P. O'Brien and J. Raftery, Dalton Trans., 2010, 39, 1460 RSC; (k) K. Ramasamy, M. A. Malik, M. Helliwell, J. Raftery and P. O'Brien, Chem. Mater., 2011, 23, 1471 CrossRef CAS.
- CSD version 5.35, update February 2015th Search PubMed.
- (a) F. D. Sokolov, D. A. Safin, N. G. Zabirov, V. V. Brusko, B. I. Khairutdinov, D. B. Krivolapov and I. A. Litvinov, Eur. J. Inorg. Chem., 2006, 2027 CrossRef CAS PubMed; (b) D. A. Safin, F. D. Sokolov, H. Nöth, M. G. Babashkina, T. R. Gimadiev, J. Galezowska and H. Kozlowski, Polyhedron, 2008, 27, 2022 CrossRef CAS PubMed; (c) D. A. Safin, M. G. Babashkina, A. Klein, M. Bolte, D. B. Krivolapov and I. A. Litvinov, Inorg. Chem. Commun., 2009, 12, 913 CrossRef CAS PubMed; (d) D. A. Safin, A. Klein, M. G. Babashkina, H. Nöth, D. B. Krivolapov, I. A. Litvinov and H. Kozlowski, Polyhedron, 2009, 28, 1504 CrossRef CAS PubMed; (e) D. A. Safin, M. G. Babashkina, A. Klein, H. Nöth, M. Bolte and D. B. Krivolapov, Polyhedron, 2010, 29, 1837 CrossRef CAS PubMed; (f) D. A. Safin, M. G. Babashkina, M. Bolte, Ł. Szyrwiel, A. Klein and H. Kozlowski, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 1739 CrossRef CAS PubMed; (g) D. A. Safin, M. G. Babashkina, M. Bolte, D. B. Krivolapov, M. L. Verizhnikov, A. R. Bashirov and A. Klein, Inorg. Chim. Acta, 2011, 366, 19 CrossRef CAS PubMed.
- (a) D. J. Birdsall, J. Green, T. Q. Ly, J. Novosad, M. Necas, A. M. Z. Slawin, J. D. Woollins and Z. Zak, Eur. J. Inorg. Chem., 1999, 1445 CrossRef CAS; (b) N. G. Zabirov, V. V. Brusko, A. Y. Verat, D. B. Krivolapov, I. A. Litvinov and R. A. Cherkasov, Polyhedron, 2004, 23, 2243 CrossRef CAS PubMed; (c) D. A. Safin, M. Bolte, M. G. Babashkina and H. Kozlowski, Polyhedron, 2010, 29, 488 CrossRef CAS PubMed; (d) M. G. Babashkina, D. A. Safin, M. Bolte and A. Klein, Polyhedron, 2010, 29, 1515 CrossRef CAS PubMed; (e) D. A. Safin, M. G. Babashkina, P. Kubisiak, M. P. Mitoraj, C. S. Le Duff, K. Robeyns and Y. Garcia, Eur. J. Inorg. Chem., 2014, 5522 CrossRef CAS PubMed.
- For example: M. A. Malik, M. Afzaal and P. O'Brien, Chem. Rev., 2010, 110, 4417 CrossRef CAS PubMed.
- (a) D. A. Safin, P. S. Mdluli, N. Revaprasadu, K. Ahmad, M. Afzaal, M. Helliwell, P. O'Brien, E. R. Shakirova, M. G. Babashkina and A. Klein, Chem. Mater., 2009, 21, 4233 CrossRef CAS; (b) M. G. Babashkina, D. A. Safin and Y. Garcia, Dalton Trans., 2012, 41, 2234 RSC; (c) M. G. Babashkina, D. A. Safin, K. Robeyns and Y. Garcia, Eur. J. Inorg. Chem., 2015, 7, 1160 CrossRef PubMed.
- (a) D. E. K. Sutherland and M. J. Stillman, Metallomics, 2011, 3, 444 RSC; (b) O. I. Leszczyszyn, H. T. Imam and C. A. Blindauer, Metallomics, 2013, 5, 1146 RSC.
- M. G. Babashkina, D. A. Safin, M. Bolte and Y. Garcia, Dalton Trans., 2012, 41, 3223 RSC.
- (a) F. D. Sokolov, V. V. Brusko, N. G. Zabirov and R. A. Cherkasov, Curr. Org. Chem., 2006, 10, 27 CrossRef CAS; (b) F. D. Sokolov, V. V. Brusko, D. A. Safin, R. A. Cherkasov and N. G. Zabirov, Coordination Diversity of N-Phosphorylated Amides and Ureas Towards VIIIB Group Cations, in Transition Metal Chemistry: New Research, ed. B. Varga and L. Kis, Nova Science Publishers Inc., Hauppauge NY, USA, 2008, p. 101 and references therein Search PubMed.
- (a) M. G. Babashkina, D. A. Safin, M. Bolte and A. Klein, Inorg. Chem. Commun., 2009, 12, 678 CrossRef CAS PubMed; (b) M. G. Babashkina, D. A. Safin, M. Bolte, M. Srebro, M. Mitoraj, A. Uthe, A. Klein and M. Köckerling, Dalton Trans., 2011, 40, 3142 RSC; (c) M. G. Babashkina, D. A. Safin, M. Srebro, P. Kubisiak, M. P. Mitoraj, M. Bolte and Y. Garcia, CrystEngComm, 2011, 13, 5321 RSC.
- H. Günter, NMR Spectroscopy: An Introduction, Wiley, Chichester, UK, 1980 Search PubMed.
- (a) A.-C. Chamayou, S. Lüdeke, V. Brecht, T. B. Freedman, L. A. Nafie and C. Janiak, Inorg. Chem., 2011, 50, 11363 CrossRef CAS PubMed; (b) A.-C. Chamayou, G. Makhloufi, L. A. Nafie, C. Janiak and S. Lüdeke, Inorg. Chem., 2015, 54, 2193 CrossRef CAS PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751 CrossRef.
- (a) L. Bain, S. Bullock, L. Harding, T. Riis-Johannessen, G. Midgley, C. R. Rice and M. Whitehead, Chem. Commun., 2010, 46, 3496 RSC.
- (a) M. Martínez-Calvo, M. J. Romero, R. Pedrido, A. M. González-Noya, G. Zaragoza and M. R. Bermejo, Dalton Trans., 2012, 41, 13395 RSC; (b) D. J. Cooke, J. M. Cross, R. V. Fennessy, L. P. Harding, C. R. Rice and C. Slater, Chem. Commun., 2013, 49, 7785 RSC.
- (a) Q. Zhu, T. Sheng, C. Tan, S. Hu, R. Fu and X. Wu, Inorg. Chem., 2011, 50, 7618 CrossRef CAS PubMed; (b) Y.-N. Zhang, P. Liu, Y.-Y. Wang, L.-Y. Wu, L.-Y. Pang and Q.-Z. Shi, Cryst. Growth Des., 2011, 11, 1531 CrossRef CAS; (c) M. V. Campian, I. Haiduc and E. R. T. Tiekink, Z. Kristallogr., 2013, 228, 204 CrossRef CAS; (d) H. Zhao, D. Jia, J. Li, G. J. Moxey and C. Zhang, Inorg. Chim. Acta, 2015, 432, 1 CrossRef CAS PubMed; (e) D.-S. Liu, W.-T. Chen, Y.-P. Xu, P. Shen, S.-J. Hu and Y. Sui, J. Solid State Chem., 2015, 226, 186 CrossRef CAS PubMed.
- (a) A. D. Watson, C. Pulla Rao, J. R. Dorfman and R. H. Holm, Inorg. Chem., 1985, 24, 2820 CrossRef CAS; (b) M. Saravanan, B. Arul Prakasam, K. Ramalingam, G. Bocelli and A. Cantoni, Z. Anorg. Allg. Chem., 2005, 631, 1688 CrossRef CAS PubMed; (c) K. Baba, T. Okamura, H. Yamamoto, T. Yamamoto and N. Ueyama, Inorg. Chem., 2008, 47, 2837 CrossRef CAS PubMed.
- For example: (a) B. J. Skinner, Am. Mineral., 1961, 46, 1399 CAS; (b) C.-Y. Yeh, Z. W. Lu, S. Froyen and A. Zunger, Phys. Rev. B: Condens. Matter, 1992, 46, 10086 CrossRef CAS.
- (a) T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1 CrossRef CAS; (b) M. Mitoraj and A. Michalak, J. Mol. Model, 2007, 13, 347 CrossRef CAS PubMed; (c) M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962 CrossRef CAS.
- (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS PubMed and references therein; (b) E. J. Baerends, J. Autschbach, D. Bashford, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo, D. P. Chong, L. Deng, R. M. Dickson, D. E. Ellis, M. van Faassen, L. Fan, T. H. Fischer, C. Fonseca Guerra, A. Ghysels, A. Giammona, S. J. A. van Gisbergen, A. W. Götz, J. A. Groeneveld, O. V. Gritsenko, M. Grüning, F. E. Harris, P. van den Hoek, C. R. Jacob, H. Jacobsen, L. Jensen, G. van Kessel, F. Kootstra, M. V. Krykunov, E. van Lenthe, D. A. McCormack, A. Michalak, M. Mitoraj, J. Neugebauer, V. P. Nicu, L. Noodleman, V. P. Osinga, S. Patchkovskii, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, J. I. Rodríguez, P. Ros, P. R. T. Schipper, G. Schreckenbach, M. Seth, J. G. Snijders, M. Solà, M. Swart, D. Swerhone, G. te Velde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo, A. L. Yakovlev and T. Ziegler, ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam Search PubMed.
- W. Gao, H. Feng, X. Xuan and L. Chen, J. Mol. Model, 2012, 18, 4577 CrossRef CAS PubMed.
- (a) D. N. Sredojević, Z. D. Tomić and S. D. Zarić, Cryst. Growth Des., 2010, 10, 3901 CrossRef; (b) S. T. Mutter and J. A. Platts, Chem. – Eur. J., 2010, 16, 5391 CrossRef CAS PubMed; (c) C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, UK, 1990 Search PubMed.
- Stoe & Cie. X-AREA, Area-Detector Control and Integration Software, Stoe & Cie, Darmstadt, Germany, 2001 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1, showing the crystal packing of the complexes, Table S1 with selected structural data, and Table S2 with a comparison of calculated and experimental bond parameters. CCDC reference numbers 1062050 ([ZnL2]) and 1062051 ([CdL2]). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02189h |
| ‡ Dedicated to Professor F. Ekkehardt Hahn on the Occasion of his 60th Birthday. |
| This journal is © The Royal Society of Chemistry 2015 |