
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bis(σ-B–H) complexes of copper(I): precursors to a heterogeneous amine–borane dehydrogenation catalyst†
Adi E.
Nako
,
Andrew J. P.
White
and
Mark R.
Crimmin
*
Department of Chemistry, Imperial College London, Exhibition Road, South Kensington, London SW7 2AZ, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 17th June 2015
Abstract
A series of bis(σ-B–H) complexes of copper(I) have been prepared by displacement of arene solvent from a β-diketiminate copper(I) complex by four-coordinate boranes, H3B–L (L = NMe3, lutidine). In the presence of the same copper arene complex, the secondary amine–borane H3B–NMe2H undergoes dehydrogenation. We provide evidence for formation of a heterogengous catalyst from decomposition of the solution species.
Since Hartwig and co-workers reported the isolation and characterisation of [Cp2Ti(η2-HBcat)2] (HBcat = catecholborane),1 our understanding of the coordination chemistry of boranes has flourished.2 Contrasting studies have investigated the interaction of 3- and 4-coordinate boranes with transition metal centres.3,4 Regardless of the environment at boron and the mode of coordination to the metal, σ-borane complexes have become synonymous with B–H bond activation. These species are invoked as intermediates in the catalytic borylation of C–H bonds,5 the hydroboration of alkenes and alkynes,6 and the dehydrogenation of amine–boranes.7 For example, Shimoi and co-workers studied the coordination of H3B–NMe3 to a series of group 6 carbonyl complexes and demonstrated dehydrogenation of H3B–NHR2 under photochemical conditions.8 In related studies, Weller, Sabo–Etienne, Aldridge, Manners, Schneider, and others have conducted extensive investigations into the coordination of H3B–NR3, H2B
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR2 and [H2B–NR2]2 fragments to a series of late transition metals, including ruthenium, rhodium and iridium complexes.7,9–12 A detailed understanding of not only the electronic structure of a clutch of σ-borane complexes but also the mechanisms of amine–borane dehydrogenation has emerged.
NR2 and [H2B–NR2]2 fragments to a series of late transition metals, including ruthenium, rhodium and iridium complexes.7,9–12 A detailed understanding of not only the electronic structure of a clutch of σ-borane complexes but also the mechanisms of amine–borane dehydrogenation has emerged.
Despite a growing interest in the catalytic applications of the 1st row transition metals, little is known about σ-complexes of copper. The coordination of σ-bonds to Cu(I) may foreshadow oxidative addition to Cu(III) and play an unappreciated role in catalysis. In line with these expectations, Bourissou and coworkers have reported the intramolecular coordination of Si–Si and Si–H bonds within carefully designed ligand frameworks to Cu(I). In the case of an Sn–Sn analogue, oxidative addition of the tin–tin bond was observed allowing isolation of the corresponding Cu(III) distannyl complex.13 Stack, Ribas and co-workers have provided EPR and computational support for an agostic interaction in a Cu(II) metallocycle.14 Recently we reported the reversible, intermolecular, coordination of Al–H and Zn–H bonds to a two-coordinate copper(I) fragment generated in situ from 12·toluene (Scheme 1).15,16 Here we disclose that amine–boranes coordinate reversibly to Cu(I), and demonstrate an effective pre-catalyst for amine–borane dehydrogenation.
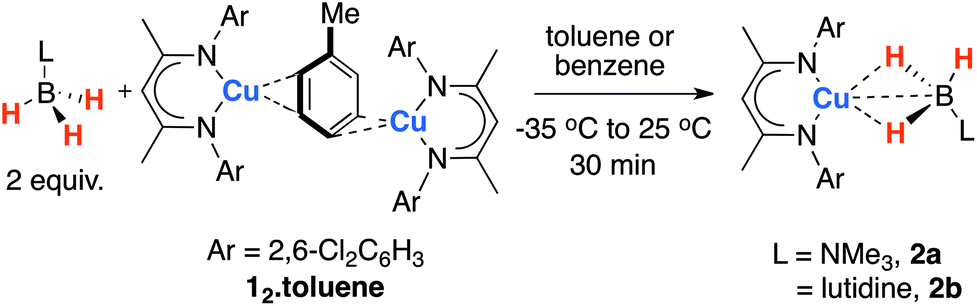 | ||
| Scheme 1 Synthesis of σ-borane complexes of copper(I). | ||
The reaction of 12·toluene15 with H3B–L (L = NMe3, lutidine) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry C6D6 resulted in a minor perturbation of the resonances of 1 and the borane as evidenced by line-broadening and chemical shift changes in both the 1H and 11B{1H} NMR spectra. Despite the weak and potentially reversible nature of the interaction, preparative scale reactions allowed the isolation of the corresponding σ-borane complexes 2a–b as yellow crystalline solids in 63–85% yield (Scheme 1).
:2 stoichiometry C6D6 resulted in a minor perturbation of the resonances of 1 and the borane as evidenced by line-broadening and chemical shift changes in both the 1H and 11B{1H} NMR spectra. Despite the weak and potentially reversible nature of the interaction, preparative scale reactions allowed the isolation of the corresponding σ-borane complexes 2a–b as yellow crystalline solids in 63–85% yield (Scheme 1).
Single crystals of 2a–b could be grown from toluene or toluene/hexane mixtures at −35 °C (Fig. 1). Compound 2a crystallises with a mirror plane that passes though the metal centre and bisects the N–Cu–N angle, necessitating that the BH3NMe3 unit be disordered (see ESI†). Due to this disorder, the hydride atoms could not be located. The Cu–B distances (2a, 2.191(6) Å; 2b, 2.152(3) Å) are longer than the 2.002(3) Å Cu–B bond of the copper–boryl complex [(IPr)Cu(Bpin)] (IPr = 1,3-bis(2,6-di-iso-propylphenyl)imidazol-2-ylidene; HBpin = pinacolborane). In contrast, the quality of data acquired on 2b allowed the location of the hydride positions from a difference electron density map. It is apparent that the ligand coordinates in an η2:η2-mode forming a bis(σ-B–H) complex. The two Cu–H distances of 1.81(2) and 1.79(2) Å are within experimental error of one another. The B–H bond lengths of the bridging hydrides (1.15(1) and 1.16(2) Å) are both significantly longer than that to the terminal hydride (1.04(2) Å).
 | ||
| Fig. 1 The crystal structure of 2a (top) and 2b (bottom). H-atoms with the exception of experimentally identified B–H units omitted for clarity. Selected bond lengths (Å) and bond angles (°) 2a, Cu–B 2.191(6), B–N(12) 1.549(11), N(1)–Cu–N(1A) 97.08(14); 2b, Cu–B 2.152(3), B–N(22) 1.590(3), N(3)–Cu–N(1) 98.58(7). | ||
The η2:η2-coordination of a borane ligand in [Ru(H)2(PCy3)2(η2:η2-H2BMes)] was reported by Sabo-Etienne and co-workers.17 There is also direct precedent for this type of interaction in amine–borane chemistry and Weller and co-workers have reported a series of rhodium, iridium and ruthenium complexes in which amine–boranes coordinate through two hydride sites.9 In contrast, Shimoi and co-workers proposed the η2:η2-mode as a transition state for the fast exchange of geminal B–H units of η2-coordinated amine–boranes within metal carbonyl complexes.18 It is worth noting that, two cationic complexes in which chelating diboranes, [H2B(PMe3)]2 and [HB(hpp)]2 (hpp = 1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]pyrimidinate), coordinate to copper(I) via vicinal B–H bonds are known.19 Inspection of the solid-state structure of 2b reveals π–π stacking between the electron-rich lutidine and electron-poor 2,6-dichlorophenyl moieties; a secondary, non-covalent, interaction that undoubtedly contributes to the stabilisation of 2b.
Infrared data are consistent with the formulation of these species as weakly coordinated boron hydrides and reveal broad B–H stretches (2a, 2423 cm−1; 2b, 2403 cm−1) in the range expected for terminal boron hydrides with no clear differentiation between the (σ-B–H) and B–H vibrations.
1H and 11B NMR spectroscopy show that, upon dissolution in toluene-d8, isolated crystalline samples of the Cu(I) σ-complexes establish an equilibrium between 2+ toluene and 1·toluene + amine–borane (Scheme 2). We have previously shown that 12·toluene forms 1·toluene by solvation, and the latter is the predominant specices in toluene solution.15
 | ||
| Scheme 2 Reversible σ-complex formation with copper(I). | ||
For complex 2b at 298 K in toluene-d8 the BH3 unit of the amine–borane is observed as a single resonance at δ = –15.5 ppm in the 11B{1H} NMR and as a broad quartet at δ = 3.27 ppm in the 1H NMR. The resonances are assigned to a time-averaged contribution from bound and unbound amine–borane; consistent with fast chemical exchange. Upon cooling, decoalescence of a series of resonances assigned to both the amine–borane and the β-diketiminate ligand occurs. At 193 K the slow exchange regime is reached and a mixure of 1·toluene, amine–borane and 2b is observed in solution (ESI, Fig. S1 and S2†).20 While at this temperature B–H resonances were apparent as broad signals in the 1H{11B} NMR between δ = 2.5 and 3.5 ppm, we have been unable to resolve terminal B–H and (σ-B–H) resonances. We suggest that even at 193 K fast exchange between the terminal B–H and (σ-B–H) units within 2b occurs.
The observation of a weak and reversible coordination of the B–H bond to Cu(I) parallels that reported for analogous Al–H and Zn–H σ-complexes.15 This fluxional process was observed for not only 2b (vide supra) but also 2a. VT NMR on toluene-d8 samples of 2a across the 193 to 353 K range allowed quantification of the equilibrium depicted in Scheme 2. Van't Hoff analysis gave ΔHrxn = –1.40(4) kcal mol−1, ΔSrxn = –5.87(2) cal K−1 mol−1 and ΔGrxn = +0.17(3) kcal mol−1. The data suggest that binding of H3B–NMe3 to 1·toluene is slightly endergonic.
In order to gain a deeper understanding of the strength and nature of the bonding within the bis(σ-B–H) complexes, a series of DFT calculations were undertaken. A minimum on the potential energy surface with a bis(σ-B–H) coordination mode was obtained for the series of complexes presented in Scheme 1. All attempts to optimise mono(σ-B–H) geometries led to this structure. The calculated B–H bond lengths are significantly longer than those determined in 2b by X-ray crystallography and range from 1.20–1.23 Å. Furthermore, across a choice of functionals, and in contrast to the X-ray data, the (σ-B–H) lengths were determined to be only slightly longer than the terminal B–H bond in these calculations (Δ = 0.03 Å). Based on the known difficulty in assigning the position of the hydrogen atoms in X-ray diffraction experiments, the calculated B–H bond lengths represent a more realistic description of the ground-state structure.
NBO calculations suggest only a minor perturbation of borane within the coordination complexes 2a and 2b. The Wiberg Bond Indices (WBIs) for the (σ-B–H) bonds are similar to that of the terminal B–H. Furthermore, both the Cu–H and Cu–B WBIs are low, suggestive of a weak interaction (Fig. 2). Second order perturbation analysis allows a quantification of the donor–acceptor interactions, donation of electrons from each of the two B–H σ-bonds occurs to the 4s orbital of copper (2a, 21.7 + 22.3 kcal mol−1: 2b, 15.6 + 16.8 kcal mol−1), significant back-donation from Cu(I) to the B–H σ*-orbitals is not recorded for either 2a or 2b.
 | ||
| Fig. 2 QTAIM electron density contour plot for 2a. Molecule presented in the {CuHB} plane. Figure annotated with selected data from QTAIM and NBO calculations. Green dots are bond critical points, red dots are ring critical points, bond critical paths are represented by thick solid lines. | ||
These data were further underscored by a quantum theory atoms-in-molecules (QTAIM) calculation on 2a which revealed bond critical points (BCPs) between the Cu/B and H atoms, but not between Cu and B. These data show a bending of the (σ-B–H) bond critical paths toward Cu and are consistent with two 3-centre,2-electron interactions (Fig. 2). In line with the NBO analysis, the QTAIM data for coordinated (ρbcp = 0.154; ∇ρbcp2 = –0.14) and non-coordinated B–H bonds (ρbcp = 0.171; ∇ρbcp2 = –0.20) within 2a suggests small changes of the bonding in the B–H bond upon coordination to Cu(I).
Further modification of the amine–borane to a substrate that contained both hydridic and acidic protons resulted in facile dehydrogenation and boron–nitrogen bond formation. While reaction of H3B–NHMe2 with 12·toluene resulted in the generation of the corresponding σ-complex, compound 2c was short-lived and only observed in situ. All attempts to isolate this latter species resulted in dehydrogenation of H3B–NHMe2 (Scheme 3).
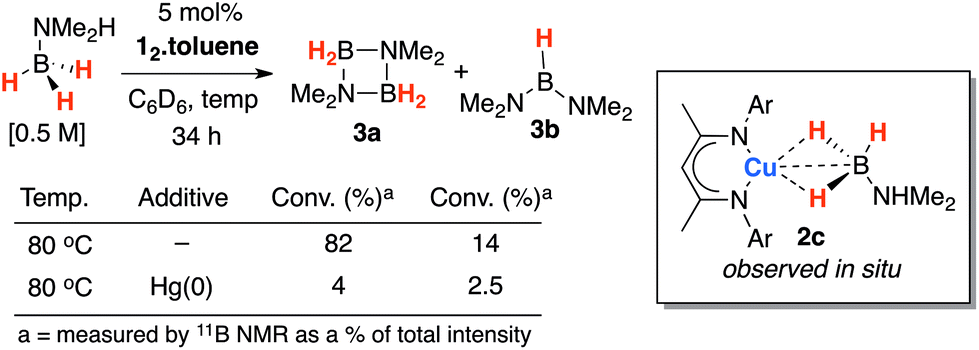 | ||
| Scheme 3 Amine–borane dehydrogenation. | ||
In line with these expectations, 12·toluene catalysed the dehydrogenation of H3B–NHMe2 in 5 mol% loading at 80 °C in C6D6 solution (Scheme 2). Notably 2c was observed as an intermediate in solution by 1H NMR spectroscopy. In this case, at 298 K the 1J11B–1H coupling can be resolved and the 1J11B–1H for the equilibrium mixture of 1·toluene, 2c and H3B–NMe2H (90.4 Hz) is slightly smaller than that of the independent amine–borane (96.4 Hz). Monitoring catalytic reactions by 1H and 11B NMR spectroscopy revealed the formation of known products [H2B–NMe2]2 (3a) and HB(NMe2)2 (3b). The reaction proceeded with concomitant formation of a Cu(0) mirror on the interior of the reaction vessel. Following a catalytic run, re-exposure of the Cu(0) mirror to the reaction conditions reestablished amine–borane dehydrogenation. An Hg(0) drop experiment resulted in a significant inhibition of catalysis. In this instance, data are consistent with 2c acting as a homogeneous precursor to a heterogeneous species.21
These data contrast those found by Philips and co-workers for the dehydrogenation of ammonia-borane catalysed by a ruthenium analogue of 1·benzene and by Bertrand and co-workers using a CAAC-stabilised copper borohydride complex.22,23
Conclusions
In summary, we have reported the first examples of isolable and crystallographically characterised σ-borane complexes of Cu(I). While in the solid-state amine–boranes coordinate via an η2:η2-mode, in solution displacement of this ligand by arene solvent is both fast and reversibile. Inclusion of both hydridic and protic hydrogen atoms on the ligand leads to a decomposition of the coordination compound and production of a heterogeneous copper catalyst that is capable of the dehydrogenation of an amine–borane.We are grateful to the Royal Society for provision of a research fellowship (MRC) and the EPSRC for project funding, including a prize research fellowship (AEN). We are grateful to Pete Haycock for his assistance with multinuclear and VT NMR experiments.
Notes and references
- J. F. Hartwig, C. N. Muhoro, X. He, O. Eisenstein, R. Bosque and F. Maseras, J. Am. Chem. Soc., 1996, 118, 10936 CrossRef CAS.
- (a) J. C. Green, M. L. H. Green and G. Parkin, Chem. Commun., 2012, 48, 11481 RSC; (b) G. Alcaraz, M. Grellier and S. Sabo-Etienne, Acc. Chem. Res., 2009, 42, 1640 CrossRef CAS PubMed.
- (a) C. N. Muhoro and J. F. Hartwig, Angew. Chem., Int. Ed., 1997, 36, 1510 CrossRef CAS PubMed; (b) C. N. Muhoro, X. He and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 5033 CrossRef CAS; (c) S. Schlecht and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 9435 CrossRef CAS; (d) M. G. Crestani, M. Muñoz-Hernández, A. Arévalo, A. Acosta-Ramírez and J. J. García, J. Am. Chem. Soc., 2005, 127, 18066 CrossRef CAS PubMed; (e) V. Montiel-Palma, M. Lumbierres, B. Donnadieu, S. Sabo-Etienne and B. Chaudret, J. Am. Chem. Soc., 2002, 124, 5624 CrossRef CAS PubMed.
- (a) M. Shimoi, S.-L. Nagai, M. Ichikawa, Y. Kawano, K. Katoh, M. Uruichi and H. Ogino, J. Am. Chem. Soc., 1999, 121, 11704 CrossRef CAS; (b) T. Kakizawa, Y. Kawano and M. Shimoi, Organometallics, 2001, 20, 3211 CrossRef CAS; (c) T. Yasue, Y. Kawano and M. Shimoi, Angew. Chem., Int. Ed., 2003, 42, 1727 CrossRef CAS PubMed; (d) Y. Kawano, M. Hashiva and M. Shimoi, Organometallics, 2006, 25, 4420 CrossRef CAS; (e) Y. Kawano, K. Yamaguchi, S.-Y. Miyake, T. Kakizawa and M. Shimoi, Chem. – Eur. J., 2007, 13, 6920 CrossRef CAS PubMed.
- C. E. Webster, Y. Fan, M. B. Hall, D. Kunz and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 858 CrossRef CAS PubMed.
- (a) X. He and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 1696 CrossRef CAS; (b) D. R. Lantero, D. L. Ward and M. R. Smith, III, J. Am. Chem. Soc., 1997, 119, 9699 CrossRef CAS.
- (a) G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 49, 7170 CrossRef CAS PubMed; (b) W. R. H. Wright, E. R. Berkeley, L. R. Alden, R. T. Baker and L. G. Sneddon, Chem. Commun., 2011, 47, 3177 RSC; (c) V. Pons and R. T. Baker, Angew. Chem., Int. Ed., 2008, 47, 9600 CrossRef CAS PubMed; (d) T. J. Clark, K. Lee and I. Manners, Chem. – Eur. J., 2006, 12, 8634 CrossRef CAS PubMed.
- (a) Y. Kawano, M. Uruichi, M. Shimoi, S. Taki, T. Kawaguchi, T. Kakizawa and H. Ogino, J. Am. Chem. Soc., 2009, 131, 14946 CrossRef CAS PubMed; (b) T. Kakizawa, Y. Kawano, K. Naganeyama and M. Shimoi, Chem. Lett., 2011, 40, 171 CrossRef CAS.
- (a) T. M. Douglas, A. B. Chaplin and A. S. Weller, J. Am. Chem. Soc., 2008, 130, 14432 CrossRef CAS PubMed; (b) R. Dallanegra, A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2009, 48, 6875 CrossRef CAS PubMed; (c) A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2010, 49, 581 CAS; (d) A. B. Chaplin and A. S. Weller, Inorg. Chem., 2010, 49, 1111 CrossRef CAS PubMed; (e) R. Dallanegra, A. B. Chaplin, J. Tsim and A. S. Weller, Chem. Commun., 2010, 46, 3092 RSC.
- (a) G. Alcaraz, L. Vendier, E. Clot and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 49, 918 CrossRef CAS PubMed; (b) G. Alcaraz, A. B. Chaplin, C. J. Stevens, E. Clot, L. Vendier, A. S. Weller and S. Sabo-Etienne, Organometallics, 2010, 29, 5591 CrossRef CAS; (c) G. Bénac-Lestrille, U. Helmstedt, L. Vendier, G. Alcaraz, E. Clot and S. Sabo-Etienne, Inorg. Chem., 2011, 50, 11039 CrossRef PubMed; (d) C. J. Wallis, H. Dyer, L. Vendier, G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 3646 CrossRef CAS PubMed.
- (a) C. Y. Tang, N. Phillips, J. I. Bates, A. L. Thompson, M. J. Gutmann and S. Aldridge, Chem. Commun., 2012, 48, 8096 RSC; (b) C. Y. Tang, A. L. Thompson and S. Aldridge, Angew. Chem., Int. Ed., 2010, 49, 921 CrossRef CAS PubMed; (c) D. Vidovic, D. A. Addy, T. Krämer, J. McGrady and S. Aldridge, J. Am. Chem. Soc., 2011, 133, 8494 CrossRef CAS PubMed; (d) C. Y. Tang, A. L. Thompson and S. Aldridge, J. Am. Chem. Soc., 2010, 132, 10578 CrossRef CAS PubMed.
- (a) A. Friedrich, M. Dress and S. Schneider, Chem. – Eur. J., 2009, 15, 10339 CrossRef CAS PubMed; (b) A. N. Marziale, A. Friedrich, I. Klopsch, M. Drees, V. R. Celinski, J. S. Günne and S. Schneider, J. Am. Chem. Soc., 2013, 135, 13342 CrossRef CAS PubMed.
- For the intramolecular coordination of Si–H and Si–Si bonds to Cu(I) see: (a) P. Gualco, A. Amgoune, K. Miqueu, S. Ladeira and D. Bourissou, J. Am. Chem. Soc., 2011, 133, 4257 CrossRef CAS PubMed; (b) P. Gualco, S. Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2011, 50, 8320 CrossRef CAS PubMed; (c) M. Joost, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Organometallics, 2013, 32, 898 CrossRef CAS; (d) N. Lassauque, P. Gualco, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13827 CrossRef CAS PubMed.
- Z. Ribas, C. Calle, A. Poater, A. Casitas, L. Gómez, R. Xifra, T. Parella, J. Benet-Buchholz, A. Schweiger, G. Mitrikas, M. Solà, A. Llobet and T. D. P. Stack, J. Am. Chem. Soc., 2010, 132, 12299 CrossRef PubMed.
- A. E. Nako, Q. W. Tan, A. J. P. White and M. R. Crimmin, Organometallics, 2014, 33, 2685 CrossRef CAS.
- For the synthesis of 12·toluene and related complexes see: (a) Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961 CrossRef CAS PubMed; (b) D. S. Laitar, C. J. N. Mathison, W. M. Davis and J. P. Sadighi, Inorg. Chem., 2003, 42, 7354 CrossRef CAS PubMed.
- (a) G. Alcaraz, E. Clot, U. Helmstedt, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2007, 129, 8704 CrossRef CAS PubMed; (b) G. Alcaraz, U. Helmstedt, E. Clot, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2008, 130, 12878 CrossRef CAS PubMed; (c) Y. Gloaguen, G. Bénac-Lestrille, L. Vendier, U. Helmstedt, E. Clot, G. Alcaraz and S. Sabo-Etienne, Organometallics, 2013, 32, 4868 CrossRef CAS; (d) Y. Gloaguen, G. Alcaraz, L. Vendier and S. Sabo-Etienne, J. Organomet. Chem., 2009, 694, 2839 CrossRef CAS PubMed.
- (a) Y. Kawano, T. Kakizawa, K. Yamaguchi and M. Shimoi, Chem. Lett., 2006, 35, 568 CrossRef CAS; (b) A. Ariafard, M. M. Amini and A. Azadmehr, J. Organomet. Chem., 2005, 690, 1147 CrossRef CAS PubMed.
- (a) M. Shimoi, K. Katoh, H. Tobita and H. Ogino, Inorg. Chem., 1990, 29, 814 CrossRef CAS; (b) A. Wagner, E. Kaifer and H.-J. Himmel, Chem. – Eur. J., 2013, 19, 7395 CrossRef CAS PubMed.
- There is an additional minor species present at these temperatures that we have been unable to assign. Chemical shift data are similar to the arene complex 1·toluene and this may be the dimer or an isotopomer of this species.
- D. van der Waals, A. Pettman and J. M. J. Williams, RSC Adv., 2014, 4, 51845 RSC.
- D. F. Schreiber, C. O'Connor, C. Grave, Y. Ortin, H. Müller-Bunz and A. D. Phillips, ACS Catal., 2012, 2, 2505 CrossRef CAS.
- X. Hu, M. Soleilhavoup, M. Melaimi, J. Chu and G. Bertrand, Angew. Chem., Int. Ed., 2015, 54, 6008 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, VT NMR data, crystallographic data, and details of the DFT calculations. CCDC 1048449–1048450. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02144h |
| This journal is © The Royal Society of Chemistry 2015 |