
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimer formation upon deprotonation: synthesis and structure of a m-terphenyl substituted (R,S)-dilithium disiloxanolate disilanol†
Daniel
Čas
a,
Natascha
Hurkes
a,
Stefan
Spirk
b,
Ferdinand
Belaj
b,
Clemens
Bruhn
a,
Gerald N.
Rechberger
c and
Rudolf
Pietschnig
*a
aUniversität Kassel, Institut für Chemie und CINSaT, Heinrich-Plett-Straße 40, 34132 Kassel, Germany. E-mail: pietschnig@uni-kassel.de
bKarl-Franzens-Universität, Institut für Chemie, NAWI Graz, Schubertstraße 1, 8010 Graz, Austria
cKarl-Franzens-Universität, Institut für Molekulare Biowissenschaften, Humboldtstraße 50, 8010 Graz, Austria
First published on 15th June 2015
Abstract
The synthesis and structural characterization of the first dilithium salt of a tetrahydroxydisiloxane, [DmpSi(OH)OLi]2O (6), is described (Dmp = 2,6-dimesitylphenyl). The solid state structure reveals the presence of a dimeric motif where two disiloxane units are linked by coordinating lithium atoms which differs from those found for the sodium and potassium analogs. The arrangement imposed by the cluster formation leads to diastereomeric silicon atoms exhibiting (R,S) configuration in the solid state. In addition, the intermediates of the reaction, monolithiated and dilithiated silanetriol could be identified by means of high-resolution mass spectrometry and the formation of 6 is discussed. Moreover, the fully protonated tetrahydroxydisiloxane, [DmpSi(OH)2]2O has been obtained as well and its solid state structures with varying hydrogen bond acceptors have been surveyed.
Introduction
The targeted synthesis and structural characterization of complex polyhedral clusters involving main group elements has been a surging topic in supramolecular chemistry.1–3 A groundbreaking area of research in this context is the field of metallasiloxanes and heterometallasiloxanes.4–7 These compounds can serve as soluble models for metal grafted silica surfaces which are widely used in catalysis e.g. V2O5/SiO2 for C–H oxidation reactions.8–11 Usually the synthesis of metallasiloxanes starts from silanols,12–14 which are treated with Lewis acids, usually metal salts to form the Si–O–M linkage. Consequently, the obtained products are mostly compounds lacking free OH-groups at silicon as described in the literature.6,15,16 By contrast, partial metallation with organometallic reagents has been shown to yield valuable building blocks suitable for the construction of polyhedral frameworks.17 Recently, we have been able to observe intermediates in the formation of siloxane polymers and cages by stabilizing them kinetically using sterically demanding substituents.18–21 It seems promising to pursue a similar strategy for metallasiloxanes, which should allow obtaining small oligomers with a remaining OH-functionality at the silicon center besides the metal-silanolate functionality. Stable partially metallated silanols should be useful precursors for either anchoring (grafting) on the surface of OH-rich substrates or the preparation of heterobimetallic metallasiloxanes.7 Herein, we describe the partial metallation of the sterically demanding DmpSi(OH)3 (Dmp = 2,6-dimesitylphenyl)18 with n-BuLi and its follow up products, i.e. the corresponding tetrahydroxy disiloxane derivatives and adducts.Results and discussion
Deprotonation of silanols occurs easily and their acidity is usually higher than that of their carbon analogs. Strong bases like n-BuLi or NaH have proven to be suitable for such purposes and with such anionic bases the proton may be removed in a neutral compound from the equilibrium (e.g. H2, n-butane).20 As a simple model system we decided to investigate the reaction of n-BuLi and silanetriol 1 in THF at −78 °C. An immediate reaction took place accompanied by a moderate warming of the reaction mixture. Upon addition of 1 eq. of butyllithium the only isolated product was, however, not the expected singly deprotonated silanetriol 2 but the doubly deprotonated dilithium disiloxanolate, 6 (Scheme 1) which could be crystallized directly from the reaction mixture. In the solid state structure of 6, the asymmetric unit contains two molecules of 6 which are connected by coordination of four lithium atoms out of which two are crystallographically independent (Fig. 1). There are three types of silicon–oxygen bonds present in 6 reflecting different bonding situations. The Si–O bonds of the lithium silanolate units exhibit the shortest bond lengths with values of 1.584(2) and 1.577(2) Å. In contrast, the other Si–O bond lengths are significantly larger with 1.6586(16)–1.6590(16) Å for the siloxane oxygen atoms and 1.670(2)–1.678(2) Å for the silanol ones.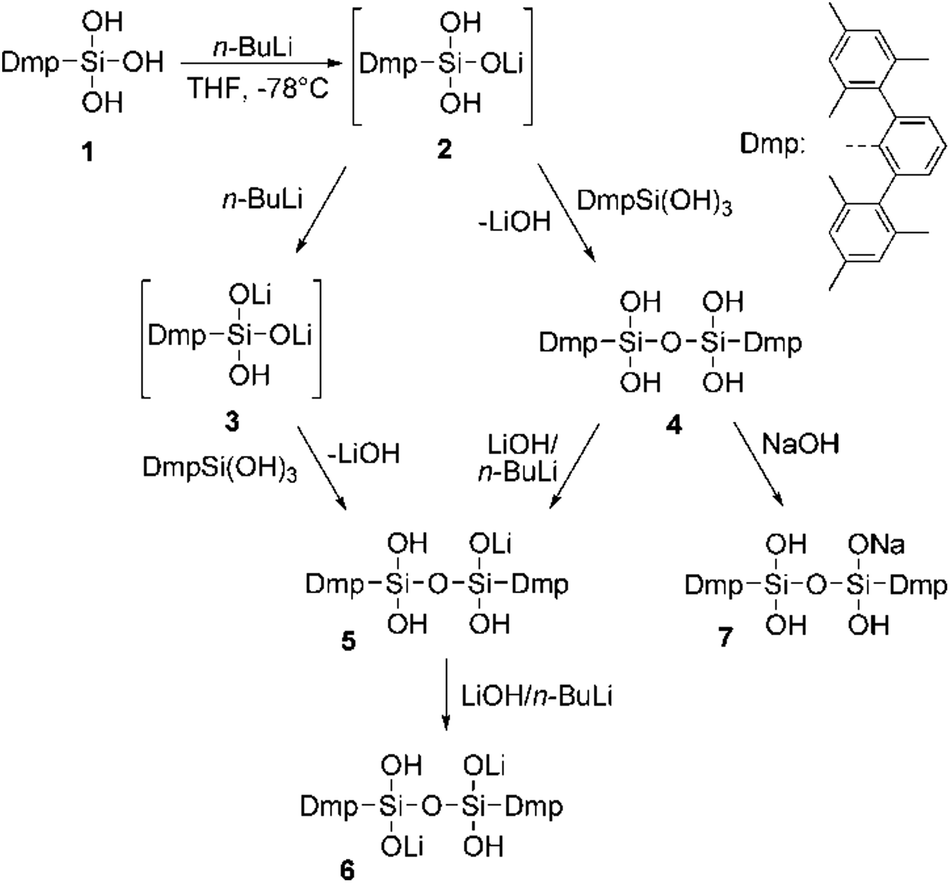 | ||
| Scheme 1 Formation of 6. | ||
 | ||
| Fig. 1 ORTEP22 plot of 6 showing the atomic numbering scheme. The probability ellipsoids are drawn at the 30% probability level and the H atoms of the phenyl rings and of the methyl groups were omitted for clarity reasons. The shortest contacts between the Li atoms and the O atoms are plotted with dashed lines. | ||
The shortened Si–O distances in the silanolate units can be explained by increased negative hyperconjugation owing to the increased negative charge at oxygen. As expected, the silanolate-oxygen atoms are better coordination sites than the silanol-oxygen atoms which is reflected in the Li–O distances and the number of coordinating lithium atoms. While for the silanol-oxygen, Li–O distances of 2.092(6) (Li2′–O2) and 2.064(6) Å (Li1′–O5) are found, coordination of two lithium atoms to each silanolate functionality is realized resulting in the formation of a dimeric motif. The distances range from 1.856(6) (Li1–O3) and 1.858(6) Å (Li2–O6′) for the stronger Li–O interactions to 1.901(6) (Li2–O3) and 1.917(6) Å (Li1–O6) indicating a slightly weaker coordination of the lithium ions. The environment around the lithium atoms can be considered as distorted trigonal planar with values for the O–Li–O angles ranging from 110.2(3) to 123.4(3)° (Σ: 343.7° for Li1 and 346.7° for Li2; deviation of Li1 and Li2 from the least-squares plane formed by the three coordinating oxygen atoms: 0.457(13) and 0.414(20) Å). Each lithium ion is coordinated to three oxygen atoms but there are also contacts to other lithium atoms with Li–Li distances of 2.660(8) (Li1–Li2′) and 2.703(8) Å (Li2–Li1′) while the distances of symmetry equivalent lithium atoms (Li1–Li1′, Li2–Li2′) are significantly longer with values of 3.357(11) and 3.283(11) Å. Even the shorter distances can however not be interpreted in terms of direct bonding interaction. Surprisingly, there is hardly any interaction between lithium ions and the π-system of the flanking mesityl rings which may originate from steric reasons (shortest Li⋯Caryl distance: 2.874(6) Å). A consequence of the steric pressure is the absence of intra- and intermolecular hydrogen bonding between silanol groups. The lack of suitable acceptors for the silanol hydrogen atoms entails coordination to the only other available coordination site, i.e. the π-system of the flanking mesityl rings. Similar to other m-terphenyl substituted silanols, the hydroxyl groups are directed towards these rings showing OH⋯Caryl minimum distances of 2.581(7) (O5–H5⋯C51) and 2.604(11) Å (O2–H2⋯C35). The Si–O–Si linkages show significant bending with values of 119.28(17)° and 119.20(17)°.
Although 6 exhibits very small Si–O–Si angles, similar values are found for related cases, e.g. in a tetrasilanolate tantalum-compound23 which has a Si–O–Si value of 113.9°. While typical Si–O–Si angles in siloxanes are in the range 135°–180° the barrier for deformation is quite low and angles near 90° have been observed for 1,3-cyclodisiloxanes.24 Beckmann and coworkers have outlined that very small Si–O–Si angles are accompanied by an increased basicity of the siloxanes oxygen atom which in turn increases the acidity of the adjacent silicon atoms.25 This increased ionic character may as well be responsible for the reduced Si–O bond lengths of the pending silanolate units.
The crystal structure reveals the presence of (R,S) diastereomers in the solid state, arising from the relative orientation of the lithiated sites per disiloxane unit. In solution, only a single resonance at −59 ppm was found in the 29Si-NMR spectra indicating the absence of other diastereomers (R,R/S,S) or a low barrier for interconversion of the diastereomers even in non-polar solvents like benzene. In agreement with this, the 1H- and 13C-NMR spectra show only one set of signals in the expected range. Additional geometric parameters can be found in the ESI.† Related to these findings, the singly deprotonated disiloxane with potassium as counter ion has been obtained starting from DmpSiF3 in the reaction with an excess of KOH.26 A major difference between these cases accounts for the different aggregation behavior of lithium compared to potassium compounds in general (and lithium silanolates) which is well documented in literature. The “softer” potassium ion is capable of interacting more efficiently with π-systems of aromatic rings and its ion potential is moreover lower than that of the smaller lithium ion. Consequently, there are plenty of K-aryl interactions documented for terphenyl substituted silicon compounds19,27 but none for lithium which is further corroborated by the structure of 6.
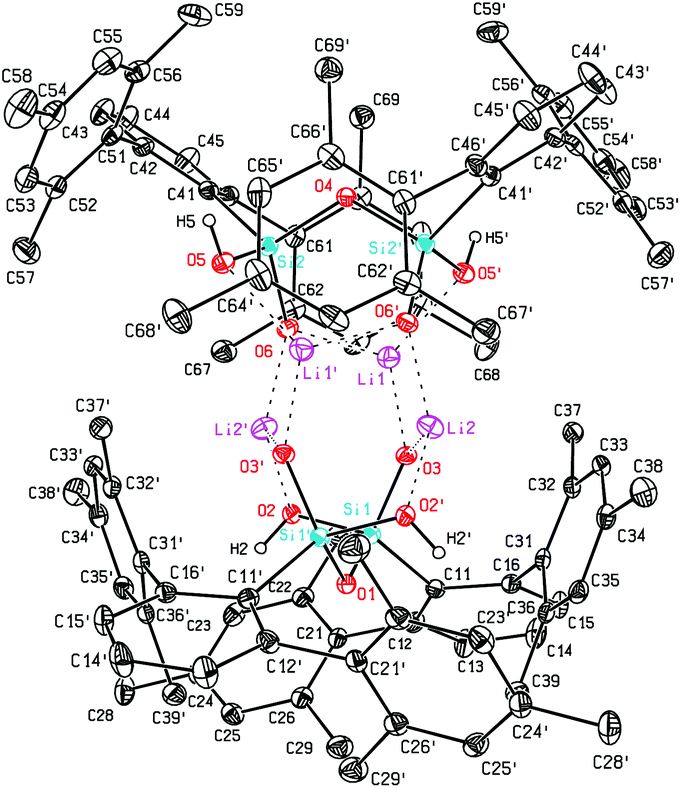
Using NaOH instead of KOH we were also able to obtain 7, the sodium analog of 5. Also for 7 crystals suitable for X-ray diffraction could be obtained (Fig. 2). The structural motif in the solid state is very comparable with that of its potassium analog.26 In addition to the Na–O interactions the dimeric cluster is held together by hydrogen bonds (O–H⋯O) with donor acceptor distances of 2.428(5) Å and 2.431(5) Å and O–H⋯O angles of 155(4)° and 158(4)°. Sodium aryl interactions are not as dominant as in the case of potassium, but distances as low as 3.025(7) Å (Na2–C44) can be observed, which is less than the sum of the corresponding van der Waals radii.
 | ||
| Fig. 2 Molecular structure of 7. | ||
This series of deprotonated tetrahydroxydisiloxanes with Dmp substituents reveals similarities in the clusters bridged by Na and K cations but marks the difference to the lithium bridged cluster 6 where Li⋯O interactions outpace hydrogen bonds.
Formation
The unexpected failure to isolate the mono-deprotonated species 2 triggered us to investigate the situation in the reaction mixture spectroscopically. Although the initial formation of such a mono-deprotonated silanetriol is plausible, no evidence could be found by means of 29Si-NMR spectroscopy. In solution, only the formation of a doubly deprotonated tetrahydroxy disiloxane is observed based on NMR spectroscopy. It seems that the initially formed monolithium silanolate readily attacks another silanetriol molecule to form a siloxane bond under elimination of lithium hydroxide which, in turn, can deprotonate one of the OH groups of the disiloxane. It needs to pointed out that data aquisition of 29Si-NMR spectra is quite time consuming since aryl silantriols show long longitudinal relaxation times28 and the other available nuclei (1H,13C, 6/7Li) are not able to distinguish between the closely related alternative products (Scheme 1).In order to get additional information on this reaction with a different technique, ESI-HiRes-MS experiments were performed. ESI-MS is known as a very soft ionization technique and therefore should be the ideal technique to identify polar compounds and to follow the aggregation behavior of lithiated silanetriols and tetrahydroxydisiloxanes in solution.27 ESI-MS measurements of the freshly prepared reaction solution revealed that the reaction mixture already contained significant amounts of lithiated disiloxane 5 (m/z = 773.3680, Δ = 1.3 ppm) after a reaction time of two hours. In addition, 2 (m/z = 399.1995, Δ = 6.8 ppm) as well as 3 (m/z = 405.2049, Δ = 0.2 ppm) could be detected and unambiguously identified. Nevertheless, it should be mentioned that 2 and 3 were observed as traces and their abundance is close to the detection limit of the used equipment. The tetrahydroxy disiloxane 4 (m/z = 767.3606, Δ = 2.3 ppm) was observed in larger quantities which is an indication that the reaction proceeds likely via2 rather than 3. Compound 6 was also characterized by HiRes mass spectrometry (m/z = 779.3752, Δ = 3.2 ppm), however, we always observed 5 as the major component in solution. As we know from our X-ray crystallographic studies, 6 is extremely sensitive towards humidity and therefore we suggest that 6 is hydrolyzed to yield 5 under the conditions used for ESI-MS.
These findings suggest that the deprotonated silanetriols 2 and 3 are initially formed but are too reactive to be isolated in substance because of their tendency to convert to disiloxanes. The latter are easily deprotonated under the reaction conditions (dropwise additon of n-BuLi) either by previously eliminated LiOH or further butyllithium added during the course of the reaction (Scheme 1).
Tetrahydroxy disiloxane
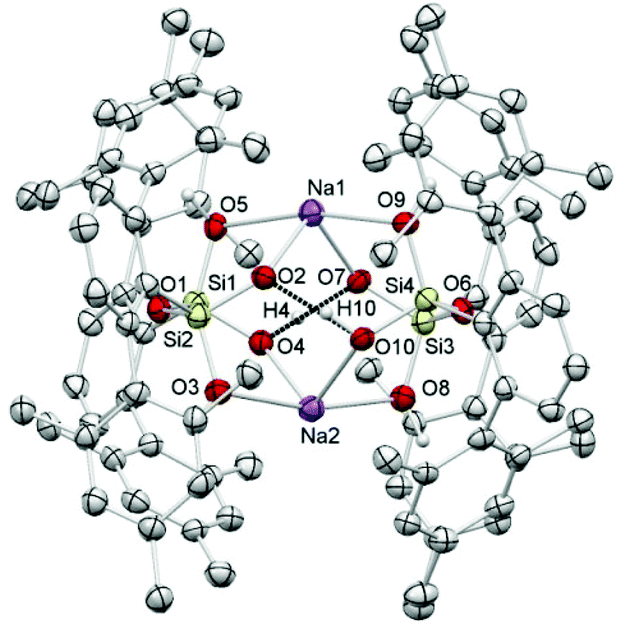
The here reported compounds 5–7 represent deprotonated versions of the parent tetrahydroxy disiloxane 4 which has not been described in the literature so far. However, the latter can be easily accessed from its deprotonated derivatives by reaction with mild acids like acetic acid or ammonium chloride. A more straightforward synthetic route starts from silanetriol 1 which on treatment with a weak nitrogen base like piperazine furnishes tetrahydroxy disiloxane 4 as well. The 29Si-NMR signal of 4 varies slightly with the solvent resonating in the range −58 ppm (C6D6) to −62 ppm (THF) which is close to the lithiated derivative 6 but more shielded compared with silanetriol 1. Also the proton resonances of the silanol units are quite variable depending on solvent and concentration which is indicative for solution equilibria as in related cases.29 In the solid state tetrahydroxy disiloxane 4 shows isolated tetrahydroxy disiloxane units without intermolecular hydrogen bonding. This structural motif differs substantially from those found for the metallated derivatives (cf. 6 and ref. 26) as well as from disiloxanes with tert-alkyl substituted tetrahydroxy disiloxanes which form extended hydrogen bonded networks.21 Nevertheless, compound 4 is prone to interaction especially with hydrogen bond acceptors like donor solvents as is evident from solid state structures from different solvents which incorporate one solvent molecule interacting with one of the silanol units of 4. Without donor solvent we find one intramolecular and no intermolecular hydrogen bond (Fig. 3). With THF as donor we find one intermolecular but no intramolecular hydrogen bond (Fig. 4).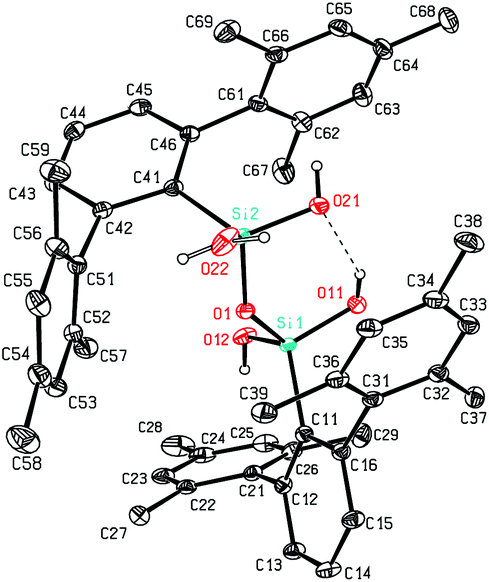 | ||
| Fig. 3 Molecular structure of 4. | ||
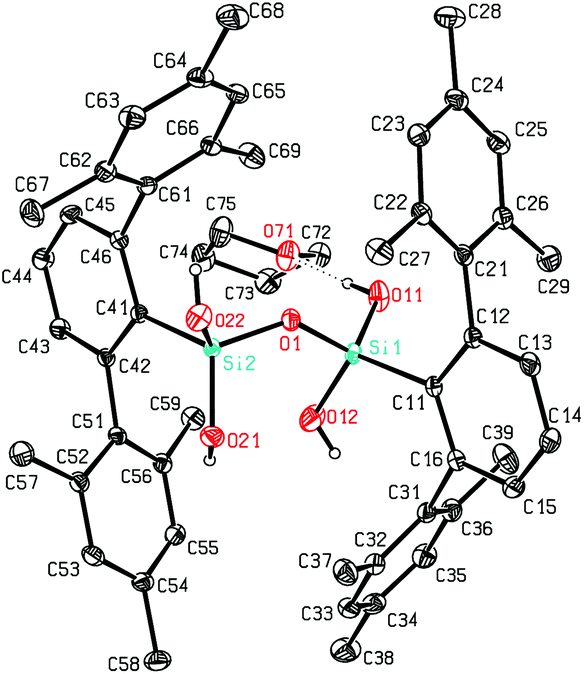 | ||
| Fig. 4 Molecular structure of 4·THF. | ||
In the case of dmf as donor we find one intramolecular and one intermolecular hydrogen bond involving the formyl oxygen atom as hydrogen bond acceptor (Fig. 5). The hydrogen bonds occurring in these three scenarios differ substantially. Thus, the hydrogen bonds involving the donor molecules are stronger than the intramolecular interactions based on their structural parameters.30 While the intermolecular O–H⋯O arrangements show angles close to 170° and H⋯O distances below 2 Å, the intramolecular interactions show smaller O–H⋯O angles and O⋯H distances beyond 2 Å (Table 1). Also the central Si–O–Si angle of the disiloxane unit is affected by the donor interaction of the adjacent silanol units. It is largest for 4 without donor and smallest for the adduct 4·dmf.
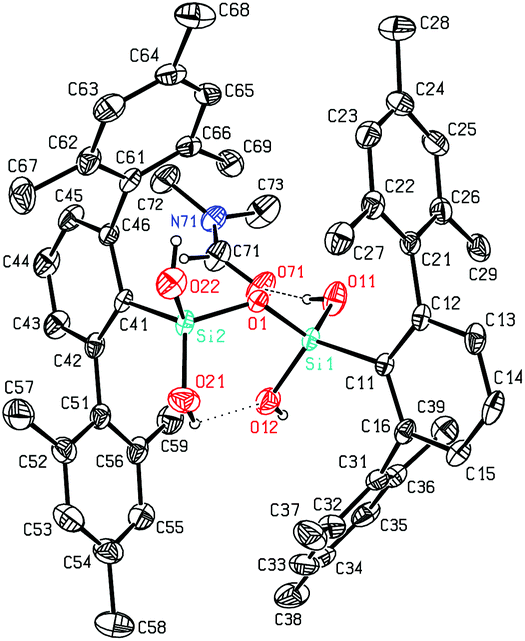 | ||
| Fig. 5 Molecular structure of 4·DMF. | ||
| Compound | D–H⋯A | d(H⋯A) | d(D⋯A) | <(DHA) | <(Si–O–Si) | Type |
|---|---|---|---|---|---|---|
| 4 | O(11)–H(11)⋯O(21) | 2.137(3) | 2.918(2) | 154.7(6) | 137.58(9) | Intra |
| 4·thf | O(11)–H(11)⋯O(71) | 1.945(2) | 2.7800(12) | 172.4(16) | 136.22(5) | Inter |
| 4·dmf | O(11)–H(11)⋯O(71) | 1.858(7) | 2.692(4) | 172(4) | 132.35(17) | Inter |
| 4·dmf | O(21)–H(21)⋯O(12) | 2.34(4) | 2.956(5) | 131(4) | 132.35(17) | Intra |
Synthetic procedures
All manipulations were carried out under strict exclusion of moisture and air in an inert argon atmosphere. All used solvents were dried over potassium or CaH2 and distilled prior to use. DmpSi(OH)3 was synthesized according to a literature procedure.311H, 13C and 29Si-NMR spectra were recorded at a Bruker Avance III at a larmor frequency of 300 MHz for 1H using TMS as reference and C6D6 as solvent. IR spectra were recorded as neat substance using an Alpha Platinum ATR spectrometer (Bruker). Electrospray mass spectra have been recorded on a Thermo LCQ-DUO ion trap mass spectrometer (Thermo, San Jose, CA, USA) equipped with a nano-ESI-source using metal coated nano-ESI-needles (Proxxeon, Odense, Denmark; outer diameter, 360 μm; inner diameter, 75 μm), employing a spray voltage of 1.5 kV and concentrations of 0.5–1.0 mg ml−1 in CHCl3. High resolution mass spectrometry was performed in direct infusion (3 μl min−1) on a LTQ-FT hybrid mass spectrometer equipped with an IonMax ESI source (Thermo). The mass spectrometer was operated at an accuracy of <3 ppm with external calibration and a resolution of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 FWHH at m/z 400. The spray voltage was set to 4500 V, capillary voltage to 35 V and the tube lens was at 150 V. Capillary temp. was at 200 °C.
000 FWHH at m/z 400. The spray voltage was set to 4500 V, capillary voltage to 35 V and the tube lens was at 150 V. Capillary temp. was at 200 °C.
X-ray diffraction measurements were performed on a Stoe 4CD or a Bruker-AXS SMART APEX 2 CCD diffractometer using graphite-monochromated Mo Kα radiation or a Stoe Stadi Vari with Dectris Pilatus 200 K detector using monochromated Cu Kα radiation. The structures were solved using direct methods (SHELXL-97) and refined by full-matrix least-squares techniques against F2 (SHELXL-2014/6).32 Details of the structure determinations and refinement for 6, 4, 4·THF, 4·DMF and 7 are summarized in Table S1 (ESI†). Supplementary crystallographic data for this paper can be obtained free of charge by quoting CCDC 1059825–1059828 and 1062439 from the Cambridge Crystallographic Data Centre.
Conclusions
The reaction of silanetriol 1 with one equivalent of n-BuLi does not lead to a stable monodeprotonated silanetriol species but to a doubly deprotonated disiloxane disilanolate 6 which can be isolated from the reaction mixture in good yields. By means of ESI-HiRes-MS a monodeprotonated silanetriol as well as a doubly deprotonated silanetriol could be observed however, and were unambiguously identified, albeit in small intensity. These findings suggest that nucleophilic attack of the lithiated silanetriol on other silanetriol molecules proceeds rapidly yielding a disiloxane which is subsequently deprotonated and lithiated under the reaction conditions. Structural comparison with the previously unknown fully protonated tetrahydroxy disiloxane indicates that the stronger O–Li⋯O interaction (compared with O–H⋯O) is responsible for the dimeric solid state structure of 6. While 4 is only prone to interaction with bases, its lithiated congener 6 provides basic binding sites itself. This makes compound 6 a promising candidate for the preparation of novel heterobimetallic metallasiloxanes or as building block for condensation or grafting to surfaces and polymer strands.Acknowledgements
This paper is dedicated to Prof. Dr Manfred Scheer on the occasion of his 60th birthday. The authors thank the Austrian BMWF for financial support within the PhD network FreCheMaterie and the Austrian Science Fund, FWF, (Grants No. P 20575-N19 and P 17882-N11) for financial support. We also thank the EU-COST network CM1302 “Smart Inorganic Polymers” (SIPs).Notes and references
- J. Bai, A. V. Virovets and M. Scheer, Science, 2003, 300, 781–783 CrossRef CAS PubMed.
- F. Dielmann, C. Heindl, F. Hastreiter, E. V. Peresypkina, A. V. Virovets, R. M. Gschwind and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 13605–13608 CrossRef CAS PubMed.
- Y. Lin, W. Massa and S. Dehnen, J. Am. Chem. Soc., 2012, 134, 4497–4500 CrossRef CAS PubMed.
- R. Murugavel, V. Chandrasekhar and H. W. Roesky, Acc. Chem. Res., 1996, 29, 183–189 CrossRef CAS.
- R. Murugavel, A. Voigt, M. G. Walawalkar and H. W. Roesky, Chem. Rev., 1996, 96, 2205–2236 CrossRef CAS PubMed.
- V. Lorenz and F. T. Edelmann, Adv. Organomet. Chem., 2005, 101–153 CAS.
- V. Lorenz, A. Fischer, S. Gießmann, J. W. Gilje, Y. Gun'ko, K. Jacob and F. T. Edelmann, Coord. Chem. Rev., 2000, 206–207, 321–368 CrossRef CAS.
- E. F. Aboelfetoh and R. Pietschnig, Catal. Lett., 2009, 127, 83–94 CrossRef CAS.
- E. F. Aboelfetoh and R. Pietschnig, Catal. Lett., 2014, 144, 97–103 CrossRef CAS.
- R. Duchateau, Chem. Rev., 2002, 102, 3525–3542 CrossRef CAS PubMed.
- R. Duchateau, T. W. Dijkstra, R. A. van Santen and G. P. A. Yap, Chem. – Eur. J., 2004, 10, 3979–3990 CrossRef CAS PubMed.
- P. D. Lickiss, Adv. Inorg. Chem., 1995, 42, 147–262 CAS.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- P. D. Lickiss, in Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley, Chichester, UK, 2001, vol. 3, pp. 695–744 Search PubMed.
- F. J. Feher and T. L. Tajima, J. Am. Chem. Soc., 1994, 116, 2145–2146 CrossRef CAS.
- F. J. Feher, K. Rahimian, T. A. Budzichowski and J. W. Ziller, Organometallics, 1995, 14, 3920–3926 CrossRef CAS.
- M. L. Montero, I. Usón and H. W. Roesky, Angew. Chem., Int. Ed. Engl., 1994, 33, 2103–2104 CrossRef PubMed.
- R. Pietschnig, F. Belaj and J. J. Tirrée, Organometallics, 2004, 23, 4897–4901 CrossRef CAS.
- R. Pietschnig and K. Merz, J. Chem. Soc., Chem. Commun., 2001, 1210–1211 RSC.
- S. Spirk, M. Nieger, F. Belaj and R. Pietschnig, Dalton Trans., 2009, 163–167 RSC.
- N. Hurkes, C. Bruhn, F. Belaj and R. Pietschnig, Organometallics, 2014, 33, 7299–7306 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–566 CrossRef CAS.
- A. I. Gouzyr, H. Wessel, C. E. Barnes, H. W. Roesky, M. Teichert and I. Uson, Inorg. Chem., 1997, 36, 3392 CrossRef CAS.
- M. J. Michalczyk, M. J. Fink, K. J. Haller, R. West and J. Michl, Organometallics, 1986, 5, 531–538 CrossRef CAS.
- S. Grabowsky, M. F. Hesse, C. Paulmann, P. Luger and J. Beckmann, Inorg. Chem., 2009, 48, 4384–4393 CrossRef CAS PubMed.
- R. Pietschnig and K. Merz, Organometallics, 2004, 23, 1373–1377 CrossRef CAS.
- S. Spirk, F. Belaj, M. Nieger, H. Köfeler, G. N. Rechberger and R. Pietschnig, Chem. – Eur. J., 2009, 15, 9521–9529 CrossRef CAS PubMed.
- S. Spirk, T. Madl and R. Pietschnig, Organometallics, 2008, 27, 500–502 CrossRef CAS.
- H. Liu, S.-i. Kondo, R. Tanaka, H. Oku and M. Unno, J. Organomet. Chem., 2008, 693, 1301–1308 CrossRef CAS PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- S. Spirk, F. Belaj, J. Baumgartner and R. Pietschnig, Z. Anorg. Allg. Chem., 2009, 635, 1048–1053 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta. Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Text, tables, figures and CIF files giving crystallographic data for 6, 7, 4, 4·THF and 4·DMF. CCDC 1059825–1059828 and 1062439. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01992c |
| This journal is © The Royal Society of Chemistry 2015 |