DOI:
10.1039/C5DT01925G
(Paper)
Dalton Trans., 2015,
44, 17209-17221
Bridged bis-BODIPYs: their synthesis, structures and properties†
Received
22nd May 2015
, Accepted 25th August 2015
First published on 25th August 2015
Abstract
A series of bis-BODIPYs 1–6 bridged via thiophene, furan, N-alkylcarbazole, triphenyl-amine, para- and meta-phenylene groups have been synthesized and characterized by various spectroscopic techniques. The change in the spectroscopic properties of bis-BODIPYs upon varying the size of spacers was studied. X-ray crystal structures of three bis-BODIPYs containing triphenylamine, para- and meta-phenylene bridges were solved. Intermolecular C(H)⋯π and π⋯π stacking interactions were observed in solid state structures of three bis-BODIPYs. The dihedral angles between the spacer unit and two boron-dipyrrin units were lower in all three compounds as compared to their corresponding monomers. This suggests increased interactions between the two boron-dipyrrin units in molecules which are in turn reflected in the anodic shifts in their reduction potentials. DFT studies indicated effective electronic interactions between spacers and two boron dipyrrin units in all the bis-BODIPYs. The calculated HOMO–LUMO gap was found to be lower for bis-BODIPY having bulky carbazole spacers and higher for bis-BODIPY having smaller furan spacers. Changing the spacer size clearly affected the spectroscopic properties of the bis-BODIPYs and red shifted absorption and emission maxima were observed for bis-BODIPYs with furan and thiophene spacers as compared to bis-BODIPYs with phenylene or bulky aromatic spacers.
Introduction
Covalently linked pairs of chromophores have been a matter of interest for the scientific community for a long time. Such chromophore dimers have been found in biology, for example, photosynthetic bacterial proteins and natural light harvesting antenna systems.1–3 Numerous artificial fluorescent dyes have been used in biosciences for cell imaging and investigation of in vivo biological processes. The boron dipyrromethenes (BODIPYs) are a well-known class of highly emissive molecules and they have been found to be extremely useful for biological applications, e.g. biochemical labelling,4–6 photosensitizers for PDT (Photo Dynamic Therapy)7 and fluorescent probes.8–10 The electronic and photophysical properties of BODIPYs can be fine-tuned for different applications by the structural modification of the boron-dipyrrin core or by making their dimers and oligomers.11 Substitution at meso positions of the boron dipyrrin core with bulky electron rich heterocycles could produce large Stokes shifts of 100–120 nm.12 β–β Linked dimers, meso–meso linked dimers and cofacial BODIPY dimers have been synthesised and studied.13–15 Such artificial BODIPY dimers possess increased Stokes shifts, broad absorption and red shifted emission for efficient light harvesting ability for solar cell and biological applications. Recently Akkaya et al. have demonstrated that meso–meso and meso-beta linked BODIPY dimers can be used as singlet oxygen photosensitizers in nonpolar organic media.16 Li and co-workers have synthesized β–β linked BODIPY dimers with triphenylsilylphenyl groups for increased solid state emission.17 Bithiophene bridged BODIPY dimers have been prepared by Ziessel and co-workers with red shifted absorption and emission for future application as NIR fluorescent probes.18 Interesting symmetry breaking ICT (intramolecular charge transfer) properties have been observed for p-phenylene bridged α-alkylated BODIPY dimers.19 Nishihara and co-workers have reported p-diethynylphenyl bridged BODIPY dimers and meso-alkynyl BODIPY with red shifted emission maxima.20 Liu et al. have synthesized triphenylamine BODIPY monomers, bridged dimers and trimers in a one pot reaction and their lasing properties in organic media were studied.21 BODIPY dimers having bipyridine spacers with interesting ECL (electrogenerated chemiluminescence) properties have been reported by Bard and co-workers.22 Benniston and co-workers have synthesized cofacial BODIPY dimers containing dibenzothiophene and dibenzofuran moieties; such dimers formed intramolecular excimers in emission studies.23 Saki et al. have designed cofacial BODIPY dimers based on the xanthene scaffold with efficient energy transfer between the two boron dipyrrin units.24 Reports on covalently linked BODIPY dimers are on the rise but still there is a need to study the photophysical properties of meso-phenylene and heterocycle bridged BODIPY dimers. In this article we present the synthesis, crystal structures, photophysical and electrochemical properties of thiophene, furan, N-butyl carbazole, triphenylamine, p- and m-phenylene bridged bis-BODIPYs. Also, DFT studies were carried out for all six bis-BODIPYs and the HOMO–LUMO energy gap was calculated.
Results and discussion
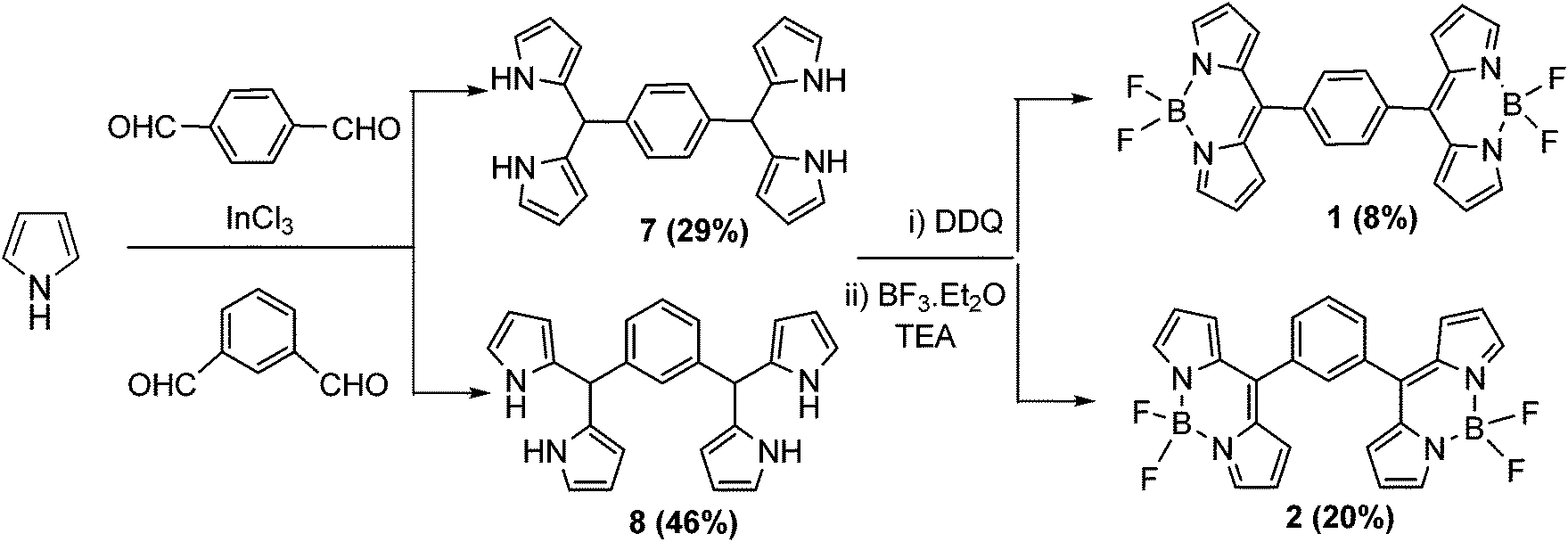
The stepwise synthesis of bis-BODIPYs 1–6 is depicted in Schemes 1–3. The corresponding key precursors such as bis-aromatic aldehydes were prepared by a Vilsmeier–Haack formylation reaction and bis-dipyrranes 7–12 were synthesized as per a reported procedure.25 Terephthalaldehyde and isophthalaldehyde were condensed with pyrrole in the presence of a catalytic amount of indium chloride to form bis-dipyrranes 7 and 8 respectively (Scheme 1). Crude dipyrranes 7 and 8 were purified by silica gel column chromatography using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4 mixture of ethyl acetate/dichloromethane/petroleum ether in 29% and 46% yields respectively. The corresponding bis-aldehydes for bis-dipyrranes 9 and 10 were prepared by a Vilsmeier–Haack formylation reaction as per a reported procedure25 (ESI†). Bis-dipyrranes 9 and 10 were synthesized by the indium chloride method25 followed by silica gel column purification in 55% and 41% yields respectively (Scheme 2). The bis-dipyrranes 7–10 were oxidized by 2.4 equivalents of DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) and the resulting bis-dipyrrins were then complexed with BF3·OEt2 by doubling the equivalents of reagents required for the standard BODIPY synthesis.26
:2:4 mixture of ethyl acetate/dichloromethane/petroleum ether in 29% and 46% yields respectively. The corresponding bis-aldehydes for bis-dipyrranes 9 and 10 were prepared by a Vilsmeier–Haack formylation reaction as per a reported procedure25 (ESI†). Bis-dipyrranes 9 and 10 were synthesized by the indium chloride method25 followed by silica gel column purification in 55% and 41% yields respectively (Scheme 2). The bis-dipyrranes 7–10 were oxidized by 2.4 equivalents of DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) and the resulting bis-dipyrrins were then complexed with BF3·OEt2 by doubling the equivalents of reagents required for the standard BODIPY synthesis.26
 |
| | Scheme 1 Synthetic route for bis-BODIPYs 1 and 2. | |
 |
| | Scheme 2 Synthetic route for bis-BODIPYs 3 and 4. | |
 |
| | Scheme 3 Synthetic route for bis-BODIPYs 5 and 6. | |
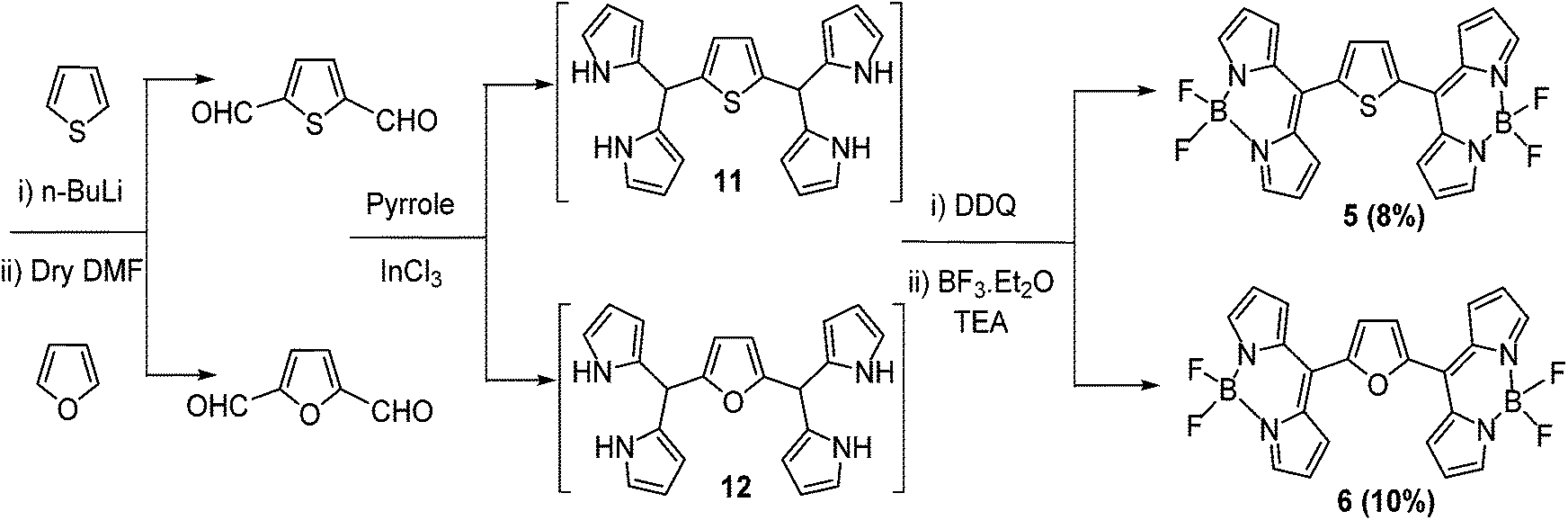
Silica gel column chromatographic purification afforded bis-BODIPYs 1–4 as solid powders in 8–33% yields (Schemes 1 and 2). In order to synthesize bis-BODIPYs 5 and 6, bis-formyl derivatives of thiophene and furan were prepared (Scheme 3) by dilithiation of thiophene and furan followed by the addition of dry DMF (N,N-dimethylformaldehyde). The bis-formylthiophene and furan were subjected to acid catalysed condensation with pyrrole to obtain the corresponding bis-dipyrranes 11 and 12. The crude bis-dipyrranes 11 and 12 were directly used further for the next step without any purification due to their instability under column conditions. The bis-dipyrranes 11 and 12 were reacted with DDQ followed by complexation with BF3·OEt2 in the presence of TEA (triethylamine, Scheme 3). Silica gel column chromatographic purification of the crude reaction mixture in a 60–70% dichloromethane/petroleum ether mixture yielded 8% of bis-BODIPY 5 as a dark orange solid and 10% of bis-BODIPY 6 as a dark brown solid.
Bis-dipyrranes 7–10 were characterized by HRMS and 1H NMR spectroscopy. Bis-BODIPYs 1–6 were characterized by HRMS, 1H, 13C, 19F and 11B spectroscopy (ESI†). Molecular ion peaks in HRMS spectra of bis-dipyrranes 7–10 and bis-BODIPYs 1–6 confirmed the formation of these compounds (ESI†). Also, single crystal X-ray structures were solved for bis-BODIPYs 1, 2 and 4. Typically in the 1H NMR spectra of bis-dipyrranes 7–10 the four N–H protons appeared around 7.8–7.9 ppm as one singlet. The four α-pyrrole protons showed up between 6.65–6.83 ppm, whereas eight β-pyrrole protons gave two separate signals around 5.92–6.14 ppm. The two protons attached to sp3 hybridized carbon (C–H protons) appeared around 5.45–5.29 ppm as one singlet (ESI†). The appearance of characteristic signals for the aromatic protons present in bridging units also confirmed the formation of bis-dipyrranes 7–10. The formation of bis-BODIPYs 1–6 was evident in their 1H NMR spectra. The representative 1H, 19F and 11B NMR spectra of bis-BODIPY 6 is presented in Fig. 1. Compared to the proton NMR spectra of their corresponding dipyrranes, two protons (C–H) attached to sp3 hybridized carbon and four N–H protons were missing in all the bis-BODIPYs 1–6. In the proton NMR spectra of bis-BODIPY 6 (Fig. 1a) all the eight β-pyrrole protons appeared as two doublets at 6.64 and 7.68 ppm and the four α-pyrrole protons appeared as a singlet at 7.99 ppm. The two β-furan protons showed up as a singlet at 7.32 ppm. As compared to the corresponding bis-dipyrranes 7–10, the four α-pyrrole proton signals were downfield shifted and appeared around 7.93–8.01 ppm in the bis-BODIPYs. In all bis-BODIPYs the eight β-pyrrole protons showed up as two separate signals between 6.61 and 7.02 ppm. In all the bis-BODIPYs the aromatic protons of bridging spacers showed up in the range of 7.29–7.59 ppm. The 19F NMR spectra of all bis-BODIPYs 1–6 were recorded in CDCl3 and fluorine resonance signals split into quartets due to the coupling with the adjacent boron atom (11B, I = 3/2).6 In 19F NMR analysis of all bis-BODIPYs 1–6, the 19F quartets appeared around −145 ppm and only one 19F signal was observed due to the symmetrical nature of the molecules (Fig. 1b). 11B NMR spectra of bis-BODIPYs 1–6 exhibited a triplet for 11B between 0.18 and 0.36 ppm (Fig. 1c).
 |
| | Fig. 1 The NMR spectra: (a) 1H, (b) 19F and (c) 11B of bis-BODIPY 6 in CDCl3. | |
Single crystal X-ray diffraction studies
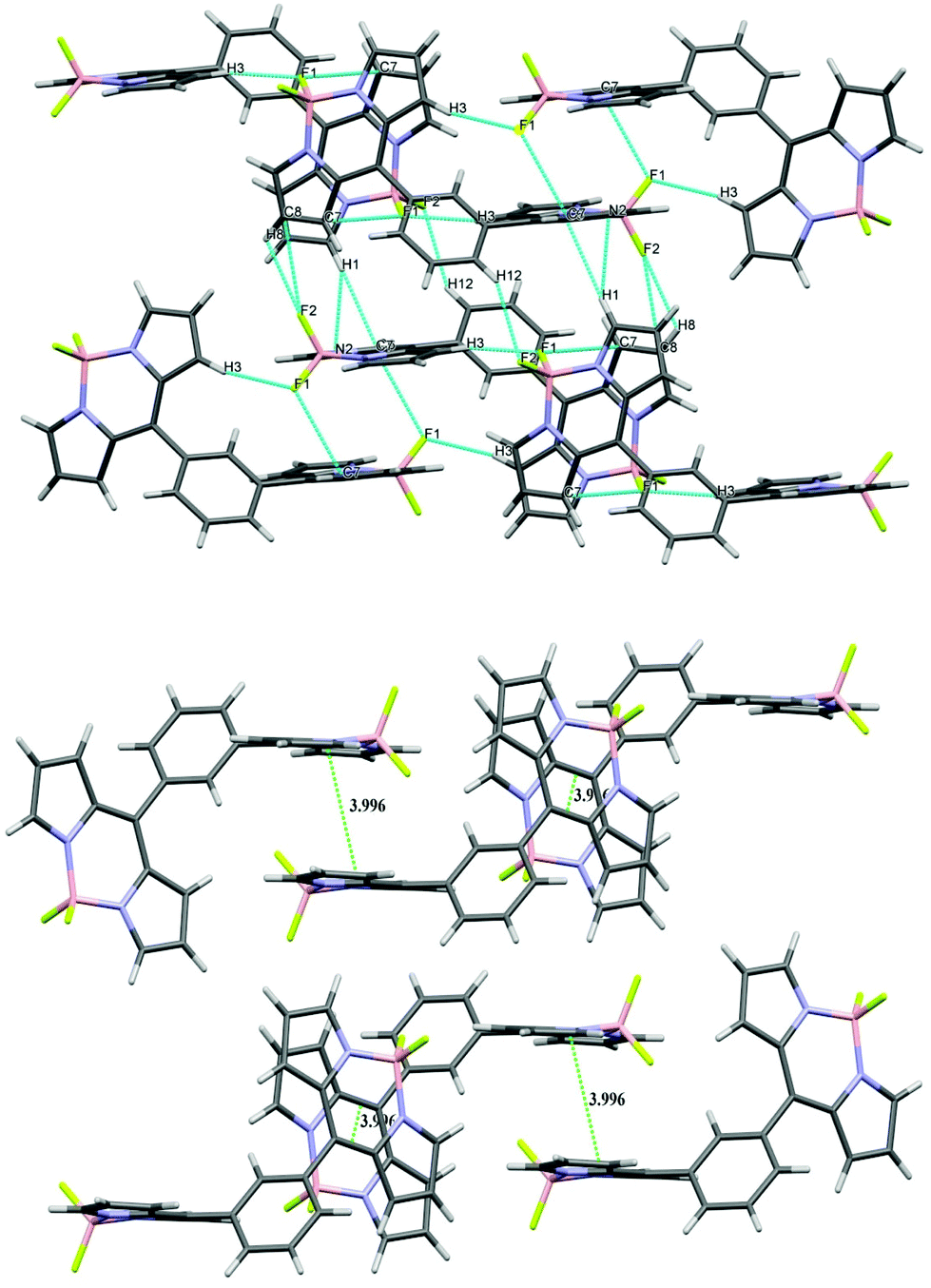
The X-ray structures of bis-BODIPYs 1, 2 and 4 were determined. ORTEP views of bis-BODIPY 1, bis-BODIPY 2 and bis-BODIPY 4 are given in Fig. 2. The single crystals of three bis-BODIPYs were obtained by slow evaporation of the chloroform/n-heptane solution over a period of two weeks. The crystal structure and data refinement parameters for 1, 2 and 4 are displayed in ESI (Table S38†). Bis-BODIPY 1 (CCDC 1006185) gave red prism shaped crystals after solvent evaporation. Bis-BODIPY 1 was crystallised in a monoclinic crystal system with P21/c space group. The values of N–B–N and F–B–F angles were 106.03° (12) and 109.12° (13) respectively. Here two boron dipyrrin units were aligned in the same plane and the bridging phenylene ring was in a different plane. The dihedral angle between the boron dipyrrin units and the phenylene ring (C4–C5–C11–C12) was 54.00° (18). The dihedral angle was comparatively lower than the meso-phenyl BODIPY27 (the reported value was 60.80°). Bis-BODIPY 2 (CCDC 1006186) gave orange rectangle shaped crystals on crystal growth. The crystals were monoclinic type with I2/a (#15) space group.
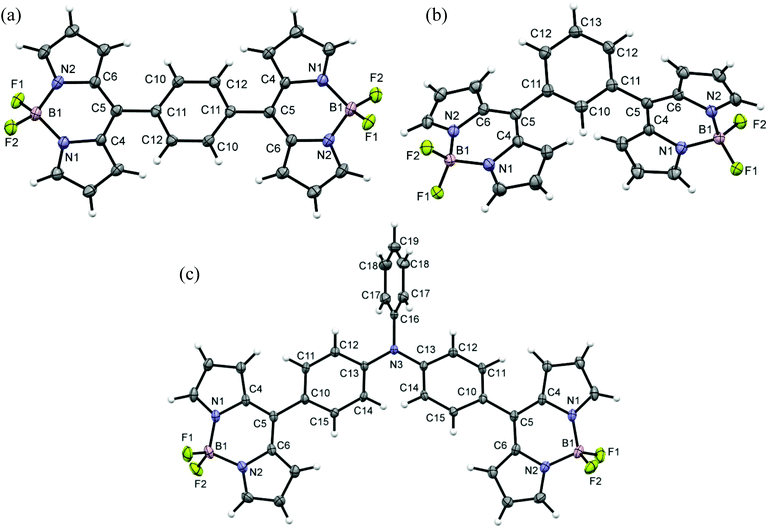 |
| | Fig. 2 ORTEP views of (a) bis-BODIPY 1, (b) bis-BODIPY 2 and (c) bis-BODIPY 4. Displacements are drawn at the 50% probability level. | |
All three units in bis-BODIPY 2 (two boron dipyrrin units and one phenylene unit) were aligned in different planes. The dihedral angle between C4–C5–C11–C10 was 52.01° (17) and that between C6–C5–C11–C12 was 54.11° (18). This suggested that the inward angle was lower than the outward angle and thus the two boron-dipyrrin units are approaching closer to each other. The values of N–B–N and F–B–F angles were 105.54° (12) and 109.42° (12) respectively. Bis-BODIPY 4 (CCDC 1028232) gave orange needle shaped crystals. The crystals were again monoclinic type with C2/c space group. The values of N–B–N and F–B–F angles were 106.21° (15) and 109.53° (17) respectively. The dihedral angles between the boron dipyrrins units and the triphenylamine unit were 44.60° (3) for C4–C5–C10–C11 and 45.50° (3) for C6–C5–C10–C15. These values were much lower than those of bis-BODIPYs 1 and 2. The N–B–N and F–B–F angles in all three bis-BODIPYs were very similar to the reported meso-aryl BODIPY monomer. All three bis-BODIPYs 1, 2 and 4 exhibited supramolecular interactions in their packing structures and the distances and angles of the interactions in the packing mode are presented in Table 1. Bis-BODIPY 1 has formed a layered structure in a 3D framework through weak hydrogen bonding (C–H⋯F) and C–H⋯π interactions (Table 1). In crystal packing, one layer was connected to the other via mutual C(2)–H (2)⋯π (pyrrolic) and C(10)–H (10)⋯π (pyrrolic) interactions (Fig. 3). In the packing diagram of bis-BODIPY 2, molecules were connected through an antiparallel head to tail interaction via two mutual C(7)⋯F(1) interactions. Two boron dipyrrin units were connected to each other through C(3)–H(3)⋯F(1) and C(3)–H(3)⋯B(1) type weak intermolecular interactions to form a stacked 3D network (Fig. 4). One layer was also connected to the other through C(1)–H(1)⋯π (pyrrolic), C(1)–H(1)⋯N(2) and C(8)–H(8)⋯F(2) interactions (Fig. 4). The metric parameters for the interactions in bis-BODIPYs 1 and 2 were C–H⋯π (pyrrolic) distances 2.69 to 2.84 Å and C–H⋯π (pyrrolic) angles 132.79° and 173.93° respectively (Table 1). In the crystal packing mode of bis-BODIPY 4, it formed a cluster of five molecules with the help of several intermolecular interactions (Fig. S37, ESI†). It included weak π⋯π interactions, hydrogen bonding and C–H⋯π interactions. These clusters were connected through C(11)–H(11)⋯F(1) to form the 3D structure of bis-BODIPY 4.
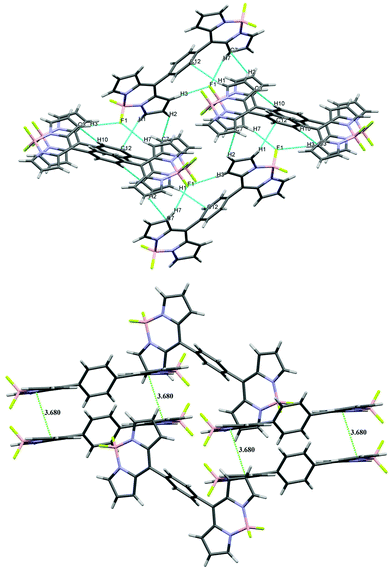 |
| | Fig. 3 Packing diagram (above) and π–π stacking diagram (below) of bis-BODIPY 1. | |
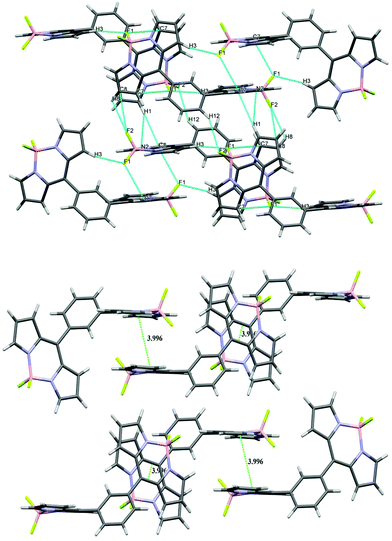 |
| | Fig. 4 Packing diagram (above) and π–π stacking diagram (below) of bis-BODIPY 2. | |
Table 1 Details of intermolecular interactions in the crystal structures of bis-BODIPYs 1, 2 and 4
| |
No. |
Interaction |
Distance (Å) |
Angle (°) |
| Bis-BODIPY 1 |
1 |
C(1)–H(1)⋯π (C(12) phenyl)) |
2.69 |
141.73 |
| 2 |
C(2)–H(2)⋯π (C(7) pyrrolic) |
2.84 |
132.79 |
| 3 |
C(3)–H(3)⋯B(1) |
3.09 |
151.41 |
| 4 |
C(3)–H(3)⋯F(1) |
2.42 |
177.11 |
| 5 |
C(7)–H(7)⋯F(1) |
2.39 |
160.80 |
| 6 |
C(10)–H(10)⋯π (C(3) pyrrolic) |
2.71 |
173.93 |
| 7 |
(C(1) pyrrolic) π⋯π (C(9) pyrrolic) |
3.56(3) |
— |
| 8 |
(C(2) pyrrolic) π⋯π (C(7) pyrrolic) |
3.55(3) |
— |
| Bis-BODIPY 2 |
1 |
C(1)–H(1)⋯π(C(6) pyrrolic) |
2.68 |
166.51 |
| 2 |
C (1)–H(1)⋯N(2) |
2.64 |
149.63 |
| 3 |
C(3)–H(3)⋯ F(1) |
2.30 |
164.40 |
| 4 |
C(3)–H(3)⋯B(1) |
3.06 |
138.83 |
| 5 |
C(7)⋯F(1) |
3.11(17) |
— |
| 6 |
C(8)–H(8)⋯F(2) |
2.58 |
117.53 |
| 7 |
C(8)⋯F(2) |
3.13(17) |
— |
| 8 |
C(12)–H(12)⋯F(2) |
2.64 |
143.28 |
| 9 |
(C(1) pyrrolic) π⋯π (C(8) pyrrolic) |
3.54(3) |
— |
| 10 |
(C(1) pyrrolic) π⋯π (C(8) pyrrolic) |
3.59(3) |
— |
| 11 |
(C(2) pyrrolic) π⋯π (C(13) phenyl) |
3.48(18) |
— |
| Bis-BODIPY 4 |
1 |
(C(1)Pyrrolic) π⋯π (C(11) phenyl) |
3.26(4) |
— |
| 2 |
C(8)–H(8)⋯F(2) |
2.59 |
134.23 |
| 3 |
C(11)–H(11)⋯F(1) |
2.30 |
169.24 |
| 4 |
C(12)–H(12)⋯F(2) |
2.55 |
131.48 |
| 5 |
(C(14) phenyl) π⋯π (C(15) phenyl) |
3.31(3) |
— |
| 6 |
(C(15) phenyl) π⋯π (C(15) phenyl) |
3.33(3) |
— |
| 7 |
C(19)–H(19)⋯π (C(14) phenyl) |
2.79 |
146.83 |
Photophysical properties
UV-vis absorption and fluorescence studies.
The UV/vis absorption, fluorescence spectra and fluorescence lifetimes of bis-BODIPYs 1–6 were examined in five different solvents. The absorption spectra of compounds 1–6 are presented in Fig. 5 and photophysical properties are summarized in Table 2. Bis-BODIPYs 1–4 showed a slight polarity dependence and exhibited a strong absorption band at around 500 nm with a shoulder at around 480 nm. The strong absorption at 500 nm is attributed to a strong S0–S1 electronic transition and the shoulder band corresponds to the S0–S1 vibrational transition. The broader and weaker band centred at 350 nm in bis-BODIPYs 1–2 and around 380 nm in bis-BODIPYs 3–4 was assigned to the higher energy S0–S2 electronic transition. The wavelength of the major absorption band in bis-BODIPYs 1–2 in toluene was comparable with that of the meso-phenyl BODIPY monomer.28 Also, the absorption maxima changed slightly (2–8 nm) upon changing the solvent polarity from toluene to acetonitrile (ESI†), reflecting the typical behavior of BODIPY chromophores.11 The wavelength of the major absorption band of bis-BODIPYs 3 and 4 was similar to the corresponding meso-carbazole BODIPY (497 nm)12 and meso-triphenylamine substituted BODIPY (500 nm).29 Bis-BODIPYs 5 and 6 exhibited a 17 nm red shifted absorption band with a reduced extinction coefficient as compared to the corresponding meso-thienyl- and meso-furyl-BODIPY monomers,30,31 shifted by 18 nm in the former. The fwhmabs (full width at half maxima of the major absorption band) of compounds 1–6 were calculated in different solvents (Table 2). Solvent polarity affected the broadening of the absorption band. Thus fwhmabs values of bis-BODIPYs 1–6 appeared as lower in the non-polar solvent (toluene) and higher in polar solvents (acetonitrile and tetrahydrofuran) for our compounds. This trend was in agreement with the meso-aryl BODIPYs. Compared to bis-BODIPYs 1–4, bis-BODIPYs 5 and 6 showed more solvent polarity dependence. As the polarity of the solvent is increased, the absorption maxima were shifted to the blue region. Among all compounds, bis-BODIPY 6 showed significantly large fwhmabs values, which suggests that significant changes occurred in bond lengths/bond angles upon excitation.32
 |
| | Fig. 5 Comparison of normalized absorption spectra of compounds 1–6 in chloroform. | |
Table 2 Photophysical data of compounds 1–6 in different solvents. The concentration used was 2.4 × 10−6 M
| Compound |
Solvent |
λ
abs (nm) |
fwhmabs (cm−1) |
Logε |
λ
emi (cm−1) |
fwhmem (cm−1) |
Δvst (cm−1) |
Φ
f
|
τ (ns) |
k
r (109 s−1) |
k
nr (109 s−1) |
|
k
r = Φf/τ and knr = (1 − Φf)/τ. |
| Bis-BODIPY 1 |
Toluene |
506 |
1510 |
4.63 |
539 |
1675 |
1210 |
0.014 |
0.08 |
0.18 |
10.75 |
| Dichloromethane |
504 |
1485 |
4.46 |
531 |
1618 |
1009 |
0.012 |
0.07 |
0.17 |
14.11 |
| Chloroform |
506 |
1516 |
4.59 |
534 |
1517 |
1036 |
0.015 |
0.08 |
0.19 |
12.31 |
| Tetrahydrofuran |
503 |
1581 |
4.69 |
532 |
1731 |
1084 |
0.0085 |
0.06 |
0.14 |
16.53 |
| Acetonitrile |
498 |
1610 |
4.75 |
526 |
1680 |
1069 |
0.0009 |
0.02 |
0.045 |
49.95 |
| |
| Bis-BODIPY 2 |
Toluene |
503 |
1497 |
4.82 |
529 |
1502 |
977 |
0.051 |
0.27 |
0.19 |
3.51 |
| Dichloromethane |
502 |
1503 |
4.34 |
525 |
1335 |
873 |
0.042 |
0.26 |
0.16 |
3.68 |
| Chloroform |
504 |
1534 |
4.89 |
527 |
1311 |
866 |
0.048 |
0.20 |
0.24 |
4.76 |
| Tetrahydrofuran |
501 |
1638 |
4.52 |
525 |
1388 |
912 |
0.031 |
0.23 |
0.14 |
4.21 |
| Acetonitrile |
496 |
1650 |
4.75 |
520 |
1428 |
931 |
0.009 |
0.04 |
0.23 |
24.78 |
| |
| Bis-BODIPY 3 |
Toluene |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
| Dichloromethane |
498 |
1630 |
4.66 |
581 |
3337 |
2869 |
0.173 |
2.90 |
0.06 |
0.29 |
| Chloroform |
499 |
1724 |
4.74 |
517 |
2115 |
698 |
0.196 |
1.08 |
0.18 |
0.74 |
| Tetrahydrofuran |
497 |
1637 |
4.91 |
569 |
3235 |
2546 |
0.18 |
0.07 |
0.07 |
0.30 |
| Acetonitrile |
— |
— |
— |
— |
— |
— |
— |
— |
— |
— |
| |
| Bis-BODIPY 4 |
Toluene |
503 |
1659 |
4.88 |
585 |
1953 |
2787 |
0.41 |
3.43 |
0.12 |
0.17 |
| Dichloromethane |
501 |
1724 |
4.69 |
680 |
2457 |
5254 |
0.018 |
2.18 |
0.008 |
0.38 |
| Chloroform |
502 |
1666 |
4.80 |
633 |
2472 |
4123 |
0.135 |
4.12 |
0.033 |
0.21 |
| Tetrahydrofuran |
500 |
1769 |
4.98 |
667 |
3981 |
5007 |
0.025 |
1.65 |
0.015 |
0.59 |
| Acetonitrile |
495 |
1844 |
4.60 |
— |
— |
— |
— |
— |
— |
— |
| |
| Bis-BODIPY 5 |
Toluene |
523 |
1796 |
4.28 |
531 |
3423 |
288 |
0.012 |
— |
— |
— |
| Dichloromethane |
520 |
1863 |
4.23 |
521 |
1977 |
37 |
0.009 |
0.03 |
0.3 |
33.03 |
| Chloroform |
522 |
1772 |
4.30 |
529 |
2943 |
253 |
0.025 |
— |
— |
— |
| Tetrahydrofuran |
517 |
1895 |
4.42 |
524 |
2393 |
528 |
0.005 |
— |
— |
— |
| Acetonitrile |
513 |
1416 |
4.32 |
526 |
1993 |
482 |
0.0002 |
— |
— |
— |
| |
| Bis-BODIPY 6 |
Toluene |
542 |
5313 |
4.18 |
633 |
— |
2652 |
0.0018 |
— |
— |
— |
| Dichloromethane |
537 |
5235 |
4.15 |
627 |
— |
2673 |
0.00087 |
0.06 |
0.0145 |
16.65 |
| Chloroform |
539 |
5612 |
4.03 |
610 |
— |
2159 |
0.0019 |
— |
— |
— |
| Tetrahydrofuran |
535 |
4983 |
4.33 |
576 |
— |
1330 |
0.0016 |
— |
— |
— |
| Acetonitrile |
529 |
4831 |
4.05 |
577 |
— |
1573 |
0.00038 |
— |
— |
— |
A comparison of emission spectra of 1–5 is shown in Fig. 6 and solvatochromism data are given in Table 2. The typical mirror-image relationship was observed for the lowest energy absorption band and emission spectra for bis-BODIPYs 1–5. The emission maxima for compounds 1 and 2 were blue shifted by 41 and 34 nm respectively as compared to that of meso-phenyl BODIPY (in chloroform). The fwhmem (full width at half maxima of the emission band) and quantum yields of compounds 1–6 were calculated in different solvents (Table 2). Solvent polarity affected the broadening of the emission bands and typically fwhmem values were lower in the non-polar solvent (toluene) and higher in polar solvents for our compounds. The fluorescence quantum yields for bis-BODIPYs 1–2 were lower than those for meso-phenyl BODIPY (0.06 in toluene).28 Usually the meso-aryl BODIPYs showed 10–15 nm Stokes shifts whereas these values were 26–33 nm for bis-BODIPYs 1 and 2. In polar media red shifted emission maxima reflect larger dipoles of the CT excited states, which was in agreement with the report by N. Boens et al.32 The fluorescence quantum yields for bis-BODIPY 3 and 4 were higher than those for the corresponding meso-carbazole- and meso-triphenylamine-BODIPY monomers.12,29 The Stokes shift values were highest for compound 4 among all the bis-BODIPYs 1–6. The emission maxima were red shifted for bis-BODIPYs 5 and 6 as compared to the corresponding meso-thienyl- and meso-furyl-BODIPY monomers. However, the quantum yields were lower for both bis-BODIPYs 5 and 6 as compared to their corresponding BODIPY monomers.30,31
 |
| | Fig. 6 Comparison of normalized emission spectra of compounds 1–5 in chloroform (λex = 488 nm). | |
Among all the bis-BODIPYs, compounds 3 and 4 showed higher quantum yields than 1–2 and 5–6. Higher quantum yield indicates the possibility of restricted rotation of the two boron-dipyrrin units in bis-BODIPYs 3 and 4 due to bulky spacers; it was supported by the fact that much lower rate constant values (knr, see Table 2) were observed for non-radiative decay processes in these two compounds. The lower quantum yields for compounds 1, 2, 5 and 6 suggest that other non-radiative decay processes (such as internal conversion or intersystem crossing from S1 to T1) might be more feasible in these compounds.27 Also, the quantum yields of bis-BODIPYs 5 and 6 being much lower than for other compounds could be associated with the electron donating ability of the heteroatom present in the bridging unit.30 The quantum yields of bis-BODIPYs 1–6 were higher in the non-polar solvent (toluene) and lower in polar solvents (tetrahydrofuran); this is due to the large dipole moment difference between the CT excited state and the ground state which in turn facilitates internal conversion in polar media.32 The time-correlated single photon counting technique was used to measure the singlet state lifetimes τ of bis-BODIPYs 1–6. The fluorescence decay of compounds 1–4 was fitted to a single exponential decay (Fig. 7). However, in the case of bis-BODIPYs 5 and 6, decay profiles fitted to three exponential decays (ESI†). The bridging phenylene unit is more rigid in bis-BODIPY 2 as compared to bis-BODIPY 1 (Table 2), thus the non-radiative decay processes are less prominent in 2. It has been reported that the meso-mesityl-BODIPY showed higher quantum yields and singlet state lifetimes than meso-phenyl BODIPY, because the hindered rotation of the bulky mesityl group leads to a drastic decrease in the internal conversion process.27 Therefore, the quantum yields and singlet state lifetimes of bis-BODIPY 2 are higher than those of bis-BODIPY 1; this was again supported by the lower value of the non-radiative decay constant for 2 than 1 (Table 2). For bis-BODIPYs 1–4 singlet state lifetimes τ were lower than those for their corresponding BODIPY monomers. For example bis-BODIPY 3 has a lifetime of 1.9 ns in CHCl3, whereas meso-carbazole BODIPY has 3.37 ns in the same solvent.12 The decrease in radiative decay constant kr and the increase in non-radiative decay constant knr were in line with the quantum yield data.
 |
| | Fig. 7 Fluorescence decay profiles of compounds 1–4 in chloroform. The excitation wavelength was 515 nm, collected at their respective emission maxima. | |
Computational details
The ground state geometry optimization of molecules 1–6 was performed with density functional theory (DFT) at the B3LYP/6-31G(d) level,33–35 using the Gaussian 09 program package.36 Frequency calculations were performed to guarantee that the optimized geometries are at energy minima on the potential energy surface. The vertical excitation energies were calculated employing the time dependent density functional theory (TD-DFT) approach at the same functional and basis sets.
Comparison of geometrical parameters
It is imperative to compare the vital structural parameters obtained as a result of geometry optimization of molecules 1, 2 and 4 with the X-ray crystallography results (ESI†). To be more precise, the largest deviations in the bond lengths of molecules 1, 2 and 4 are 0.017 Å, 0.025 Å and 0.021 Å respectively. The deviations from the X-ray geometry are reflected from the bond angles of the molecules (1, 2 and 4) and are found to be 2.59°, 2.29° and 1.43° respectively. Moreover, the maximum deviations of the optimized dihedral angle in 1, 2 and 4 are 6.14°, 12.54° and 8.87° respectively. The computed results compare well with the experimental data, thereby providing reliable support for the use of the selected level of theory for structural and spectroscopic studies of the rest of the molecules.
Absorption studies
To gain detailed insights into the roots of absorption bands the singlet–singlet transitions of BODIPY molecules were simulated using the TD-DFT/B3LYP level of theory at the valence double-ζ 6-31G(d) basis set. The gas phase determined electronic spectra exhibit a number of absorption peaks, and the peaks with maximum wavelength absorption (λmax) were selected for the comparison. The effect of solvent polarities has not been taken into account as is obvious from the experimental results. The ineffectiveness of the solvent polarity on λmax excludes any ground state intramolecular charge transfer (ICT) process of the molecules under consideration.37 The lowest energy absorption maxima of molecules 1–6 are summarised in Table 3. The λmax of 1, 2, 3 and 4 were observed at around 500 nm and for 5 and 6, the red-shifted bands at 522 and 539 nm respectively indicate a lower HOMO–LUMO gap, which is supported by DFT studies. The delocalization of frontier molecular orbitals (FMOs) of compounds 1–6 indicated a significant electronic conjugation between the boron dipyrrin units and the spacers. Interestingly the HOMO–LUMO gap for 6 (2.513 eV) is less than that of 1 (2.877 eV), 2 (3.040 eV), 3 (3.050 eV), 4 (2.812 eV) and 5 (2.770 eV). Though DFT produced HOMO–LUMO gaps do not directly corroborate the experimentally observed (UV-Vis) energy gap values, almost the same trend of band gap increment (3 > 2 > 1 > 4 > 5 > 6) is reflected. For bis-BODIPY 3, in spite of the highest HOMO energy level, the most destabilized LUMO resulted in its highest HOMO–LUMO gap. On the other hand, for bis-BODIPY 6, the destabilization of the LUMO is considerably lower because of approximately planar arrangement of the furan spacer increasing the level of effective conjugation, resulting in a HOMO–LUMO gap that is lower than the rest of molecules 1, 2, 3, 4 and 5.
Table 3 Absorption data of bis-BODIPYs 1–6
| |
Experimental λmax (nm) |
Log∈ |
Calculated λmax (nm) |
(F) |
| ∈ is the molar extinction coefficient and F is the oscillator strength. |
| Bis-BODIPY 1 |
506 |
4.59 |
472.02 |
0.0631 |
| Bis-BODIPY 2 |
504 |
4.89 |
466.98 |
0.0029 |
| Bis-BODIPY 3 |
499 |
4.74 |
445.05 |
0.2490 |
| Bis-BODIPY 4 |
502 |
4.80 |
520.17 |
0.0411 |
| Bis-BODIPY 5 |
522 |
4.30 |
493.65 |
0.0390 |
| Bis-BODIPY 6 |
539 |
4.03 |
541.08 |
0.0440 |
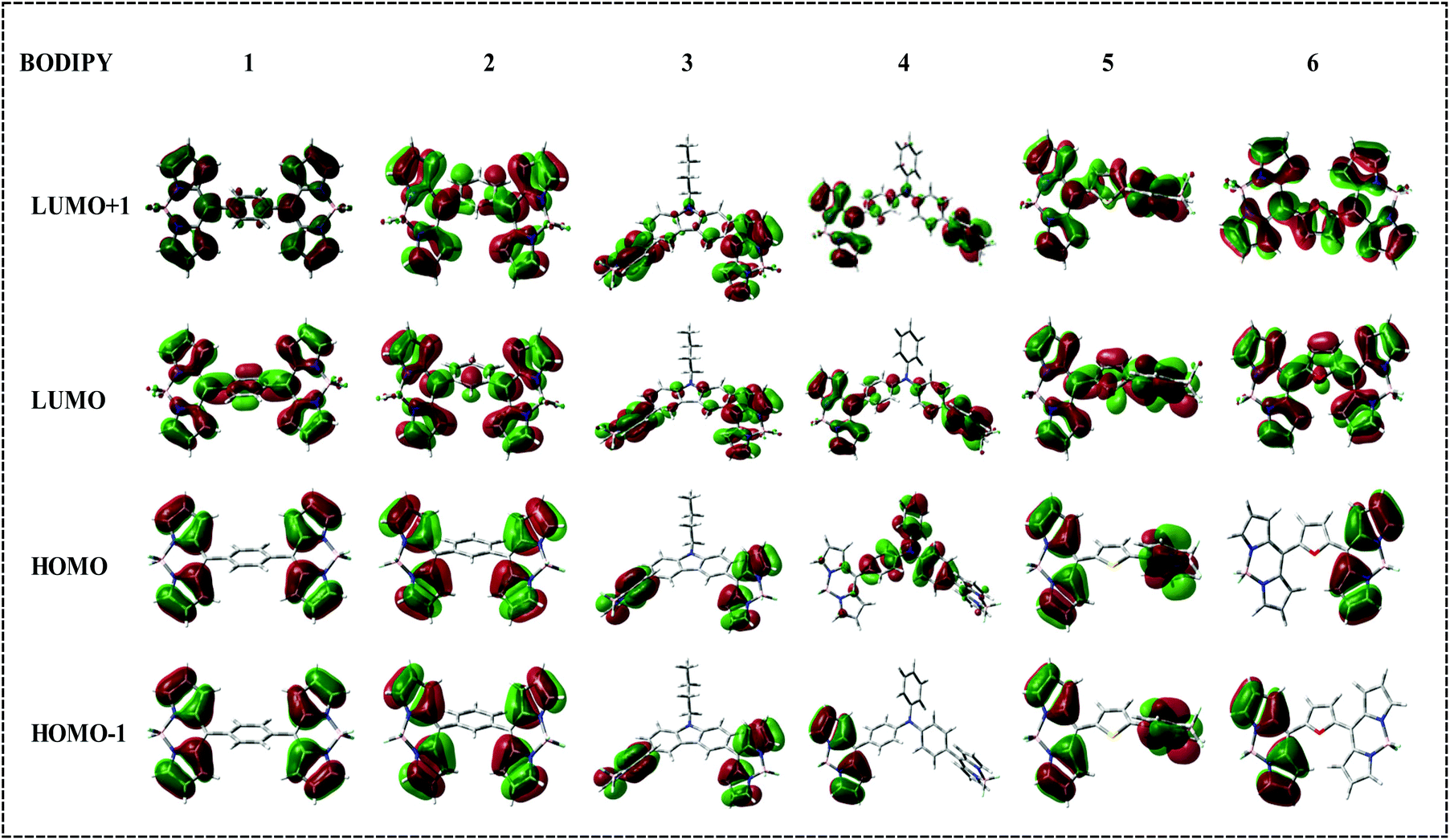
The frontier molecular orbitals of molecules 1–6 are displayed in Fig. 8. The distribution of HOMO and LUMO reflects the strong donor–acceptor interactions. In bis-BODIPYs 1, 2, 3 and 5, the HOMO was localized on the BODIPY moiety and the LUMO was shifted to the corresponding spacer groups. On the other hand in 4 and 6, the HOMO was localized on the spacer moieties whereas the LUMO was budged to the BODIPY moiety. The major absorption band in the electronic absorption spectra of molecules 1–6 is related to the BODIPY absorption which reflects that the origin of major absorption peaks is from HOMO energy levels. It can be seen that the TD-DFT calculations were in good agreement with the experimental results and unveil that the main transitions for 1, 2, 3, 4, 5 and 6 are from HOMO → LUMO, HOMO → LUMO+1, HOMO → LUMO+1, HOMO → LUMO, HOMO → LUMO and HOMO → LUMO+1 respectively.
 |
| | Fig. 8 Frontier molecular orbitals of bis-BODIPYs 1–6 at the B3LYP/6-31G(d) level for all the atoms. | |
Electrochemical studies
The cyclic voltammetry measurements of bis-BODIPYs 1–6 were carried out at a scan rate of 50 mV s−1 using tetrabutylammonium perchlorate (TBAP) as a supporting electrolyte. The redox potential data and a comparison of reduction waves are summarised in Table 4 and Fig. 9 respectively. All the bis-BODIPYs showed only reduction peaks. Bis-BODIPY 1 showed two reduction peaks. The first reduction peak was chemically reversible at −0.49 V and the other one was chemically irreversible around −1.75 V. Bis-BODIPY 2 showed three reduction waves, two reversible and one irreversible.
 |
| | Fig. 9 Comparison of cyclic voltammograms of compounds 1–6 in dichloromethane, containing 0.1 M TBAP as the supporting electrolyte recorded at 50 mV s−1 scan speed (V vs. SCE). | |
Table 4 Electrochemical redox data (V) of compounds 1–6 in dichloromethane containing 0.1 M TBAP as the supporting electrolyte recorded at 50 mV s−1 scan speed
| |
E
red (V vs. SCE) |
| I |
II |
III |
| Bis-BODIPY 1 |
−0.49 |
−1.75 |
— |
| Bis-BODIPY 2 |
−0.55 |
−0.67 |
−1.67 |
| Bis-BODIPY 3 |
−0.66 |
−1.72 |
— |
| Bis-BODIPY 4 |
−0.64 |
−1.36 |
— |
| Bis-BODIPY 5 |
−0.17 |
— |
— |
| Bis-BODIPY 6 |
−0.17 |
— |
— |
The reported meso-tolyl BODIPY monomer showed two reduction peaks at −0.788 and −1.81 V. As compared to the reduction potentials of meso-tolyl BODIPY,31 the first reduction potential of both bis-BODIPYs 1 and 2 exhibited an anodic shift, i.e. they were much easier to reduce compared to the monomer meso-tolyl BODIPY. Bis-BODIPY 3 also showed two reduction waves. The first reduction potential of bis-BODIPY 3 was shifted towards the less negative side by 50 mV as compared to its corresponding monomer.12 This reflects that compound 3 was much easier to reduce compared to the monomer. Bis-BODIPY 4 also showed two reduction waves with a reduction potential of −0.64 V and −1.36 V. Bis-BODIPYs 5 and 6 showed single reduction waves with a reduction potential of around −0.17 V. The reported first reduction potential for monomeric meso-furyl BODIPY was −0.66 V.31 The electrochemical study of compounds 1–6 suggests that compared to their corresponding BODIPY monomers, the reduction potentials of 1–6 were low and they were very easy to reduce.
Conclusion
In conclusion, we have synthesized and characterized six boron-dipyrrins containing a bridging arene or heterocyclic ring. Their absorption, emission, electrochemical and time resolved florescence studies were carried out. Changing the spacer size from bulky carbazole to phenylene to smaller thiophene and furan rings affected the spectroscopic properties of bis-BODIPYs. The absorption and emission maxima were blue shifted for bis-BODIPYs having bulky aromatic spacers and red shifted for bis-BODIPYs having smaller thiophene/furan spacers. Also, the X-ray crystal structures of three bis-BODIPYs were solved. The reduced dihedral angle between the bridging ring and the dipyrrin core in all three crystal structures indicated better interactions between the two constituting boron dipyrrin units. Also, phenyl bridged bis-BODIPYs showed C–H⋯π (pyrrolic) and π⋯π interactions in 3D structures. DFT calculations showed the largest HOMO–LUMO gap for 3 and the lowest one for 6. Also, gas phase absorption maxima calculated for 1–6 were closer to the experimental values. The observed anodic shifts in the reduction potentials of all six bis-BODIPYs suggested increased electronic interactions between two boron dipyrrin units within the molecule. Also, as compared to their corresponding monomer BODIPYs all six bis-BODIPYs were easier to reduce.
Experimental
General methods and reagents
Unless otherwise mentioned, all the reagents and solvents were purchased from Aldrich, Acros Organics or Merck and used without further purification. Pyrrole was distilled under vacuum prior to use. Silica gel (60–120 mesh size) used for column chromatography were procured from Merck. The solution NMR spectra of the compounds were recorded with a Bruker Avance III 500 MHz NMR spectrometer at IIT Gandhinagar. Absorption spectra were recorded with a Shimadzu UV-1700 and fluorescence emission studies were performed using a Horiba-Jobin Yvon Fluorolog-3 spectrometer at IIT Gandhinagar. The fluorescence quantum yields (Φf) were estimated from the emission and absorption spectra by a comparative method at the excitation wavelength of 488 nm using Rhodamine B (Φf = 0.49) in ethanol as the standard. The time-resolved fluorescence decay measurements were carried out at IIT Gandhinagar using a pico-second-diode-laser-based, time-correlated, single-photon counting (TCSPC) fluorescence spectrometer from Edinburgh Instruments Ltd, LifespecII (Livingston, UK). Cyclic voltammetric (CV) studies were carried out with an electrochemical system utilizing the three electrode configuration consisting of a glassy carbon (working electrode), platinum wire (auxiliary electrode) and saturated calomel (reference electrode) electrodes. The experiments were done in dry dichloromethane using 0.1 M tetrabutylammonium perchlorate as the supporting electrolyte. All potentials were calibrated vs. a saturated calomel electrode by the addition of ferrocene as an internal standard, taking E½ (Fc/Fc+) = 0.51 V vs. SCE.
Synthesis of bis-dipyrrane 7.
To a stirred solution of terephthalaldehyde (500 mg, 3.72 mmol) and pyrrole (38.79 mL, 559.14 mmol), acid catalyst (InCl3, 82.49 mg, 0.37 mmol) was added at room temperature. The reaction mixture was allowed to stir for 2 h at room temperature under a N2 atmosphere. After stirring for 2 h, excess pyrrole was removed by distillation under reduced pressure. The crude residue was dissolved in a small amount of dichloromethane and purified by column chromatography on silica gel (ethyl acetate/dichloromethane/hexane = 1:2:4) to give 7 as a pale yellow powder. Yield: 29% (400 mg). 1H NMR (500 MHz, CDCl3, δ ppm): 7.921 (bs, 4H), 7.170 (s, 4H), 6.695–6.692 (d, J = 1.5 Hz, 4H), 6.160–6.143 (m, 4H), 5.914 (s, 4H), 5.45 (s, 2H). HRMS [ESI]: calcd for C24H21N4 [(M − H)+]: m/z 365.1766, obsvd: m/z 365.1769.
Synthesis of bis-dipyrrane 8.
Isophthalaldehyde (600 mg, 4.47 mmol) and pyrrole (150 eq., 46.5 mL, 670 mmol) were taken in a 100 mL round bottom flask and stirred for 5 minutes under nitrogen at room temperature. After stirring for 5 min InCl3 (0.1 eq., 98.91 mg, 0.44 mmol) was added and the reaction mixture was allowed to stir for another 2 h. By vacuum distillation, excess pyrrole was removed. The crude mixture was subjected to silica gel column chromatography (ethyl acetate/dichloromethane/hexane = 1:2:4) to isolate the desired compound 8 as a sticky yellow solid in 46% yield (750 mg). 1H NMR (500 MHz, CDCl3, δ ppm): 7.862 (bs, 4H), 7.263–7.233 (t, J = 6 Hz 1H), 7.121 (s, 1H), 7.089–7.074 (dd, 2H), 6.654–6.651 (d, J = 1.5 Hz, 4H) 6.140–6.123 (m, 4H), 5.864 (s, 4H), 5.367 (s, 2H). HRMS [ESI]: calcd for C24H21N4 [(M − H)+]: m/z 365.1766, obsvd: m/z 365.1769.
Synthesis of bis-dipyrrane 9.
N-Butylcarbazole-bis-aldehyde (500 mg, 1.78 mmol) was dissolved in pyrrole (150 eq., 18.63 mL, 268.51 mmol) under a nitrogen atmosphere. After 5 min InCl3 (0.1 eq. 39.49 mg, 0.18 mmol) was added and the reaction mixture was allowed to stir for 2 h. Excess pyrrole was removed by vacuum distillation. The desired compound 9 was isolated via column chromatography (ethyl acetate/dichloromethane/hexane = 5:20:75) in 55% yield (550 mg). 1H NMR (500 MHz, CDCl3, δ ppm):7.94 (s, 4H), 7.32 (s, 4H), 6.68 (s, 4H), 6.16 (d, J = 3 Hz, 4H), 5.96 (s, 4H), 5.62 (s, 2H), 4.26 (t, J = 7 Hz, 2H), 1.82 (m, 2H), 1.41 (m, 2H), 0.93 (m, 3H). HRMS [ESI]: calcd for C34H32N5 [(M − H)+]: m/z 510.2658, obsvd: m/z 510.2657.
Synthesis of bis-dipyrrane 10.
To a stirred solution of triphenylamine-bis-aldehyde (700 mg, 2.32 mmol) and pyrrole (24.12 mL, 347.68 mmol), InCl3 (51 mg, 0.23 mmol) was added. The reaction mixture was protected from light and kept under stirring under nitrogen for 8 h at room temperature. After verifying the formation of the compound by TLC, excess pyrrole was vacuum distilled and the crude compound was subjected to silica gel column chromatography. The desired compound 10 was eluted with 60% dichloromethane/hexane. Yield: 41% (510 mg). 1H NMR (500 MHz, CDCl3, δ ppm): 7.93 (s, 4H), 7.07 (m, 7H), 7.00 (d, J = 8 Hz, 6H), 6.70 (s, 4H), 6.16 (d, J = 2.5 Hz, 4H), 5.93 (s, 4H), 5.41 (s, 2H). HRMS [ESI]: calcd for C36H32N5 [(M + H)+]: m/z 534.2658, obsvd: m/z 534.2664.
Synthesis of bis-BODIPY 1.
Bis-dipyrrane 7 (200 mg, 0.54 mmol) was dissolved in dichloromethane (75 mL) and oxidized with DDQ (297 mg, 1.3 mmol, 2.4 eq.) at RT under air. The reaction mixture was allowed to stir at room temperature for 1 h. Triethylamine (6.1 mL, 80 eq.) followed by BF3·Et2O (6.80 mL, 100 eq.) was added to the reaction mixture successively without any time delay. The stirring was continued at room temperature for an additional 30 min, the reaction mixture was evaporated and the crude product was purified by silica gel column chromatography and eluted with a ethyl acetate/dichloromethane/petroleum ether (5:15:80) mixture to afford bis-BODIPY 1 as an orange powder in 8% yield (20 mg). 1H NMR (500 MHz, CDCl3, δ ppm): 8.008 (s, 4H), 7.761 (s, 4H), 7.002–6.994 (d, J = 3.5 Hz, 4H), 6.615–6.609 (d, J = 3 Hz). 13C NMR (125.7 MHz, CDCl3, δppm): 145.44, 144.96, 136.15, 134.77, 131.47, 130.49, 119.03. 19F NMR (470.4 MHz, CDCl3, δppm): −145.03 (q, 4F). 11B NMR (160 MHz, CDCl3, δ ppm): 0.29 (t, 2B). HRMS [ESI]: calcd for C24H16B2F3N4 [(M − F)+]: m/z 439.1513, obsvd: m/z 439.1520.
Synthesis of bis-BODIPY 2.
Bis-dipyrrane 8 (200 mg, 0.54 mmol) was dissolved in dichloromethane (75 mL) and oxidized with DDQ (297 mg, 1.3 mmol, 2.4 eq.) at RT under air. The reaction mixture was allowed to stir at room temperature for 1 h. Triethylamine (6.1 mL, 80 eq.) and BF3·Et2O (6.8 mL, 100 eq.) were added to the reaction mixture without any time delay. The stirring was continued at room temperature for another 30 min; after this the reaction mixture was evaporated and the crude product was purified by silica gel column chromatography. The column was eluted with an ethyl acetate/dichloromethane/petroleum ether (5:15:80) mixture to afford bis-BODIPY 2 as an orange powder in 20% yield (50 mg). 1H NMR (500 MHz, CDCl3, δ ppm): 7.988 (s, 4H), 7.817–7.790 (t, J = 7.5 Hz, 3H), 7.745–7.715 (d, J = 7.5 Hz, 1H), 6.944–6.938 (d, J = 3 Hz, 4H), 6.593–6.589 (d, J = 2 Hz 4H). 13C NMR (125.7 MHz, CDCl3, δppm): 145.22, 145.01, 134.84, 134.27, 132.42, 131.87, 131.31, 128.76, 119.11. 19F NMR (470.4 MHz, CDCl3, δppm): −145.01 (q, 4F). 11B NMR (160 MHz, CDCl3, δ ppm): 0.26 (t, 2B). HRMS [ESI]: calcd for C24H16B2F3N4 [(M − F)+]: m/z 439.1513, obsvd: m/z 439.1520.
Synthesis of bis-BODIPY 3.
Bis-Dipyrrane 9 (200 mg, 0.39 mmol) was dissolved in dichloromethane (54 mL) and oxidized with DDQ (213 mg, 0.94 mmol, 2.4 eq.) at RT under air. The reaction mixture was allowed to stir at room temperature for 1 h. Triethylamine (4.3 mL, 80 eq.) followed by BF3·Et2O (4.91 mL, 100 eq.) was added to the reaction mixture without any time delay. The stirring was continued at room temperature for an additional 30 min, the reaction mixture was evaporated and the crude product was purified by silica gel column chromatography and eluted with a dichloromethane/petroleum ether (80:20) mixture to afford bis-BODIPY 3 as an orange powder in 33% yield (80 mg). 1H NMR (500 MHz, CDCl3, δ ppm): 8.385 (s, 2H), 7.954 (s, 4H), 7.804–7.787 (d, J = 8.5 Hz 2H), 7.637–7.620 (d, J = 8.5 Hz, 2H), 7.012–7.006 (d, J = 3 Hz, 4H), 6.579–6.575 (d, J = 2 Hz, 4H) 4.487–4.459 (t, J = 7 Hz, 2H), 2.016–1.987 (t, J = 7.5 Hz, 2H), 1.422–1.393 (t, J = 7 Hz, 2H), 1.067–1.038 (t, J = 7 Hz, 3H). 13C NMR (125.7 MHz, CDCl3, δ ppm): 143.40, 142.56, 135.19, 131.66, 129.53, 125.78, 123.63, 118.44, 109.29, 43.22, 31.21, 20.65, 13.89. 19F NMR (470.4 MHz, CDCl3, δ ppm): −145.10 (q, 4F). 11B NMR (160 MHz, CDCl3, δ ppm): 0.36 (t, 2B). HRMS [ESI]: calcd for C34H27B2F4N5 [(M − F)+]: m/z 584.2405, obsvd: m/z 584.2444.
Synthesis of bis-BODIPY 4.
Bis-dipyrrane 10 (500 mg, 0.94 mmol) was dissolved in 75 mL of dichloromethane and oxidised with DDQ (511 mg, 2.27 mmol, 2.4 eq.) at room temperature under air. The reaction mixture was allowed to stir at RT for 1 h. After 1 h triethylamine (10.4 mL, 75 mmol, 80 eq.) followed by BF3·Et2O (8.13 mL, 94 mmol, 100 eq.) was added to the reaction mixture one by one without much time lag. The reaction mixture was allowed to stir at room temperature for 30 min; after this the solvent was evaporated and the crude product was purified by silica gel column chromatography. The desired compound bis-BODIPY 4 was eluted with an ethyl acetate/hexane (15:85) mixture in 30% yield (176 mg, reddish powder). 1H NMR (500 MHz, CDCl3, δ ppm): 7.937 (s, 4H), 7.557–7.540 (d, J = 8.5 Hz, 4H), 7.467–7.436 (t, J = 8.5 Hz, 2H), 7.306–7.275 (m, 7H), 7.063–7.057 (d, J = 3 Hz, 4H), 6.577–6.573 (d, J = 2 Hz, 4H). 13C NMR (125.7 MHz, CDCl3, δ ppm): 149.65, 146.89, 145.88, 143.51, 134.71, 132.30, 131.19, 130.19, 128.32, 126.94, 125.96, 122.55, 118.32. 19F NMR (470.4 MHz, CDCl3, δ ppm): −145.13 (q, 4F). 11B NMR (160 MHz, CDCl3, δ ppm): 0.29 (t, 2B). HRMS [ESI]: calcd for C36H25B2F3N5 [(M − F)+]: m/z 606.2248, obsvd: m/z 606.2269.
Synthesis of bis-BODIPY 5.
Bis-dipyrrane 11 (400 mg, 0.28 mmol) and pyrrole (30 mL, 428 mmol) were stirred under nitrogen for 5 min. InCl3 (6 mg, 0.03 mmol) was added and the reaction mixture was allowed to stir for a further 2 h. After 2 h excess pyrrole was vacuum distilled and the crude reaction mixture was dissolved in 50 mL of dichloromethane and oxidised with DDQ (292 mg) and allowed to stir for 1 h at RT. After 1 h, triethylamine (10.4 mL, 75 mmol, 80 eq.) and BF3·Et2O (8.13 mL, 94 mmol, 100 eq.) were added to the reaction mixture. The stirring was continued at room temperature for a further 30 min, and then reaction mixture was evaporated and the crude product was subjected to silica gel column chromatography. The desired compound was eluted in a dichloromethane/hexane (70:30) mixture in 8% yield (25 mg, dark orange powder). 1H NMR (500 MHz, CDCl3, δ ppm): 7.999 (s, 4H), 7.689 (s, 4H), 7.331–7.323 (d, J = 4 Hz, 4H), 6.652–6.645 (d, J = 3.5 Hz, 4H). 13C NMR (125.7 MHz, CDCl3, δ ppm): 145.16, 139.38, 137.26, 134.21, 132.86, 131.43, 119.22. 19F NMR (470.4 MHz, CDCl3, δ ppm): −145.13 (q, 4F). 11B NMR (160 MHz, CDCl3, δ ppm): 0.20 (t, 2B). HRMS [ESI]: calcd for C22H14B2F3N4S [(M − F)+]: m/z 445.1077, obsvd: m/z 445.1107.
Synthesis of bis-BODIPY 6.
The synthesis of bis-BODIPY 6 was performed by following a similar procedure to that used for bis-BODIPY 5. Yield: 10% (25 mg) 1H NMR (500 MHz, CDCl3, δ ppm):7.976 (s, 4H), 7.452–7.445 (d, J = 3.5 Hz, 4H), 7.413 (s, 2H), 6.642–6.637 (d, J = 2.5 Hz, 4H). 13C NMR (125.7 MHz, CDCl3, δ ppm): 151.75, 144.70, 130.84, 121.23, 119.37. 19F NMR (470.4 MHz, CDCl3, δ ppm): −145.49 (q, 4F). 11B NMR (160 MHz, CDCl3, δ ppm): 0.18 (t, 2B). HRMS [ESI]: calcd for C22H14B2F3N4O [(M − F)+]: m/z 429.1306, obsvd: m/z 429.1277.
Acknowledgements
IG gratefully acknowledges DST (New Delhi), CSIR (New Delhi) and IIT Gandhinagar for financial support. IG thanks the Chemistry Department, IIT Bombay for CV studies and Prof. H. Furuta, Kyushu University for X-ray measurements. PEK and SD thank IIT Gandhinagar for their fellowship. PCJ acknowledges CUG for providing basic computational facilities and UGC, New Delhi for start-up grants. MYL thanks UGC for fiscal assistance.
References
- L. L. Shipman, T. M. Cotton, J. R. Norris and J. J. Katz, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1791 CrossRef CAS.
- J. Deisenhofer, O. Epp, R. Miki, R. Huber and H. Michel, J. Mol. Biol., 1984, 180, 385 CrossRef CAS.
- J. Deisenhofer, O. Epp, R. Miki, R. Huber and H. Michel, Nature, 1985, 318, 618 CrossRef CAS PubMed.
- M.-C. Yee, S. C. Fas, M. M. Stohlmeyer, T. J. Wandless and K. A. Cimprich, J. Biol. Chem., 2005, 280, 29053 CrossRef CAS PubMed.
- M. Fa, F. Bergstrom, P. Hagglof, M. Wilczynska, L. B. A. Johansson and T. Ny, Structure, 2000, 8, 397 CrossRef CAS.
- J. Karolin, L. B. A. Johansson, L. Strandberg and T. Ny, J. Am. Chem. Soc., 1994, 116, 7801 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC.
- Y. Wu, X. Peng, B. Guo, J. Fan, Z. Zhang, J. Wang, A. Cui and Y. Gao, Org. Biomol. Chem., 2005, 3, 1387 CAS.
- Z. N. Sun, H. L. Wang, F. Q. Liu, Y. Chen, P. Kwong, H. Tam and D. Yang, Org. Lett., 2009, 11, 1887 CrossRef CAS PubMed.
- R. J. Middleton, S. J. Briddon, Y. Cordeaux, A. S. Yates, C. L. Cale, M. W. George, J. G. Baker, S. J. Hill and B. Kellam, J. Med. Chem., 2007, 50, 782 CrossRef CAS PubMed.
- K. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef PubMed.
- P. E. Kesavan and I. Gupta, Dalton Trans., 2104, 43, 12405 RSC.
- R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611 RSC.
- Y. Hayashi, S. Yamaguchi, W. Y. Cha, D. Kim and H. Shinokubo, Org. Lett., 2011, 13, 2992 CrossRef CAS PubMed.
- W. Pang, X. F. Zhang, J. Y. C. Zhou, A. Haoa and L. Jiao, Chem. Commun., 2012, 48, 5437 RSC.
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937 CrossRef CAS PubMed.
- L. Gai, B. Lu, B. Zou, G. Lai, Z. Shen and Z. Li, RSC Adv., 2012, 2, 8840 RSC.
- A. Poirel, A. D. Nicola, P. Retailleau and R. Ziessel, J. Org. Chem., 2012, 11, 7512 CrossRef PubMed.
- M. T. Whited, N. M. Patel, S. T. Roberts, K. Allen, P. I. Djurovich, S. E. Bradforth and M. E. Thompson, Chem. Commun., 2012, 48, 284 RSC.
- S. Kusaka, R. Sakamoto, Y. Kitagawa, M. Okumura and H. Nishihara, Chem. – Asian J., 2013, 8, 723 CrossRef CAS PubMed.
- V. Yang, L. Li, B. Zhang, L. Zhanga and X. Liu, RSC Adv., 2013, 3, 16933 Search PubMed.
- H. Qi, J. J. Teesdale, R. C. Pupillo, J. Rosenthal and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 13558 CrossRef CAS PubMed.
- A. V. Benniston, G. Copley, A. Harriman, D. Howgego, R. S. Harrington and W. Clegg, J. Org. Chem., 2010, 75, 2018 CrossRef CAS PubMed.
- N. Saki, T. Dinc and E. U. Akkaya, Tetrahedron, 2006, 62, 2721 CrossRef CAS PubMed.
- H. Zhao, J. Liao, D. Yang, Y. Xie, Y. Xu, H. Wang and B. Wang, Aust. J. Chem., 2013, 66, 972 CrossRef CAS.
- R. W. Wagner and J. S. Lindsey, Pure Appl. Chem., 1996, 68, 1373 CrossRef CAS.
- H. L. Kee, C. Kirmaier, L. Yu, P. Thamyongkit, W. J. Younblood, M. E. Calder, L. Ramos, B. C. Noll, D. B. Bocian, R. Scheidt, R. R. Brige, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 109, 20433 CrossRef CAS PubMed.
- C. Yu, L. Jiao, H. Yin, J. Zhou, W. Pang, Y. Wu, Z. Wang, G. Yang and E. Hao, Eur. J. Org. Chem., 2011, 5460 CrossRef CAS PubMed.
- E. Lager, J. Liu, A. Aguilar-Aguilar, B. Z. Tang and P. E. Cabrera, J. Org. Chem., 2009, 74, 2053 CrossRef CAS PubMed.
- K. Kim, C. Jo, S. Eswaramoorthi, J. Sung, D. H. Kim and D. G. Churchill, Inorg. Chem., 2010, 49, 4881 CrossRef CAS PubMed.
- T. K. Khan, S. K. Jana, M. R. Rao, M. S. Shaikh and M. Ravikanth, Inorg. Chim. Acta, 2012, 383, 257 CrossRef CAS PubMed.
- W. Qin, M. Baruah, M. V. D. Auweraer, F. C. D. Schryver and N. Boens, J. Phys. Chem. A, 2005, 109, 7371 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1998, 38, 3098 CrossRef.
- A. D. Becke, J. Chem. Phys., 1986, 84, 4524 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucos, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09 Revision A 02, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- A. S. P. Chinna, M. Sunoj and T. Pakkirisamy, Inorg. Chem., 2014, 53, 4813 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The synthetic procedures of some starting materials, characterization data like HRMS, 1H, 13C, 19F, and 11B NMR spectra, fluorescence decay profiles, DFT calculation and X-ray structure details of reported compounds. CCDC 1006185, 1006186 and 1028232. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01925g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence