
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and unique reversible splitting of 14-membered cyclic aminomethylphosphines on to 7-membered heterocycles†
Elvira I.
Musina
*a,
Tatiana I.
Fesenko
a,
Igor D.
Strelnik
a,
Fedor M.
Polyancev
a,
Shamil K.
Latypov
a,
Peter
Lönnecke
b,
Evamarie
Hey-Hawkins
b,
Andrey A.
Karasik
a and
Oleg G.
Sinyashin
a
aA.E. Arbuzov Institute of Organic and Physical Chemistry of KSC RAS, Arbuzov str. 8, Kazan, 420088, Russian Federation. E-mail: elli@iopc.ru; Fax: +(007)8432732253; Tel: +(007)8432734893
bInstitute of Inorganic Chemistry, Leipzig University, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de; Fax: +(0049)341973319; Tel: +(0049)3419736151
First published on 16th June 2015
Abstract
A novel type of 14-membered cyclic polyphosphine, namely 1,8-diaza-3,6,10,13-tetraphosphacyclotetradecanes 2a–4a has been synthesized by the condensation of 1,2-bis(phenylphosphino)ethane, formaldehyde and alkylamines (isopropylamine, ethylamine and cyclohexylamine) as a RRRR/SSSS-stereoisomer. The structure of macrocycle 2a was investigated by NMR-spectroscopy and X-ray crystal structure analysis. The unique reversible processes of macrocycles 2a–4a splitting onto the corresponding rac- (2b–4b) and meso- (2c–4c) stereoisomers of 1-aza-3,6-diphosphacycloheptanes were discovered.
Introduction
The development of phosphorus-containing macrocyclic mixed-donor ligands has emerged as an important subject in coordination chemistry, because they provide characteristic chelating sites that are difficult to construct with their acyclic analogues.1–3 Such hybrid macrocyclic donors are highly promising for designing reactive and multifunctional transition metal catalysts, as, firstly, the metal center can be supported and stabilized by multidentate macrocyclic platforms, secondly, various coordination properties, such as coordination numbers, oxidation state, and geometry of the metal center are available by changing the components of the heteroatom donors, and thirdly, a conformational mobility of the ligands is controllable by a suitable choice of the bridging atoms/groups between donor atoms. The macrocyclic multidentate phosphine ligands as formal analogues of the crown ethers hold promise as incredibly stable ligands for applications requiring robust complexes, such as radioactive metal complexes for use as radiopharmaceuticals,4 for the stable Ni(II) complexes with direct metal-borohydride coordination as potential hydrogenation catalysts,5 and Cr(II) complexes as reducing agents for the catalytic production of hydrazine and/or ammonia from nitrogen.6It has been over 35 years since the first macrocyclic phosphine ligands were synthesized and few strategies have been designed for their synthesis. However, macrocyclic phosphine ligands have been difficult to synthesize stereoselectively in a good yield and only a handful of synthetic methods have shown broad applicability in terms of the ring sizes, functional groups, and metal complexes that can be obtained.1,3
The convenient route to macrocyclic polydentate P,N-ligands by the condensation of primary phosphines or secondary bisphosphines with formaldehyde and primary amines or diamines was developed in our laboratory. This method for the design of macrocyclic polyphosphines was based on the principles of covalent self-assembly and was successfully used for the stereoselective synthesis of macrocyclic aminomethylphosphines: 16-, 18-, 20-membered corands and 28-, 36- and 38-membered cyclophanes.7 The distinctive feature of the covalent self-assembly processes is their ability to self-correct, when the “incorrect” intermediate is able to decompose into starting compounds due to the reversibility of the reaction. These compounds react further to give a more thermodynamically stable “correct” product. The P–CH2–N fragments of aminomethylphosphines are labile enough for the realization of self-assembly processes. It was shown that lability of aminomethylphosphines plays a key role for the stereoconversion between RS- and RR/SS isomers of 1-aza-3,6-diphosphacycloheptanes8 and 16-membered macrocycles as well as alternative formation of only RSSR or RRRR/SSSS isomer in the row of 16-, 18- and 20-membered cyclic aminomethylphosphines.7,9
The interaction of 1,2-bis(hydroxymethylphenylphosphino)ethane with primary aryl- and benzylamines gives only 7-membered 1-aza-3,6-diphosphacycloheptanes as thermodynamically more stable products8 in contrast to hydroxymethyl derivatives of 1,3-, 1,4- and 1,5-bisphosphines giving 16-, 18- and 20-membered cyclic tetraphosphines.7 However the formation of a 14-membered macrocycle was suggested as an intermediate of interconversion of rac- and meso-isomers of 1-aza-3,6-diphosphacycloheptanes.8b
Here we reported the synthesis and crystal structure of novel 14-membered aminomethylphosphines and their unique ability to undergo a reversible splitting on to two 7-membered 1-aza-3,6-diphosphacycloheptanes in solutions.
Results and discussion
Synthesis
It has been shown that sufficient basicity of primary amines is one of the key requirements for the realization of self-assembly during Mannich type condensation. The macrocycles 2a–4a were obtained by Mannich-like condensation of an equimolar mixture of racemic RR/SS- and RS-isomers of 1,2-bis(hydroxymethylphenylphosphino)ethane 1 with highly basic isopropylamine, ethylamine, and cyclohexylamine respectively in DMF (Scheme 1). It should be underlined that less basic aryl- or benzylamines in ethanol gave only 7-membered cycles as a result of analogous condensations.8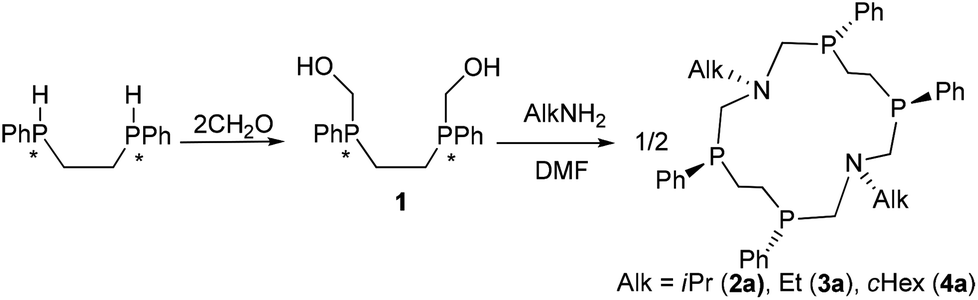 | ||
| Scheme 1 Synthesis of macrocycles 2a–4a. | ||
In all cases 31P NMR spectra of the reaction mixtures in DMF (after ca. 1–2 hours) show three main signals in the region −27 to −37 ppm which were assigned to the macrocyclic corands 2a–4a and to rac- (2b–4b) and meso- (2c–4c) isomers of the corresponding 1-aza-3,6-diphosphacycloheptanes. However the signals of a few acyclic half-products were also registered.
After heating the reaction mixture at 60–70 °C followed by cooling white crystalline products in the yields of 59%, 26% and 20% for 2a, 3a and 4a respectively were isolated. The isolated macrocycles 2a–4a are insoluble in DMF and acetone, but are soluble in benzene, toluene and chloroform. The ESI-MS spectra of these compounds display the peaks, the m/z values and isotopic patterns fitted completely with the stoichiometry of macrocycles 2a–4a. In the 1H and 31P NMR spectra of 2a–4a recorded immediately after the dissolution in C6D6 only one set of signals was registered evidencing the formation of only one isomer.
NMR data
Complete structure elucidation of the title compounds was accomplished by a variety of 1D/2D correlation NMR experiments (COSY, HSQC, 1H–13C/1H–15N/1H–31P HMBC).10,11 2D DOSY12,13 and 1D DPFGNOE14 techniques were used to measure self-diffusion coefficients and NOEs, respectively. The efficiency of the NMR approach for total macrocyclic tetraphosphine structure elucidation has been demonstrated recently.15 Related 1D/2D NMR spectra can be obtained in the ESI (Fig. S1–S29†).The chemical structure of the titled 14-membered macrocycles can be established practically “directly” through a variety of internuclear NMR connectivities starting from P- and N-substituents (e.g. for 2aFig. 1). Characteristic NOE's between CH (i-Pr) and NCHax2P, Mein (i-Pr) and NCHax2P, and Meout (i-Pr) and NCHeq2P allow unequivocal assignment of in- and out-orientation towards macrocycle methyl groups of the isopropyl substituent.
 | ||
| Fig. 1 1H (a) and 31P (b) NMR spectra of 2a in C6D6 at T = 303 K; (c) schematic representation of the macrocycle conformations in exchange and the main 1H–15N (blue)/1H–31P (red) HMBC correlations. | ||
One singlet in 31P spectra and only one set of signals for each type of proton in 1H spectra of 2a–4a suggest high overall symmetry of their 3D structures, viz. there are four magnetically equivalent fragments that can be superimposed by symmetry operation (Fig. 1c). Such high symmetry can be well explained by the fast exchange (on the NMR time scale) between two degenerate conformations that occur through intramolecular rotations around P–CH2–N–CH2–P bonds with synchronous inversions of both nitrogen's LPs (Fig. 1c). It is interesting to note that the magnetic environments of each i-Pr's methyl group do not change during this process, e.g. one methyl (in) is always oriented inside while another (out) is always outside with respect to the macrocycle cavity. Thus configurations of phosphorus atoms of both P–CH2–N–CH2–P fragments have to be the same otherwise i-Pr's methyls would be dynamically equivalent and resonate as a single doublet. On the other hand the P–CH2–CH2–P moiety resonates in 1H spectra as the AA′BB′MM′ spin system with large vicinal spin–spin couplings (JAB′ = ca. 14 Hz, JA′B = ca. 15 Hz) that suggests trans orientation of two ending phosphorus and opposite directions of their LP's with respect to the macrocycle plane. Thus, NMR spectra of 2a correspond to the SPSPSPSP (RPRPRPRP) isomer with three symmetry C2 axis (passing through two nitrogens, through the middle of CH2–CH2 bonds of the P–CH2–CH2–P moiety and orthogonal to the macrocycle plane, Fig. 1c).
Similar NMR characteristics of 2a, 3a and 4a (ESI, Fig. S1–S29†) let us to conclude that other macrocycles (3a, 4a) were also isolated as one enantiomeric pair rac- (SPSPSPSP (RPRPRPRP)) isomers. The formation of SPSPSPSP/RPRPRPRP isomers obeys the recently formulated rule – “If the phosphorus atoms in the macrocycle are linked by an aliphatic chain consisting of an even number of methylene groups, the SPSPSPSP/RPRPRPRP isomer is formed”.9
X-Ray structure
X-ray analysis of the isolated product 2a indicates that the SPSPSPSP/RPRPRPRP isomer of the 14-membered corand was formed in the course of the reaction (Fig. 2).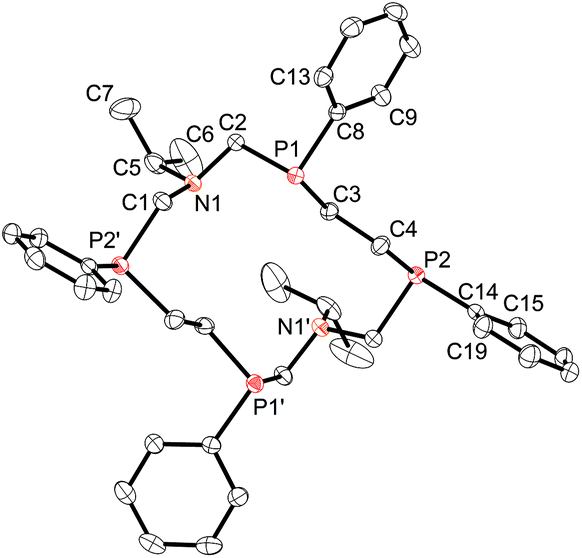 | ||
| Fig. 2 Molecular structure of 2a. | ||
Two enantiomers of 2a, being arranged as separate columns, form a true racemic mixture. Conformation of the cycle is similar to that of the SPSPSPSP/RPRPRPRP isomer of 18-membered 1,10-diaza-3,8,12,17-tetraphosphacyclooctadecane9 and differs only by the length of the hydrocarbon bridge between phosphorus atoms (4.477 Å and 6.928 Å for 14- and 18-membered macrocycles respectively) (Fig. 3). The P⋯P-distances between phosphorus atoms bridged by the CH2–N–CH2 fragment have similar values (4.738 and 4.525 Å) for 14- and 18-membered macrocycles respectively.
 | ||
| Fig. 3 Conformation of 18-membered (a) and 14-membered (b) macrocycles. | ||
According to Dale's nomenclature the conformation of the macrocycle 2a can be designated as the non-diamond lattice biangular [77]. In the biangular [77] conformations as well as in the [99] conformations for the 18-membered macrocycle, one half of a macrocycle,–CH2P(CH2)nPCH2N–, is the rotated analogue (by 2π/2 radians) of the other with the same configurations at the asymmetric phosphorus atoms (SSSS or RRRR) and opposing orientations of their lone pair of electrons relative to each other. The substituents at the nitrogen atoms in 2a are located on the one side of the macrocyclic plane. The nitrogen atoms are trigonal pyramidal (the sum of angles is 335.75°). The phenyl substituents on the phosphorus atoms are located equatorial and alternating relative to the macrocycle plane.
Reversible splitting of 2a–4a in solutions
The lability of the P–CH2–N-fragment is a key property causing the self-assembly and stereoconversion processes of the cyclic aminomethylphosphines. Recently the interconversions between the stereoisomers of 1,9-diaza-3,7,11,15-tetraphosphacyclohexadecanes9 and rac- and meso-isomers of seven-membered 1-aza-3,7-diphosphacycloheptanes8 in solutions were reported. The 14-membered heterocycle was supposed to be the intermediate of the conversion of seven-membered heterocycles.8bIt has been shown that the titled 14-membered macrocycles (2a–4a) are not stable in the solution. After half an hour two additional sets of signals appeared in the NMR spectra (e.g.Fig. 4). Intensities of new sets of signals increased with time while the signals ascribed to the 14-membered macrocycle diminished. Finally, after ca. 14 days the system changed significantly. 1H and 31P spectra (Fig. 4a and b) corresponded to the mixture of the three compounds with integral intensities as ca. 75 (2b–4b)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22 (2c–4c):3 (2a–4a). The quantity of macrocycles 2a–4a in the equilibrium mixture is only 2–6%. The isomers 2b–4b noticeably prevailed in the equilibrium (73–83%), and the amount of 2c–4c isomers is 15–25%.
:22 (2c–4c):3 (2a–4a). The quantity of macrocycles 2a–4a in the equilibrium mixture is only 2–6%. The isomers 2b–4b noticeably prevailed in the equilibrium (73–83%), and the amount of 2c–4c isomers is 15–25%.
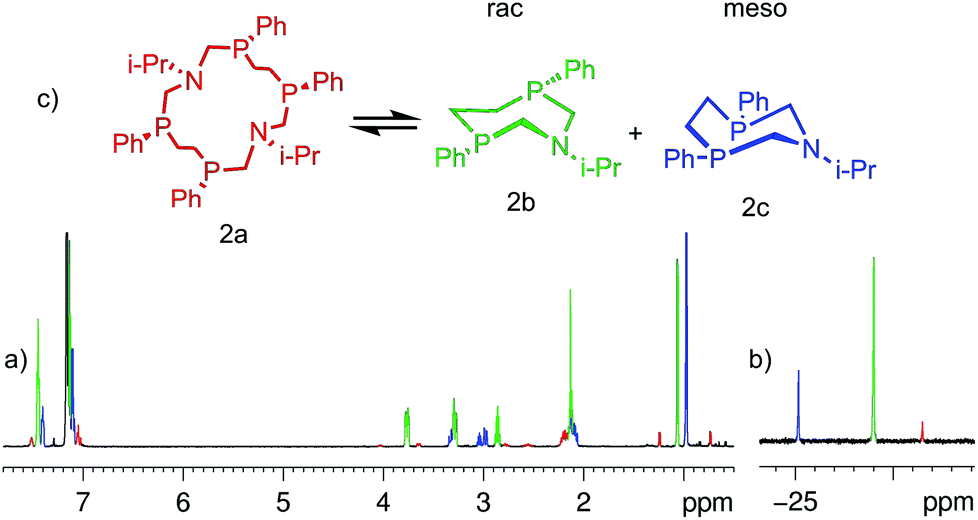 | ||
| Fig. 4 1H (a) and 31P (b) NMR spectra of 2a, 2b and 2c (correspondingly colored) in C6D6 at T = 303 K after ca. 14 days. (c) Schematic representation of the macrocycle and structures of the 7-membered isomers. The signals on the spectra are colored respectively. | ||
The signals of two new components in 1H and 31P spectra are situated in the same regions and have the same intensity and multiplicity relationships as described in 7-membered heterocycles8 suggesting that they have chemically close structures. So, it can be well hypothesized that in these cases the 14-membered macrocycles are disassembled onto the corresponding halves to give two molecules of 1-aza-3,6-diphosphacycloheptanes in solution (Fig. 4c).
1H–13C/1H–15N/1H–31P HMBC correlation experiments allow us to unequivocally ascribe signals in NMR spectra to each of the components of the mixture (ESI Fig. S7–S29†).
Indeed, according to 2D DOSY spectra for 2 self-diffusion coefficients’ (SDC) of new species in solution are ca. equal and notably higher (1.78 × 10−9 m2 s−1) than that for the initial 14-membered macrocycle (1.42 × 10−9 m2 s−1) (ESI Fig. S13†). Thus by taking into account Einstein–Stokes relationship between SDC and effective molecular volume,16 it can be figured out that weight (or volume) of new products is ca. two times less than that of the initial 14-membered macrocycle. Thus based upon these data we can conclude that new signals correspond to the 7-membered heterocycles. Similar relationships for SDC of different isomers were also observed for 3 (ESI Fig. S21†).
Unequivocal assignment of rac- and meso-isomers can be done on symmetry consideration. For example, two i-Pr's methyls of 2 are equivalent in the meso-form but they should resonate separately in the rac-isomer like in the 14-membered macrocycle. Thus in C6D6 solution the rac-isomer dominates (δP = −29.0 ppm), the meso-form (δP = −25.2 ppm) being less populated. In a similar manner for 3 and 4, equivalence/non-equivalence of N–CH2–CH3 and N-cyclohexyl protons was used to differentiate meso- versus rac-isomers.
This assignment is also strongly supported by GIAO 31P chemical shift (CS) data. According to calculations (Table 1) for 2 and 4 macrocycles (2a, 4a) should resonate at a higher field while the meso-isomers (2c, 4c) are expected to be at a lower field than rac-isomers (2b, 4b). In the case of 3 a reverse relationship is expected due to slightly different conformational preference around the N–R bond. All this is in full agreement with experimental findings (Table 1).
| Compound | |||
|---|---|---|---|
| 2 | 3 | 4 | |
| δ calc/δexpa | δ calc /δexp | δ calc/δexp | |
| a Experimental 31P NMR CSs of 2a,b,c–4a,b,c in C6D6 at 303 K. b For 3 in rac- and meso-isomers CSs are averaged between two almost isoenergetical conformations around the N–R bond. | |||
| 14-Memb. (a) | −33.5/−31.5 | −33.7/−31.5 | −33.3/−31.7 |
| rac (b) | −32.3/−29.0 | −43.7/−36.5 | −32.3/−30.0 |
| meso (c) | −22.8/−25.2 | −36.9/−33.5 | −23.1/−26.2 |
The NMR monitoring of the interconversion showed that two processes take place: the splitting of the macrocycle onto RR/SS- and RS-isomers of the 7-membered heterocycle and the stereoconversion between those isomers (Fig. 5).
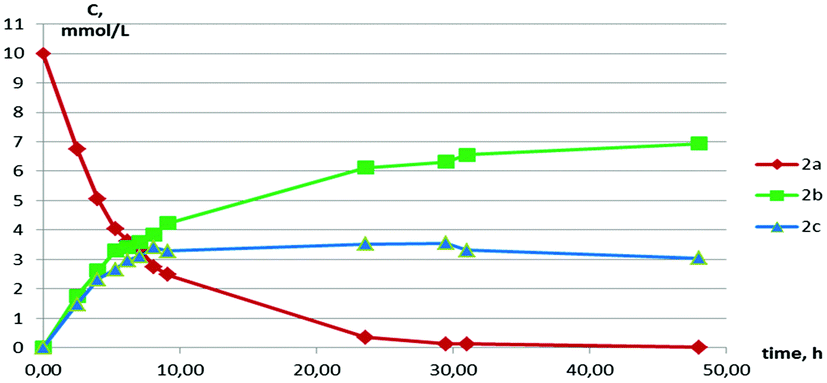 | ||
| Fig. 5 A plot of the concentration of 2a, 2b and 2c on the time of the standing in the solution (initial concentration of 2a is 10 mmol L−1, T = 343 K, solvent – C6D6). | ||
The rate of both the processes is increased by the increase of the initial concentration of the macrocycle (from 10 mmol L−1 to 40 mmol L−1, ESI Tables S4 and S5†) and temperature (295.5 K or 343 K) (ESI Tables S5 and S6†). Moreover, the presence of 10% of p-toluenesulfonic acid as a proton source accelerates the dissociation and the stereoconversion processes (ESI, Table S7†). The equilibrium is established during ca. 30 h in contrast to ca. 340 h for acid free mixtures with the same initial concentration and temperature (ESI Tables S5 and S7†). The relative content of the products a, b and c in the final equilibrium mixtures does not change in all cases. So, we suppose that the found interconversion processes are catalyzed by protons from acid or water traces.
Unexpectedly, in spite of prevalence of 2b–4b (73–83%) only crystals of macrocycles 2a–4a were obtained in nearly quantitative yields after the slow evaporation of solvent from equilibrium mixtures. Moreover, only the signal of the 14-membered cycle was registered in the NMR spectra of the sample that was obtained after the solvent removal from the equilibrated mixture predominantly containing 7-membered cycles and the subsequent addition of the fresh solvent. These results indicate that the splitting processes are reversible and the difference between formation energies of the 14-membered and 7-membered heterocycles is not significant and crystal packing energy could cause the back formation of the 14-membered heterocycle, whereas the stabilization of seven-membered forms in solution may be well explained by the impact of solvent effects and entropic contribution.
Conclusions
In summary, we have demonstrated the effectiveness of the covalent self-assembly approach for the stereoselective synthesis of 14-membered macrocyclic tetrakisphosphines as the SPSPSPSP/RPRPRPRP stereoisomer thereby expanding the row of 16-, 18- and 20-membered macrocyclic aminomethylphosphines and showing the versatility of Mannich-like condensation reaction between α,ω-bis(arylphosphino)alkanes, formaldehyde and primary amines. In contrast to the higher macrocycles the 14-membered aminomethylphosphines undergo the unusual process of the reversible cycle splitting onto two molecules of 1-aza-3,6-diphosphacycloheptanes. The splitting ability of 14-membered cycles should be taken into account for further design of transition metal complexes and catalytic active systems on their basis, e.g. catalysts for electrochemical hydrogen transformations.17Experimental
All manipulations were carried out using standard high-vacuum and dry-nitrogen techniques. Solvents were dried and degassed prior to use and stored under a nitrogen atmosphere.NMR spectroscopy
All NMR experiments were performed with a Bruker AVANCE-600 spectrometer (14.1 T) equipped with a 5 mm diameter gradient inverse broad band probehead and a pulsed gradient unit capable of producing magnetic field pulse gradients in the z-direction of 53.5 G cm−1. Frequencies are 600.13 MHz in 1H NMR, 242.94 MHz in 31P NMR, 150.90 MHz in 13C NMR, and 60.81 MHz in 15N NMR experiments. For 1H–13C correlations the HSQC experiment was optimized for J = 165 Hz. For 1H–13C long range correlations the HMBC experiment was optimized for J = 8 Hz. For 1H–31P long range correlations the HMBC experiment was optimized for J = 8 Hz. For 1H–15N long range correlations the HMBC experiment was optimized for J = 6 Hz. DOSY experiments were performed with ledbpgp2s, using a stimulated echo sequence and two spoil gradients. NOE experiments were performed using 1D DPFGNOE techniques. Samples (ca. 1 mg) were prepared by dissolving in 0.6 mL of the corresponding solvent (C6D6 99.5% D (Sigma-Aldrich, Germany)) under an inert atmosphere (Ar) and were placed in standard NMR tubes (Norell, USA). Chemical shifts are reported on the δ (ppm) scale relative to the 1H and 13C signals of tetramethylsilane (TMS) (0.00 ppm). 15N and 31P chemical shifts were referenced to the 15N signal of CH3CN (235.5 ppm) and the 31P signal of 85% H3PO4 (0.00 ppm), respectively.The Fourier transform pulsed-gradient spin-echo (FTPGSE) experiments12,13 were performed by using a BPP-STE-LED (bipolar pulse pair-stimulated echo-longitudinal eddy current delay) sequence. Data were acquired with a 50.0 or 120.0 ms diffusion delay, with bipolar gradient pulse duration from 2.2 to 6.0 ms (depending on the system under investigation), a 1.1 ms spoil gradient pulse (30%) and a 5.0 ms eddy current delay. The bipolar pulse gradient strength was varied incrementally from 0.01 to 0.32 T m−1 in 16 steps.
Calculations
The quantum chemical calculations were performed using the Gaussian 03 software package.18 Full geometry optimizations have been carried out within the framework of the DFT (PBE1PBE) method using 6-31+G(d) basis sets. As recommended1931P CSs were calculated at the PBE1PBE/6-311G(2d,2p) level of theory. 31P CSs were referred at H3PO4. A linear scaling procedure was applied for correcting systematic errors.19The ESI mass spectra were obtained on a Bruker Esquire 3000 Plus. The melting points were determined on a Boetius apparatus and are uncorrected.
Synthesis of starting 1,2-bis(phenylphosphino)ethane was carried out by the described method. All other reagents were purchased from commercial sources and used as received.
Compound 1 was synthesised by the described method from the corresponding 1,2-bis(phenylphosphino)ethane and formaldehyde.8
After 14 days of standing of 2a in C6D6 the signals of dissociation products rac- (2b) and meso- (2c) isomers of 1-isopropyl-3,6-diphenyl-1-aza-3,6-diphosphacycloheptane prevailed.
Rac-isomer of 1-isopropyl-3,6-diphenyl-1-aza-3,6-diphosphacycloheptane (2b). 1H NMR (600.13 MHz; C6D6; ppm): 7.45 (4 H, dd, 3JHH 7.0, 3JPH 6.7, o-Ph), 7.13 (6 H, t, 3JHH 7.0, m,p-Ph), 3.77 (2 H, ddd, 2JHH 13.6, 2JPH 4.4, 4JHH 4.4, PCH2N), 3.28 (2 H, dd, 2JHH 13.6, 4JPH 3.1, PCH2N), 2.86 (1 H, o, 3JHH 6.7, 3JHH 6.4, CH3CH), 2.04–2.25 (4 H, m, PCH2), 1.06 (3 H, d, 3JHH 6.4, CH3CH), 0.97 (3 H, d, 3JHH 6.7, CH3CH). 13C{1H} NMR (150.90 MHz, C6D6; ppm): 140.7 (dd, 1JPC 10.7, 4JPC 2.9, i-Ph), 133.1 (dd, 2JPC 9.1, 5JPC 5.7, o-Ph), 129.4 (m, m-Ph), 129.3 (m, p-Ph), 57.7 (m, AA′X spin system, 1JPC −15.1, 3JPC 10.5, 3JPP 11.5, PCH2N), 57.4 (t, 3JPC 3.6, CH3CH), 27.7 (m, AA′X spin system, 1JPC −15.9, 2JPC 14.1, 3JPP 11.5, PCH2), 20.4 (s, CH3CH). 31P{1H} NMR (242.94 MHz; C6D6; ppm): −29.0. 15N (60.81 MHz, C6D6, ppm): 42.3.
Meso-isomer of 1-isopropyl-3,6-diphenyl-1-aza-3,6-diphosphacycloheptane (2c). 1H NMR (600.13 MHz; C6D6; ppm): 7.40 (4 H, dd, 3JHH 7.4, 3JPH 6.3, o-Ph), 7.11 (6 H, t, 3JHH 7.4, m,p-Ph), 3.26–3.35 (2 H, m, PCH2N), 3.04 (1 H, p, 3JHH 6.6, CH3CH), 2.98 (2 H, dd, 2JHH 14.0, 2JPH 4.2, PCH2N), 2.04–2.25 (4 H, m, PCH2), 0.97 (6 H, d, 3JHH 6.7, CH3CH). 13C{1H} NMR (150.90 MHz, C6D6; ppm): 141.1 (dd, 1JPC 10.7, 4JPC 5.3, i-Ph), 132.7 (dd, 2JPC 9.0, 5JPC 8.0, o-Ph), 129.4 (m, m-Ph), 129.3 (m, p-Ph), 56.9 (m, AA′X spin system, 1JPC −9.3, 3JPC 2.1, 3JPP 19.5, PCH2N), 56.4 (t, 3JPC 5.9, CH3CH), 23.1 (br, AA′X spin system, 1JPC −23.0, 3JPC 15.1, 3JPP 19.5, PCH2), 20.1 (s, CH3CH). 31P{1H} NMR (242.94 MHz; C6D6; ppm): −25.2. 15N (60.81 MHz, C6D6, ppm): 41.3.
After 7 days of standing of 3a in C6D6 the signals of dissociation products rac- (3b) and meso- (3c) isomers of 1-ethyl-3,6-diphenyl-1-aza-3,6-diphosphacycloheptane 3 prevailed.
Rac-isomer of 1-ethyl-3,6-diphenyl-1-aza-3,6-diphosphacycloheptane (3b). 1H NMR (600.13 MHz; C6D6; ppm): 7.34–7.42 (4 H, br, o-Ph), 7.08–7.15 (6 H, m, m,p-Ph), 3.83 (2 H, ddd, 2JHH 13.7, 4JHH 4.5, 2JPH 4.5, PCH2N), 3.20–3.32 (2 H, m, PCH2N), 2.60–2.79 (2 H, m, NCH2CH3), 2.00–2.20 (4 H, m, PCH2), 0.98 (3 H, t, 3JHH 7.1, CH2CH3). 13C{1H} NMR (150.90 MHz, C6D6; ppm): 140.2 (dd, 1JCP 8.2, 4JCP 3.9, i-Ph), 133.0 (dd, 2JCP 15.8, 5JCP 6.1, o-Ph), 129.4 (m, m-Ph), 129.0 (s, p-Ph), 61.4 (m, AA′X spin system, 1JPC −14.1, 3JPC 11.0, 3JPP 13.5, PCH2N), 53.9 (t, 3JCP 6.0, NCH2CH3), 28.8 (m, AA′X spin system, 1JPC −13.8, 2JPC 11.9, 3JPP 13.5, PCH2), 13.9 (s, CH2CH3). 31P{1H} NMR (242.94 MHz; C6D6; ppm): −36.5.
Meso-isomer of 1-ethyl-3,6-diphenyl-1-aza-3,6-diphosphacycloheptane (3c). 1H NMR (600.13 MHz; C6D6; ppm): 7.34–7.42 (4 H, br, o-Ph), 7.08–7.15 (6 H, m, m,p-Ph), 3.20–3.32 (4 H, m, PCH2N), 2.94 (2 H, q, 3JHH 7.1, NCH2CH3), 2.00–2.20 (4 H, m, PCH2), 1.03 (3 H, t, 3JHH 7.1, NCH2CH3). 13C{1H} NMR (150.90 MHz, C6D6; ppm): 140.9 (dd, 1JCP 8.5, 4JCP 5.5, i-Ph), 132.8 (dd, 2JCP 18.3, 5JCP 9.2, o-Ph), 129.4 (m, m-Ph), 129.1 (s, p-Ph), 60.8 (dd, 1JCP 11.7, 3JCP 8.9, P–CH2–N), 51.3 (t, 3JCP 10.2, NCH2CH3), 24.2 (m, AA′X spin system, 1JPC −22.1, 2JPC 17.9, 3JPP 19.5, PCH2), 13.8 (s, CH2CH3). 31P{1H} NMR (242.94 MHz; C6D6; ppm): −33.5. MS (ESI+), m/z, (Irel, %): 316 (100) [M + H]+, 332 (203) [M + O + H]+.
After 5 days of standing of 4a in C6D6 the signals of dissociation products rac- (4b) and meso- (4c) isomers of 1-cyclohexyl-3,6-diphenyl-1-aza-3,6-diphosphacycloheptane 4 prevailed.
Rac-isomer of 1-cyclohexyl-3,6-diphenyl-1-aza-3,6-diphosphacycloheptane (4b). 1H NMR (600.13 MHz; C6D6; ppm): 7.47 (4 H, br, o-Ph), 7.07–7.14 (6 H, m,p-Ph), 3.87 (2 H, ddd, 2JHH 13.7, 2JPH 4.2, 4JHH 4.2, PCH2N), 3.38 (2 H, d, 2JHH 13.7, PCH2N), 2.52 (1 H, m, 1-H (cHex)), 2.08–2.24 (4 H, m, P–CH2), 2.00–2.08 (1 H, m, 2-H (cHex)), 1.85–1.98 (1 H, m, 2′-H (cHex)), 1.57–1.71 (2 H, m, 3,3′-H (cHex)), 1.45 (1 H, br, 4-H (cHex)), 1.04–1.32 (4 H, m, 2,2′,3,3′-H (cHex)), 0.90–1.02 (1 H, m, 4-H (cHex)). 13C{1H} NMR (150.90 MHz, C6D6; ppm): 140.6 (dd, 1JCP 9.7, 4JCP 4.3, i-Ph), 133.1 (ddd, 2JCP 18.7, 5JCP 9.8, o-Ph), 129.4 (m, m-Ph), 129.0 (m, p-Ph), 66.0 (s, C-1 (cHex)), 58.1 (m, AA′X spin system, 1JPC −14.2, 3JPC 10.8, 3JPP 12.9, PCH2N), 31.19 (s, C-2 (cHex)), 31.14 (s, C-2′ (cHex)), 28.0 (m, AA′X spin system, 1JPC −14.2, 2JPC 12.2, 3JPP 12.9, PCH2), 27.1 (s, C-4 (cHex), 26.7 (s, C-3 (cHex)), 26.6 (s, C-3′ (cHex)). 31P{1H} NMR (242.94 MHz; C6D6; ppm): −30.0.
Meso-isomer of 1-cyclohexyl-3,6-diphenyl-1-aza-3,6-diphosphacycloheptane (4c). 1H NMR (600.13 MHz; C6D6; ppm): 7.41 (4 H, br, o-Ph), 7.07–7.14 (6 H, m,p-Ph), 3.45 (2 H, m, PCH2N), 3.05 (2 H, d, 2JHH 14.2, PCH2N), 2.78 (1 H, m, 1-H (cHex)), 2.08–2.24 (4 H, m, PCH2), 1.85–1.98 (2 H, m, 2-H (cHex)), 1.57–1.71 (2 H, m, 3-H (cHex)), 1.45 (1 H, br, 4-H (cHex)), 1.04–1.32 (2 H, m, 3-H (cHex)), 0.90–1.02 (1 H, m, 4-H (cHex)). 13C{1H} NMR (150.90 MHz, C6D6; ppm): 141.2 (dd, 1JCP 10.7, 4JCP 6.5, i-Ph), 132.7 (dd, 2JCP 8.7, 5JCP 5.5, o-Ph), 129.4 (m, m-Ph), 129.0 (m, p-Ph), 64.0 (s, C-1 (cHex)), 57.5 (dd, 1JCP −8.7, 3JCP 5.1, PCH2N), 30.8 (s, C-2 (cHex)), 27.0 (s, C-4 (cHex)), 26.5 (s, C-3 (cHex)), 23.4 (m, AA′X spin system, 1JPC −22.2, 2JPC 15.2, 3JPP 20.1, PCH2). 31P{1H} NMR (242.94 MHz; C6D6; ppm): −26.2. MS (ESI+), m/z, (Irel, %): 370 (100) [M + H]+.
X-ray crystallography data
The crystals of 2a suitable for X-ray diffraction were hand selected from precipitates that separated from the reaction mixtures. The data of 2a were collected on a Gemini diffractometer (Agilent Technologies) using MoKα radiation (λ = 0.71073 Å) and ω-scan rotation. Data reduction was performed with CrysAlis-Pro20 including the program SCALE3 ABSPACK for empirical absorption correction. The structure was solved by direct methods (SIR92)21 and the refinement was performed with SHELXL-2014.22 The molecule is located at a special position (C2 axis). All non-hydrogen atoms were refined with anisotropic thermal parameters. A difference-density Fourier map was used to locate all hydrogen atoms in the final stage of the structure refinement. Fig. 2 was generated with Diamond.23 CCDC 1060031 contains the supplementary crystallographic data for this paper.Acknowledgements
This work was supported by the Russian Foundation for Basic Researches (No. 13-03-00169, 15-43-02292), President's of RF Grant for the support of leading Scientific schools No. 4428.2014.3 and Programs of Russian Academy of Sciences.Notes and references
- A. A. Karasik and O. G. Sinyashin, in Phosphorus Compounds Advanced Tools in Catalysis and Material Sciences, ed. M. Peruzzini and L. Gonsalvi, Springer, Netherlands, 2011, ch. 37, vol. 12, pp. 375–444 Search PubMed.
- (a) A.-M. Caminade and J. P. Majoral, Chem. Rev., 1994, 94, 1183 CrossRef CAS; (b) A.-M. Caminade and J. P. Majoral, Synlett, 1996, 1019 CrossRef CAS PubMed; (c) M. Pabel and B. Wild, Compr. Heterocycl. Chem. II, 1996, 947 CAS; (d) I. Bauer, W. D. Habicher, I. S. Antipin and O. G. Sinyashin, Russ. Chem. Bull., 2004, 53, 1402 CrossRef CAS; (e) V. Simulescu and G. Ilia, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 3 CrossRef CAS.
- C. D. Swor and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 2860 CrossRef CAS PubMed.
- (a) C. Toulhoat, M. Vidal and M. Vincens, Phosphorus, Sulfur Silicon Relat. Elem., 1992, 71, 127 CrossRef CAS PubMed; (b) B. Lambert and J. F. Desreux, Synthesis, 2000, 1668 CrossRef CAS.
- A. J. Churchard, E. Banach, A. Borgschulte, R. Caputo, J.-C. Chen, D. Clary, K. J. Fijalkowski, H. Geerlings, R. V. Genova, W. Grochala, T. Jaroń, J. C. Juanes-Marcos, B. Kasemo, G.-J. Kroes, I. Ljubić, N. Naujoks, J. K. Nørskov, R. A. Olsen, F. Pendolino, A. Remhof, L. Románszki, A. Tekin, T. Vegge, M. Zäch and A. Züttel, Phys. Chem. Chem. Phys., 2011, 13, 16955–16972 RSC.
- M. T. Mock, S. Chen, M. O'Hagan, R. Rousseau, W. G. Dougherty, W. S. Kassel and R. M. Bullock, J. Am. Chem. Soc., 2013, 11493–11496 CrossRef CAS PubMed.
- (a) A. A. Karasik, A. S. Balueva and O. G. Sinyashin, C. R. Chim., 2010, 13, 1151 CrossRef CAS PubMed; (b) D. V. Kulikov, A. A. Karasik, A. S. Balueva, O. N. Kataeva, I. A. Livinov, E. Hey-Hawkins and O. G. Sinyashin, Mendeleev Commun., 2007, 17, 195 CrossRef CAS PubMed; (c) A. S. Balueva, D. V. Kulikov, R. M. Kuznetsov, A. T. Gubaidullin, L. Ricard, A. A. Karasik and O. G. Sinyashin, J. Inclusion Phenom. Macrocyclic Comp., 2008, 60, 321 CrossRef CAS; (d) R. N. Naumov, A. A. Karasik, K. B. Kanunnikov, A. V. Kozlov, S. K. Latypov, K. V. Domasevitch, E. Hey-Hawkins and O. G. Sinyashin, Mendeleev Commun., 2008, 18, 80 CrossRef CAS PubMed; (e) R. N. Naumov, A. A. Karasik, O. G. Sinyashin, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2004, 357 RSC; (f) A. A. Karasik, D. V. Kulikov, A. S. Balueva, S. N. Ignat'eva, O. N. Kataeva, P. Lönnecke, A. V. Kozlov, S. K. Latypov, E. Hey-Hawkins and O. G. Sinyashin, Dalton Trans., 2009, 490 RSC; (g) R. N. Naumov, A. V. Kozlov, K. B. Kanunnikov, S. Gomez-Ruiz, E. Hey-Hawkins, S. K. Latypov, A. A. Karasik and O. G. Sinyashin, Tetrahedron Lett., 2010, 51, 1034 CrossRef CAS PubMed; (h) A. A. Karasik, D. V. Kulikov, R. M. Kuznetsov, A. S. Balueva, S. N. Ignat'eva, O. N. Kataeva, P. Lönnecke, O. G. Sharapov, S. K. Latypov, E. Hey-Hawkins and O. G. Sinyashin, Macroheterocycles, 2011, 4, 324 CrossRef CAS.
- (a) A. A. Karasik, A. S. Balueva, E. I. Moussina, R. N. Naumov, A. B. Dobrynin, D. B. Krivolapov, I. A. Litvinov and O. G. Sinyashin, Heteroat. Chem., 2008, 9, 125–132 CrossRef PubMed; (b) E. I. Musina, A. A. Karasik, A. S. Balueva, I. D. Strelnik, T. I. Fesenko, A. B. Dobrynin, T. P. Gerasimova, S. A. Katsyuba, O. N. Kataeva, P. Lönnecke, E. Hey-Hawkins and O. G. Sinyashin, Eur. J. Inorg. Chem., 2012, 1857 CrossRef CAS PubMed; (c) T. I. Fesenko, I. D. Strelnik, E. I. Musina, A. A. Karasik and O. G. Sinyashin, Russ. Chem. Bull., 2012, 61, 1792 CrossRef CAS PubMed.
- (a) R. N. Naumov, E. I. Musina, K. B. Kanunnikov, T. I. Fesenko, D. B. Krivolapov, I. A. Litvinov, P. Lönnecke, E. Hey-Hawkins, A. A. Karasik and O. G. Sinyashin, Dalton Trans., 2014, 43, 12784–12789 RSC; (b) A. A. Karasik, R. N. Naumov, K. B. Kanunnikov, D. B. Krivolapov, I. A. Litvinov, P. Lönnecke, A. S. Balueva, E. I. Musina, E. Hey-Hawkins and O. G. Sinyashin, Macroheterocycles, 2014, 7, 181–188 CrossRef CAS.
- A. E. Derome, Modern NMR Techniques for Chemistry Research, Pergamon, Cambridge, U.K., 1988 Search PubMed.
- Atta-ur-Rahman, One and Two Dimensional NMR Spectroscopy, Elsevier, Amsterdam, 1989 Search PubMed.
- W. S. Price, Concepts Magn. Reson., 1997, 9, 299–336 CrossRef CAS; W. S. Price, Concepts Magn. Reson., 1998, 10, 197–237 CrossRef.
- C. S. Johnson, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef CAS.
- K. Stott, J. Stonehouse, J. Keeler, T. L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1995, 117, 4199–4200 CrossRef CAS.
- (a) Sh. K. Latypov, A. V. Kozlov, E. Hey-Hawkins, A. S. Balueva, A. A. Karasik and O. G. Sinyashin, J. Phys. Chem. A, 2010, 114, 2588–2596 CrossRef CAS PubMed; (b) S. K. Latypov, A. G. Strelnik, S. N. Ignatieva, E. Hey-Hawkins, A. S. Balueva, A. A. Karasik and O. G. Sinyashin, J. Phys. Chem. A, 2012, 116, 3182–3193 CrossRef CAS PubMed.
- S. V. Kharlamov and S. K. Latypov, Russ. Chem. Rev., 2010, 79, 635–653 CrossRef CAS PubMed.
- (a) M. P. Stewart, M.-H. Ho, S. Wiese, M. L. Lindstrom, C. E. Thogerson, S. Raugei, R. M. Bullock and M. L. Helm, J. Am. Chem. Soc., 2013, 135, 6033–6046 CrossRef CAS PubMed; (b) M. H. Helm, M. P. Stewart, R. M. Bullock, M. Rakowski DuBois and D. L. Dubois, Science, 2011, 333, 863–8662 CrossRef CAS PubMed; (c) M. L. Reback, G. W. Buchko, B. L. Kier, B. Ginovska-Pangovska, Y. Xiong, S. Lense, J. Hou, J. A. S. Roberts, C. M. Sorensen, S. Raugei, T. C. Squier and W. J. Shaw, Chem. – Eur. J., 2014, 20, 1510 CrossRef CAS PubMed; (d) R. M. Galimullina, M. I. Valitov, Y. S. Spiridonova, E. I. Musina, S. A. Krasnov, M. K. Kadirov, A. A. Karasik, Y. G. Budnikova and O. G. Sinyashin, Russ. J. Phys. Chem., 2011, 85, 2214 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision A.1), Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed.
- S. K. Latypov, F. M. Polyancev, D. G. Yakhvarov and O. G. Sinyashin, Phys. Chem. Chem. Phys., 2015, 17, 6976–6987 RSC.
- CrysAlis-Pro: Data collection and data reduction software package, Agilent Technologies.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- K. Brandenburg, DIAMOND (Version 3.2k), Crystal Impact GbR, Bonn, Germany Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D/2D NMR spectra of macrocycle 2a; 1H–13C/1H–15N/1H–31P HMBC correlation experiments; kinetics of splitting of 2a; X-ray crystallography. CCDC 1060031. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01910a |
| This journal is © The Royal Society of Chemistry 2015 |