
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ferrocenylmethylation reactions with a phosphinoferrocene betaine†
Martin
Zábranský
,
Ivana
Císařová
and
Petr
Štěpnička
*
Department of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 128 40 Prague, Czech Republic. E-mail: stepnic@natur.cuni.cz
First published on 8th July 2015
Abstract
A phosphinoferrocene betaine, N-{[1′-(diphenylphosphino)ferrocenyl]methyl}-N,N-dimethyl-3-sulfo-1-propanaminium, inner salt, Ph2PfcCH2NMe2(CH2)3SO3 (2; fc = ferrocene-1,1′-diyl), was prepared by alkylation of Ph2PfcCH2NMe2 (1) with 1,3-propanesultone, and was studied as a ferrocenylmethylation agent. The treatment of 2 with NaOH in hot water–dimethyl sulfoxide produced phosphinoalcohol Ph2PfcCH2OH (3) in a 64% yield, whereas a similar reaction with MeONa in dimethylsulfoxide–methanol furnished the corresponding ether, Ph2PfcCH2OMe (4), in a 47% yield. In subsequent experiments, betaine 2 was employed in the synthesis of phosphinoferrocene sulfones, Ph2PfcCH2SO2R, where R = Me (6a), Ph (6b), and 4-tolyl (6c). Compounds 6a–c and some by-products of the ferrocenylmethylation reactions, namely alcohol 3, 1′-(diphenylphosphino)-1-methylferrocene (5), and 1-{[diphenyl(2,4-cyclopentadien-1-ylidene)phosphoranyl]methyl}-1′-(diphenylphosphino)ferrocene (7) structurally characterised. Reactions of 6a as the representative with ZnX2/NaX (X = Br and I) afforded unique coordination polymers [ZnNaX3(6a)(CH3OH)]n featuring tetrahedral Zn(II) and octahedral Na(I) centres bridged by halide ions, solvating methanol and the sulfone ligands. The reaction of 6a with ZnBr2/KBr produced an analogous product, [ZnKBr3(6a)(CH3OH)]n, while that with ZnBr2/LiBr furnished a different, pseudodimeric complex [Zn2Li2Br6(6a)2(CH3OH)4(H2O)]·CH3OH, featuring tetrahedrally coordinated Zn(II) and Li(I) centres bridged by 6a. Reactions of 6a with ZnBr2/MBr (M = Rb, Cs) and NaCl/ZnCl2 did not yield similar products because of an easy precipitation (low solubility) of the respective alkali metal halides.
Introduction
Shortly after the discovery of ferrocene1 and the elucidation of its real structure2 in the early 1950s, ferrocenylmethylation was recognised as a powerful method for the preparation of various ferrocene derivatives.3 This reaction typically utilises stable starting materials capable of serving as precursors of the stabilised ferrocenylmethylium cation,4 which is allowed to react with nucleophiles (Nu) to afford derivatives of the type FcCH2Nu (Fc = ferrocenyl). The most often employed ferrocenylmethylation reagents are undoubtedly FcCH2NMe2 and [FcCH2NMe3]I, with which the reaction was developed, though other compounds, e.g., [FcCH2PPh3]I or FcCH2OH (the latter in combination with an acid), have also found practical applications.3In the synthesis of phosphinoferrocene donors,5 ferrocenyl-methylation reactions have been applied only rarely because of competitive reactions affecting the phosphine moieties (alkylation).6 Therefore, the [1′-(diphenylphosphino)ferrocene-1-yl]methyl derivatives have typically been synthesised either indirectly (e.g., via late-stage lithiation/phosphinylation7) or from P-protected building blocks, such as Ph2P(S)fcCH2OH8 or the borane adduct Ph2PfcCH2OH·BH3.9
In view of our recent work focusing on the preparation and coordination properties of 1′-(diphenylphosphino)-1-[(dimethylamino)methyl]ferrocene (1)10,11 and other 1′-functionalised phosphinoferrocene derivatives possessing an inserted methylene group,9,12 we wanted to extend the hitherto unexplored synthetic chemistry of the former compound, which led us to attempt the preparation of ammonium salts derived from 1 and study their prospective synthetic applications. In this contribution, we describe the selective synthesis and structural characterisation of the phosphinoferrocene betaine Ph2PfcCH2N+Me2(CH2)3SO3− (2; fc = ferrocene-1,1′-diyl) and its utilisation in the preparation of the known and some new 1′-functionalised phosphinoferrocene donors such as 1′-[(diphenylphosphino)ferrocenyl]methyl sulfones Ph2PfcCH2SO2R. Furthermore, we report on the reactions of the representative ligand, Ph2PfcCH2SO2Me, with zinc(II) and alkali metal halides, leading to structurally unique mixed-metal coordination polymers.
Results and discussion
Synthesis of betaine 2 and initial reaction tests
Betaine 2 was synthesised similarly to its non-phosphinylated analogue (Scheme 1)13 by the reaction of phosphinoamine 110 with 1,3-propanesultone (1,2-oxathiolane-2,2-dioxide) in dry benzene. The compound was isolated by column chromatography, resulting as an air-stable orange solid in an 81% yield (at the 10 mmol scale).14 Crystallisation from methanol–tetrahydrofuran–diethyl ether afforded the stoichiometric solvate 2·CH3OH, which was structurally characterised by single-crystal X-ray diffraction analysis (vide infra). | ||
| Scheme 1 Synthesis and model reactions of betaine 2. | ||
The 1H and 13C NMR spectra of 2 combine the signals due to the phosphinoferrocenyl moiety with those of the NMe2 group and the propane-1,3-diyl bridge. The 31P NMR resonance is observed at δP −18.1 ppm, suggesting that the phosphine moiety remained intact. In its IR spectrum, betaine 2 shows strong signals attributable to the vibrations of the terminal sulfonate moiety (νs 1037 cm−1 and νas 1189 cm−1), whereas the electrospray ionisation (ESI) mass spectrum reveals signals of the pseudomolecular ions [M + X]+, where M = H, Na, and K, and of the characteristic fragment15 ions due to the substituted ferrocenylmethylium cation [Ph2PfcCH2]+ at m/z 383.
The possible synthetic applications of betaine 2 were first examined by its conversion into the known alcohol 312a and the corresponding methyl ether 49via reactions with the respective nucleophiles (Scheme 1). These ferrocenylmethylation reactions were carried out similarly to the literature13,16 but carefully optimised. For solubility reasons, dimethyl sulfoxide was chosen as the solvent, and the reactions were performed at temperatures above 100 °C since lower reaction temperatures markedly reduced the yield of the substitution product (N.B. unreacted 2 could be recovered from the reaction mixture in such cases). The reaction time was maintained at minimum (typically 1 h) in order to prevent decomposition and oxidation of the phosphine moiety.
For the preparation of alcohol 3, the best reaction conditions were found to consist of refluxing the solution of betaine 2 in a mixture of DMSO and 2 M aqueous NaOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; the concentration of NaOH in the resulting solution was 1 M) for 1 h. Product 3 was isolated by extraction and purified by column chromatography, resulting in a 64% yield (at the 2 mmol scale). A small amount of a less polar side-product was also isolated, being identified as 1′-(diphenylphosphino)-1-methyl-ferrocene, Ph2PfcMe (5; typically ca. 5%). This rather unexpected product probably results via “quenching” of the intermediate cation Ph2PfcCH2+ upon attack of other C–H bonds (acid–base equilibria) rather than by interaction with any proton source in the reaction system.
:1; the concentration of NaOH in the resulting solution was 1 M) for 1 h. Product 3 was isolated by extraction and purified by column chromatography, resulting in a 64% yield (at the 2 mmol scale). A small amount of a less polar side-product was also isolated, being identified as 1′-(diphenylphosphino)-1-methyl-ferrocene, Ph2PfcMe (5; typically ca. 5%). This rather unexpected product probably results via “quenching” of the intermediate cation Ph2PfcCH2+ upon attack of other C–H bonds (acid–base equilibria) rather than by interaction with any proton source in the reaction system.
The etherification reaction was similarly performed in a mixture of dimethyl sulfoxide and methanolic MeONa (1 M MeONa in the reaction system) at lower temperatures (but still under reflux conditions) for 2 h, affording phosphinoether 4 in a 47% isolated yield.
The crystal structures of 2·CH3OH and 5
The solvate 2·CH3OH, isolated after crystallisation by liquid-phase diffusion of tetrahydrofuran and diethyl ether into a solution of the betaine in methanol, crystallises with the symmetry of the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Its structure is presented in Fig. 1, and the relevant geometric data are summarised in Table 1.
. Its structure is presented in Fig. 1, and the relevant geometric data are summarised in Table 1.
 | ||
| Fig. 1 PLATON plot of the phosphinobetaine molecule in the structure of 2·CH3OH showing the atom-labelling scheme and displacement ellipsoids at a 30% probability level. | ||
| Distances | Angles | ||
|---|---|---|---|
| a Note: parameters pertaining to the ferrocene moiety as well as the N–C–N angles are discussed in the main text. | |||
| S–O1 | 1.463(1) | C6–P–C12 | 102.25(6) |
| S–O2 | 1.450(1) | C6–P–C18 | 101.92(6) |
| S–O3 | 1.454(1) | C12–P–C18 | 102.45(6) |
| S–C28 | 1.777(2) | O1–S–O2 | 112.85(7) |
| N–C11 | 1.525(2) | O1–S–O3 | 113.21(7) |
| N–C24 | 1.499(2) | O2–S–O3 | 113.00(7) |
| N–C25 | 1.497(2) | C28–S–O1 | 103.67(7) |
| N–C26 | 1.518(2) | C28–S–O2 | 106.38(7) |
| P–C6 | 1.811(1) | C28–S–O3 | 106.84(7) |
| P–C12 | 1.834(1) | C1–C11–N–C26 | −177.8(1) |
| P–C18 | 1.837(1) | C11–N–C26–C27 | −54.8(1) |
| C1–C11 | 1.489(2) | N–C26–C27–C28 | −172.0(1) |
| C1S–O1S | 1.412(2) | C26–C27–C28–S | 171.39(9) |
The ferrocene unit in the structure of 2 shows similar Fe–C distances (2.019(1)–2.051(1) Å) and, accordingly, practically negligible tilting (the dihedral angle of the least-squares planes of the cyclopentadienyl ring is 1.40(8)°). The substituents attached to the ferrocene moiety adopt a nearly ideal synclinal eclipsed conformation, as evidenced by the torsion angle C1–Cg1–Cg2–C6 of −73.49(9)° (cf. the ideal value of 72°).
The carbons surrounding the positively charged nitrogen atom in 2 constitute a regular tetrahedral environment, with the C–N distances and associated bond angles (C–N–C) in the range of 1.497(2)–1.525(2) Å and 106.9(1)–111.0(1)°, respectively. The environment of the sulfur atom is somewhat distorted, presumably because of the different sizes of the bonded atoms and, also, a repulsion of the oxygen atoms (S–C > S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and OSO > C–SO; see parameters in Table 1).
O and OSO > C–SO; see parameters in Table 1).
The individual molecules constituting the crystals of 2·CH3OH assemble into dimers of inversion-related molecules through charge-supported hydrogen bonds between two oxygen atoms of the negatively charged sulfonate group and the CH3 hydrogens polarised by the positively charged nitrogen (Fig. 2). The solvating methanol forms an O–H⋯O hydrogen bond with the remaining sulfonate oxygen. Additional C–H⋯O interactions further interconnect the (2)2(CH3OH)2 units into columnar stacks oriented along the crystallographic a-axis.
 | ||
| Fig. 2 View of the hydrogen bonded dimers in the structure of 2·CH3OH. For clarity, only the pivotal atoms of the phenyl rings are shown. The hydrogen bond parameters are as follows: C11–H11B⋯O2i: C11⋯O2 = 3.417(2) Å, angle at H11 = 166°; C25–H25B⋯O3i: C25⋯O3 = 3.396(2) Å, angle at H25B = 167°; O1S–H1S⋯O1: O1S⋯O1 = 2.719(2) Å, angle at H1S = 172°; (i) (1 − x, 1 − y, 2 − z). | ||
The solid-state structure of 5 (Fig. 3) resembles that of the corresponding borane adduct 5·BH3.9 Whereas the C1–C11 bond lengths (1.497(2) Å) in both compounds are practically identical (within the three-sigma level), the P–C bonds in 5 (P–C6 1.810(2), P–C12 1.841(2), and P–C18 1.834(2) Å) are slightly but statistically significantly longer (by ca. 0.02 Å) than those in the mentioned reference compound, reflecting the electronic changes associated with the adduct formation (R3P → BH3). The Fe–C distances in the molecule of 5 span a narrow range of 2.037(1)–2.051(2) Å, which is in turn reflected in an insignificant tilting of the cyclopentadienyl rings (the tilt angle is as low as 1.1(1)°). As indicated by the torsion angle C1–Cg1–Cg2–C6 of −77.8(1)°, the ferrocene unit has a synclinal eclipsed conformation, which is also similar to that of the aforementioned borane adduct.
 | ||
| Fig. 3 PLATON plot of the molecular structure of 5 showing 30% displacement ellipsoids. | ||
Synthesis and characterisation of phosphine-sulfones 6
Aiming at the preparation of new phosphinoferrocene ligands via ferrocenylmethylation, betaine 2 was subsequently reacted with sodium sulfinates to give the respective phosphino-ferrocene sulfones 6 (Scheme 2). The reactions were performed with an excess of the sulfinate salts (2:RSO2Na = 1:2.5) in refluxing DMSO–water for 2 h, similarly to the synthesis of ferrocenylmethyl sulfones FcCH2SO2R from simple ferrocenyl-methylation agents.13,17
 | ||
| Scheme 2 Preparation of phosphinoferrocenyl sulfones 6. | ||
The yields of the sulfones after chromatographic purification were ca. 30% for 6a and approximately 50% for the compounds bearing the aromatic substituents (6b and 6c), which are less than in the reactions leading to FcSO2R. Therefore, we sought for other reaction products to gain more detailed information regarding the course of these particular ferrocenylmethylation reactions.
In the case of the reaction of betaine 2 with MeSO2Na, a careful chromatographic purification of the reaction mixture led to the isolation of alcohol 3 (2%) and phosphorane 7 (Scheme 3; 12%). Together with 6a, compounds 3 and 7 account for nearly 45% of the starting material. The reactions leading to aryl sulfones 6b and 6c are more selective (isolated yields: ca. 50%) but afford identical by-products (isolated yields of 3 and 7 are ca. 3% and 10–15%, respectively). Apparently, the cation Ph2PfcCH2+ generated in situ from 2 enters into reactions with all other available nucleophiles, including OH− or phosphines. Interaction with the latter provides cationic products (i.e., phosphonium salts arising from “self-alkylation” of the parent 2 with Ph2PfcCH2+) and, consequently, also their decomposition products such as 7. The fact that no 7 could be detected in the reaction mixtures obtained after treatment of 2 with NaOH and NaOMe (vide supra) can well reflect the higher relative amounts of these nucleophilic reagents, that suppress the competing reactions with other nucleophiles. Attempts to isolate the anticipated cationic (and hence more polar) side products or to recover unreacted 2 during the course of chromatographic purification of crude sulfones 6 failed.
 | ||
| Scheme 3 Canonical forms of compound 7. | ||
The formulation of 6a–c and 7 was inferred from NMR and IR spectra, ESI mass spectra, and elemental analysis and was unequivocally confirmed by single-crystal X-ray diffraction. The NMR spectra of sulfones 6 comprise the signals due to the (diphenylphosphino)ferrocenyl unit and its attached methylene linker (CH2: δH/δC 3.59/56.65 for 6a, and ca. 3.68/58.3 for 6b and 6c). The signals of the sulfone substituents, as well as the 31P NMR resonances (δP ≈ −17 ppm), are observed in the usual ranges. The IR spectra of the sulfones display the characteristic strong bands of the sulfone moieties centred at approximately 1310 (νas) and 1145 cm−1 (νs).17a
The 1H and 13C NMR spectra of the by-product 7 contain signals of the 1,1′-disubstituted ferrocene moiety and two sets of resonances of the non-equivalent PPh2 groups, which is also reflected in the 31P NMR spectrum showing two singlets at δP −16.9 and 10.1. The presence of the cyclopentadienylidene unit18 in 7 is manifested by a pair of multiplets at δH 6.12 and 6.40 and a pair of doublets at δC 113.92 and 115.83 in the 1H and 13C NMR spectra, respectively. The 13C NMR signal due to Cipso in the PC5H4 moiety is observed at δC 78.21 as a phosphorus-coupled doublet (1JPC = 110 Hz). The signals of the connecting methylene group are found at δH 3.59 (doublet with 2JPH = 12.7 Hz) and δC 30.01 (dd, 1JPC = 53, JPC = 1 Hz).
Crystallisation of 6a from ethyl acetate–hexane provided crystals of a triclinic modification (denoted as 6a). Crystals of another polymorph, 6a′, were serendipitously isolated during an attempted preparation of Zn(II) complexes, i.e., upon crystallization of a 6a/ZnBr2 mixture from methanol–diethyl ether (vide infra). The polymorphs differ by the symmetry of the crystal lattice (6a: triclinic, P; 6a′: monoclinic, P21/c) and by the overall conformation of the molecules constituting their crystals (Fig. 4 and Table 2).
 | ||
| Fig. 4 PLATON plots of the molecular structures of the triclinic (top; 6a) and monoclinic (bottom; 6a′) polymorphs of 1′-(diphenylphosphino)-1-[(methylsulfonyl)methyl]ferrocene. The displacement ellipsoids enclose the 30% probability level. | ||
| Parametera | 6a | 6a′ | 6b | 6c |
|---|---|---|---|---|
| a Definitions: tilt stands for the dihedral angle of the least-squares planes of the cyclopentadienyl rings. The torsion angle C1–Cg2–Cg2–C6 reflects the mutual conformation of the substituents at the ferrocene unit; Cg1 and Cg2 are the centroids of the cyclopentadienyl rings C(1–5) and C(6–10), respectively. | ||||
| Fe–C | 2.031(2)–2.060(2) | 2.035(2)–2.054(2) | 2.026(1)–2.058(2) | 2.040(2)–2.056(2) |
| Tilt | 2.98(9) | 3.3(1) | 2.64(9) | 1.9(1) |
| C1–Cg2–Cg2–C6 | −79.1(1) | 162.5(1) | 128.2(1) | 73.3(1) |
| P–C6 | 1.817(2) | 1.821(2) | 1.812(2) | 1.815(2) |
| P–C12 | 1.838(2) | 1.835(2) | 1.832(2) | 1.835(2) |
| P–C18 | 1.839(2) | 1.832(2) | 1.834(2) | 1.834(2) |
| C1–C11 | 1.494(2) | 1.494(3) | 1.488(2) | 1.489(3) |
| C11–S | 1.786(2) | 1.781(2) | 1.784(2) | 1.792(2) |
| S–O1 | 1.435(1) | 1.432(2) | 1.446(1) | 1.440(2) |
| S–O2 | 1.439(1) | 1.433(2) | 1.441(1) | 1.445(2) |
| S–C24 | 1.764(2) | 1.754(2) | 1.765(2) | 1.770(2) |
| C1–C11–S | 111.5(1) | 110.9(1) | 113.0(1) | 111.8(1) |
| O1–S–O2 | 117.80(8) | 117.6(1) | 118.60(7) | 118.42(9) |
| C11–S–C24 | 103.23(7) | 103.2(1) | 103.72(7) | 106.67(8) |
| C1–C11–S–C24 | 176.3(1) | −178.6(1) | −55.3(1) | 71.8(1) |
Thus, whereas the molecular structures of the two polymorphs are expectedly very similar in terms of interatomic distances and angles, they differ in the mutual orientation of the substituted cyclopentadienyl rings, which are nearly synclinal eclipsed in 6a and exactly halfway between anticlinal eclipsed and antiperiplanar staggered in 6a′ (compare the C1–Cg1–Cg2–C6 angles in Table 1).19 Another less pronounced difference can be observed in the orientation of the PPh2 units resulting from different rotations along the pivotal C6–P bond and from the tilting of the phenyl rings.
The geometry of the (methylsulfonyl)methyl moiety in 6a and 6a′ agrees well with that of, e.g., phenyl methyl sulfone, 4-methoxyphenyl methyl sulfone,20 and (benzylsulfonyl)-methanol.21 Similar to these compounds, the O1–S–O2 angle is the most opened and the C11–S–C24 angle is the most acute among the bond angles around the sulfur atoms in 6a and 6a′, most likely due to an electrostatic repulsion of the electronegative oxygen atoms.
The main difference between the molecular structures of sulfones 6b and 6c (Fig. 5 and Table 2) can also be found in the conformation of the 1,1′-disubstituted ferrocene unit, which is intermediate between anticlinal staggered and anticlinal eclipsed for 6b and synclinal eclipsed for 6c. In both cases, the CH2SO2Ar (Ar is an aryl) pendants extend away from the ferrocene core but adopt different orientations as indicated by the torsion angle C1–C11–S–C24 (see Table 2) and the dihedral angles of the C(1–5) and C(24–29) ring planes of 45.93(8)° and 33.5(1)° for 6b and 6c, respectively. On the other hand, the compounds comprise regular ferrocene moieties (tilt angles below ca. 3°) and show similar individual interatomic distances and angles that do not depart substantially from those of 6a/6a′ and benzylsulfones of the type ArCH2SO2Ar (Ar = an aryl).22
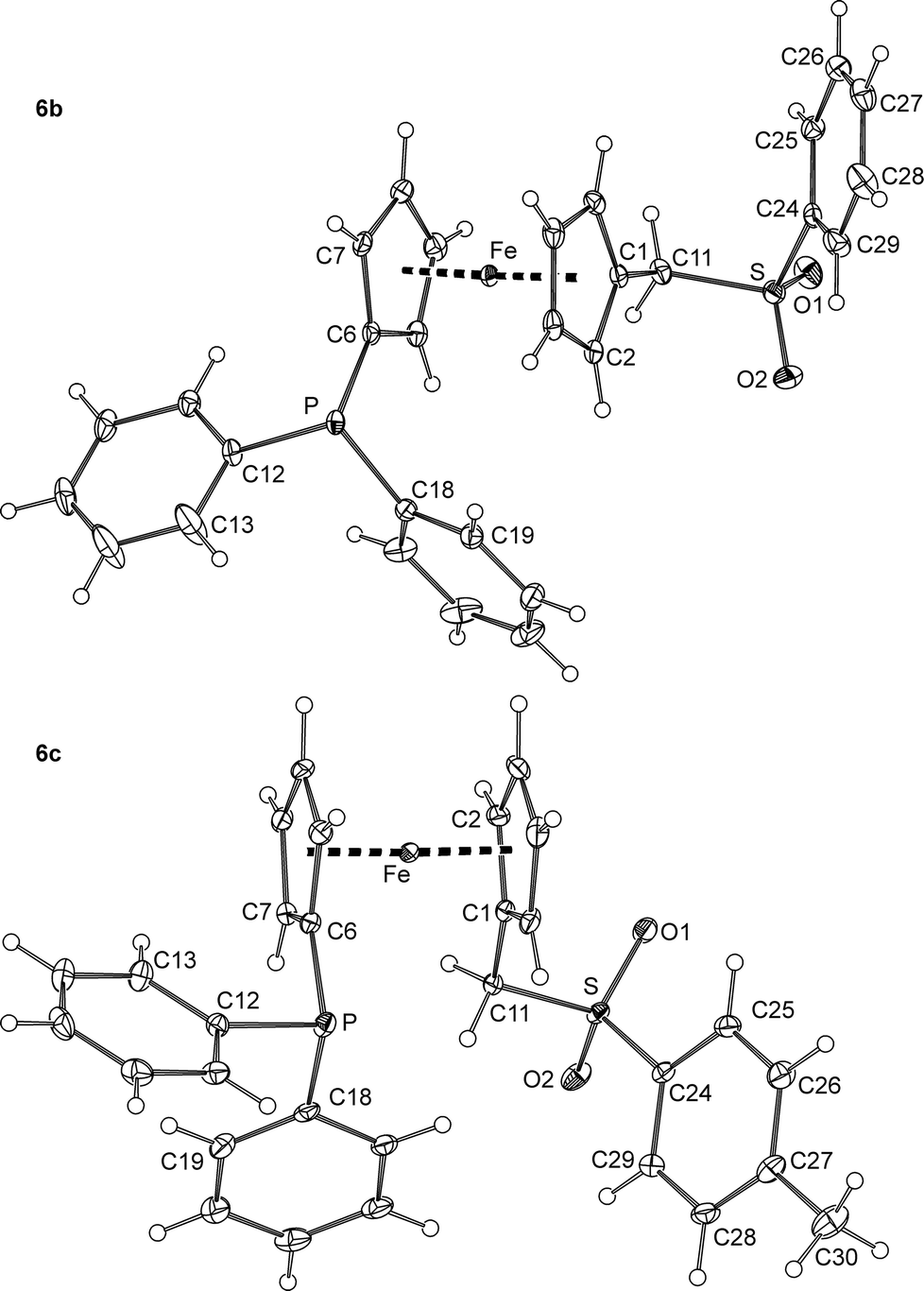 | ||
| Fig. 5 PLATON plots of the molecular structure of the phosphinoferrocene sulfones 6b and 6c at the 30% probability level. | ||
The ferrocene cyclopentadienyls in the structure of by-product 7 (Fig. 6) are tilted by 2.6(1)° and assume a conformation near synclinal eclipsed, as evidenced by the C1–Cg1–Cg2–C6 torsion angle of 79.3(1)°. The individual Fe–C distances are in the range of 2.026(2)–2.050(2) Å. The cyclopentadienylidene substituent at the phosphorus P2 exhibits only moderate bond alternation (C31–C32 1.425(3), C32–C33 1.382(3), C33–C34 1.406(3), C34–C35 1.382(3), and C35–C31 1.414(3) Å). Its pivotal P2–C31 bond (1.722(2) Å) is of similar length to those in MePh2PC5R4 (R = H: 1.728(2) and 1.727(2) Å;18 R = Me: 1.720(2) and 1.717(2) Å23). As such, it is shorter than the P1–C6 bond in 7 (1.811(2) Å) and the P+–C5H4 bond in [Fe(η5-C5H4P+Ph2Me){η5-C5H4Cr−(CO)6}] (1.769(5) Å)24 but longer than the PCH2 bond in the archetypal phosphorane Ph3PCH2 (1.662(8) and 1.659(8) Å).25 These features suggest compound 7 to possess an intermediate structure between two extreme canonical forms, viz. cyclopenta-2,4-dien-1-ylidene phosphorane (7a) and zwitterionic phosphonium cyclopentadienide (7b), as depicted in Scheme 3. It is also noteworthy that the P–Ph bonds are significantly longer for P1 (P1–C12 1.839(2), P1–C18 1.841(2) Å) than for P2 (P2–C36 1.801(2), P2–C42 1.810(2) Å), which bears a positive charge, most likely due to an electron density transfer from the aromatic rings to the partly positively charged P2. In contrast, the length of the P2–C11 bond (1.817(2) Å) agrees well with that of the similar bond in the phosphonium salt [FcPPh2(CH2Ph)]Cl (1.819(3) Å).26
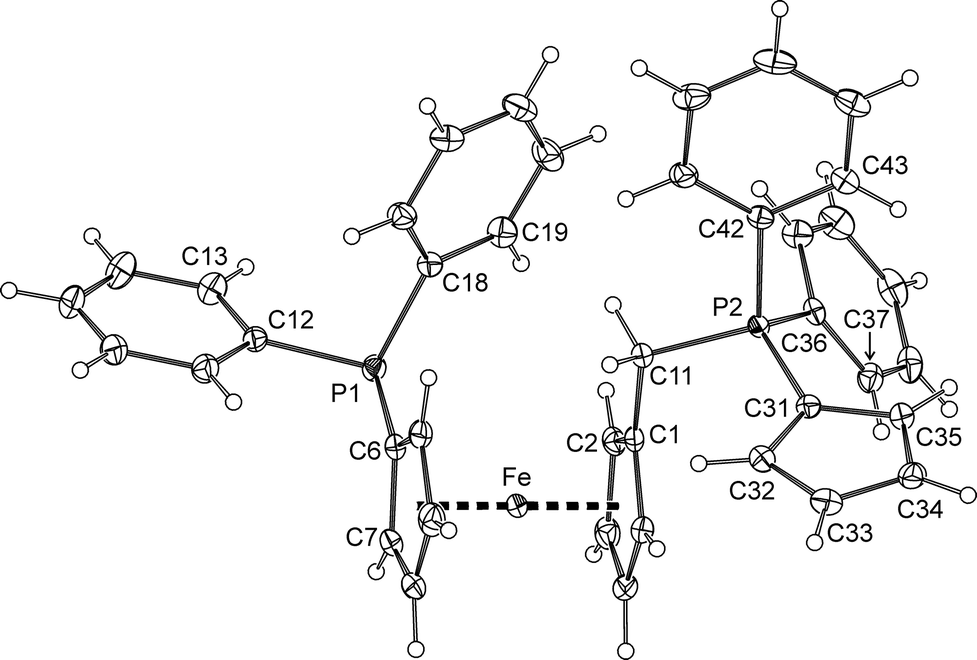 | ||
| Fig. 6 PLATON plot of the molecular structure of 7 (30% displacement ellipsoids). | ||
Preparation and structure of mixed-cation complexes with 6a
Coordination preferences of the newly prepared phosphino-ferrocene sulfones were studied in reactions of 6a as the model representative, with zinc(II) halides. These salts were chosen mainly due to the position of the Zn(II) ion at the borderline between hard and soft metal ions27 and due to its closed (d10) coordination sphere, which makes it structurally variable because of the absence of crystal-field stabilisation.28To our disappointment, repeated experiments aiming at the preparation of defined products by co-crystallisation of zinc(II) halides with 6a were unsuccessful. In one case, the crystallisation of a ZnBr2-6a mixture in methanol/diethyl ether provided crystals of monoclinic 6a′. Eventually, the attempted preparation of a Zn(II) complex by reacting ZnBr2 and 6a at a 1:1 molar ratio in methanol–chloroform, followed by evaporation and crystallisation from CHCl3/methyl tert-butyl ether, yielded a few crystals of 8b, which were used directly for X-ray structure determination.29 The intriguing structure of this rather unexpected product (vide infra) led us to study the formation of such complexes systematically by changing the halide in ZnX2/NaX (NaCl–NaBr–NaI) and then also the alkali metal cation in ZnBr2/MBr (M = Li, Na, K, Rb, and Cs).
Attempts to isolate any mixed-cation compound from the 6a/ZnCl2/NaCl system failed, presumably because of the ionic nature of NaCl, which limits its solubility in organic solvents. The experiments furnished only crystals of uncoordinated 6a. On the other hand, the reactions of 6a with NaX/ZnX2, where X = Br and I (all in equimolar amounts), in methanol followed by crystallisation upon layering the reaction mixture with methyl tert-butyl ether produced the respective coordination polymers 8a (X = Br) and 8b (X = I) as orange, nicely crystalline, and air-stable solids in good yields (Scheme 4).
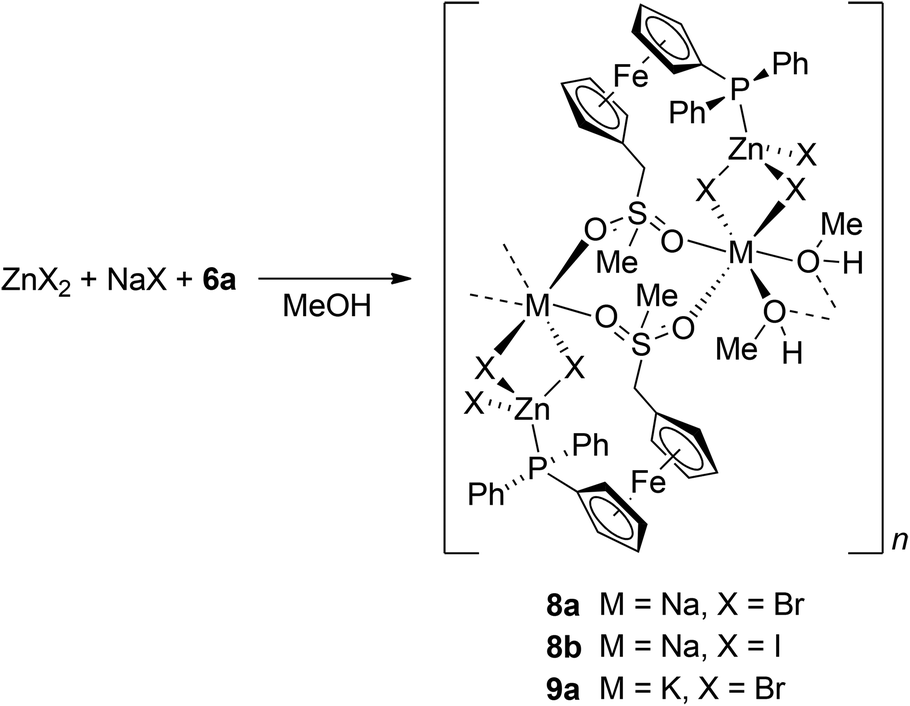 | ||
| Scheme 4 Preparation of the mixed-metal complexes 8a, 8b and 9a. | ||
The reactions of 6a with ZnBr2 with other alkali metal bromides were performed similarly to the preparation of the mentioned Na–Zn complexes but in solvent mixtures with an optimised chloroform/methanol ratio to ensure sufficient solubility of the alkali metal halide and not suppress separation of the product after the addition of methyl tert-butyl ether.30 Attempted reactions with RbBr and CsBr did not afford any M–Zn complex because these salts separated from the reaction mixture, whereas the analogous reaction of 6a with ZnBr2 and KBr produced K–Zn complex 9a (Scheme 4), which adopts the structure of its sodium congener. In contrast, the reaction 6a with ZnBr2 and LiBr produced a Zn–Li complex [Zn2Li2Br6(6a)2(CH3OH)4(H2O)]·CH3OH (10a·CH3OH). In its structure, the phosphinoferrocene ligands coordinate the terminal ZnBr3 units via their phosphine groups (similarly to the mentioned Na–Zn and K–Zn complexes) and further bind solvated Li+ ions via the sulfone oxygens to form a discrete pseudodimeric assembly (Scheme 5).31
 | ||
| Scheme 5 Preparation of the Li–Zn complex 10a. | ||
The M–Zn complexes (M = alkali metal cation) disintegrate upon dissolving in donor solvents. This was evidenced by the 1H and 31P{1H} NMR spectra recorded for solutions of crystalline 8a in CD3OD that reveal the exclusive presence of uncoordinated 6a in solution (δP −16.3 ppm) and also by the ESI mass spectra showing only signals due to 6a and its fragments (see Experimental). The same applies to the elusive 6a-ZnBr2 complexes (intermediates) as similar features have been observed in the NMR and ESI MS spectra of a residue obtained by evaporation of a 1:1 mixture of 6a-ZnBr2. Hence, the characterisation of the mixed-metal complexes had to be confined to solid-state techniques. Unfortunately, the IR spectra were of little diagnostic value because of their relative complexity (see ESI, Fig. S1†) and only marginal shifts of the diagnostic bands upon coordination (e.g., the bands due to the sulfone moiety as compared to uncoordinated 6a). Nonetheless, the IR spectra of 8a, 8b and 9a were very similar, suggesting analogous structures for these compounds, and displayed additional broad bands attributable to νOH vibrations at ca. 3470–3490 cm−1, attributable to the “solvating” methanol. Nonetheless, unequivocal structural information was gained from single-crystal X-ray diffraction analysis.
Compounds 8a and 8b are essentially isostructural, and the minor differences in the lattice parameters and atomic coordinates are associated with the different sizes of the halide anions. Compound 9a also has practically the same structure, albeit described by different cell parameters because even a small variation in the cell angles near 90° as in this particular case can result in another reduced triclinic cell setting.32 The structure of 8a is depicted in Fig. 7, and the displacement ellipsoid plots for all three compounds are presented in the ESI (Fig. S2–S4†). Pertinent geometric parameters are given in Table 3.
 | ||
| Fig. 7 Section of the infinite coordination chain in the structure of 8a (for a conventional displacement ellipsoid plot, see the ESI†). For clarity, the CH hydrogens are omitted, and the CH3OH⋯Br hydrogen bond is indicated by a red dotted line (O1S⋯Br1 = 3.599(2) Å; the corresponding distance in the structures of 8b and 9a are as follows: 8b, O1S⋯I1 = 3.828(3) Å; 9a, O1S⋯Br1 = 3.540(2) Å). | ||
| Parameter | 8a (M/X = Na/Br) | 8b (M/X = Na/I) | 9a (M/X = K/Br) |
|---|---|---|---|
| a For definitions, see the footnote to Table 2. Symmetry codes: (i) 2 − x, −y, −z, (ii) 1 − x, −y, −z, (iii) 1 − x, 1 − y, −z; (iv) −x, 1 − y, −z. | |||
| Zn–P | 2.4304(8) | 2.445(1) | 2.4310(6) |
| Zn–X1 | 2.3965(4) | 2.6030(4) | 2.4065(4) |
| Zn–X2 | 2.3980(4) | 2.5956(4) | 2.4049(4) |
| Zn–X3 | 2.4517(4) | 2.6610(4) | 2.4440(3) |
| M–X2 | 3.089(1) | 3.362(2) | 3.3218(6) |
| M–X3 | 3.010(1) | 3.244(2) | 3.2865(6) |
| M–O1 | 2.512(2) | 2.485(3) | 2.725(2) |
| M–O2i/iii | 2.260(2)i | 2.264(3)i | 2.576(2)iii |
| M–O1S | 2.616(2) | 2.686(3) | 2.829(2) |
| M–O1Sii/iv | 2.387(2)ii | 2.399(3)ii | 2.730(2)iv |
| Fe–C | 2.024(3)–2.051(3) | 2.034(3)–2.043(4) | 2.027(2)–2.056(2) |
| Tilt | 4.1(2) | 4.1(2) | 5.1(1) |
| C1–Cg1–Cg2–C6 | −85.6(2) | −84.7(2) | −85.4(1) |
| S–O1 | 1.446(2) | 1.450(3) | 1.444(2) |
| S–O2 | 1.439(2) | 1.437(3) | 1.442(2) |
Compounds 8a and 8b are one-dimensional coordination polymers in which the ZnX3(6a-κP) units coordinate the Na(I) ions via two halide ions (bridging X2 and X3) and the sulfone oxygen O1. The coordination sphere of the Na(I) ion is completed by the sulfone O2 located in an adjacent ZnX3(6a-κP) moiety, related by crystallographic inversion, and also by a methanol molecule and its inversion-related counterpart. The solvating methanol further stabilises the structure through a hydrogen bond with the Zn-bound halide (O1S–H1O⋯X1, see Fig. 7).
The Zn(II) ions in the structures of 8a and 8b have the usual, albeit distorted, tetrahedral donor environment (donor set: PX3), where two Zn–X distances are somewhat shorter than the remaining one (Zn–X1/2 < Zn–X3). The associated interligand angles increase in the following order: X1–Zn–X3 < X2–Zn–P ≈ X2–Zn–X3 < X1–Zn–P ≈ X1–Zn–X2 < X3–Zn–P (cf. the ranges of 102.64(2)–114.50(2)° and 101.61(1)–114.68(3)° for 8a and 8b, respectively). In contrast, the sodium cation, as the second metal centre in the structures, has an unsymmetric octahedral coordination (donor set: cis-X2O4), wherein the extreme Na-donor distances differ by ca. 0.8 and 1.1 Å and the interligand angles (cis-only) span the ranges of 78.25(7)–101.12(8)° and 77.1(1)–104.7(1)° for 8a and 8b, respectively. In both cases, the Na atoms appear displaced from the geometrical centre of the octahedron towards O1Sii and O2i in heavily twisted pseudoequatorial planes {Na, X2, X3, O1Sii, O2i}. The observed angular distortions of the coordination spheres around both the Zn(II) and Na(I) ions appear to result from an interplay between the unlike metal–donor distances, steric requirements of the individual donors, constraints imposed by the doubly bridged fragments (Zn(μ-Br)2Na and Na(μ-CH3OH)2Na), and the hydrogen-bond interactions.
As indicated above, the structure of 9a is very similar to its Na congener 8a, with the observed differences reflecting the larger size of the alkali metal cation present in the structure. The geometry of the PZnBr3 moiety remains virtually unchanged upon going from 8a to 9a; the interligand angles span the range 102.97(1)–113.11(2)° and follow the trend described for 8a though not with the same differences between the individual values. Although the overall coordination environment of the K+ ion also remains seemingly the same (cis-interligand angles: 74.02(1)–107.97(5)°), the coordination sphere is expanded because of the longer potassium-donor bonds (by approximately 0.21–0.34 Å in the respective pairs).
The structure of 10a·CH3OH (Fig. 8, parameters in Table 4) reveals that the replacement of Na+ with the Li+ ion, which is smaller and prefers a tetrahedral coordination environment, results in an opening of the polymeric structure and incorporation of solvent molecules as additional donors into the structure. One of the Li+ cations (Li1) is coordinated by a sulfonate oxygen (O2) and three methanol molecules (OnS, n = 1–3) constituting a tetrahedral donor set. The other Li+ cation (Li2) has a similar coordination, binding two sulfonate oxygens (from different molecules of 6a), methanol (O4S) and water molecule (O1W). It is noteworthy that the asymmetric environment of the sulfur atoms renders the structure chiral.
 | ||
| Fig. 8 Perspective drawing of the structure of 10a·CH3OH. The molecule of solvating methanol and CH hydrogens are omitted for clarity. For a displacement ellipsoid plot, see the ESI (Fig. S5†). | ||
| a For definitions, see the footnote to Table 2. | |||
|---|---|---|---|
| Zn1–P1 | 2.437(1) | Zn2–P2 | 2.442(1) |
| Zn1–Br1 | 2.4174(8) | Zn2–Br4 | 2.3763(8) |
| Zn1–Br2 | 2.4016(8) | Zn2–Br5 | 2.4056(8) |
| Zn1–Br3 | 2.4034(8) | Zn2–Br6 | 2.4362(8) |
| Li1–O2 | 1.98(1) | Li2–O1 | 1.917(8) |
| Li1–O1S | 1.94(1) | Li2–O4 | 1.907(9) |
| Li1–O2S | 1.911(9) | Li2–O4S | 1.916(9) |
| Li1–O3S | 1.962(9) | Li2–O1W | 1.92(1) |
| O–Li1–O | 100.9(4)–123.8(5) | O–Li2–O | 99.1(4)–114.4(4) |
| Fe1–C | 2.028(6)–2.052(5) | Fe2–C | 2.020(6)–2.053(6) |
| Tilt | 5.2(3) | Tilt | 5.5(3) |
| C1–Cg1–Cg2–C6 | −81.6(4) | C1–Cg1–Cg2–C6 | 82.2(4) |
| S1–O1 | 1.433(3) | S2–O3 | 1.443(4) |
| S1–O2 | 1.451(4) | S2–O4 | 1.454(3) |
On the other hand, the halide anion is transferred to the zinc(II) centre, which thus gains a distorted tetrahedral PBr3 coordination as observed for the Na/K–Zn complexes discussed above. Both ZnBr3 units in the structure of 10a assume a sterically loose staggered orientation with respect to their bonding PC3 moieties and do not exert any pronounced angular distortion (cf. the interligand angles ranging 105.64(4)–114.28(4)° for Zn1 and 104.54(3)–112.79(4)° for Zn2).
Conclusion
Alkylation of phosphinoamine 1 with 1,3-propanesultone proceeds selectively under alkylation of the hard nitrogen group to afford the quaternary ammonium salt 2. Betaine 2, possessing an intact phosphine substituent, is an attractive functionalised starting material for ferrocenylmethylation reactions with nucleophiles, which was demonstrated in this paper by model reactions leading to 3 and 4, and by the synthesis of the phosphinosulfones 6 representing new entries among hybrid33 phosphinoferrocene donors possessing flexible methylene spacers.6–11 Compound 6a, chosen as a representative phosphinosulfone donor, was shown to form unprecedented34 alkali metal–Zn coordination polymers of the general formula [ZnMX3(6a)(CH3OH)]n (M/X = Na/Br, Na/I, and K/Br), in which the ferrocene-based ligand coordinates the softer Zn(II) ion via its phosphine substituent and the alkali metal cation through the sulfone oxygen atoms, while the methanol molecules complete octahedral coordination around the alkali metal ions. These compounds are zwitterions combining negatively charged ZnBr3 units with cationic centres represented by the alkali metal cations in their structures. In the case of ZnBr2–LiBr, the analogous reaction with 6a provides [Br3Zn(6a)Li2(CH3OH)4(H2O)(6a)ZnBr3] (methanol solvate), a pseudodimeric complex comprising two chemically different, tetrahedral Li+ centres coordinated by the sulfonate oxygens, solvating methanol and a water molecule, and the phosphine-coordinated ZnBr3 units.Experimental
Materials and methods
All reactions were performed under an argon atmosphere by standard Schlenk techniques. Amine 1 was prepared as reported previously.10 Benzene and chloroform were dried by standing over sodium metal and CaH2, respectively, and distilled under argon. Dimethyl sulfoxide was distilled under vacuum. Methanol was dried with an in-house PureSolv MD5 solvent-drying system (Innovative Technology, USA). A solution of sodium methoxide was prepared by dissolving the appropriate amount of sodium metal in anhydrous methanol. Other chemicals and solvents were obtained from commercial suppliers (Sigma-Aldrich or Lachner, Czech Republic) and were used without any additional purification.NMR spectra were recorded at 25 °C on a Varian Unity INOVA spectrometer operating at 399.95 MHz for 1H, 100.58 MHz for 13C, and 161.92 MHz for 31P. The chemical shifts (δ in ppm) are given relative to internal tetramethylsilane (1H and 13C) or to external 85% aqueous H3PO4 (31P). IR spectra were obtained on a Nicolet Magna 6700 FTIR spectrometer in the range of 400–4000 cm−1. Electrospray ionisation mass spectra (ESI MS) were acquired with a Bruker Esquire 3000 spectrometer using samples dissolved in HPLC-grade methanol.
Syntheses
:1) to remove the less polar impurities. The polarity of the eluent was then increased (dichloromethane–methanol 3:1) to elute a salt containing the protonated amine 1 as the cation, very likely hydrochloride [1H]Cl.35 Finally, the eluent was changed to dichloromethane–methanol 1:1, which removed the major orange band of the desired product. Following evaporation and drying under vacuum over sodium hydroxide, betaine 2 was isolated as an air-stable orange solid (4.476 g, 81%). An analytical sample, in the form of the defined solvate 2·CH3OH, was obtained upon crystallisation from methanolic solution layered with tetrahydrofuran and diethyl ether.
Analytical data for betaine 2. 1H NMR (DMSO-d6): δ 1.99 (m, 2 H, NCH2CH2CH2SO3), 2.45 (t, 3JHH = 7.1 Hz, 2 H, NCH2CH2CH2SO3), 2.78 (s, 6 H, N(CH3)2), 3.21 (m, 2 H, NCH2CH2CH2SO3), 4.04 (s, 2 H, C5H4CH2), 4.16 (vq, J′ = 1.9 Hz, 2 H, PC5H4), 4.22 (vt, J′ = 1.9 Hz, 2 H, CH2C5H4), 4.44 (vt, J′ = 1.9 Hz, 2 H, CH2C5H4), 4.53 (vt, J′ = 1.9 Hz, 2 H, PC5H4), 7.30–7.43 (m, 10 H, PPh2). 31P{1H} NMR (DMSO-d6): δ −18.1 (s). 13C{1H} NMR (DMSO-d6): δ 18.89 (s, NCH2CH2CH2SO3), 47.63 (s, NCH2CH2CH2SO3), 48.86 (s, NMe2), 62.03 (s, NCH2CH2CH2SO3), 63.04 (s, C5H4CH2), 71.04 (s, CH of CH2C5H4), 71.95 (d, 3JPC = 4 Hz, β-CH of PC5H4), 72.86 (s, CH of CH2C5H4), 73.35 (s, Cipso of CH2C5H4), 73.48 (d, 2JPC = 15 Hz, α-CH of PC5H4), 76.68 (d, 1JPC = 9 Hz, Cipso of PC5H4), 128.33 (d, 3JPC = 7 Hz, CHmeta of PPh2), 128.71 (s, CHpara of PPh2), 133.00 (d, 2JPC = 20 Hz, CHortho of PPh2), 138.33 (d, 1JPC = 10 Hz, Cipso of PPh2). IR (Nujol): νmax 3420 br m, 2723 vw, 2669 vw, 1656 m, 1649 m, 1643 m, 1584 w, 1568 vw, 1308 m, 1189 br vs (νa(SO3)), 1094 m, 1069 m, 1037 vs (νs(SO3)), 998 m, 920 w, 889 m, 842 m, 820 m, 743 s, 727 m, 697 s, 634 w, 619 w, 602 m, 570 w, 548 w, 523 m, 501 m, 489 m, 456 m, 408 vw cm−1. MS (ESI+): m/z 383 ([Ph2PfcCH2]+), 550 ([Ph2PfcCH2NMe2(CH2)3SO3 + H]+), 572 ([Ph2PfcCH2NMe2(CH2)3SO3 + Na]+), 588 ([Ph2PfcCH2NMe2(CH2)3SO3 + K]+). Anal. calc. for C28H32FeNO3PS·MeOH (581.5): C 59.90, H 6.24, N 2.41%. Found: C 59.66, H 5.84, N 2.39%.
:10) resulted in a yellow band that contained compound 5, which was isolated in a 6% yield (44 mg) after evaporation. Subsequent elution with diethyl ether–hexane 2:1 led to the development of a major orange band due to alcohol 3, which was isolated as slowly crystallising orange oil upon evaporation under vacuum. Yield of 3: 511 mg (64%). The compound was identified by NMR spectroscopy.12a
Analytical data for 5. 1H NMR (CDCl3): δ 1.81 (s, 3 H, CH3), 3.92 (vt, J′ = 1.8 Hz, 2 H), 3.99 (vt, J′ = 1.8 Hz, 2 H), 4.00 (vt, J′ = 1.9 Hz, 2 H) and 4.29 (vt, J′ = 1.9 Hz, 2 H) (4 × CH of C5H4), 7.28–7.40 (m, 10 H, P(C6H5)2) ppm. 31P{1H} NMR (CDCl3): δ −16.0 (s) ppm. 13C{1H} NMR (CDCl3): δ 14.40 (s, CH3), 68.41 (s, CH of MeC5H4), 70.23 (s, CH of MeC5H4), 71.66 (d, 3JPC = 4 Hz, CH of PC5H4), 73.45 (d, 2JPC = 15 Hz, CH of MeC5H4), 84.67 (s, Cipso of MeC5H4), 128.07 (d, 3JPC = 7 Hz, CHmeta in PPh2), 128.40 (s, CHpara in PPh2), 133.51 (d, 2JPC = 20 Hz, CHortho in PPh2), 139.22 (d, 1JPC = 10 Hz, Cipso in PPh2) ppm. The signal of Cipso of PC5H4 is probably obscured by the solvent resonance. MS (ESI+): m/z 200 ([FcCH3]+˙), 384 ([Ph2PfcCH3]+˙). Anal. calc. for C23H21FeP (400.2): C 71.89, H 5.51%. Found: C 71.72, H 5.41%.
:1 as the eluent. The major band due to the product was collected and evaporated under vacuum to yield ether 4 as a yellow-orange solid. Yield: 388 mg (47%). The NMR spectra of the product were identical with those reported in the literature.9
:1 to elute the main orange band of 6a and a small tailing band containing alcohol 3. The mobile phase was then changed to dichloromethane–methanol 20:1, which led to the development of an additional broad yellow band containing the side product 7. The complete separation of 3 and 6a was achieved through additional chromatography over silica gel using ethyl acetate–hexane (1:2) as the eluent (note: the alcohol elutes first under such conditions) or, alternatively, via crystallisation from ethyl acetate/hexane. Yields: 6a – yellow solid (272 mg, 29%); 3 – orange, slowly crystallising oil (14 mg, 2%); and 7 – rusty brown oil (74 mg, 12%).
Analytical data for 6a. 1H NMR (CDCl3): δ 2.62 (s, 3 H, SO2Me), 3.59 (s, 2 H, C5H4CH2), 4.10 (vq, J′ = 1.8 Hz, 2 H), 4.19 (vt, J′ = 1.9 Hz, 2 H), 4.26 (vt, J′ = 1.9 Hz, 2 H) and 4.40 (vt, J′ = 1.8 Hz, 2 H) (4 × CH in fc); 7.31–7.41 (m, 10 H, PPh2). 31P{1H} NMR (CDCl3): δ −17.0 (s). 13C{1H} NMR (CDCl3): δ 38.43 (SO2Me), 56.65 (s, C5H4CH2), 70.59 (s, CH of CH2C5H4), 71.07 (s, CH of CH2C5H4), 71.73 (d, 3JPC = 4 Hz, β-CH of PC5H4), 74.03 (d, 2JPC = 15 Hz, α-CH of PC5H4), 74.88 (s, Cipso of CH2C5H4), 128.31 (d, 3JPC = 7 Hz, CHmeta of PPh2), 128.80 (s, CHpara of PPh2), 133.50 (d, 2JPC = 20 Hz, CHortho of PPh2), 138.67 (d, 1JPC = 10 Hz, Cipso of PPh2). The resonance of ferrocene C–P was not found. IR (Nujol): νmax 3015 m, 2725 vw, 2282 vw, 1583 w, 1567 vw, 1435 s, 1419 w, 1412 w, 1397 w, 1322 m, 1304 vs (νas(SO2)), 1286 m, 1242 w, 1228 w, 1192 w, 1161 m, 1139 vs (νs(SO2)), 1122 m, 1099 m, 1084 vw, 1069 w, 1058 vw, 1038 m, 1023 m, 998 w, 963 s, 928 m, 903 vw, 843 m, 835 m, 788 s, 750 s, 742 vs, 704 s, 697 vs, 630 w, 618 vw, 603 w, 569 vw, 529 m, 510 m, 486 s, 479 vs, 455 s, 441 m, 406 vw cm−1. MS (ESI+): m/z 383 ([Ph2PfcCH2]+), 463 ([6a + H]+), 485 ([6a + Na]+), 501 ([6a + K]+). Anal. calc. for C24H23FeO2PS (462.3): C 62.35, H 5.01%. Found: C 62.59, H 4.82%.
Analytical data for 7. 1H NMR (CDCl3): δ 3.59 (d, 2JPH = 12.7 Hz, 2 H, PCH2), 3.83 (d of vt, J = 1.9, 0.8 Hz, 2 H), 3.94 (vt, J′ = 1.9 Hz, 2 H), 4.02 (vt, J′ = 1.9 Hz, 2 H) and 4.33 (vt, J′ = 1.9 Hz, 2 H) (4 × CH in fc); 6.12 (m, 2 H, α-CH of P
C5H4), 6.40 (m, 2 H, β-CH of PC5H4), 7.25–7.35 (m, 10 H, fcPPh2), 7.41–7.47 (m, 8 H, CHortho and CHmeta of CH2PPh2), 7.55–7.61 (m, 2 H, CHpara of CH2PPh2). 31P{1H} NMR (CDCl3): δ −16.9 (s, fcPPh2), 10.1 (s, CH2PC5H4). 13C{1H} NMR (CDCl3): δ 30.01 (dd, 1JPC = 53 Hz, JPC = 1 Hz, PCH2), 69.50 (s, CH of fc), 71.49 (s, CH of fc), 71.77 (d, 3JPC = 4 Hz, CH of fc), 73.89 (d, 2JPC = 15 Hz, CH of fc), 77.42 (d, 2JPC = 2 Hz, Cipso of fc), 78.21 (d, 1JPC = 110 Hz, Cipso of PC5H4), 113.92 (d, 2JPC = 18 Hz, α-CH of PC5H4), 115.83 (d, 3JPC = 15 Hz, β-CH of PC5H4), 125.49 (d, 1JPC = 86 Hz, Cipso of CH2PPh2), 128.19 (d, 3JPC = 7 Hz, CHmeta of CH2PPh2), 128.59 (d, 2JPC = 12 Hz, CHortho of fcPPh2), 128.63 (s, CHpara of fcPPh2), 132.54 (d, 4JPC = 3 Hz, CHpara of CH2PPh2), 133.37 (s, CHmeta of fcPPh2), 133.52 (d, 2JPC = 10 Hz, CHortho of CH2PPh2), 138.85 (d, 1JPC = 10 Hz, Cipso of fcPPh2). MS (ESI+): m/z 383 ([Ph2PfcCH2]+), 633 ([7 + H]+). HRMS (APCI+) calc. for C40H35FeP2 ([M + H]+): 633.1558, found: 633.1561. Anal. calc. for C40H34FeP2·1/8CHCl3 (647.4): C 74.44, H 5.31%. Found: C 74.30, H 5.38%.
:1 produced a major orange band due to sulfone 6, followed by a minor band of alcohol 3. Subsequent elution with dichloromethane–methanol 20:1 led to the development of a broad yellow band containing crude 7. Compound 3 was further purified as described above (see the preparation of 3). Particular details are as follows.
The reaction of 2 (1.099 g, 2.0 mmol) with sodium phenylsulfinate (0.838 g, 5.0 mmol) as described above yielded 6b (yellow solid; 524 mg, 50%), 3 (14 mg, 2%), and 7 (61 mg, 10%). Starting with 2 (1.095 g, 2.0 mmol) and sodium 4-toluenesulfinate (0.937 g, 5.0 mmol), an analogous procedure produced 6c (yellow solid; 570 mg, 53%), 3 (25 mg, 3%), and 7 (98 mg, 16%).
Analytical data for 6b. 1H NMR (CDCl3): δ 3.68 (s, 2 H, C5H4CH2), 3.94 (vt, J′ = 1.9 Hz, 2 H), 4.02 (vq, J′ = 1.8 Hz, 2 H), 4.05 (vt, J′ = 1.9 Hz, 2 H) and 4.32 (vt, J′ = 1.8 Hz, 2 H) (4 × CH of fc), 7.28–7.35 (m, 10 H, PPh2), 7.42–7.47 (m, 2 H, SO2Ph), 7.57–7.62 (m, 3 H, SO2Ph). 31P{1H} NMR (CDCl3): δ −16.9 (s). 13C{1H} NMR (CDCl3): δ 58.23 (s, C5H4CH2), 70.16 (s, CH of CH2C5H4), 71.30 (s, CH of CH2C5H4), 71.52 (d, 3JPC = 4 Hz, β-CH of PC5H4), 73.81 (d, 2JPC = 15 Hz, α-CH of PC5H4), 74.68 (s, Cipso of CH2C5H4), 76.79 (d, 1JPC = 7 Hz, Cipso of PC5H4), 128.24 (d, 3JPC = 7 Hz, CHmeta of PPh2), 128.69 (s, CHortho and CHmeta of SO2Ph + CHpara of PPh2), 133.44 (d, 2JPC = 20 Hz, CHortho of PPh2), 133.51 (s, CHpara of SO2Ph), 137.84 (s, Cipso of SO2Ph), 138.73 (d, 1JPC = 10 Hz, Cipso of PPh2). IR (Nujol): νmax 2723 vw, 2667 vw, 1583 vw, 1568 vw, 1400 w, 1311 s, 1304 vs (νas(SO2)), 1288 m, 1263 w, 1240 vw, 1223 vw, 1149 vs, (νs(SO2)), 1110 w, 1086 m, 1068 w, 1058 vw, 1036 w, 1029 m, 999 vw, 974 vw, 927 w, 888 vw, 880 vw, 871 vw, 834 m, 761 m, 749 s, 708 m, 700 m, 686 m, 633 w, 614 w, 571 s, 549 vw, 531 w, 503 s, 490 s, 458 m, 432 vw, cm−1. MS (ESI+): m/z 383 ([Ph2PfcCH2]+), 525 ([6b + H]+), 547 ([6b + Na]+), 563 ([6b + K]+). Anal. calc. for C29H25FeO2PS (524.4): C 66.42, H 4.81%. Found: C 66.20, H 4.80%.
Analytical data for 6c. 1H NMR (CDCl3): δ 2.43 (s, 3 H, C6H4CH3), 3.67 (s, 2 H, C5H4CH2), 3.95 (vt, J′ = 1.9 Hz, 2 H), 4.02 (vq, J′ = 1.8 Hz, 2 H), 4.06 (vt, J′ = 1.9 Hz, 2 H) and 4.32 (vt, J′ = 1.8 Hz, 2 H) (4 × CH of fc), 7.23–7.26 (m, 2 H, C6H4), 7.29–7.35 (m, 10 H, PPh2), 7.46–7.50 (m, 2 H, C6H4). 31P{1H} NMR (CDCl3): δ −16.9 (s). 13C{1H} NMR (CDCl3): δ 21.64 (s, C6H4CH3), 58.28 (s, C5H4CH2), 70.14 (s, CH of CH2C5H4), 71.33 (s, CH of CH2C5H4), 71.50 (d, 3JPC = 4 Hz, β-CH of PC5H4), 73.78 (d, 2JPC = 14 Hz, α-CH of PC5H4), 74.86 (s, Cipso of CH2C5H4), 76.77 (d, 1JPC = 7 Hz, Cipso of PC5H4), 128.24 (d, 3JPC = 7 Hz, CHmeta of PPh2), 128.69 (s), 128.71 (s) and 129.32 (s) (2 × CH of C6H4 + CHpara of PPh2), 133.45 (d, 2JPC = 20 Hz, CHortho of PPh2), 134.96 (s, CSO2 of C6H4), 138.75 (d, 1JPC = 10 Hz, Cipso of PPh2), 144.43 (s, CCH3 of C6H4). IR (Nujol): νmax 2726 vw, 2669 vw, 1594 w, 1436 m, 1403 w, 1319 vs (νas(SO2)), 1301 m, 1289 m, 1259 m, 1221 m, 1194 vw, 1183 vw, 1148 vs (νs(SO2)), 1117 w, 1106 m, 1086 s, 1070 w, 1053 vw, 1047 vw, 1038 w, 1027 m, 998 vw, 926 w, 888 w, 875 m, 868 w, 848 vw, 837 m, 824 m, 813 m, 799 w, 750 s, 698 s, 633 m, 611 w, 552 m, 513 s, 497 s, 485 m, 469 w, 453 m, 440 vw, 431 vw cm−1. MS (ESI+): m/z 383 ([Ph2PfcCH2]+), 539 ([6c + H]+), 561 ([6c + Na]+). Anal. calc. for C30H27FeO2PS (538.4): C 66.92, H 5.06%. Found: 66.62, H 5.00%.
IR (Nujol): νmax 3495 br m, 3090 m, 2724 vw, 2670 vw, 1618 w, 1433 m, 1410 w, 1354 w, 1317 m, 1303 vs, 1269 m, 1248 vw, 1228 w, 1193 vw, 1169 m, 1145 s, 1119 s, 1096 w, 1065 vw, 1052 vw, 1043 vw, 1030 m, 1022 m, 1006 m, 971 m, 953 w, 931 w, 904 vw, 890 m, 857 w, 847 w, 828 w, 792 w, 751 s, 705 w, 694 m, 629 w, 610 w, 571 vw, 543 w, 518 m, 490 s, 457 m, 434 vw cm−1. MS (ESI+): m/z 383 ([Ph2PfcCH2]+), 463 ([6a + H]+), 485 ([6a + Na]+), 501 ([6a + K]+). Anal. calc. for C25H27Br3FeNaO3PSZn (822.4): C 36.51, H 3.31%. Found: C 36.64, H 3.37%.
IR (Nujol): νmax 3490 br m, 3099 w, 3091 m, 2724 vw, 2670 vw, 1618 w, 1435 m, 1407 w, 1314 m, 1299 vs, 1267 m, 1227 w, 1192 w, 1166 m, 1143 s, 1118 m, 1107 m, 1095 m, 1090 m, 1062 w, 1052 vw, 1042 w, 1030 m, 1021 m, 1000 m, 970 m, 953 w, 931 w, 887 m, 879 w, 854 w, 847 w, 834 w, 828 w, 790 w, 774 vw, 749 s, 704 w, 692 m, 627 w, 608 w, 570 w, 541 w, 517 m, 499 m, 490 s, 457 m, 432 vw cm−1. MS (ESI+): m/z 383 ([Ph2PfcCH2]+), 463 ([6a + H]+), 485 ([6a + Na]+), 501 ([6a + K]+). Anal. calc. for C25H27FeI3NaO3PSZn (963.4): C 31.17, H 2.82%. Found: C 31.44, H 2.86%.
IR (Nujol): νmax 3475 br m, 3089 m, 1619 w, 1433 m, 1407 w, 1318 m, 1293 s, 1267 m, 1228 w, 1193 w, 1167 m, 1146 s, 1118 m, 1107 m, 1091 m, 1064 w, 1052 vw, 1043 w, 1030 m, 1021 m, 1013 s, 971 m, 960 m, 931 w, 905 w, 890 m, 881 w, 857 w, 846 w, 826 w, 790 w, 748 s, 704 w, 692 s, 627 w, 610 w, 543 w, 517 m, 499 s, 489 s, 459 s, 409 w cm−1. Anal. calc. for C25H27Br3FeKO3PSZn (838.5): C 35.81, H 3.25%. Found: C 35.61, H 3.29%.
IR (Nujol): νmaxca. 3270–3650 m composite, 3086 w, 1708 w, 1623 br s, 1403 w, 1318 m, 1306 w, 1283 s, 1263 m, 1247 w, 1228 w, 1193 vw, 1168 m, 1143 s, 1116 s, 1116 s, 1095 m, 1062 w, 1041 w, 1030 m, 1020 m, 973 m, 930 w, 888 s, 857 w, 876 w, 833 vw, 828 vw, 790 m, 748 vs, 705 w, 692 w, 627 w, 609 w, 542 w, 517 m, 489 br s, 459 s cm−1. Anal. calc. for Li2Zn2Br6(6a)2(MeOH)5(H2O)3 (1762.9): C 36.11, H 4.12%. Found: C 35.88, H 3.79% (the sample is slightly hygroscopic).
X-ray crystallography
The diffraction data (θmax = 27.5°; data completeness ≥99.3%) were recorded with a Nonius Kappa diffractometer equipped with an APEX-II CCD detector (Bruker) and a Cryostream Cooler (Oxford Cryosystems) at 150(2) K using graphite monochromatized Mo Kα radiation (λ = 0.71073 Å) and were corrected for absorption using routines included in the diffractometer software.The structures were solved by the direct methods (SHELXS97) and refined by full-matrix least squares routines based on F2 (SHELXL97).36 The non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms residing on the oxygen atoms (OH protons) in the structures of 2·CH3OH, 8a, 8b, and 9a were identified on the difference electron density maps and refined as riding atoms with Uiso(H) set to 1.2-times Ueq(O). In the case of 10a·CH3OH, they were placed into positions suitable for the formation of hydrogen bonds and refined similarly. Hydrogen atoms residing on the carbon atoms were included in their theoretical positions and refined analogously.
Description of the crystallization experiments and a listing of relevant crystallographic data and structure refinement parameters are available in the ESI (Table S1†). Geometric data as well as all structural drawings were obtained with a recent version of the PLATON program.37 All numerical values are rounded with respect to their estimated standard deviations (ESDs) given with one decimal. Parameters pertaining to atoms in constrained positions are presented without ESDs.
Acknowledgements
Results reported in this paper were obtained with financial support from the Czech Science Foundation (project no. 15-11571S) and the Grant Agency of Charles University in Prague (project no. 8415).Notes and references
- (a) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039 CrossRef CAS; (b) S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632 RSC.
- (a) G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125 CrossRef CAS; (b) E. O. Fischer and W. Pfab, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1952, 7, 377 Search PubMed.
- (a) Methods of Organoelement Compounds, volume Organoiron Compounds, Ferrocene, ed. E. G. Perevalova, M. D. Reschetova and K. I. Grandberg, Nauka, Moscow, 1983 (in Russian) Search PubMed; (b) V. I. Boev, L. V. Snegur, V. N. Babin and Y. S. Nekrasov, Usp. Khim., 1997, 66, 677 CAS; (c) Ferrocenes, ed. A. Togni and T. Hayashi, VCH, Weinheim, 1995, p. 677 ff Search PubMed; (d) B. W. Rockett and G. Marr, J. Organomet. Chem., 1991, 416, 327 CrossRef CAS and previous “Annual surveys of ferrocene chemistry”.
- R. Gleiter, C. Bleiholder and F. Rominger, Organometallics, 2007, 26, 4850 CrossRef CAS.
- (a) Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, 2008 Search PubMed; (b) R. C. J. Atkinson, V. C. Gibson and N. J. Long, Chem. Soc. Rev., 2004, 33, 313 RSC; (c) R. Goméz Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef PubMed.
- A proper choice of the alkylation reagent can sometimes eliminate this problem. For an example, see: P. Štěpnička and I. Císařová, Organometallics, 2003, 22, 1728 CrossRef.
- M. Widhalm, U. Nettekoven and K. Mereiter, Tetrahedron: Asymmetry, 1999, 10, 4369 CrossRef CAS.
- (a) A. Labande, J.-C. Daran, E. Manoury and R. Poli, Eur. J. Inorg. Chem., 2007, 1205 CrossRef CAS; (b) S. Gülcemal, A. Labande, J.-C. Daran, B. Çetinkaya and R. Poli, Eur. J. Inorg. Chem., 2009, 1806 CrossRef; (c) A. Labande, N. Debono, A. Sournia-Saquet, J.-C. Daran and R. Poli, Dalton Trans., 2013, 42, 6531 RSC.
- P. Štěpnička and I. Císařová, Dalton Trans., 2013, 42, 3373 RSC.
- P. Štěpnička, M. Zábranský and I. Císařová, ChemistryOpen, 2012, 1, 71 CrossRef PubMed.
- For an alternative synthesis of 1, see: M. E. Wright, Organometallics, 1990, 9, 853 CrossRef CAS.
- (a) P. Štěpnička and T. Baše, Inorg. Chem. Commun., 2001, 4, 682 CrossRef; (b) P. Štěpnička, I. Císařová and J. Schulz, Organometallics, 2011, 30, 4393 CrossRef; (c) P. Štěpnička, J. Schulz, T. Klemann, U. Siemeling and I. Císařová, Organometallics, 2010, 29, 3187 CrossRef; (d) U. Siemeling, T. Klemann, C. Bruhn, J. Schulz and P. Štěpnička, Dalton Trans., 2011, 40, 4722 RSC; (e) P. Štěpnička and I. Císařová, J. Organomet. Chem., 2012, 716, 110 CrossRef.
- V. I. Boev, Zh. Obshch. Khim., 1991, 61, 1174 CAS.
- In addition to the betaine, the reaction of 1 with the sultone gives rise to other products, namely salt(s) with protonated amines (presumably hydrochloride), which are less polar than 2, and some P-alkylated products (δP 26.7; more polar than 2).
- (a) W. Henderson, A. G. Oliver and A. J. Downard, Polyhedron, 1996, 15, 1165 CrossRef CAS; (b) Y. S. Nekrasov, R. S. Skazov, A. A. Simenel, L. V. Snegur and I. V. Kachala, Russ. Chem. Bull., 2006, 55, 1368 CrossRef CAS.
- J. K. Lindsay and C. R. Hauser, J. Org. Chem., 1957, 22, 355 CrossRef CAS.
- (a) M. Uher and Š. Toma, Collect. Czech. Chem. Commun., 1971, 36, 3056 CrossRef CAS; (b) J. B. Evans and G. Marr, J. Chem. Soc., Perkin Trans. 1, 1972, 2502 RSC; (c) V. I. Boev and A. V. Dombrovskii, Zh. Obshch. Khim., 1984, 54, 1863 CAS.
- J. H. Brownie, M. C. Baird and H. Schmider, Organometallics, 2007, 26, 1433 CrossRef CAS.
- K.-S. Gan and T. S. A. Hor, in Ferrocenes: Heterogeneous Catalysis, Organic Synthesis, Materials Science, VCH, Weinheim, 1995, ch. 1.3, p. 19 Search PubMed.
- P. A. Chaloner, R. M. Harrison, P. B. Hitchcock and R. T. Pedersen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 717 CrossRef.
- P. C. Healy, W. A. Loughlin, B. M. Sweeney and I. D. Jenkins, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, o137 CAS . The distances and angles were calculated from coordinates deposited in the Cambridge Crystallographic Database (refcode: MIFSID).
- F. A. M. Rudolph, A. L. Fuller, A. M. Z. Slawin, M. Bühl, R. A. Aitken and J. D. Woollins, J. Chem. Crystallogr., 2010, 40, 253 CrossRef CAS.
- C. Lichtenberg, N. S. Hillesheim, M. Elfferding, B. Oelkers and J. Sundermeyer, Organometallics, 2012, 31, 4259 CrossRef CAS.
- I. R. Butler, W. R. Cullen, F. W. B. Einstein and A. C. Willis, Organometallics, 1985, 4, 603 CrossRef CAS.
- J. C. J. Bart, J. Chem. Soc. B, 1969, 350 RSC.
- W. E. McEwen, C. E. Sullivan and R. O. Day, Organometallics, 1983, 2, 420 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
- R. H. Prince, Zinc and Cadmium, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, ch. 56.1, vol. 5, pp. 925–1097 Search PubMed.
- The Na+ necessary for the formation of 8a seems to have come from the commercial ZnBr2 sample, which was shown to contain 0.04 μg Na per 1 mg by atomic absorption spectrometry, or simply from the solvents and/or the glassware.
- Increasing the chloroform-to-methanol ratio can result in a separation of the alkali metal halide (MX) from the reaction mixture (full or partial), which in turn reduces the yield of the mixed-metal complex or even fully prevents its formation. On the other hand, a low chloroform-to-methanol ratio (i.e., more polar reaction mixture) can significantly reduce the yield of the desired mixed-metal complex by increasing its solubility.
- We feel that compound 10a need not necessarily be the only product possibly arising in the 6a/LiBr/ZnBr2 system as the reaction can, in principle, afford also other, differently solvated species.
- See, for instance: (a) A. Santoro and A. D. Mighell, Acta Crystallogr., Sect. A: Found. Crystallogr., 1970, 26, 124 CrossRef CAS; (b) V. Balashov and H. D. Ursell, Acta Crystallogr., 1957, 10, 582 CrossRef CAS; (c) J. D. H. Donnay, Am. Mineral., 1943, 28, 507 CAS.
- (a) A. Bader and E. Lindner, Coord. Chem. Rev., 1991, 108, 27 CrossRef CAS; (b) C. S. Slone, D. A. Weinberger and C. A. Mirkin, Prog. Inorg. Chem., 1999, 48, 233 CrossRef CAS.
- The search for structurally characterised compounds combining octahedral Na(I) and tetrahedral Zn(II) sites in the Cambridge Structural Database, version 5.36 of November 2014 with updates from November 2014, resulted in 42 hits. These compounds were mostly coordination polymers featuring various O-donors. None of them contained phosphine donors or motifs similar to that encountered in 8a and 8b.
- Dissolution of this by-product in dichloromethane, washing with 5% aqueous NaOH, and subsequent chromatographic purification recovered pure 1.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A comparison of the IR spectra of 6a, 8a, and 8b in the fingerprint region, additional structural drawings for 8a and 8b, and a summary of the crystallographic data and structure refinement parameters (Table S1). CCDC 1401233–1401241, 1410159 and 1410160. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01877c |
| This journal is © The Royal Society of Chemistry 2015 |