
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aza-macrocyclic complexes of the Group 1 cations – synthesis, structures and density functional theory study†
John
Dyke
a,
William
Levason
a,
Mark E.
Light
a,
David
Pugh
a,
Gillian
Reid
*a,
Hanusha
Bhakhoa
b,
Ponnadurai
Ramasami
b and
Lydia
Rhyman
b
aChemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
bComputational Chemistry Group, Department of Chemistry, Faculty of Science, University of Mauritius, Réduit 80837, Mauritius
First published on 18th June 2015
Abstract
The Group 1 complexes, [M(Me6[18]aneN6)][BArF] (M = Li–Cs; Me6[18]aneN6 = 1,4,7,10,13,16-hexamethyl-1,4,7,10,13,16-hexaazacyclooctadecane; BArF = tetrakis{3,5-bis(trifluoromethyl)-phenyl}borate), are obtained in high yield by reaction of the macrocycle with M[BArF] in anhydrous CH2Cl2 solution, and characterised spectroscopically (1H, 13C{1H}, 7Li, 23Na, and 133Cs NMR), by microanalysis and, for M = Li, K, and Rb, by single crystal X-ray analysis. The structures show N6-coordination to the metal ion; the small ionic radius for Li+ leads to a puckered conformation. In contrast, the K+ ion fits well into the N6 plane, with the [BArF]− anions above and below, leading to two K+ species in the asymmetric unit (a hexagonal planar [K(Me6[18]aneN6)]+ cation and a ‘[K(Me6[18]aneN6)(κ1-BArF)2]− anion’, with long axial K⋯F interactions). The Rb+ ion sits above the N6 plane, with two long axial Rb⋯F interactions in one cation and two long, mutually cis Rb⋯F interactions in the other. The unusual sandwich cations, [M(Me3tacn)2]+ (M = Na, K; distorted octahedral, N6 donor set) and half-sandwich cations [Li(Me3tacn)(thf)]+ (distorted tetrahedron, N3O donor set), [Li(Me4cyclen)(OH2)]+, and [Na(Me4cyclen)(thf)]+ (both distorted square pyramids with N4O donor sets) were also prepared (Me3tacn = 1,4,7-trimethyl-1,4,7-triazacyclononane, Me4cyclen = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane). Density functional theory (DFT) calculations, using the BP86 and B3LYP functionals, show that the accessibility of the [M(Me3tacn)2]+ sandwich cations depends strongly on the M+ ionic radius, such that it is sufficiently large to avoid steric clashing between the Me groups of the two rings, and small enough to avoid very acute N–M–N chelate angles. The calculations also show that coordination to the Group 1 cation involves significant donation of electron density from the p-orbitals on the N atoms of the macrocycle, rather than purely electrostatic interactions.
Introduction
The coordination chemistry of Group 1 cations with neutral ligands is dominated by oxygen-donor ligands such as alcohols, ethers, and water, including the ubiquitous crown ethers and cryptands which are frequently used as ligands towards Group 1 cations.1,2 The corresponding chemistry with the less electronegative neutral polyaza macrocycles has been much less studied. We recently reported the preparation of several unusual complexes based upon coordination of Na+ to Me3tacn (1,4,7-trimethyl-1,4,7-triazacyclononane) and Me4cyclam (1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane), including structural characterisation of the sandwich cation salt, [Na(Me3tacn)2][BArF] (BArF = tetrakis{3,5-bis(trifluoromethyl)phenyl}borate) and [Na(Me4cyclam)(thf)][BArF], containing tetradentate coordination of the macrocycle.3 The Me3tacn complexes were initially isolated serendipitously from reactions of Na[BArF] with SiCl4 in the presence of the N-donor macrocycles, towards preparation of potential precursors for the supercritical fluid electrodeposition (SCFED) of semiconductor materials.4 The Na+ preferentially coordinated to the N-donor macrocycle, and we attributed this unusual behaviour to the low lattice energy of Na[BArF], coupled with the high solubility of the [BArF]− salts in non-competitive, very weakly coordinating solvents, such as CH2Cl2 and toluene.Neutral diamines (e.g. N,N,N′,N′-tetramethylethylenediamine) are often used as complexation agents to increase the reactivity of alkyl lithiums5 and to increase the solubility of heavier Group 1 alkyls.6 Whilst a small number of pendant arm functionalised N-donor macrocycles are known to form complexes with alkali metal cations,7 complexes of neutral N-donor macrocycles with these cations have been very little studied. Earlier work has shown that N-methylated aza macrocycles are more stable towards electrides compared to the analogous crown ethers.8 Other than the two Na+-aza macrocyclic complexes that we reported,3 four examples involving Me3tacn coordinated to lithium are known: two lithium-based electrides, a BzLi complex with Me3tacn and a lithium enolate stabilised by Me3tacn.9 Two sodium complexes with Me3tacn and Me3cyclen (1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane) were structurally characterised.10 No examples of neutral tri- and tetra-aza macrocycles are known for K+, Rb+, or Cs+ and there are very few examples with the N-methylated aza analogue of 18-crown-6, Me6[18]aneN6 (1,4,7,10,13,16-hexamethyl-1,4,7,10,13,16-hexaazacyclooctadecane).8,11 The crystal structures of two electrides, [K(Me6[18]aneN6)]+Na− and [Cs(Me6[18]aneN6)]+Na−, were determined by Dye and co-workers,11a who also reported NMR spectroscopic evidence for [Rb(Me6[18]aneN6)]+Na−, [Cs(Me6[18]aneN6)]+, [Cs(Me6[18]aneN6)2]+, and two [Cs(Me6[18]aneN6)(crown)]+ complexes.11b,c,8
We report here the first systematic study of the synthesis, spectroscopic, and structural characterisation of Group 1 cation (Li+ to Cs+) complexes involving tri-, tetra- and hexa-aza macrocycles. Density functional theory (DFT) with the BP86 and B3LYP functionals has been used to investigate the electronic structure and bonding present in the resulting cations.
Experimental
All preparations were carried out under a dry dinitrogen atmosphere using standard Schlenk and glove box techniques. [Li(thf)4][BArF] and Na[BArF]·2thf were synthesised using a slight modification of the literature procedure.12 The lithium salt was isolated as [Li(OH2)4][BArF], which can be converted to the thf adduct by stirring in thf for 16 h over 4 Å molecular sieves. Filtration and removal of solvents affords the thf adduct. Crude Na[BArF] was recrystallised from thf/n-hexane, resulting in the isolation of Na[BArF]·2thf. K[BArF], Rb[BArF], and Cs[BArF] were synthesised via cation exchange of Na[BArF]·2thf in water at 95 °C with excess (5 mol. equiv.) KNO3, RbNO3, or CsNO3, respectively.13 Me3tacn was synthesised by a literature procedure.14 Me4cyclen was obtained by methylation of cyclen (Sigma) using formic acid/formaldehyde.15 Me6[18]aneN6 (Sigma) was used as received. CH2Cl2 was dried by distillation from CaH2, toluene distilled from Na, and n-hexane distilled from Na/K alloy. 1H and 13C{1H} NMR spectra were recorded in CD2Cl2 solution at 298 K using a Bruker AV II-400 spectrometer and are referenced to the residual CH2Cl2 resonance. 7Li, 23Na, and 133Cs NMR spectra were obtained on a Bruker AV II-400 spectrometer at 298 K (unless otherwise stated) and referenced to a 0.1 mol dm−3 solution of LiCl, NaCl, or CsNO3 in D2O, respectively. Microanalyses were undertaken at London Metropolitan University.X-ray crystallography
Crystals were obtained as described below. Details of the crystallographic data collection and refinement are in Table S1.† Diffractometer: Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum rotating anode generator (λ1 = 0.71073 Å) with VHF Varimax optics (70 μm focus). Cell determination and data collection: CrystalClear-SM Expert 3.1 b27, data reduction, cell refinement, and absorption correction: CrystalClear-SM Expert 2.1 b29.16 Structure solution and refinement were carried out using WinGX and software packages within.17 Disorder in the CF3 groups of the [BArF]− anions was present in all of the structures, which is often observed in compounds containing [BArF]−,18 and this was satisfactorily modelled using DFIX, DELU, and SIMU restraints. Positional disorder was also present in the –CH2CH2– linkers of the macrocycle in [Na(Me4cyclen)(thf)][BArF] and was modelled similarly. Two locations for the water molecule in [Li(Me4cyclen)(H2O)][BArF] were observed, with the combined electron density consistent with one fully-occupied water molecule. Attempts to model this as other moieties (e.g. methoxide) were unsuccessful. H-atoms were placed in geometrically-assigned positions with C–H distances of 0.95 Å (CH), 0.98 Å (CH2), or 0.99 Å (CH3) and refined using a riding model with Uiso(H) = 1.2Ueq.(C) (CH, CH2) or 1.5Ueq.(C) (CH3). enCIFer was used to prepare material for publication.19 CCDC reference numbers 1051099–1051104 contain crystallographic data in CIF format.Preparations
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2). 13C{1H} NMR (298 K, CD2Cl2): δ = 162.35 (C, q, JC–B = 49.9 Hz, BArF C1), 135.39 (CH, BArF C2/6), 129.48 (C, qq, 2JC–F = 31.5, 2.9 Hz, BArF C3/5), 125.20 (C, q, JC–F = 272 Hz, CF3), 118.08 (CH, septet, 3JC–F = 4.0 Hz, BArF C4), 68.95 (OCH2), 54.99 (NCH2), 45.99 (NMe), 26.02 (OCH2
2). 13C{1H} NMR (298 K, CD2Cl2): δ = 162.35 (C, q, JC–B = 49.9 Hz, BArF C1), 135.39 (CH, BArF C2/6), 129.48 (C, qq, 2JC–F = 31.5, 2.9 Hz, BArF C3/5), 125.20 (C, q, JC–F = 272 Hz, CF3), 118.08 (CH, septet, 3JC–F = 4.0 Hz, BArF C4), 68.95 (OCH2), 54.99 (NCH2), 45.99 (NMe), 26.02 (OCH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2). 7Li NMR (298 K, CD2Cl2): δ = +1.47.
2). 13C{1H} NMR (298 K, CD2Cl2): δ = 162.36 (C, q, JC–B = 49.9 Hz, BArF C1), 135.41 (CH, BArF C2/6), 129.48 (C, qq, 2JC–F = 31.5, 2.9 Hz, BArF C3/5), 125.21 (C, q, JC–F = 272 Hz, CF3), 118.09 (CH, septet, 3JC–F = 4.0 Hz, BArF C4), 68.52 (OCH2), 53.71 (NCH2), 43.47 (NMe), 26.14 (OCH2C2). 23Na NMR (298 K, CD2Cl2): δ = +12.7.
H2). 7Li NMR (298 K, CD2Cl2): δ = +1.47.
2). 13C{1H} NMR (298 K, CD2Cl2): δ = 162.36 (C, q, JC–B = 49.9 Hz, BArF C1), 135.41 (CH, BArF C2/6), 129.48 (C, qq, 2JC–F = 31.5, 2.9 Hz, BArF C3/5), 125.21 (C, q, JC–F = 272 Hz, CF3), 118.09 (CH, septet, 3JC–F = 4.0 Hz, BArF C4), 68.52 (OCH2), 53.71 (NCH2), 43.47 (NMe), 26.14 (OCH2C2). 23Na NMR (298 K, CD2Cl2): δ = +12.7.
Computational details
DFT calculations were performed on the isolated cations [M(Me3tacn)]+, [M(Me3tacn)2]+, and [M(Me6[18]aneN6)]+, with M = Li, Na, K, Rb, and Cs, using the BP86 and B3LYP functionals with a 6-311G(d,p) basis set. Further details are given in the ESI.†Results and discussion
In view of the limited literature examples of aza-macrocyclic complexes with Group 1 cations, their expected lability in solution, and the lack of diagnostic spectroscopic signatures for these complexes, X-ray crystallographic data provide the key characterisation method. We therefore describe the solid state data first, comparing these with computed structures obtained using DFT, followed by their solution NMR spectroscopic data.DFT calculations were carried out using the BP86 and B3LYP functionals. In general, these functionals perform equally well in replicating bond distances and angles in the cations investigated (Tables S2, S3, S7, S8 and S12–S23†). The results from the BP86 functional are discussed in the main manuscript, while those computed with the B3LYP functional are collected in the ESI.†
Hexa-aza macrocyclic complexes
The reaction of [Li(thf)4][BArF] with Me6[18]aneN6 in CH2Cl2 led to the isolation of a white crystalline solid (Scheme 1) which was formulated as [Li(Me6[18]aneN6)][BArF] on the basis of microanalytical, 1H, 13C{1H}, and 7Li NMR spectroscopic data. The crystal structure (Fig. 1) shows the [Li(Me6[18]aneN6)]+ cation contains an N6 donor set (without retention of thf) and the macrocycle is in a puckered conformation; the distortion from an ideal octahedron is severe, as seen by two of the ‘trans’ N–Li–N angles which show a large deviation from 180°, whilst the Li–N bond lengths vary by ca. 0.3 Å. They are longer than the sum of ionic radii for Li (6-coordinate) and N (2.39 Å),20 but significantly shorter than the sum of van der Waals radii (3.78 Å),21 consistent with the mismatch between the small Li+ cation and the 18-membered ring. The 1H NMR spectrum is broad at 298 K, indicating dynamic behaviour in solution, while the 7Li NMR spectrum shows a singlet at −0.30 ppm.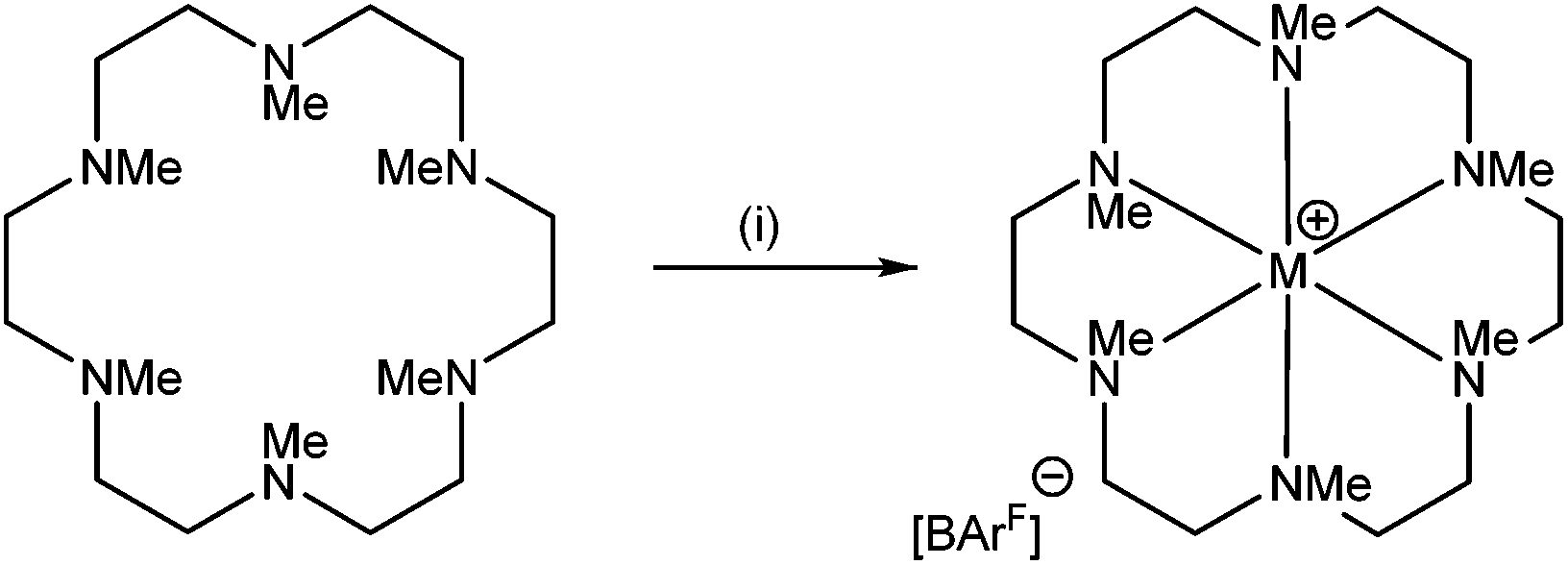 | ||
| Scheme 1 Synthesis of Me6[18]aneN6 complexes. Conditions: (i) M = Li: [Li(thf)4][BArF], CH2Cl2; M = Na: Na[BArF]·2thf, CH2Cl2; M = K, Rb, Cs: M[BArF], CH2Cl2. | ||
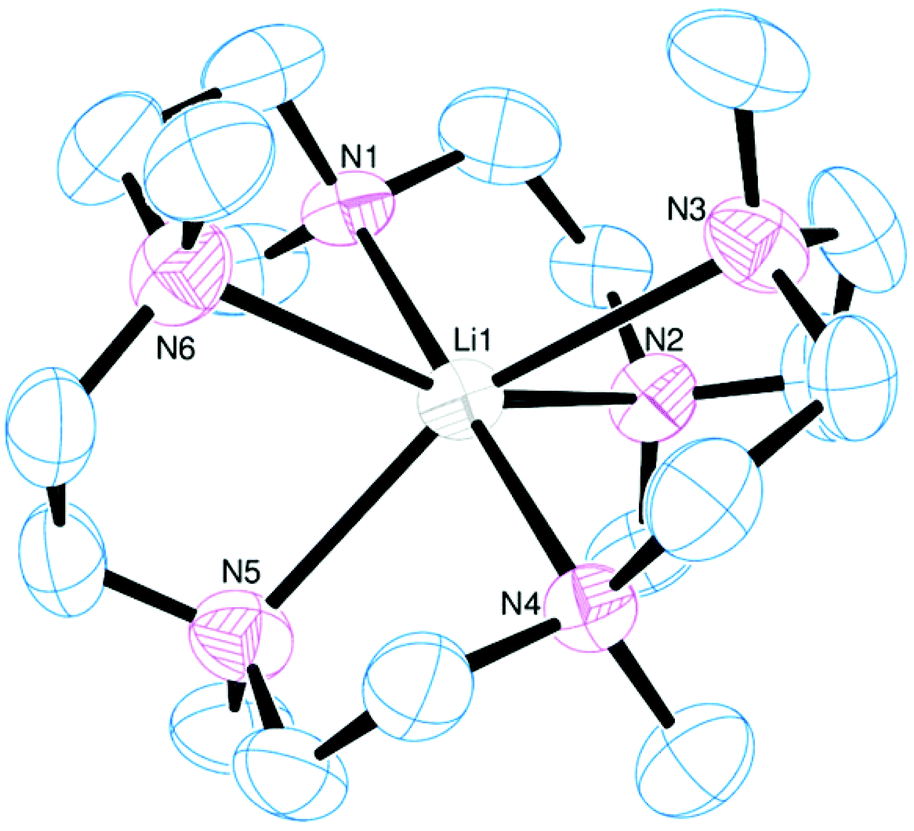 | ||
| Fig. 1 View of the cation in [Li(Me6[18]aneN6)][BArF]. Thermal ellipsoids are drawn at 50% probability. H atoms and the [BArF]− anion are omitted for clarity. Selected bond lengths (Å) and angles (°): Li–N1 2.274(9), Li–N2 2.384(9), Li–N3 2.587(9), Li–N4 2.315(9), Li–N5 2.45(1), Li–N6 2.554(9); N1–Li–N2 77.3(3), N2–Li–N3 73.1(3), N3–Li–N4 74.3(3), N4–Li–N5 77.7(3), N5–Li–N6 73.5(3), N6–Li–N1 75.9(3). | ||
The computed minimum energy geometry of the isolated [Li(Me6[18]aneN6)]+ cation (Fig. 2) has C2 symmetry. The macrocycle is puckered as in the crystal structure, and overall the computed geometric parameters are also in good agreement with the experimental values (Tables S2 and S3†).
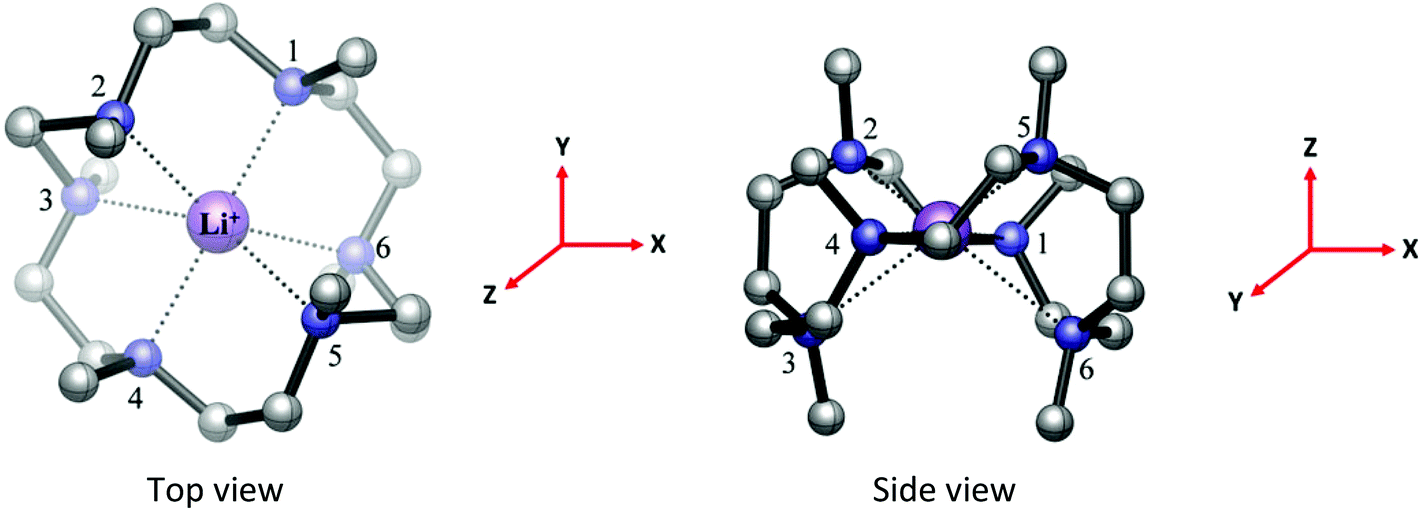 | ||
| Fig. 2 The optimised BP86 DFT geometry of the [Li(Me6[18]aneN6)]+ cation with C2 symmetry and 1A electronic ground state. The reference axes are shown (in red). | ||
Fig. 3 shows the isodensity plots of the frontier molecular orbitals (FMOs) of [Li(Me6[18]aneN6)]+. The results show that the highest occupied molecular orbital (HOMO) is centred on the six N atoms of the Me6[18]aneN6 ring, and is composed of the N 2p lone pairs with their associated lobes oriented in the direction of the Li+ ion. Population analysis shows that the low-lying HOMOs (HOMO−1 to HOMO−5) are also dominated by the N 2p lone pairs, however, an increasing contribution of Li+ is observed from HOMO−1 to HOMO−5 (i.e. HOMO−1, HOMO−3, HOMO−4, and HOMO−5 have 3%, 6%, 6%, and 10% contributions from the 2pz, 2px, 2py, and 2s orbitals of Li+, respectively). LUMO and LUMO+1 are antibonding molecular orbitals (ABMOs) and correspond predominantly to the 2s and 2p orbitals of the Li+ centre.
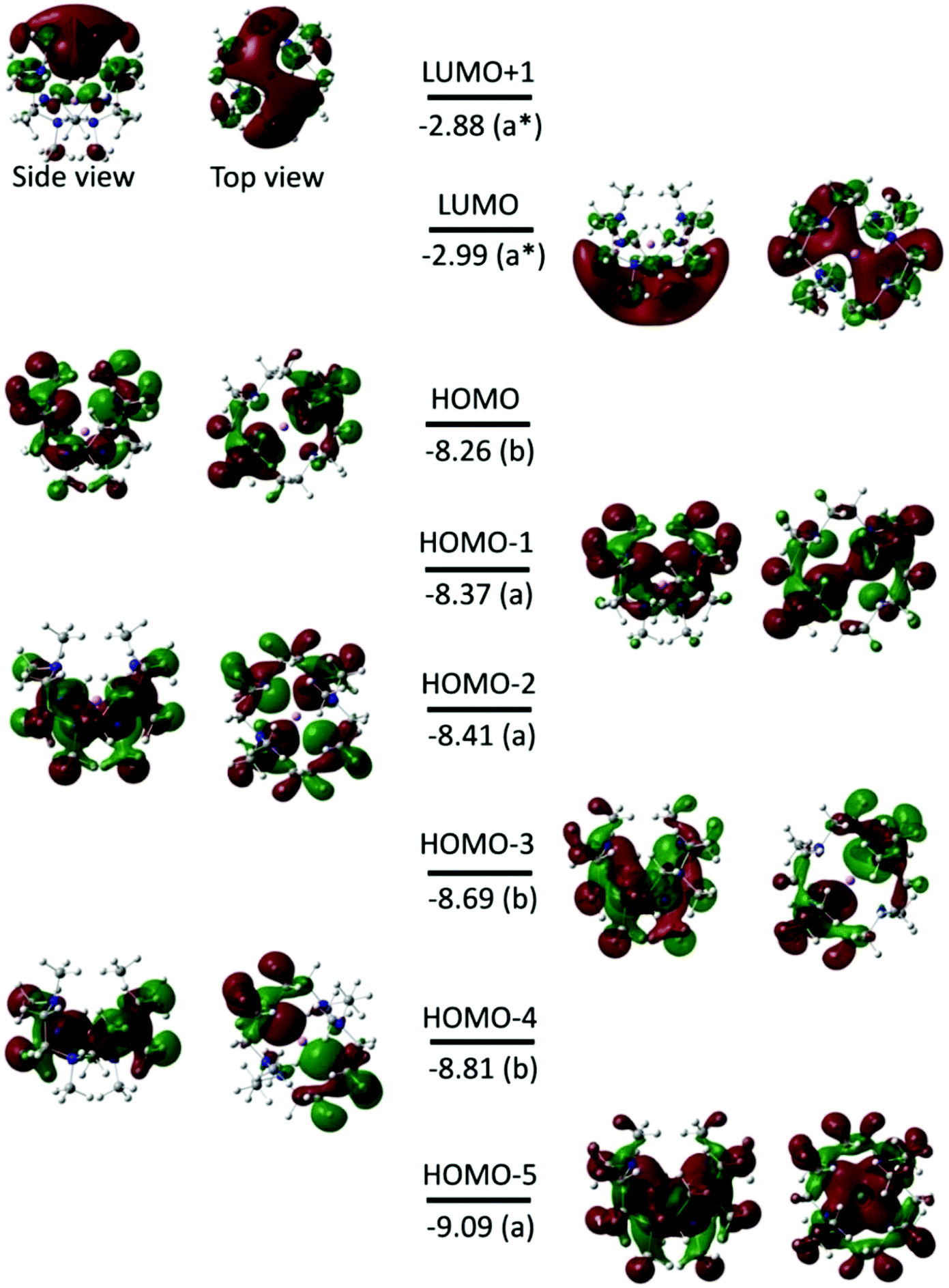 | ||
| Fig. 3 Frontier molecular orbitals (isovalue of 0.02 electrons Bohr−3) for the [Li(Me6[18]aneN6)]+ complex obtained from BP86 DFT calculations. The energy and symmetry of the molecular orbitals are given in eV and in parentheses, respectively. The dark red (+) and green (−) lobes represent the signs of the angular part of the AO contributing to a MO. | ||
[Na(Me6[18]aneN6)][BArF] was also isolated as a white solid from the reaction of Na[BArF]·2thf with Me6[18]aneN6 and similarly characterised. Whilst we were unable to grow crystals suitable for X-ray analysis, hexadentate coordination of the macrocycle is likely. Two possible minimum energy structures of [Na(Me6[18]aneN6)]+ (1 and 2 in Fig. 4) were located in the BP86 DFT calculations. Structure 1 (C2 symmetry) is analogous to the computed minimum energy structure of [Li(Me6[18]aneN6)]+, while structure 2 (D3d symmetry) has a geometry similar to its K+ and Rb+ counterparts (vide infra). Structure 1 is more stable than 2 by 28.2 kJ mol−1 in the gas phase (Tables S12, S13, S18 and S19†). It is important to note that the presence of the [BArF]− anions in the crystal will influence the relative stabilities of structures 1 and 2 in the isolated solids.
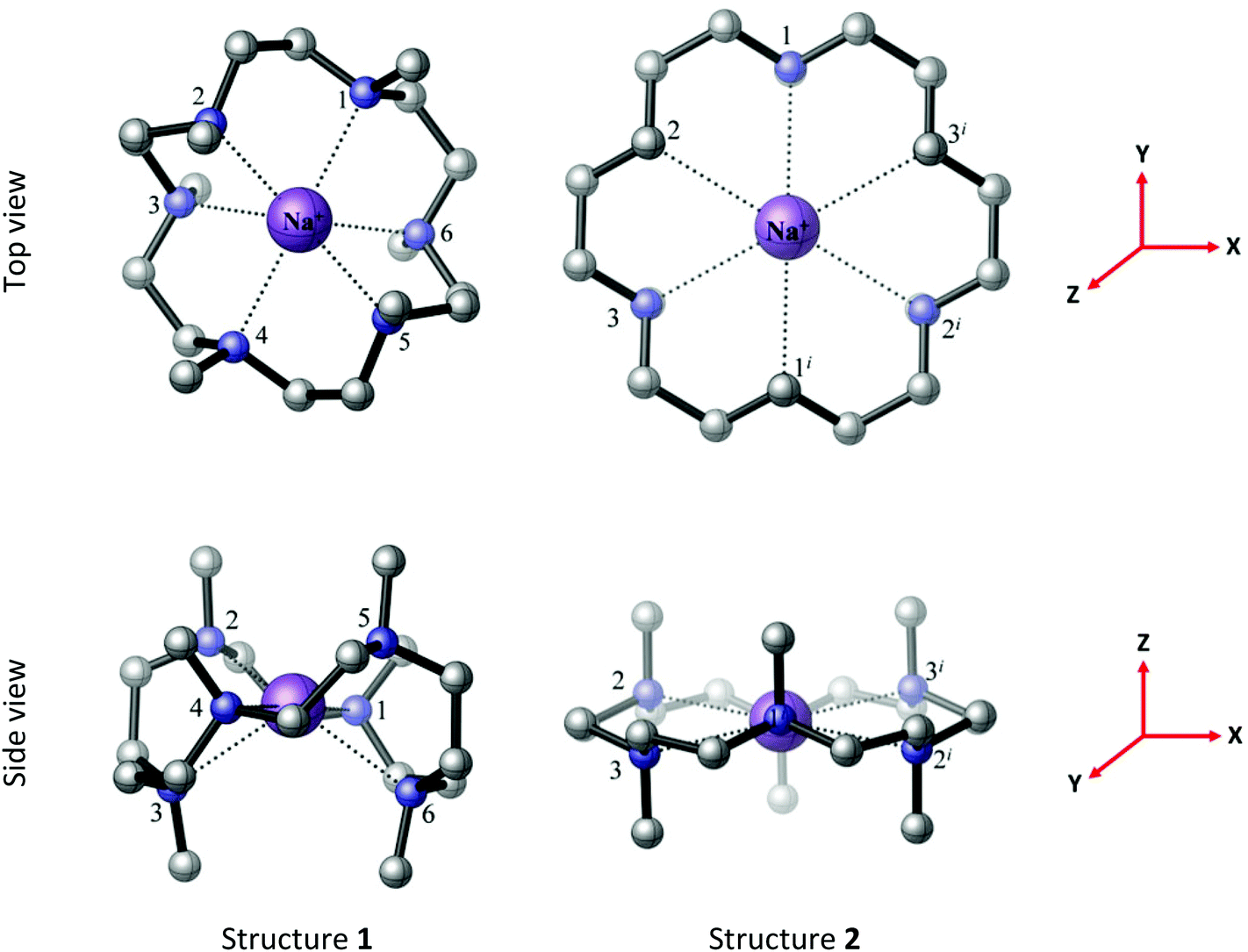 | ||
| Fig. 4 The BP86 DFT optimised geometries of [Na(Me6[18]aneN6)]+ structures 1 (C2 symmetry) and 2 (D3d symmetry). The reference axes are shown (in red). Structure 1 is computed to be more stable than structure 2 by 28.2 kJ mol−1. | ||
[K(Me6[18]aneN6)][BArF] was isolated and characterised similarly. Its X-ray structure (Fig. 5) showed two half-cations in the asymmetric unit, with the K+ ions occupying crystallographic inversion centres. [K(Me6[18]aneN6)]+ shows hexagonal planar N6 coordination at K+, with the NMe groups alternating ‘up-down’ around the ring. The difference between the two K+ cations in the asymmetric unit is found in the long-range interactions to the [BArF]− anions. There are two K2⋯F1 interactions of 3.658(1) Å perpendicular to the N6 plane (ΣvdW = 4.19 Å),22 which gives K2 an overall hexagonal bipyramidal arrangement (Fig. 5a). The other potassium cation (K1) contains no K⋯F interactions and is therefore hexagonal planar (Fig. 5b).
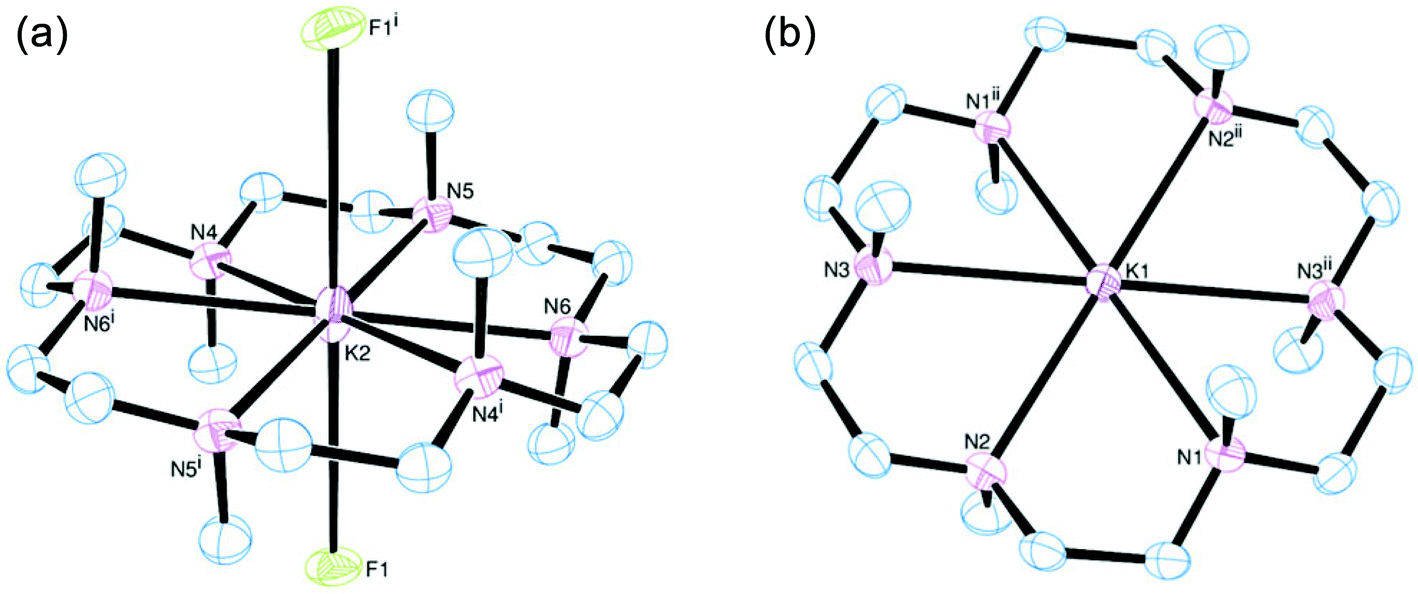 | ||
| Fig. 5 (a) View of the K2 centred cation (left) and (b) the K1 cation (right) of [K(Me6[18]aneN6)][BArF]. Thermal ellipsoids are drawn at 50% probability. H atoms and the [BArF]− anions (bar F1) are omitted for clarity. Selected bond lengths (Å) and angles (°): K1–N1 2.883(2), K1–N2 2.896(2), K1–N3 2.911(2), K2–N4 2.918(2), K2–N5 2.866(2), K2–N6 2.911(2); N1–K1–N2 61.86(5), N2–K1–N3 61.35(5), N1–K1–N3ii 60.67(5), N4–K2–N5 61.79(5), N5–K2–N6 61.11(5), N4–K1–N6i 61.04(5). Symmetry codes: (i) 1 − x, −y, 1 − z; (ii) −x, 1 − y, −z. | ||
[Rb(Me6[18]aneN6)][BArF] also crystallises with two Rb+ cations in the asymmetric unit, with the Rb+ ion sitting out of the N6 plane by 0.561(3) Å (Rb1) and 0.382(3) Å (Rb2). For Rb1, this has the effect that one Rb⋯F interaction is much shorter than the other. The Rb1–F12 distance of 3.235(2) Å is much shorter than the sum of van der Waals radii (4.67 Å) (Fig. 6a),21 and is also significantly shorter than the corresponding K⋯F distance. The Rb1⋯F12i distance (4.128(1) Å) is also within the sum of van der Waals radii. Rb2 has two long Rb⋯F distances of 3.951(1) and 4.233(1) Å, both of which are on the same side of the N6 ring (Fig. 6b).
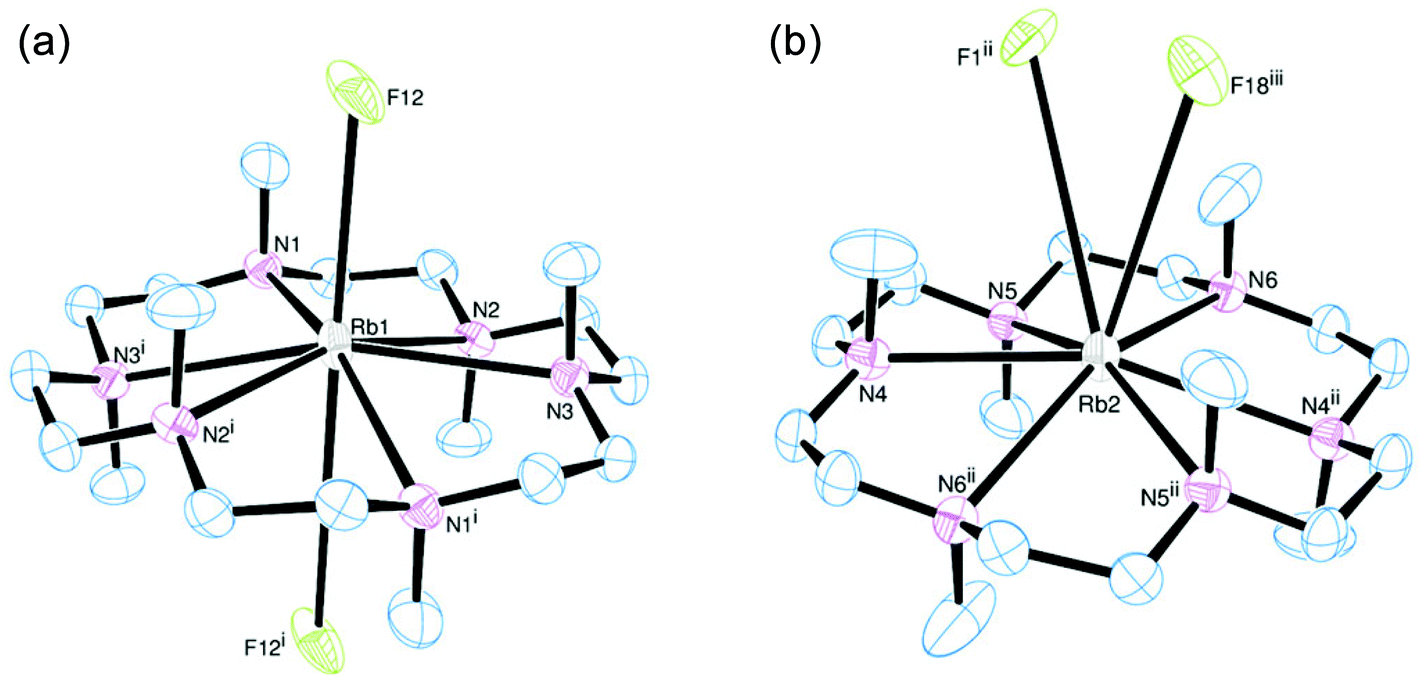 | ||
| Fig. 6 (a) View of the cation based on Rb1 (left) and (b) view of the cation based on Rb2 (right) of [Rb(Me6[18]aneN6)][BArF] in the asymmetric unit. Thermal ellipsoids are drawn at 50% probability. H atoms and the [BArF]− anion (bar coordinating fluorines) are omitted for clarity. Selected bond lengths (Å) and angles (°): Rb1–N1 2.899(2), Rb1–N2 2.980(2), Rb1–N3 2.948(2), Rb1⋯F12 3.235(2), Rb1⋯F12i 4.128(1), Rb2–N4 2.921(2), Rb2–N5 2.924(2), Rb2–N6 2.902(2), Rb2⋯F18iii 3.951(1), Rb2⋯F1ii 4.233(1); N1–Rb1–N2 61.78(6), N2–Rb1–N3 60.09(6), N1–Rb1–N3i 60.95(6), N4–Rb2–N5 60.04(6), N5–Rb2–N6 60.50(6), N4–Rb2–N6ii 59.50(6). Symmetry code: (i) −x, 2 − y, −z; (ii) 1 − x, 1 − y, 1 − z; (iii) x + 1, y, z. | ||
The BP86 DFT calculations satisfactorily reproduce the experimental X-ray structures of [M(Me6[18]aneN6)]+ (M = K and Rb) with D3d symmetry (Fig. 7), which contrasts with C2 symmetry identified for the Li+ counterpart. In the computed structures both metal cations lie within the plane of the Me6[18]aneN6 ring, with six equivalent bond distances (K–N = 2.984 Å; Rb–N = 3.062 Å), indicating that despite the [BArF]− anion being large and usually considered as very weakly interacting, in some cases it still influences the experimentally determined geometry.
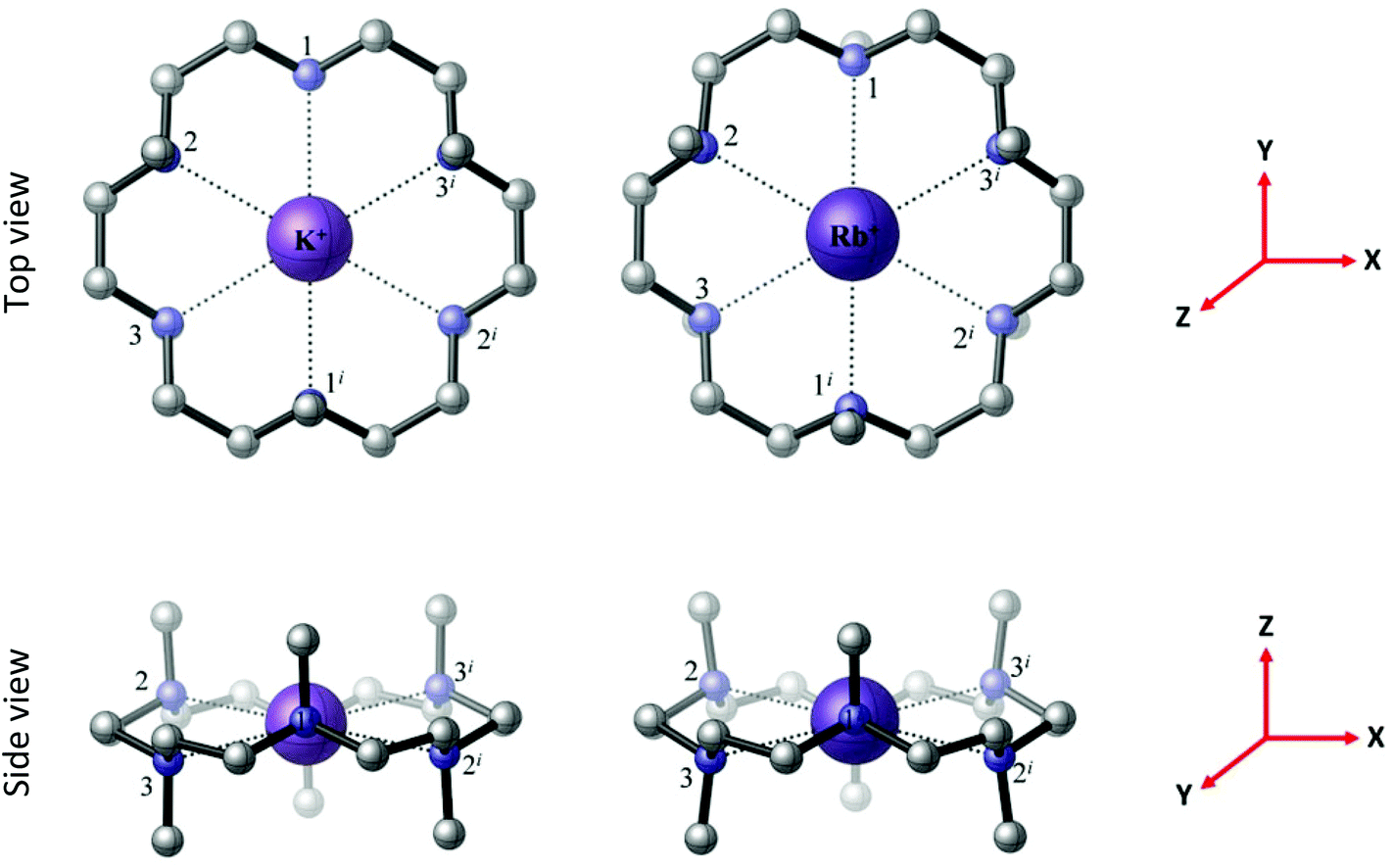 | ||
| Fig. 7 The BP86 DFT optimised geometries of the [M(Me6[18]aneN6)]+ complexes, M = K, Rb, both with D3d symmetry and a 1A1g electronic ground state. The reference axes are shown (in red). | ||
Fig. 8 and S3† show the isodensity plots of the frontier MOs (FMOs) of the [M(Me6[18]aneN6)]+ (M = K or Rb, respectively), which are very similar. As for the Li+ complex discussed above, the HOMO corresponds mainly to the 2p orbitals of N atoms, and the LUMO (and LUMO+1) correspond predominantly to the ns (np) orbitals of K+ and Rb+ respectively; the main metal contribution is in the lower HOMO−3 to HOMO−5 orbitals.
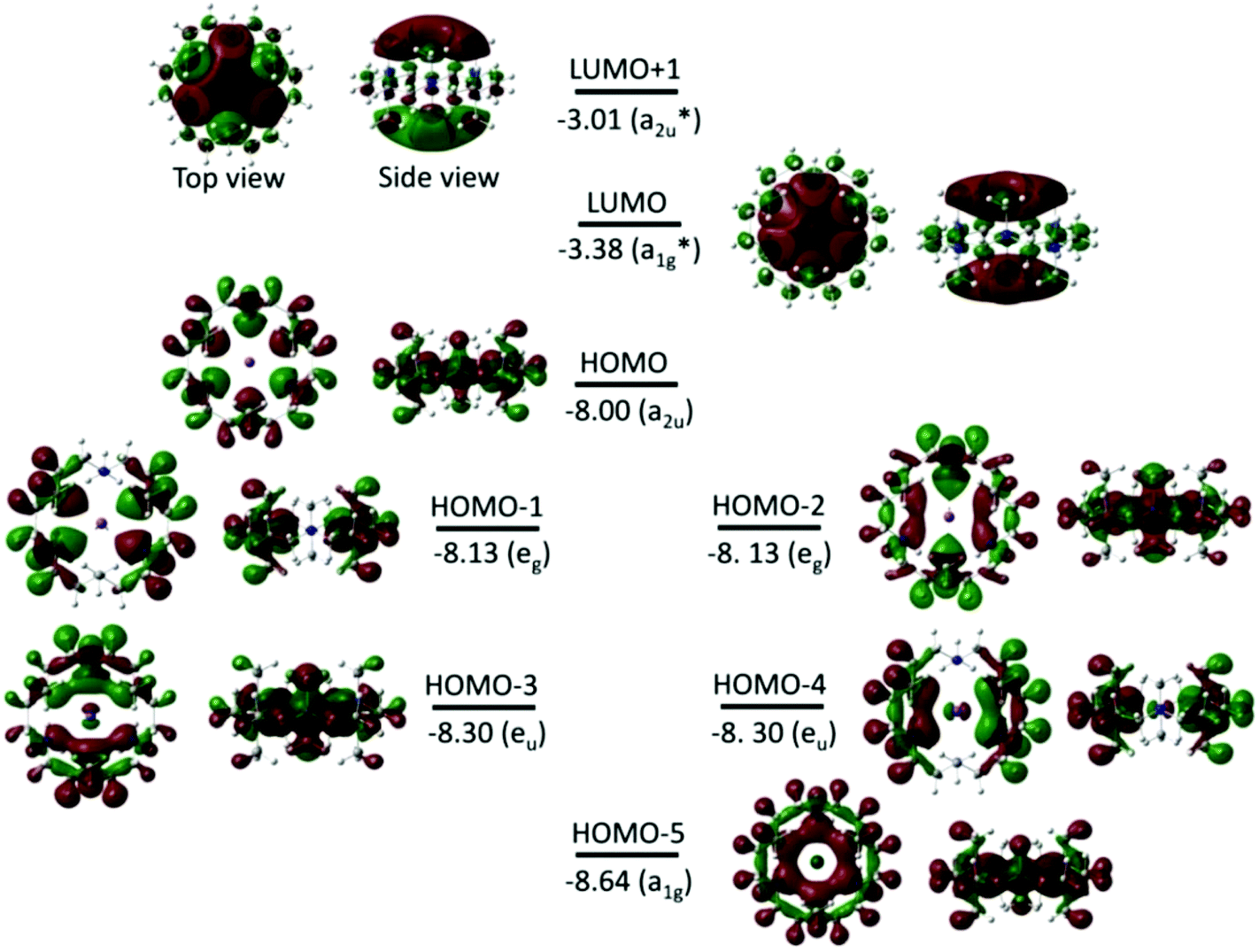 | ||
| Fig. 8 Frontier molecular orbitals (FMOs) (isovalue of 0.02 electrons Bohr−3) for [K(Me6[18]aneN6)]+ obtained from BP86 DFT calculations. The energy and symmetry of the MOs are given in eV and in parentheses, respectively. The dark red (+) and green (−) lobes represent the signs of the angular part of the AO contributing to a MO. | ||
The relevant charge densities for the complex cations are shown in Table 1, together with those for Me6[18]aneN6 itself (which has two conformers of very similar energy, structures 3 and 4, Fig. S2†). This shows that electron density is transferred from the bonding σ C–H and σ C–N orbitals and N 2p non-bonding orbitals of [M(Me6[18]aneN6)]+ to the ns and np valence orbitals of the metal. The metal ion in the [M(Me6[18]aneN6)]+ species is more positively charged on going from Li+ → K+ → Rb+, consistent with more electron density being retained on the ligand as Group 1 is descended.
| Centres | M | N |
|---|---|---|
| a The structures of the optimised geometry of Me6[18]aneN6, labelled as 3 and 4, are provided in Fig. S2. | ||
| Me6[18]aneN6 (structure 3)a | — | −0.503 (N3/6), −0.512 (N2/5), −0.515 (N1/4) |
| Me6[18]aneN6 (structure 4)a | — | −0.493 |
| [Li(Me6[18]aneN6)]+ | 0.522 | −0.529 (N3/6), −0.544(N2/5), −0.548 (N1/4) |
| [K(Me6[18]aneN6)]+ | 0.734 | −0.531 |
| [Rb(Me6[18]aneN6)]+ | 0.934 | −0.543 |
Crystals of [Cs(Me6[18]aneN6)][BArF] all showed severe disorder, although microanalytical and spectroscopic data were consistent with the formulation [Cs(Me6[18]aneN6)][BArF]. Two possible minimum energy structures for the [Cs(Me6[18]aneN6)]+ complex, 5 and 6, are located by the BP86 DFT computations, with structure 5 lower in energy than structure 6 by 43.1 kJ mol−1 (Fig. 9). In 5, the Cs+ ion lies out of the N6 plane by 0.869 Å, similar to the experimentally derived [Rb(Me6[18]aneN6)]+ structure. Notably, the reported [Cs(Me6[18]aneN6)]+ is structurally very different.11a
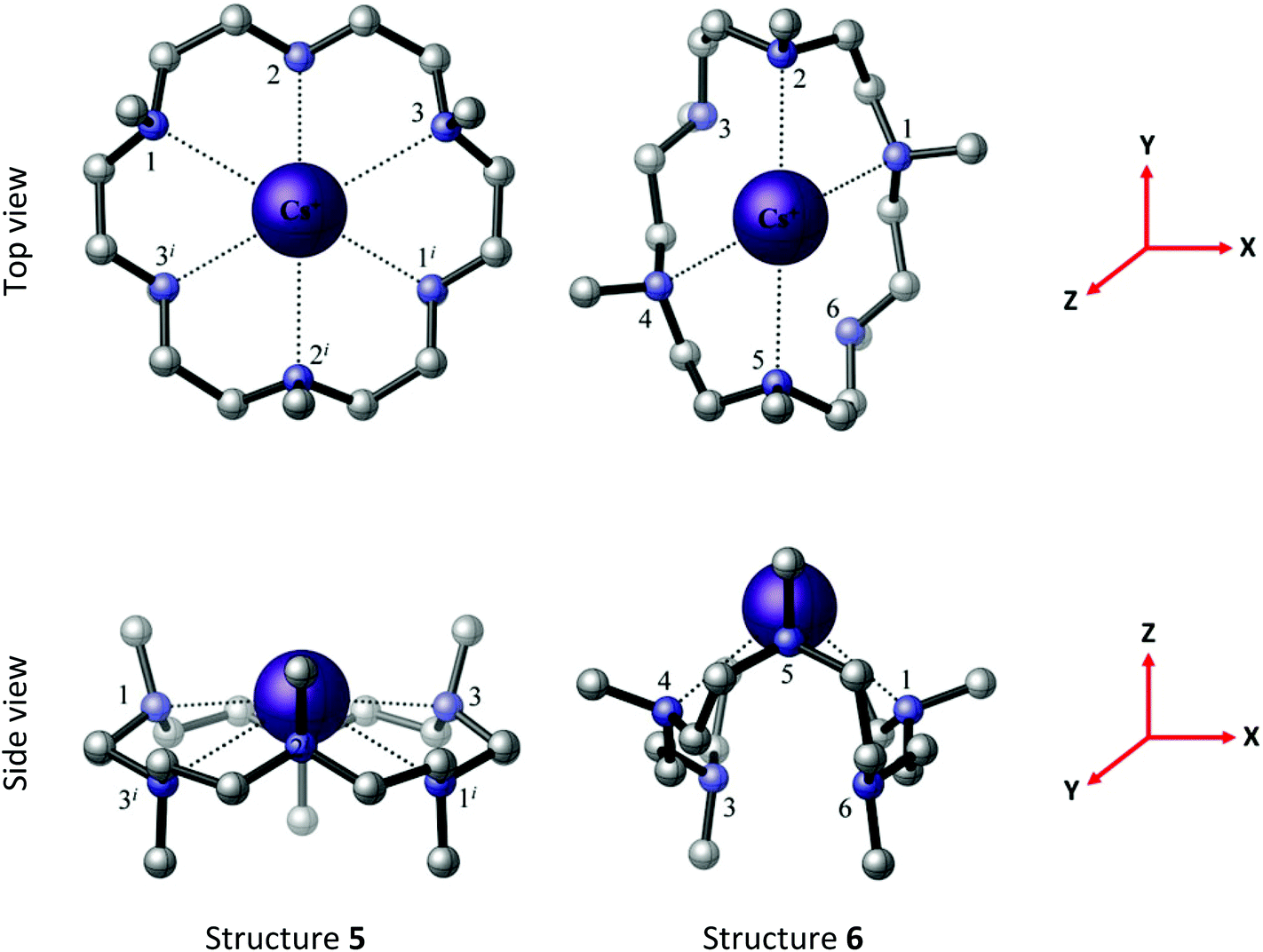 | ||
| Fig. 9 The BP86 DFT optimised geometries of [Cs(Me6[18]aneN6)]+ structures 5 (C3v symmetry) and 6 (C2 symmetry). The reference axes are shown (in red). Structure 5 is computed to be 43.1 kJ mol−1 lower than structure 6. | ||
The bond dissociation energies calculated for [M(Me6[18]aneN6)]+ → M+ + Me6[18]aneN6 (1) (Table 2) decrease on going from Li+ to Cs+, as expected.
Tri-aza macrocyclic complexes
Based upon our earlier work on Na-Me3tacn complexes,3 we also attempted to extend the chemistry of Me3tacn to the other members of Group 1 (Scheme 2).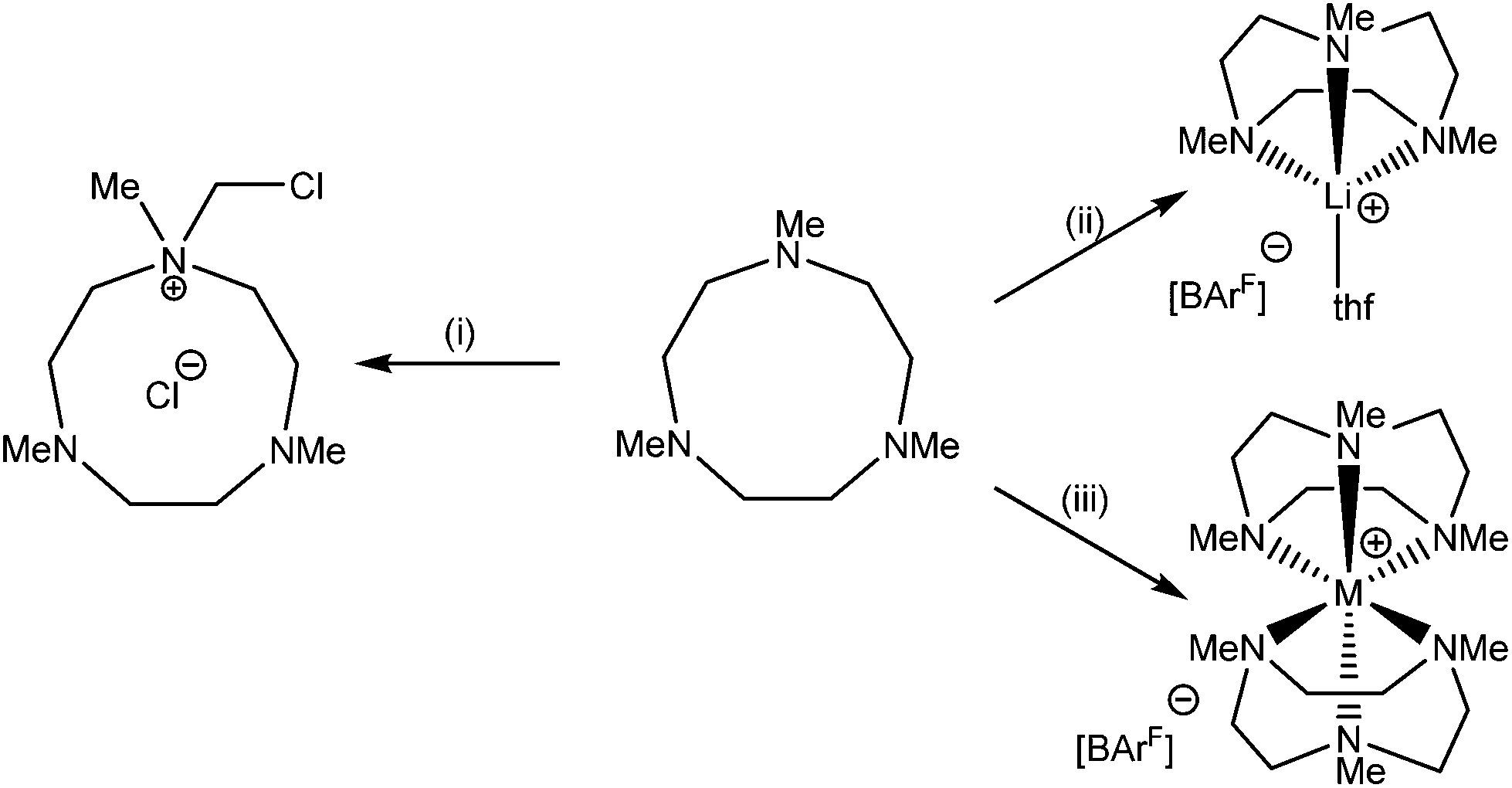 | ||
| Scheme 2 Products from the reaction of M[BArF] with 2 mol. equiv. of Me3tacn (M = Li, Na, K). Conditions: (i) 0.5 mol. equiv. [Li(thf)4][BArF], CH2Cl2; (ii) 0.5 or 1 mol. equiv. [Li(thf)4][BArF], toluene; (iii) M = Na: 0.5 mol. equiv. Na[BArF]·2thf, CH2Cl2; M = K: 0.5 mol. equiv. K[BArF], CH2Cl2. | ||
Using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of [Li(thf)4][BArF]:Me3tacn in toluene resulted in the isolation of the half-sandwich complex [Li(Me3tacn)(thf)][BArF] in good yield. This contrasts with the analogous reaction with Na+, for which both the sandwich and half-sandwich complexes were obtained depending upon the molar ratio of metal:ligand used. Further attempts to form the sandwich [Li(Me3tacn)2]+ cation from [Li(thf)4][BArF] and 2 mol. equiv. of Me3tacn in CH2Cl2 instead led to a quaternary ammonium cation as its chloride salt (Scheme 2), which was identified by 1H NMR spectroscopy and from comparison with the literature unit cell.22 No evidence for the [Li(Me3tacn)2]+ cation was observed.
:2 molar ratio of [Li(thf)4][BArF]:Me3tacn in toluene resulted in the isolation of the half-sandwich complex [Li(Me3tacn)(thf)][BArF] in good yield. This contrasts with the analogous reaction with Na+, for which both the sandwich and half-sandwich complexes were obtained depending upon the molar ratio of metal:ligand used. Further attempts to form the sandwich [Li(Me3tacn)2]+ cation from [Li(thf)4][BArF] and 2 mol. equiv. of Me3tacn in CH2Cl2 instead led to a quaternary ammonium cation as its chloride salt (Scheme 2), which was identified by 1H NMR spectroscopy and from comparison with the literature unit cell.22 No evidence for the [Li(Me3tacn)2]+ cation was observed.
Although attempts to crystallise the [Li(Me3tacn)(thf)][BArF] were unsuccessful, its identity is clear from the spectroscopic and analytical data. DFT calculations on the Me3tacn complexes with Na+ and Li+ were undertaken to probe their relative stabilities. The computed bond distances and angles at Na+ in [Na(Me3tacn)2]+ (Fig. 10, Tables S7 and S8†) agree well with the experimental values reported earlier.3 For Li+, the calculations show that the Li–N bond in [Li(Me3tacn)2]+ (S6 symmetry) is very long (2.460 Å), whereas in the half-sandwich cation [Li(Me3tacn)]+ the Li–N distance of 2.029 Å (Fig. 10) is more akin to the reported Li–N bond distances.9,23 This strongly suggests that the much smaller Li+ ion can comfortably accommodate only one Me3tacn (consistent with the isolation of the [Li(Me3tacn)(thf)][BArF] half-sandwich), but introducing a second Me3tacn is much less favourable, due to steric clashing between the Me groups on the two rings. As noted earlier,3 the isolation of the bis-Me3tacn sandwich cation is unusual; the large ionic radius of Na+ apparently permitting its formation (the Ag+ analogue is the only other structurally authenticated example).24
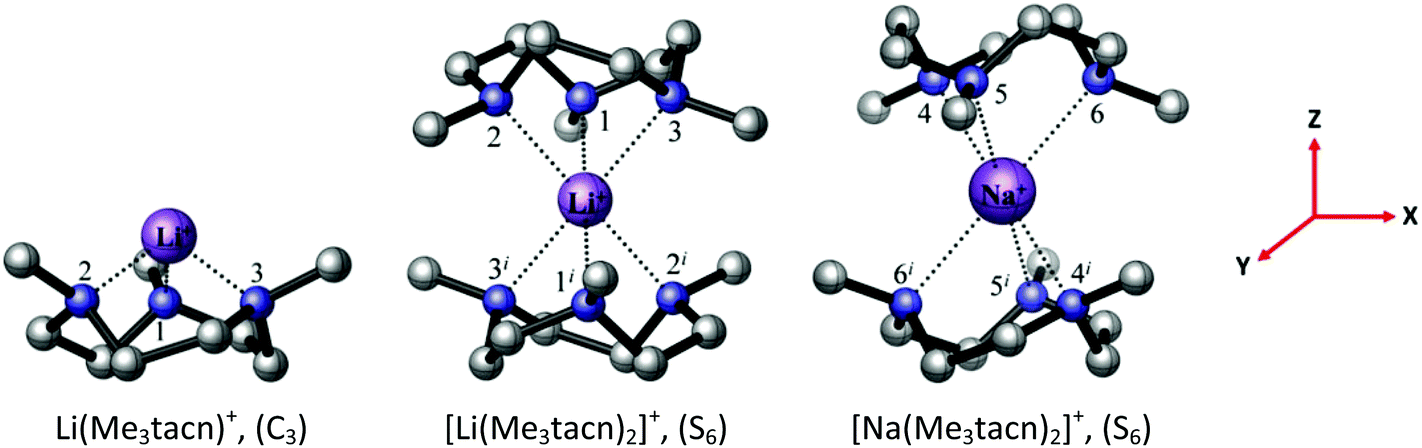 | ||
| Fig. 10 The BP86 DFT optimised geometry of the [Li(Me3tacn)]+ and [M(Me3tacn)2]+ complexes, M = Li, Na. The symmetry of each complex is given in parentheses. The reference axes are shown (in red). | ||
[K(Me3tacn)2][BArF] was successfully prepared and isolated similarly to the Na+ analogue and is isostructural (Fig. 11), forming a centrosymmetric very distorted octahedral cation. This contrasts with the hexagonal planar coordination present in [K(Me6[18]aneN6)]+, although the bond distances and angles at K+ are actually quite similar.
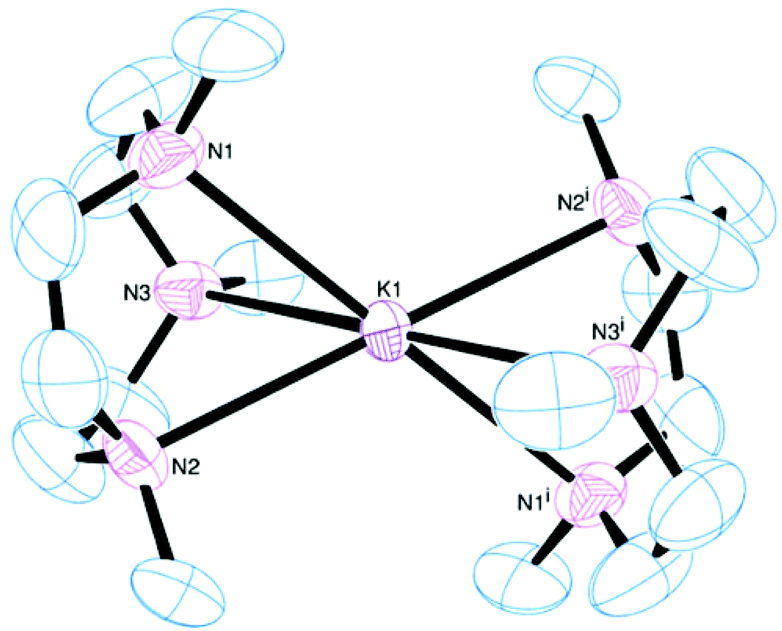 | ||
| Fig. 11 View of the cation based on K1 in the asymmetric unit of [K(Me3tacn)2][BArF]. Thermal ellipsoids are drawn at 50% probability. H atoms, the [BArF]− anion, and the other half cation are omitted for clarity. Selected range of bond lengths (Å) and angles (°): K1–N1 2.815(6), K1–N2 2.768(5), K1–N3 2.881(6), K2–N4 2.768(6), K2–N5 2.821(6), K2–N6 2.808(6); N1–K1–N2 63.2(2), N2–K1–N3 62.8(2), N3–K1–N1 63.2(2), N4–K2–N5 64.2(2), N5–K2–N6 63.9(2), N6–K2–N4 64.2(2). Symmetry code: (i) 1 − x, −y, 1 − z. | ||
The K–N bonds are 0.2–0.3 Å longer than the corresponding Na–N bonds, while the intra-macrocyclic N–K–N angles are more acute by 8–9°. These differences may reflect a poorer match between the large K+ cation and the small nine-membered Me3tacn ring, although as we have noted, they are similar to the angles in [K(Me6[18]aneN6)]+. As shown in Fig. 11, the Me groups of the two rings are well separated.
In contrast to the BP86 DFT optimised [Li(Me3tacn)2]+ and [Na(Me3tacn)2]+ complexes (S6 symmetry), [K(Me3tacn)2]+ is computed to have D3 symmetry (Fig. 12). The computed K–N bond distances are 2.915 Å; slightly longer than the experimental values (2.765(5)–2.880(5) Å), while the calculated N–K–N bond angles within the five-membered chelate rings (63.0°) are closely comparable to the crystallographic values.
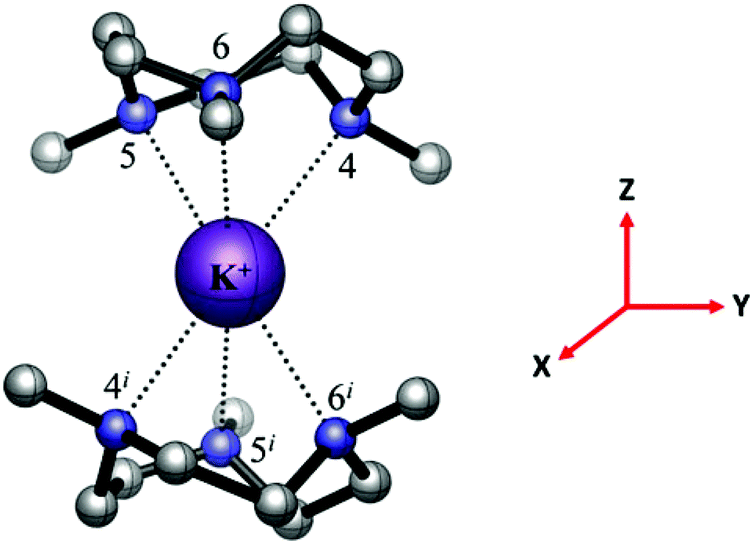 | ||
| Fig. 12 The BP86 DFT optimised geometry of the [K(Me3tacn)2]+ complex with D3 symmetry and 1A1 electronic ground state. The reference axes are shown (in red). | ||
The calculations on [K(Me3tacn)2]+ show that the HOMO and HOMO−1 are doubly degenerate (as are HOMO−2 and HOMO−3) (Fig. 13), and they are mainly located on the N atoms of the Me3tacn ring, corresponding to N 2p valence orbitals. The LUMO, LUMO+1 and LUMO+2 are mainly localised on K+. The LUMO corresponds to the 4s orbital of K+, while LUMO+1 and LUMO+2 correspond mainly to the 4px and 4py orbitals, respectively. The FMOs of the [Na(Me3tacn)2]+ analogue follow similar trends (Fig. S5†).
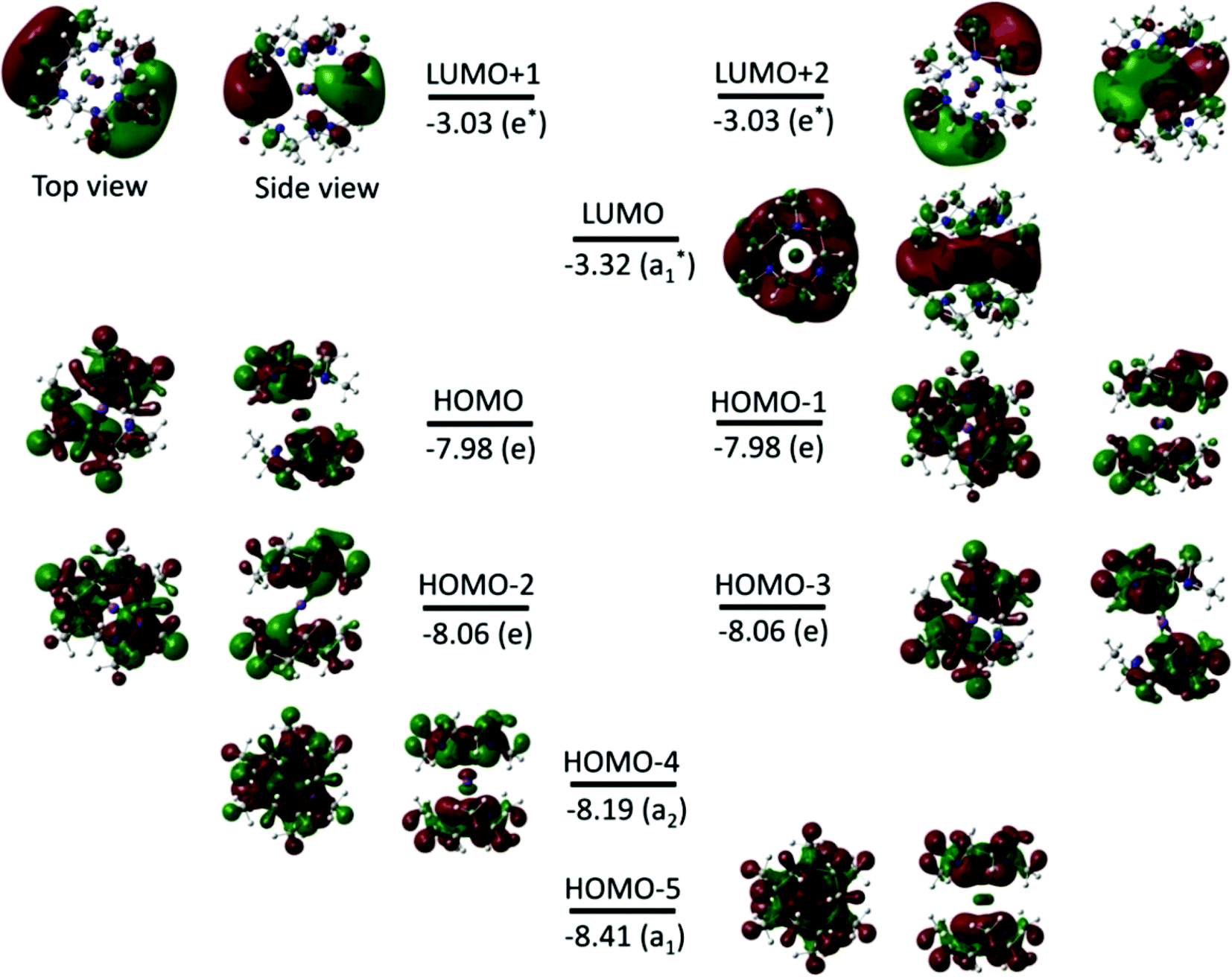 | ||
| Fig. 13 FMOs (isovalue of 0.02 electrons Bohr−3) for the [K(Me3tacn)2]+ complex obtained from BP86 DFT calculations. The energy and symmetry of the MOs are given in eV and in parentheses, respectively. The dark red (+) and green (−) lobes represent the signs of the angular part of the AO contributing to a MO. | ||
Table 3 shows the charge densities on the metal and N atoms of Me3tacn, [M(Me3tacn)]+ and [M(Me3tacn)2]+, M = Na and K. The natural charge on K+ is lower than the formal charge of +1, showing a significant electron density transfer from the ligands. The charge densities on the N atoms for [M(Me3tacn)]+ and [M(Me3tacn)2]+ are more negative than in the ‘free’ ligand, because electron density is withdrawn from σ C–H and σ C–N orbitals of the ligand.
| M = Na | M = K | |||
|---|---|---|---|---|
| Centres | Na | N | K | N |
| Me3tacn | — | −0.499 | — | −0.499 |
| [M(Me3tacn)]+ | +0.867 | −0.587 | +0.913 | −0.575 |
| [M(Me3tacn)2]+ | +0.551 | −0.549 | +0.680 | −0.542 |
As in the [M(Me6[18]aneN6)]+ analogues, the charge densities on M show that less electron density is transferred to the metal in the K+ case, suggesting a relatively weaker interaction between the K+ and the Me3tacn ligand(s), and correspondingly longer K–N distances, than in the Na+ case.
Attempts to prepare analogous complexes with Rb+ and Cs+ ions failed; the optimised minimum energy structures of [M(Me3tacn)2]+ (M = Rb, Cs) are presented in Tables S16, S17, S22 and S23.†
Zero-point and BSSE corrected bond dissociation energies (kJ mol−1) for the dissociation [M(Me3tacn)]+ → M+ + Me3tacn (2), M = Li, Na, K, Rb, and Cs, decrease on going down Group 1 (Table 4), correlating with the increase in the ionic radii, and the poorer fit with the small triaza macrocycle. A similar trend in zero-point corrected bond dissociation energies is observed for the dissociation of [M(Me3tacn)2]+ → [M(Me3tacn)]+ + Me3tacn (3), except for the dissociation energy of the [Li(Me3tacn)2]+ complex which is much lower. This appears to be mainly a consequence of the inability of the small lithium ion to accommodate six-coordination because the presence of the Me substituents on one macrocycle sterically impedes the approach of the second.
| M | (2) | (3) |
|---|---|---|
| Li | 350.4 | 63.9 |
| Na | 234.0 | 104.5 |
| K | 165.5 | 86.5 |
| Rb | 122.0 | 77.4 |
| Cs | 96.2 | 63.2 |
It seems clear, therefore, that isolation of the [M(Me3tacn)2]+ cations is a fine balance between the metal ion being sufficiently large to avoid significant steric clashing between the Me groups of the two rings, but small enough to avoid very acute N–M–N chelate angles.
Tetra-aza macrocyclic complexes
Changing the ligand from Me3tacn to Me4cyclen has the effect of both increasing the denticity and expanding the macrocyclic binding cavity. Reaction of [Li(OH2)4][BArF] with Me4cyclen in CH2Cl2 (Scheme 3) led to the formation of [Li(Me4cyclen)(OH2)][BArF].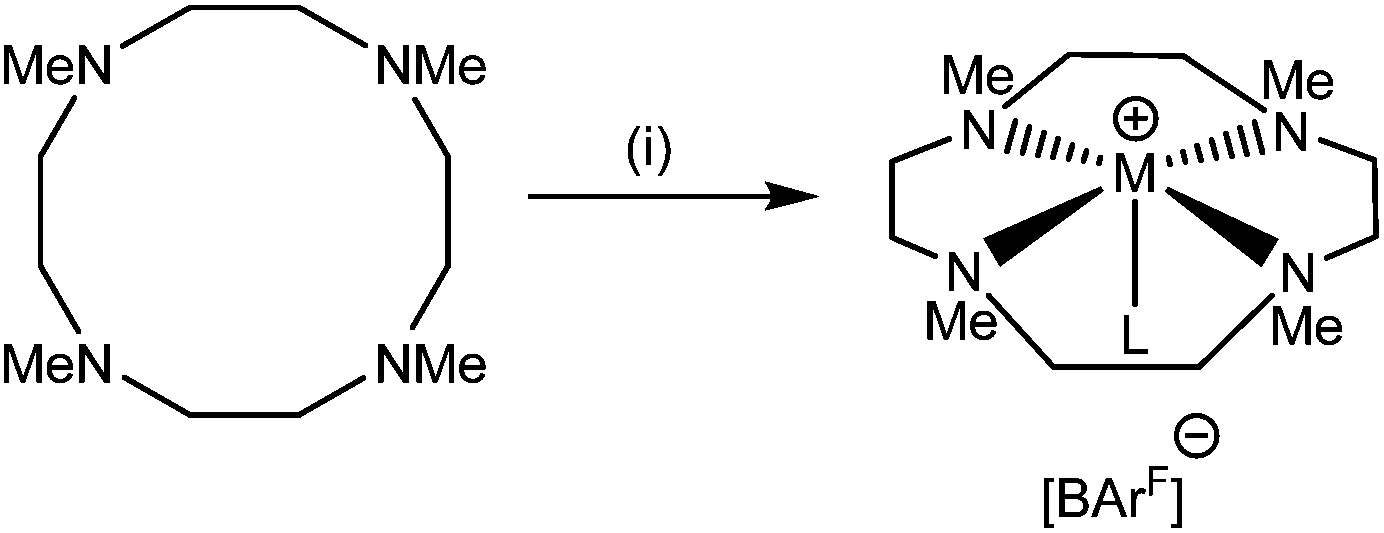 | ||
| Scheme 3 Reaction of M[BArF] with Me4cyclen. Conditions: (i) M = Li: [Li(OH2)4][BArF], CH2Cl2, L = H2O; M = Na: Na[BArF]·2thf, CH2Cl2, L = thf. | ||
The crystal structure shows a five-coordinate square pyramidal (τ = 0.02)25 cation with the macrocycle tetradentate and one apical water molecule (Fig. 14). The NMe groups all lie on the same side of the N4 plane as the metal, with the Li cation displaced out of the N4 plane by 0.758(7) Å. The same product is obtained irrespective of the ratio of Me4cyclen:Li[BArF] used.
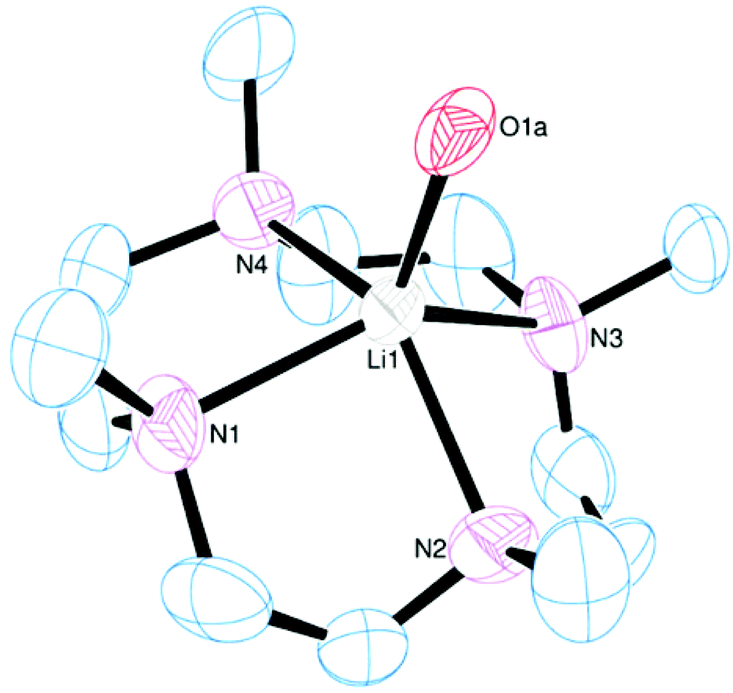 | ||
| Fig. 14 View of the cation in [Li(Me4cyclen)(OH2)][BArF]. Thermal ellipsoids are drawn at 50% probability. H atoms and the [BArF]− anion are omitted for clarity. Selected bond lengths (Å) and angles (°): Li1–N1 2.186(8), Li1–N2 2.206(8), Li1–N3 2.179(8), Li1–N4 2.154(9), Li1–O1a 1.98(1); N1–Li1–N2 82.8(3), N2–Li1–N3 82.1(3), N3–Li1–N4 84.7(3), N1–Li1–N4 = 82.6(3). | ||
The reaction between Me4cyclen and Na[BArF]·2thf in CH2Cl2 yielded [Na(Me4cyclen)(thf)][BArF], with the thf ligand apical (Fig. 15). The τ value of 0.00 confirms an ideal square-based pyramidal geometry and the sodium cation is out of the N4 plane by 1.225(2) Å.
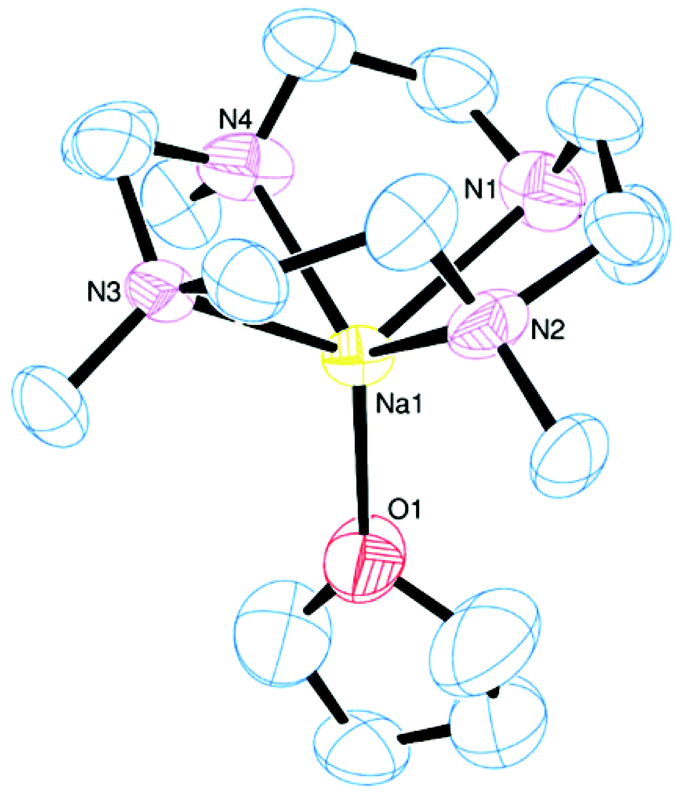 | ||
| Fig. 15 View of the cation present in [Na(Me4cyclen)(thf)][BArF]. Thermal ellipsoids are drawn at 50% probability. H atoms, the [BArF]− anion, and disorder of the ethylene linkers within the Me4cyclen ring are omitted for clarity. Selected bond lengths (Å) and angles (°): Na–N1 2.463(4), Na–N2 2.461(3), Na–N3 2.453(4), Na–N4 2.444(4), Na–O1 2.244(3); N1–Na–N2 75.4(1), N2–Na–N3 75.6(1), N3–Na–N4 76.0(1), N1–Na–N4 75.5(2). | ||
Although sandwich complexes of the Group 1 cations with 12-crown-4, e.g. [Na(12-crown-4)2]+, are well known,1 over the course of our studies we found no evidence for [M(Me4cyclen)2]+ sandwich cations. Attempts to synthesise the half-sandwich [M(Me4cyclen)]+ salts with the heavier Group 1 cations (K+, Rb+, Cs+) resulted only in the isolation of [Me4cyclenH][BArF].
Solution multinuclear NMR spectroscopy
The 1H and 13C{1H} NMR spectra (Experimental) each show the presence of the coordinated aza-macrocycle. The 1H NMR spectra of Me6[18]aneN6 complexes of K+, Rb+, and Cs+ salts at ambient temperatures show broad CH3 and CH2 resonances with no resolved coupling evident, indicative of exchanging species. On cooling the solutions the resonances broaden and then split, the effects being most evident in the Cs+ salt, but even at 185 K the resonances are still broad and lack resolved couplings, showing that the low temperature limiting spectra have not been reached. These effects are reversible with temperature. Upon cooling the [Cs(Me6[18]aneN6)][BArF] solution, the broad signal for the CH2 protons resolved into complex multiplets and the resonance for the methyl protons split into sharp singlets. This behaviour is most likely a result of two major stereoisomers (arising from the different orientations of the Me groups: ‘all-up’ and alternating ‘up-down’) for which the separate resonances are evident at low temperature as their rate of interconversion slows.The Group 1 elements offer several nuclei,‡ suitable for NMR spectroscopic studies.26 Lithium-7 NMR spectra on the aza complexes each show singlet resonances close to δ = 0 (see Experimental). Like the Na+-aza macrocyclic cations in our earlier work,3 the 23Na NMR spectra of the new complexes are sharp resonances with chemical shifts to high frequency of Na+ in water (δ = 0): [Na(Me6[18]aneN6)][BArF] +3.1; [Na(Me4cyclen)(thf)][BArF] +12.7; [Na(Me4cyclam)(thf)][BArF] +11.4; [Na(Me3tacn)(thf)][BArF] +3.7; [Na(Me3tacn)2][BArF] +6.2. Although the cations are expected to be exchanging rapidly in solution, these values can be compared with the Na+-crown ether cations which have chemical shifts to low frequency of 0 ppm, and therefore indicate that the aza macrocycle coordination is retained in CH2Cl2 solution.
Solutions of the potassium complexes in CH2Cl2 solution do not show a 39K resonance at room temperature (295 K) or on cooling to 185 K. Studies of K+-crown ether cations over a range of temperatures and K : crown ether ratios in a range of donor solvents show that often only the ‘free’ K+ resonance is seen. The [K(crown)]+ is often not observed most likely due to fast quadrupolar relaxation in the low symmetry environment.26c,d In [K(Me6[18]aneN6)]+ and [K(Me3tacn)2]+ it is probable that fast ligand exchange in solution produces low symmetry environments and fast quadrupolar relaxation (the corresponding 1H NMR spectra show only broad singlets for the macrocyclic CH2 groups). For similar reasons,26e no 85Rb resonance was observed at any temperature between 298 and 185 K.
The Q value for the 133Cs nucleus is small, hence NMR spectra are readily observed. The VT 133Cs NMR data for [Cs(Me6[18]aneN6)][BArF] show a singlet at 54.1 ppm (298 K) and this splits into two singlets at 73.3 and 58.7 ppm at 183 K, which we attribute to the slowing of dynamic processes and the presence of two significant stereoisomers (‘all up’ and alternating ‘up–down’). These chemical shifts are also significantly to high frequency of those typically observed in crown ether adducts.26f
Conclusions
By taking advantage of the lower lattice energies associated with M[BArF] precursors, which leads to increased solubility in very weak donor solvents such as CH2Cl2, the unusual Group 1 cation complexes [M(Me6[18]aneN6)][BArF] can be obtained in good yield for all members from M = Li to Cs. Structural characterisation of several of these (M = Li, K, and Rb) allows comparisons down the Group, as well as with the rarely observed bis-Me3tacn sandwich cations, [M(Me3tacn)2][BArF], isolated for M = Na and K. The combined experimental and DFT study indicates that the isolation of the [M(Me3tacn)2]+ cations requires a fine balance of, on one hand, the metal ion being sufficiently large to avoid significant steric clashing between the Me groups of the two rings, and on the other, the ion being sufficiently small to avoid extremely acute N–M–N chelate angles. In contrast, the very small Li+ ion forms only the half-sandwich [Li(Me3tacn)(thf)]+ cation. These complex cations show significant structural differences which correlate closely with the trends in ionic radii down Group 1 and the available macrocyclic binding cavity. The [BArF]− anions also show quite significant M⋯F interactions, particularly towards the larger metal ions (K+ and beyond), despite being large, diffuse ions with delocalised charge.DFT calculations show very good agreement with the experimentally determined structures and confirm significant donation of electron density from the N atoms of the ligand upon complexation, which is accompanied by transfer of some electron density to N from the σ C–H and σ C–N bonding orbitals. The nature of the FMOs show that contributions from the metal orbitals are only significant in the lower energy valence occupied orbitals, while the HOMO itself is dominated by the N 2p orbitals. The calculations also show that coordination of the aza macrocycle to the Group 1 cation in these complexes involves significant donation of electron density from the p-orbitals on the N atoms, rather than purely electrostatic interactions.
Acknowledgements
We thank the EPSRC for supporting the SCFED project through a Programme Grant (EP/I033394/1). The SCFED Project (http://www.scfed.net) is a multidisciplinary collaboration of British universities investigating the fundamental and applied aspects of supercritical fluids. The authors also acknowledge the use of the EPSRC UK National Service for Computational Chemistry Software (NSCCS). This work was also supported by funding provided by the Mauritius Tertiary Education Commission (TEC).Notes and references
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, 2nd edn, 2009, p. 116 Search PubMed.
- (a) T. Hashimoto and K. Maruoka, Asymmetric Phase Transfer Catalysis, ed. K. Maruoka, Wiley, Weinheim, 2008, p. 1 Search PubMed; (b) S. Chun, S. V. Dzyuba and R. A. Bartsch, Anal. Chem., 2001, 73, 3737 CrossRef CAS PubMed; (c) J. K. Choi, S. H. Kim, J. Yoon, K.-H. Lee, R. A. Bartsch and J. S. Kim, J. Org. Chem., 2006, 71, 8011 CrossRef CAS PubMed; (d) G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723 CrossRef CAS PubMed; (e) B. P. Hay and R. D. Hancock, Coord. Chem. Rev., 2001, 212, 61 CrossRef CAS; (f) J. W. Steed, Coord. Chem. Rev., 2001, 215, 171 CrossRef CAS.
- M. Everett, A. Jolleys, W. Levason, D. Pugh and G. Reid, Chem. Commun., 2014, 50, 5843 RSC.
- P. Bartlett, D. A. Cook, M. W. George, A. L. Hector, J. Ke, W. Levason, G. Reid, D. Smith and W. Zhang, Phys. Chem. Chem. Phys., 2014, 16, 9202 RSC.
- J. Clayden, Organolithiums: Selectivity for Synthesis, Elsevier Science Ltd., Oxford, 2002 Search PubMed.
- W. Clegg, B. Conway, A. R. Kennedy, J. Klett, R. E. Mulvey and L. Russo, Eur. J. Inorg. Chem., 2011, 721 CrossRef CAS.
- (a) G. W. Gokel, Chem. Soc. Rev., 1992, 21, 39 RSC; (b) R. S. Dhillon, S. E. Madbak, F. G. Ciccone, M. A. Buntine, S. F. Lincoln and K. P. Wainwright, J. Am. Chem. Soc., 1997, 119, 6126 CrossRef CAS; (c) H. Tsukube, J. Chem. Soc., Perkin Trans. 1, 1989, 1537 RSC.
- (a) J. L. Dye, Prog. Inorg. Chem., 1984, 32, 327 CrossRef CAS; (b) J. Kim, M. Shamsipur, S. Z. Huang, R. H. Huang and J. L. Dye, J. Phys. Chem. A, 1999, 103, 5615 CrossRef CAS.
- (a) R. H. Huang, D. L. Ward and J. L. Dye, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1835 CrossRef; (b) J. L. Dye and R. H. Huang, Pure Appl. Chem., 1993, 65, 435 CrossRef CAS; (c) J. Arnold, V. Knapp, J. A. R. Schmidt and A. Shafir, J. Chem. Soc., Dalton Trans., 2002, 3273 RSC; (d) K. J. Kolonko, I. A. Guzei and H. J. Reich, J. Org. Chem., 2010, 75, 6163 CrossRef CAS PubMed.
- (a) H.-H. Giese, T. Habereder, H. Nöth, W. Ponikwar, S. Thomas and M. Warchhold, Inorg. Chem., 1999, 38, 4188 CrossRef CAS; (b) S. Standfuss, T. P. Spaniol and J. Okuda, Eur. J. Inorg. Chem., 2010, 2987 CrossRef CAS.
- (a) M. E. Kuchenmeister and J. L. Dye, J. Am. Chem. Soc., 1989, 111, 935 CrossRef CAS; (b) A. S. Ellaboudy, C. J. Bender, J. Kim, D.-H. Shin, M. E. Kuchenmeister, G. T. Babcock and J. L. Dye, J. Am. Chem. Soc., 1991, 113, 2347 CrossRef CAS; (c) J. Kim, J. L. Eglin, A. S. Ellaboudy, L. E. H. McMills and S. Huang, J. Phys. Chem., 1996, 100, 2885 CrossRef CAS.
- M. Brookhart, B. Grant and A. F. Volpe Jr., Organometallics, 1992, 11, 3920 CrossRef CAS.
- I. Mon, D. A. Jose and A. Vidal-Ferran, Chem. – Eur. J., 2013, 19, 2720 CrossRef CAS PubMed.
- K. Wieghardt, P. Chaudhuri, B. Nuber and J. Weiss, Inorg. Chem., 1982, 21, 3086 CrossRef CAS.
- S. H. Pine and B. L. Sanchez, J. Org. Chem., 1971, 36, 829 CrossRef CAS.
- (a) CrystalClear-SM Expert 3.1 b27, Rigaku Corporation, Tokyo, Japan, 2012 Search PubMed; (b) CrystalClear-SM Expert 2.1 b29, Rigaku Corporation, Tokyo, Japan, 2013 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- A. B. Chaplin and A. S. Weller, Eur. J. Inorg. Chem., 2010, 5124 CrossRef CAS.
- F. H. Allen, O. Johnson, G. P. Shields, B. R. Smith and M. Towler, J. Appl. Crystallogr., 2004, 37, 335 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751 CrossRef.
- S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC.
- R. D. Ernst, B. G. Harvey, O. J. Oteri and A. M. Arif, Z. Kristallogr. - New Cryst. Struct., 2005, 220, 373 CAS.
- (a) S. Standfuss, T. P. Spaniol and J. Okuda, Eur. J. Inorg. Chem., 2010, 2987 CrossRef CAS; (b) Y. Habata, C. Okazaki, K. Ogura, S. Akabori, X. X. Zhang and J. S. Bradshaw, Inorg. Chem., 2007, 46, 8264 CrossRef CAS PubMed.
- C. Stockheim, K. Wieghardt, B. Nuber, J. Weiss, U. Flörke and H.-J. Haupt, J. Chem. Soc., Dalton Trans., 1991, 1487 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- (a) Multinuclear NMR, ed. J. Mason, Plenum Press, NY, 1987 Search PubMed; (b) NMR and the Periodic Table, ed. R. K. Harris and B. E. Mann, Academic Press, NY, 1978 Search PubMed; (c) M. Shporer and Z. Luz, J. Am. Chem. Soc., 1975, 97, 665 CrossRef CAS; (d) J.-S. Shih and A. I. Popov, Inorg. Chem., 1979, 19, 1689 CrossRef; (e) S. Khazaeli, J. L. Dye and A. I. Popov, Spectrochim. Acta, Part A, 1983, 39, 19 CrossRef; (f) A. Vendilo, K. Popov, M. Lajunen, V. Chistov, D. Djigailo, H. Rönkkömäki, V. Privalov and I. Pletnev, Polyhedron, 2014, 81, 341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray crystallographic parameters and cif files for the complexes reported (Table S1), crystallographic data in CIF format, together with full DFT computational details using both the BP86 and B3LYP functionals (Tables S2–S30, Fig. S1–S5). CCDC 1051099–1051104. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01865j |
| ‡ NMR properties:- 7Li: I = 3/2, N = 92.6%, Rc = 1.54 × 103, Q = −3.7 × 10−30 m2, Ξ = 38.87 MHz; 23Na: I = 3/2, N = 100%, Rc = 5.24 × 102, Q = 0.10 × 10−28, Ξ = 26.43; 39K: I = 3/2, N = 93.3%, Rc = 2.69, Q = 5.5 × 10−30, Ξ = 4.67; 85Rb: I = 5/2, N = 72.1%, Rc = 43.4, Q = 0.25 × 10−28, Ξ = 9.69; 133Cs: I = 7/2, N = 100%, Rc = 2.75 × 102, Q = −0.3 × 10−30, Ξ = 13.21. |
| This journal is © The Royal Society of Chemistry 2015 |