
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing a pyrazole/imidazole based hybrid cyclophane: a hydrogen bond sensor and binucleating ligand precursor†
Philipp J.
Altmann
,
Christian
Jandl
and
Alexander
Pöthig
*
Catalysis Research Center & Department of Chemistry, Technische Universität München, Ernst-Otto-Fischer-Str. 1, 85747 Garching bei München, Germany. E-mail: alexander.poethig@tum.de
First published on 18th May 2015
Abstract
A novel cyclophane consisting of methylene-bridged imidazolium- and pyrazole-moieties (calix[4]imidazolium[2]pyrazole) and two of its applications are presented. First, supramolecular recognition of an acetonitrile molecule by hydrogen bonding in the solid state is examined. Second, the capability to act as a ligand precursor is shown by the synthesis and characterisation of the respective dinuclear NHC-nickel(II) complex.
Macrocyclic poly-NHC ligands are the focus of current research.1 Very recently, cyclophanes featuring exclusively alkylene bridged NHC moieties have been successfully applied in the respective iron complexes,2 which showed high stability and reactivity e.g. towards oxygen and CH activation. Furthermore, their respective ligand precursors are excellent receptors for anion binding due to the remarkable hydrogen bond donor abilities of the acidic imidazolium moieties.3 Various macrocyclic imidazolium/N-donor hybrid systems have also been shown to function as NHC ligand precursors and H-bond donors.1a,4 For open-chain poly-NHC ligands, the introduction of a pyrazole-bridge between two NHC moieties allowed for the synthesis of dinuclear complexes of a variety of transition metals;5 the respective Cu6 and Ru5c complexes showed electronic coupling through the ligand which enabled access to intermediate oxidation states, a feature that is crucial for specific catalytic reactions.
Our presented approach combines the concept of macrocyclic poly-NHCs with that of bridging pyrazole moieties. The resulting macrocycle is both capable of acting as a multiple hydrogen bond donor and, upon deprotonation, of coordinating two metal centres in close proximity within a rigid framework. For this purpose, we first improved the accessibility of the literature-known pyrazole-bridge precursor 2,7 which was subsequently treated with sodium imidazolide to yield bisimidazole 3 (Scheme 1, for experimental details, see the ESI†). The latter then underwent a macrocyclisation protocol utilising methylene bistriflate,2b leading to a mixture of differently sized macrocycles, from which ligand precursor 4-PF6 was selectively isolated after workup and subsequent anion exchange. The 1H NMR spectra of both 4-OTf and 4-PF6 suggest an overall symmetric behaviour in DMSO solution as only one set of signals was observable for chemically equivalent protons. In the case of 4-PF6 exactly one equivalent of acetonitrile was found to be present in the thoroughly dried sample as verified by elemental analysis and NMR spectroscopy (Fig. 1). Elemental analysis further proved the existence of four anions and therefore the neutral charge of the pyrazole moieties in both compounds. Mass spectra of 4-OTf and 4-PF6 solely feature signals for combinations of the macrocycle in different protonation states of the pyrazoles and the counterions PF6− and OTf−, respectively. Single crystals of 4-OTf and 4-PF6 suitable for diffraction analysis were grown by slow diffusion of diethyl ether into a solution in acetonitrile. The solid state structures of the cyclophane are shown in Fig. 2.
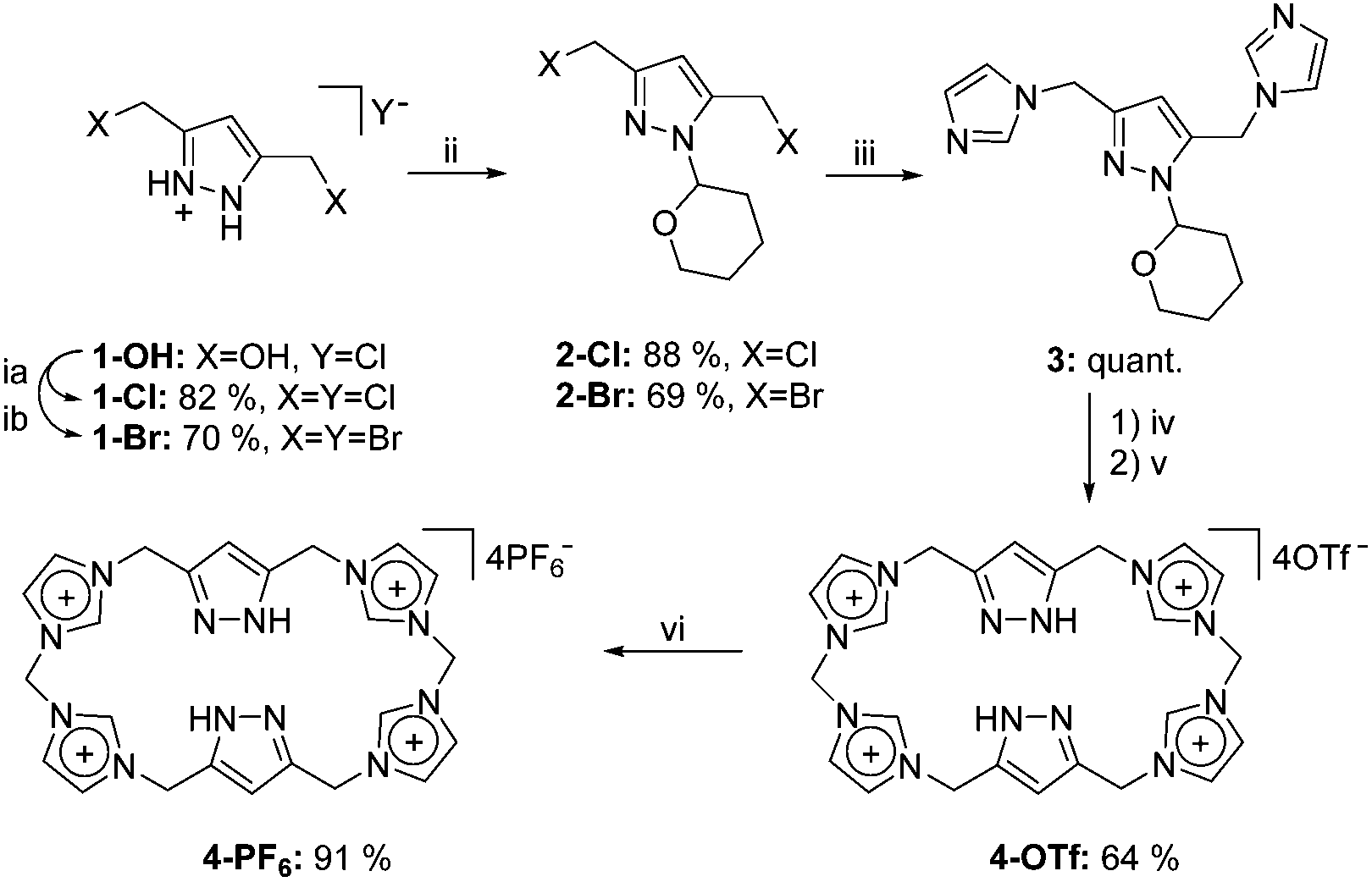 | ||
| Scheme 1 Synthesis of hydrogen bond donors 4-OTf and 4-PF6: (ia) SOCl2 (neat), 95 °C, 30 min; (ib) SOBr2 (neat), 0–40 °C, 2 h; (ii) 3,4-dihydro-2H-pyran, (DCM), r.t., 16 h; (iii) NaIm, (MeCN), r.t., 16 h; (iv) Me(OTf)2, (MeCN), −40 °C-r.t., 16 h; (v) Tf2O/H2O, (EtOH), r.t., 30 min; (vi) NH4PF6, (H2O), r.t., 30 min. | ||
 | ||
| Fig. 1 1H NMR spectrum of 4-PF6 containing exactly one equivalent of MeCN. | ||
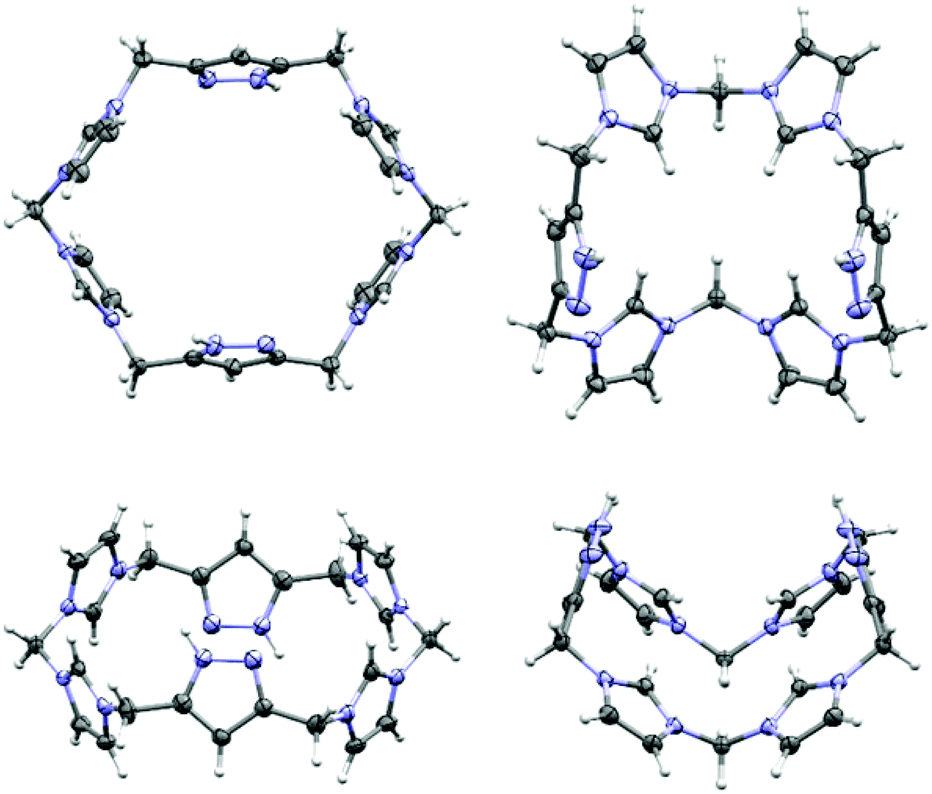 | ||
| Fig. 2 Different conformations of 4 in the solid state (top and side perspectives). Ellipsoids are shown at 50% probability. Counterions and solvent molecules are omitted for clarity. Left: molecular structure of the cation of 4-OTf showing an unstrained conformation of alternatingly oriented five-membered heterocycles (one out of two independent molecules of the asymmetric unit is shown). Right: molecular structure of the cation of 4-PF6 showing a strained bowl-shape conformation. | ||
The simple anion exchange has a tremendous effect on the molecular structure of 4 in the crystal. Whereas in 4-OTf the cation forms almost perfectly unstrained hexagons with alternatingly oriented five-membered rings (Fig. 2, left), in 4-PF6 the conformation changes drastically producing a bowl-shaped cavity (Fig. 2, right). As expected, Hirshfeld analysis8 of the cations shows that the majority of interactions in both compounds originate from H-atoms of 4 interacting with H-bond acceptors (77%, see the ESI†). In the case of 4-OTf the H⋯O contacts to different triflate anions can be assigned as the dominant interaction (37%), followed by less directed H⋯F contacts (22%). This way, all different types of H-atoms, namely those at the imidazolium, pyrazole and methylene bridge moieties, are involved in these contacts and do not show a systematic tendency towards specific acceptor atoms.
By contrast, for 4-PF6 a strong selectivity of the different protons towards the acceptors can be observed in the crystal structure. The C2-protons of all four imidazolium moieties are selectively pointing towards the nitrogen atom of an acetonitrile molecule (Fig. 3, N7⋯H2(C2) 2.307(3.249) Å, N7⋯H10(C10) 2.446 (3.141) Å), which corresponds to 9% of the interaction of the cation with other molecules. Consequently, the acetonitrile molecule perfectly fits into the cavity, rationalising why one equivalent of the solvent could not be removed in vacuo.
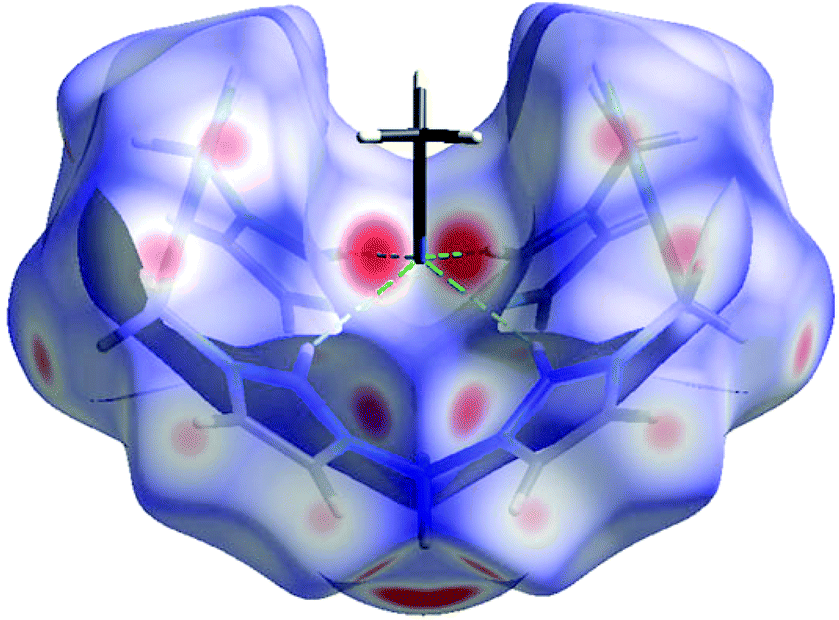 | ||
| Fig. 3 Supramolecular recognition of MeCN by the cation of 4-PF6 in the solid state shown by Hirshfeld surface analysis.8 Directed hydrogen bonding (green) from all four imidazolium C2 protons towards a MeCN nitrogen acceptor. | ||
The biggest share consists of H⋯F contacts to the PF6− anions (49%), which are formed by the pyrazole, the methylene bridge and the backbone protons of the imidazolium units. This hydrogen bonding may stabilise the bowl shaped conformation of the cation of 4-PF6, which is less stable by 28.5 kcal mol−1 in the gas phase compared to the unstrained conformation in 4-OTf according to single-point DFT calculations. This is a remarkable difference certainly caused by the repulsive Coulomb interactions in the bowl-shaped conformation of the highly charged macrocycle. However, in the solid state, this intramolecular repulsive effect apparently is overcompensated by attractive interactions with the anions and acetonitrile molecules, such that this conformation can be experimentally observed. The fact that both 4-OTf and 4-PF6 were crystallised from the same solvent mixtures underlines the effect of the counterion. At the moment, we are systematically evaluating the influence of other anions and solvents and whether a recognition of hydrogen bond acceptors can also be observed in the liquid phase.
We further tested whether the N-heterocyclic moieties can be deprotonated and subsequently coordinated to a set of two metal centres. Out of the different metalation strategies, we chose the reaction with a metal precursor upon deprotonation by an external base. Treatment of 4-PF6 with caesium carbonate in the presence of bis(triphenylphosphine)nickel(II) chloride produced the dinuclear Ni complex 5-PF6 which was obtained in good yields after workup (Scheme 2). The overall yield of 5-PF6 for the multistep synthesis starting from 1-OH is up to 27% for the 1-Cl route. Compound 5-PF6 gives rise to seven independent sets of signals in the 1H NMR spectrum (see the ESI†). Most notable is the splitting of the protons of the bridging methylene groups indicating a reduction of the molecular symmetry in solution compared to 4-PF6. No change of the signal shapes was observed up to 80 °C suggesting an overall rigid and robust ligand framework (Fig. 4). As expected, the 13C NMR spectrum exhibits seven signals (see the ESI†).
 | ||
| Fig. 4 Variable temperature 1H NMR spectra of dinuclear complex 5-PF6 (blank areas are left out intentionally for clarity; for full spectra see the ESI†). No significant change of the signal shapes at high temperatures is observable, suggesting a rigid ligand framework. | ||
 | ||
| Scheme 2 Synthesis of dinuclear NHC-nickel(II) complex 5-PF6. | ||
Interestingly, the carbene carbon resonance at 157.6 ppm appears as a rather intense signal, which is unusual because of the commonly poor relaxation processes for carbene carbon atoms. This finding might point towards cooperative electronic effects involving the two Ni(II) centres connected via the pyrazolate moieties. The mass spectrum of 5-PF6 shows two characteristic signals, one at m/z = 297.4 for (5-PF6–2PF6−)2+ and the other at 739.2 for (5-PF6–PF6−)+. Furthermore, the elemental analysis indicates the presence of two equivalents of hexafluorophosphate anions per macrocycle, further supporting the sixfold deprotonation of the ligand precursor during the synthesis.
Single crystals suitable for X-ray diffraction were grown by slow diffusion of diethyl ether into a solution in dimethyl formamide. The molecular structure of 5-PF6 is shown in Fig. 5 and is in accordance with the results obtained by prior analysis methods. The two nickel ions are coordinated in a slightly distorted square planar fashion by two Ccarbene and two N donors each. The Ni–C bond lengths between 1.844(4) and 1.870(4) Å (two molecules in the asymmetric unit) are within literature-reported values and a little shorter than the Ni–N bonds of 1.897(3)–1.912(3) Å.1b,5b,9 In addition, the C–Ni–C angles (85.4(2)–86.7(2)°) are smaller than the N–Ni–N angles (94.4(1)–95.0(1)°). The molecular structure is best described as bowl-shaped with the pyrazolate rings at the bottom and the NHC moieties bent upwards to each side which places the two Ni ions at distances of 3.5957(8) and 3.6043(8) Å, respectively. The two mean planes established by each Ni ion and its coordinating atoms form opening angles of 122–123° which leaves wide open cavities occupied by hexafluorophosphate ions in the solid state.
 | ||
| Fig. 5 Solid state structure of dinuclear complex 5-PF6. View from two perspectives showing the bowl-shaped arrangement of the complex. The asymmetric unit contains two fragments out of which one was chosen. ADPs are shown at the 50% probability level, and hydrogen atoms and hexafluorophosphate counterions are omitted for clarity. | ||
The bowl-shaped structure perfectly explains the splitting pattern in the NMR spectra. In addition, the rather rigid structure causes the protons in the bridges to split up as there is no possibility of a ring flip in the complex that would result in magnetically equivalent protons in solution. The bent structure of the complex forms promising binding pockets for small molecules that might facilitate their activation after reducing the complex chemically or electrochemically. Similar reactivities have recently been demonstrated for mononuclear nickel NHC and non-NHC complexes.10
The presented results are the fundamentals of a divergent chemistry, which we are currently pursuing. First, the promising hydrogen bond donor (and acceptor) capabilities of cation 4 towards acetonitrile will be further evaluated by applying a variety of other hydrogen bond acceptors. Second, Ni complex 5-PF6 as the first example of an NHC complex bearing this macrocyclic ligand is a promising molecular catalyst for several catalytic transformations and will therein be tested and characterised in greater detail. Lastly, we aim at expanding the scope of the ligand by applying it to other (transition) metals and intensively studying its coordination chemistry and the properties of the resulting complexes.
Acknowledgements
We thank Prof. W. A. Herrmann and Prof. F. E. Kühn for their mentorship and support. The CRC, the Faculty of Chemistry (TUM) as well as the Fonds der Chemischen Industrie (FCI, Sachkostenzuschuss A.P.) are very much acknowledged for funding this work. Tobias Schirmer is acknowledged for practical support.Notes and references
- (a) P. G. Edwards and F. E. Hahn, Dalton Trans., 2011, 40, 10278 RSC and references cited therein; (b) Z. Lu, S. A. Cramer and D. M. Jenkins, Chem. Sci., 2012, 3, 3081 RSC; (c) C. Schulte to Brinke, T. Pape and F. E. Hahn, Dalton Trans., 2013, 42, 7330 RSC.
- (a) S. Meyer, I. Klawitter, S. Demeshko, E. Bill and F. Meyer, Angew. Chem., Int. Ed., 2013, 52, 901 CrossRef CAS PubMed; (b) M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2015, 54, 3797 CrossRef CAS PubMed.
- (a) H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406 CrossRef CAS PubMed; (b) C. J. Serpell, J. Cookson, A. L. Thompson and P. D. Beer, Chem. Sci., 2011, 2, 494 RSC; (c) Y. Chun, N. J. Singh, I. C. Hwang, J. W. Lee, S. U. Yu and K. S. Kim, Nat. Commun., 2013, 4, 1797 CrossRef PubMed.
- I. Klawitter, M. R. Anneser, S. Dechert, S. Meyer, S. Demeshko, S. Haslinger, A. Pöthig, F. E. Kühn and F. Meyer, Organometallics, 2015 DOI:10.1021/acs.organomet.5b00103.
- (a) U. J. Scheele, M. John, S. Dechert and F. Meyer, Eur. J. Inorg. Chem., 2008, 373 CrossRef CAS PubMed; (b) Y. Zhou, Z. Xi, W. Chen and D. Wang, Organometallics, 2008, 27, 5911 CrossRef CAS; (c) S. A. Reindl, A. Pöthig, M. Drees, B. Bechlars, E. Herdtweck, W. A. Herrmann and F. E. Kühn, Organometallics, 2013, 32, 4082 CrossRef CAS.
- B. Liu, B. Liu, Y. Zhou and W. Chen, Organometallics, 2010, 29, 1457 CrossRef CAS.
- (a) J. C. Röder, F. Meyer, M. Konrad, S. Sandhöfner, E. Kaifer and H. Pritzkow, Eur. J. Org. Chem., 2001, 4479 CrossRef; (b) J. C. Röder, F. Meyer and H. Pritzkow, Organometallics, 2001, 20, 811 CrossRef.
- M. A. Spackman and D. Jayatilakaa, CrystEngComm, 2009, 11, 19 RSC.
- (a) M. V. Baker, B. W. Skelton, A. H. White and C. C. Williams, Organometallics, 2002, 21, 2674 CrossRef CAS; (b) H. M. Bass, S. A. Cramer, A. S. McCullough, K. J. Bernstein, C. R. Murdock and D. M. Jenkins, Organometallics, 2013, 32, 2160 CrossRef CAS.
- (a) N. J. Findlay, S. R. Park, F. Schoenebeck, E. Cahard, S.-Z. Zhou, L. E. A. Berlouis, M. D. Spicer, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2010, 132, 15462 CrossRef CAS PubMed; (b) J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932 CrossRef CAS PubMed; (c) M. H. Reineke, M. D. Sampson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2015, 54, 3211 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1057574–1057576. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01775k |
| This journal is © The Royal Society of Chemistry 2015 |