
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel photofunctional multinuclear complexes using a coupling reaction†
Yasuomi
Yamazaki
a,
Tatsuki
Morimoto‡
a and
Osamu
Ishitani
*ab
aDepartment of Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1-NE-1, O-okayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: ishitani@chem.titech.ac.jp; Fax: +81-3-5734-2284; Tel: +81-5734-2240
bCREST, Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi-shi, Saitama 332-0012, Japan
First published on 14th May 2015
Abstract
Various photofunctional metal complexes with functional groups, i.e. bromo and vinyl groups, were integrated into hetero-multinuclear complexes using the Mizoroki–Heck reaction. The obtained trinuclear complexes absorb a wide range of visible light and have a long excited state lifetime and the photocatalytic ability to obtain CO2 reduction.
Introduction
Multinuclear transition-metal complexes with photofunctional properties, many of which are constructed using the same repeated metal complexes, have become of interest in recent years because of their potential use in dye-sensitised solar cells1 and light-harvesting systems.2 Hetero-multinuclear metal complexes constructed using different functional units have recently been found to be efficient photocatalysts for the oxidation of water,3,4 evolution of hydrogen,5–7 and CO2 reduction.8–13 For example, Ru(II)–Re(I) dinuclear complexes, in which the Ru(II) unit works as a photosensitiser and the Re(I) unit works as a catalyst, are effective photocatalysts for CO2 reduction because of rapid intramolecular electron transfer from the photochemically-reduced Ru(II) unit to the Re(I) unit.10Most of these multinuclear metal complexes have been synthesised using stepwise methods with several complexation steps.14 A mononuclear complex with a multidentate ligand having one chelating moiety already coordinated to the metal centre and another able to coordinate is synthesised first, which can then react with another metal centre to afford a dinuclear complex. Various multinuclear complexes have been synthesised by repeating these processes; however, there are some limitations with respect to the product selectivity achieved using this method. For example, a bridging ligand with a number of different diimine moieties can react with a metal complex (or complexes) to give a mixture of products with different numbers of metal complexes and/or with one or more metal complexes connected to different diimine moieties.
There are several reports of one-pot syntheses of hetero-dinuclear complexes using an asymmetric bridging ligand with two different coordination sites.15 This synthetic method requires very different rates of coordination reactions of each metal ion to each coordination site for high production selectivity of one heteronuclear complex.
Homo-coupling reactions of metal complexes have recently been applied for the synthesis of asymmetric dinuclear complexes, though the isolated yields were relatively low (5.4%–8.5%).16 It has also been reported that cross-coupling reactions (Sonogashira coupling17–19 and Suzuki–Miyaura coupling20–22) were used to synthesise multinuclear complexes. In this method, each complex possessing functional groups such as a bromo group, an ethynyl group, or a boronic-acid substituent as building blocks can be connected to give a heteronuclear complex with a relatively high degree of selectivity.
Herein we report the first use of the Mizoroki–Heck reaction23,24 in a coupling reaction with different types of emissive metal complexes (Ru(II), Ir(III) and Re(I) complexes with one or more diimine ligands) to give multinuclear complexes with various photofunctional properties.
Results and discussion
Synthesis of a Re(I) dinuclear complex using the Heck reaction
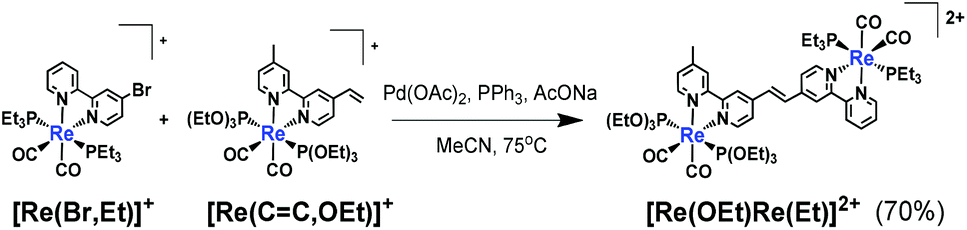
| (1) |
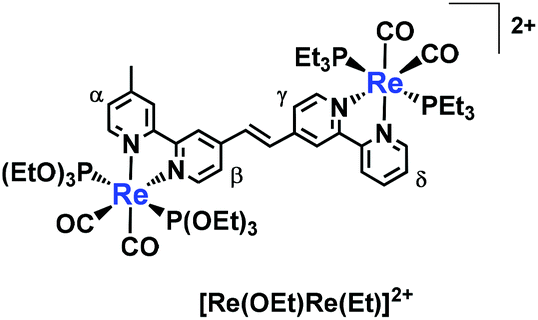
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C,OEt)]+, 23 μmol), was heated in the presence of palladium acetate (Pd(OAc)2, 4.5 μmol), triphenylphosphine (PPh3, 9.2 μmol) and sodium acetate (AcONa, 140 μmol) at 75 °C under an Ar atmosphere for one day. Ar was pumped into the reaction mixture during synthesis. The solvent was evaporated once the reaction reached completion. An MeCN solution (4 mL) of Pd(OAc)2 (4.5 μmol) and PPh3 (9.2 μmol) was then added to the residue along with a small amount of air; this addition of air was needed to achieve a good yield.25 The solution was heated for one day. Electrospray ionisation mass spectrometry (ESI-MS) analysis of the crude products indicated three major peaks attributed to [Re(OEt)Re(Et)]2+ as the main product, [Re(CC,OEt)]+ and [Re(bpy)(CO)2(PEt3)2]+ (bpy = 2,2′-bipyridine), which should be produced via debromination of [Re(Br,Et)]+ (Fig. 1). The target dinuclear Re(I) complex ([Re(OEt)Re(Et)]2+) was chromatographically isolated using silica gel and SP Sephadex as adsorbents, giving an isolated yield of 70%. Note that the reaction was much slower under anaerobic conditions, as a small amount of O2 was required to accelerate the reaction.
C,OEt)]+, 23 μmol), was heated in the presence of palladium acetate (Pd(OAc)2, 4.5 μmol), triphenylphosphine (PPh3, 9.2 μmol) and sodium acetate (AcONa, 140 μmol) at 75 °C under an Ar atmosphere for one day. Ar was pumped into the reaction mixture during synthesis. The solvent was evaporated once the reaction reached completion. An MeCN solution (4 mL) of Pd(OAc)2 (4.5 μmol) and PPh3 (9.2 μmol) was then added to the residue along with a small amount of air; this addition of air was needed to achieve a good yield.25 The solution was heated for one day. Electrospray ionisation mass spectrometry (ESI-MS) analysis of the crude products indicated three major peaks attributed to [Re(OEt)Re(Et)]2+ as the main product, [Re(CC,OEt)]+ and [Re(bpy)(CO)2(PEt3)2]+ (bpy = 2,2′-bipyridine), which should be produced via debromination of [Re(Br,Et)]+ (Fig. 1). The target dinuclear Re(I) complex ([Re(OEt)Re(Et)]2+) was chromatographically isolated using silica gel and SP Sephadex as adsorbents, giving an isolated yield of 70%. Note that the reaction was much slower under anaerobic conditions, as a small amount of O2 was required to accelerate the reaction.
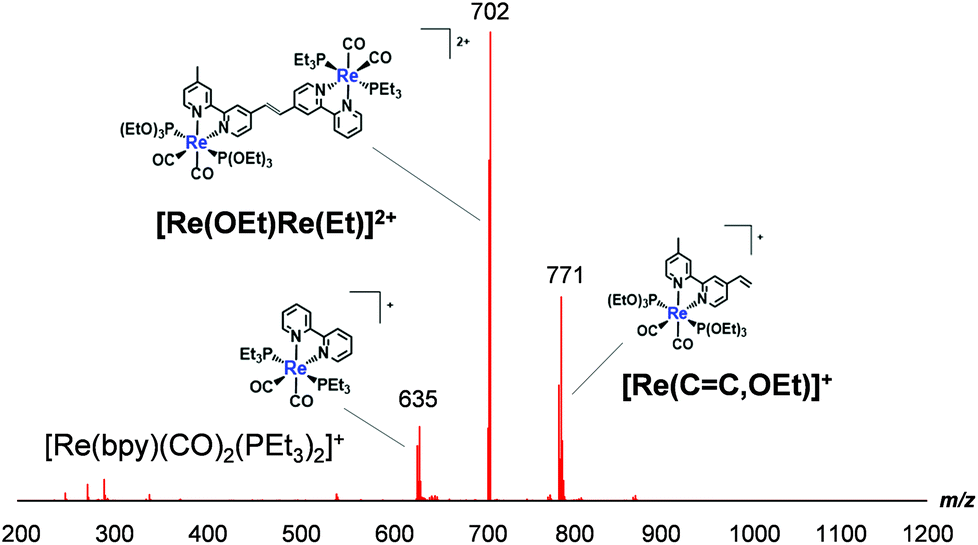 | ||
| Fig. 1 ESI-MS spectrum of crude products recorded after the synthesis of [Re(OEt)Re(Et)]2+. The mobile phase was MeCN. | ||
A single peak attributed to olefinic protons was observed in the 1H NMR spectrum of [Re(OEt)Re(Et)]2+ (Fig. 2). This suggests that either the cis or the trans isomer was formed. The product [Re(OEt)Re(Et)]2+ was assumed to be the trans isomer because the Re(I) units on either side were bulky. Such a low-field resonance of the olefinic protons (δ = 8.07) was similar to the resonance seen for trans-1,2-bis(4-(4′-methyl)-2,2′-bipyridine)ethene.26 The high resolution mass spectrum and 31P NMR spectrum clearly show that neither the P(OEt)3 nor the PEt3 ligands on the Re(I) units have been substituted with the coupling reagents.
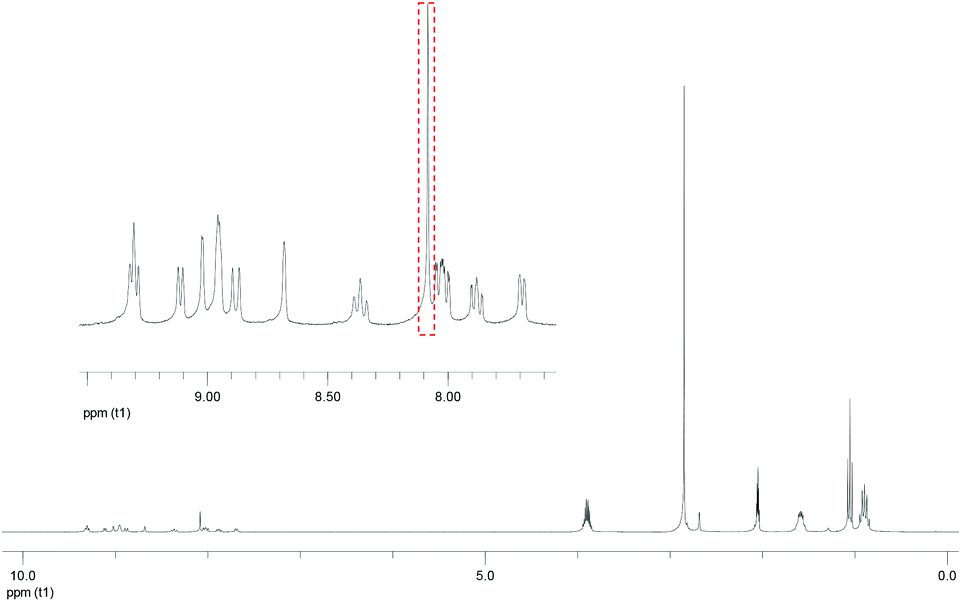 | ||
| Fig. 2 1H NMR spectrum of the dinuclear complex [Re(OEt)Re(Et)]2+. The solvent was acetone-d6. The red square emphasises the peak attributed to olefin protons. | ||
It should be emphasised that the selective synthesis of only one type of dinuclear complex with an asymmetric bridging ligand, such as [Re(OEt)Re(Et)]2+, is difficult using ordinary methods, which possibly results in a mixture of four types of dinuclear complexes, i.e. two types of complexes with identical phosphine ligands in both units and two structural isomers of complexes with two different phosphine ligands in each unit.
Synthesis of various trinuclear complexes using the Heck reaction
A similar reaction could successfully be used to synthesise various trinuclear complexes. At the centre of each of these products was a Re(I) complex with a bpy ligand and two other metal complexes as substituents (eqn (2)). A Re(I) bis-carbonyl complex with two bromo groups, [ReI(4,4-dibromo-bpy)(CO)2(PPh3)2]+ ([Re(Br2,Ph)]+), reacted with various photofunctional complexes possessing a vinyl group, i.e.[Re(CC,OEt)]+, [RuII(bpy)2(vbpy)]2+ ([Ru(CC,bpy)]2+, vbpy = 4-methyl-4′-vinyl-bpy), [RuII(dmb)2(vbpy)]2+ ([Ru(CC,dmb)]2+, dmb = 4,4′-dimethyl-bpy) and [IrIII(ppy)(vbpy)]+ ([Ir(CC,ppy)]+, ppy = 2-phenyl-pyridine). This was performed under the same reaction conditions as were used to synthesise [Re(OEt)Re(Et)]2+, with the exception of repeating both the heating and addition of the MeCN solution of Pd(OAc)2 and PPh3 twice, in the case of [Ru(bpy)2Re(Ph)]5+ and [Ir(ppy)2Re(Ph)]3+. The trinuclear complexes could be separated using size exclusion chromatography (SEC) or ion-exchange chromatography. The trinuclear complexes with Ru(II) or Re(I) in both edge units, i.e.[Re(OEt)2Re(Ph)]3+, [Ru(bpy)2Re(Ph)]5+ and [Ru(dmb)2Re(Ph)]5+, were obtained in 31%, 41% and 32% yields, respectively. ESI-MS analysis of the solution after the reaction between [Re(4,4′-dibromo-bpy)(CO)2{P(p-FC6H4)3}2]+ ([Re(Br2,FPh)]+) and [Ru(CC,dmb)]2+ was performed and clearly showed that the phosphine ligands on the Re(I) unit were not substituted by the coupling reagents. The isolated yield of [Ru(dmb)2Re(FPh)]5+ was 25%. The isolated yield was relatively lower (8%) for [Ir(ppy)2Re(Ph)]3+, which had Ir(III) in both edge units, as the reaction between [Ir(CC,ppy)]+ and [Re(Br2,Ph)]+ was much slower than the other reactions; therefore, conversion of the starting complexes was low (Fig. S5, ESI†).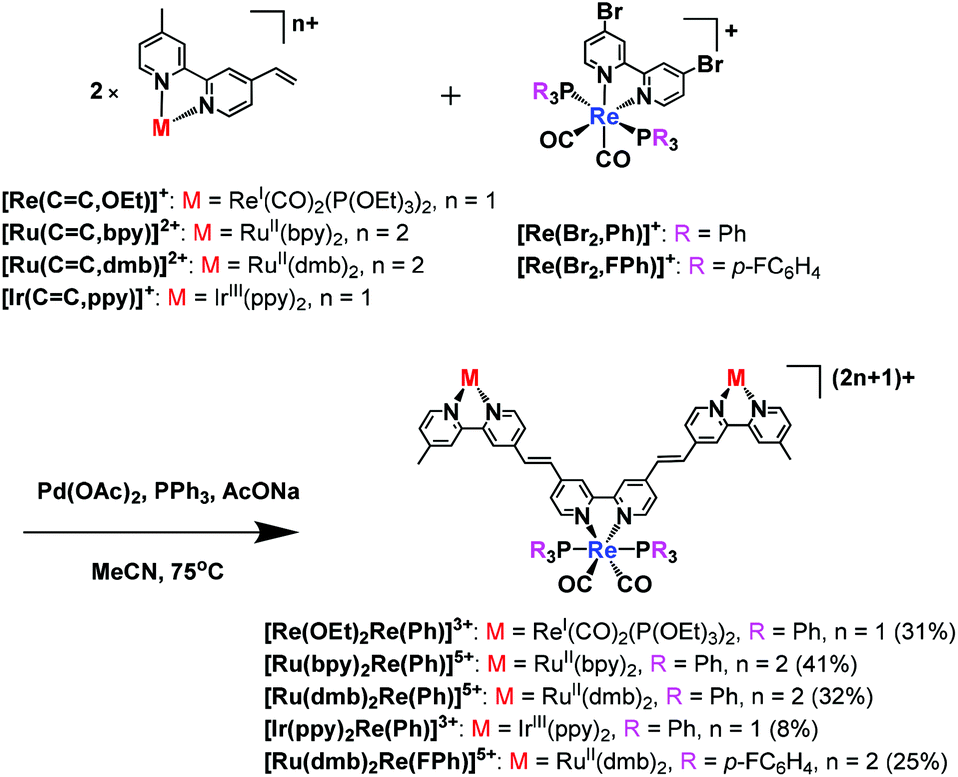 | (2) |
Attempts were also made to use the Sonogashira coupling reaction to synthesise a trinuclear complex from a Ru(II) complex with an ethynyl group at the 4-position of the bpy moiety and [Re(Br2,FPh)]+, as shown in eqn (3). An N,N-dimethylformamide (DMF, 5 mL) solution containing the Ru(II) complex (38 μmol), [Re(Br2,FPh)]+ (19 μmol), PdCl2(PPh3)2 (0.62 μmol), CuI (2.9 μmol) and diisopropylamine (1 mL) was heated at 50 °C for 12 h under Ar. The reaction mixture was analysed by SEC and ESI-MS, which indicated that the target trinuclear complex with triple bonds between the Ru(II) and Re(I) units was successfully synthesised. Unfortunately, the trinuclear complex decomposed during the SEC purification process, most likely due to instability in the mobile phase (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of methanol and MeCN containing 0.15 M CH3COONH4).27
:1 (v/v) mixture of methanol and MeCN containing 0.15 M CH3COONH4).27
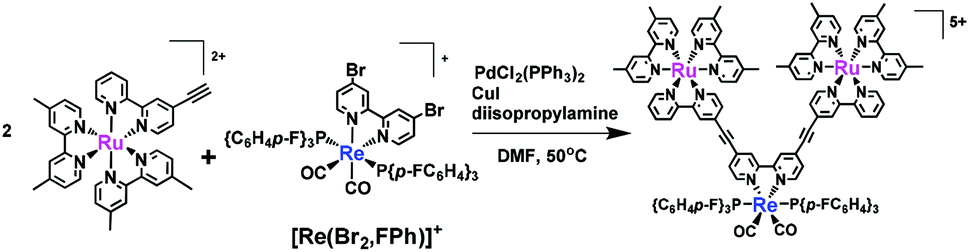 | (3) |
As described above, the complexes that were synthesized in this research are not obtained selectively using ordinary stepwise methods because mixtures of isomers with the metals in different units are produced. Isolating the target complex from such a mixture is often difficult because of the similarities between the molecular sizes, charges, and solubility of the isomeric complexes.
Photophysical properties of the trinuclear complexes
As typical examples, UV-Vis absorption spectra of the synthesised trinuclear complexes [Ru(dmb)2Re(Ph)]5+, [Re(OEt)2Re(Ph)]3+ and [Ir(ppy)2Re(Ph)]3+ are shown with those of the corresponding mononuclear complexes in Fig. 3 (those of other complexes are shown in Fig. S4, ESI†). The 1MLCT absorption band of [Ru(dmb)2Re(Ph)]5+ was observed between 370 nm and 570 nm and was red-shifted compared to the 1MLCT absorption band of [Ru(CC,dmb)]2+. This was most likely caused by extension of the π conjugation in the bridging ligand, decreasing the energy level of the π* orbital.28 In addition, the molar extinction coefficient of the 1MLCT absorption band was higher for [Ru(dmb)2Re(Ph)]5+ than for the corresponding mononuclear complexes. This suggests that the transition has more π–π* character. The π–π* absorption band of [Ru(dmb)2Re(Ph)]5+ with λmax = 288 nm had a shoulder at longer wavelengths, which was not observed in the summation spectra of the corresponding mononuclear complexes (a dotted line in Fig. 3a). This shoulder can be attributed to the π–π* transition in the bridging ligand with a wide π conjugation system. In the case of [Re(OEt)2Re(Ph)]3+ (Fig. 3(b)) and [Ir(ppy)2Re(Ph)]3+ (Fig. 3(c)), similar phenomena were observed, i.e. red-shifts of both the π–π* and 1MLCT absorption bands and increase of the molar extinction coefficients of the 1MLCT absorption bands (Table 1).
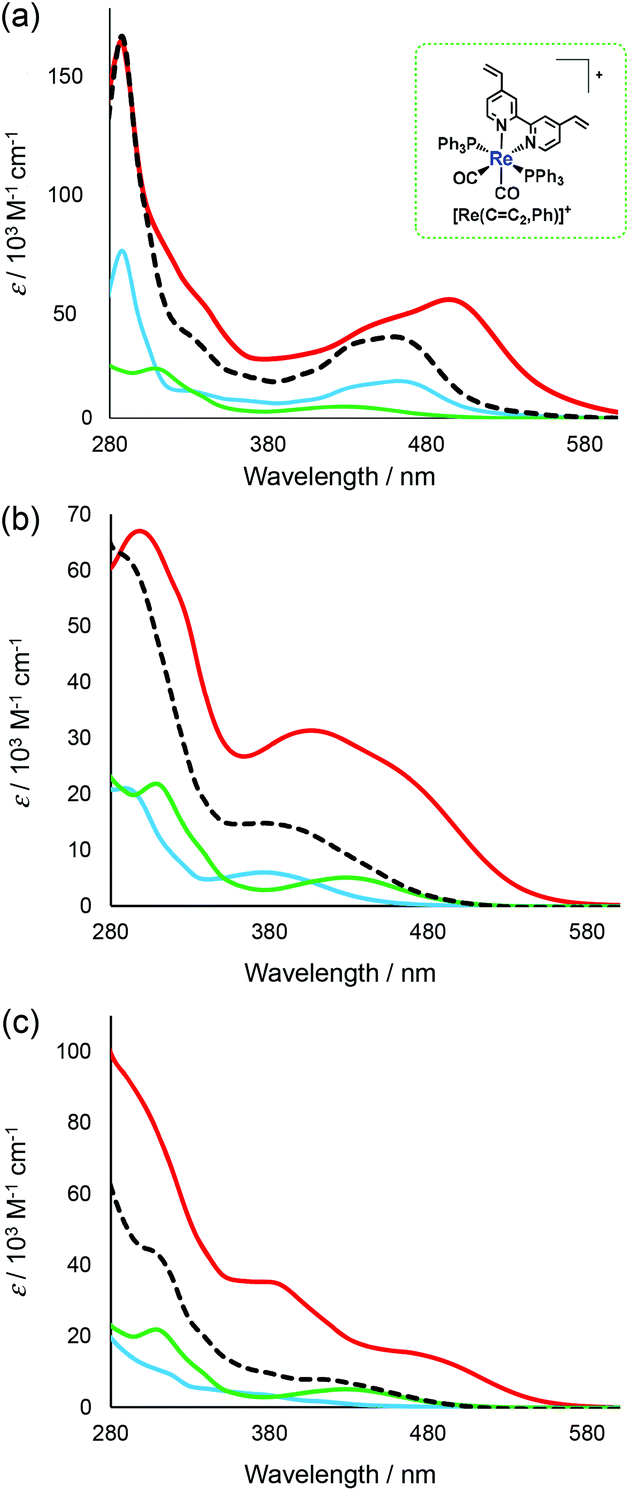 | ||
| Fig. 3 UV-Vis absorption spectra of trinuclear complexes obtained and corresponding mononuclear complexes. The solvent was MeCN. (a) [Ru(dmb)2Re(Ph)]5+ (red), [Ru(CC,dmb)]2+ (blue), [Re(CC2,Ph)]+ (green) and the 2:1 summation spectrum of [Ru(CC,dmb)]2+ and [Re(CC2,Ph)]+ (dotted line). (b) [Re(OEt)2Re(Ph)]3+ (red), [Re(CC,OEt)]+ (blue), [Re(CC2,Ph)]+ (green) and the 2:1 summation spectrum of [Re(CC,OEt)]+ and [Re(CC2,Ph)]+ (dotted line). (c) [Ir(ppy)2Re(Ph)]3+ (red), [Ir(CC,ppy)]+ (blue), [Re(CC2,Ph)]+ (green) and the 2:1 summation spectrum of [Ir(CC,ppy)]+ and [Re(CC2,Ph)]+ (dotted line). | ||
| Entry | Complex | λ abs/nm (ε /103 M−1 cm−1) | λ maxem/nm | Φ em | τ 1 /nsd (A/%) | τ 2 /nsd (A/%) |
|---|---|---|---|---|---|---|
| a Measured in MeCN at 25 °C. b Excitation wavelength: 456 nm. c Excitation wavelength: 444 nm. d Detected at each wavelength of the emission maximum. | ||||||
| 1 | [Re(OEt)2Re(Ph)]3+ | 406 (31.3), 476 (sh, 20.8), 299 (65.4) | 714c | 0.01c | 140 (56)c | 771 (44)c |
| 2 | [Ru(bpy)2Re(Ph)]5+ | 481 (50.6), 341 (sh, 47.3), 287 (162) | 710b | 0.02b | 674 (34)b | 1424 (66)b |
| 3 | [Ru(dmb)2Re(Ph)]5+ | 494 (52.1), 341 (sh, 48.9), 288(150) | 738b | 0.02b | 610 (18)b | 1310 (82)b |
| 4 | [Ir(ppy)2Re(Ph)]3+ | 474 (14.9), 383 (35.0), 257 (120) | 685b | 0.01c | 104 (82)c | 784 (18)c |
| 5 | [Ru(dmb)2Re(FPh)]5+ | 495 (52.3), 341 (sh, 50.7), 287 (156) | 743b | 0.03b | 471 (6)b | 1112 (94)b |
| 6 |
[Re(CC,OEt)]+
|
376 (5.97), 290 (20.4) | 635c | 0.07c | 326 (100)c | — |
| 7 |
[Ru(CC,bpy)]2+
|
456 (17.0), 289 (82.1) | 636c | 0.14b | 1551 (100)b | — |
| 8 |
[Ru(CC,dmb)]2+
|
462 (16.3), 288 (73.1) | 645c | 0.10b | 1376 (100)b | — |
| 9 |
[Ir(CC,ppy)]+
|
384 (3.26), 304 (11.5) | 617c | 0.14c | 200 (100)c | — |
| 10 |
[Re(CC2,Ph)]+
|
428 (5.38), 308 (21.9) | 647c | 0.08c | 1561 (100)c | — |
| 11 |
[Re(CC2,FPh)]+
|
424 (4.29), 300 (22.2) | 642c | 0.07c | 1409 (100)c | — |
All trinuclear complexes displayed emission (Fig. 4 and S5 (ESI†)). The emission for each trinuclear complex was red-shifted relative to the corresponding mononuclear complexes, and the emission quantum yield (Φem) for each trinuclear complex was much lower than those of the corresponding mononuclear complexes (Table 1).
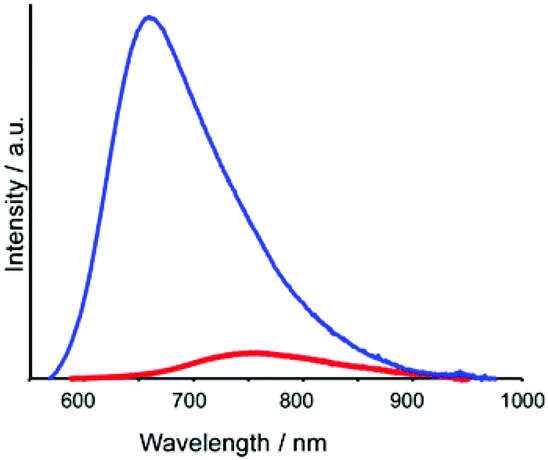 | ||
| Fig. 4 Emission spectra of [Ru(CC,dmb)]2+ (blue) and [Ru(dmb)2Re(Ph)]5+ (red) recorded in MeCN (excitation wavelength: 456 nm). | ||
The emission decay for each trinuclear complex could be fitted using a double exponential function (Fig. S6, ESI†). The emission lifetimes (τem) were similar to the corresponding mononuclear complexes, though the Φem were much lower; therefore, the radiative decay of the excited states of the trinuclear complexes should be slower than those of the mononuclear complexes with respect to the following equations, which show the relationships among Φem, τem and the radiative and nonradiative decay constants (kr and knr: eqn (4) and (5)).
| Φem = kr/(kr + knr) | (4) |
| τem = 1/(kr + knr) | (5) |
This also indicates that the lowest 3MLCT excited states of the trinuclear complexes contained higher π–π* characteristics than those of the mononuclear complexes. It has been reported that increase in π–π* character of the 3MLCT excited states of transient metal complexes induces slower radiative decay.26,29
For a more detailed investigation of the excited states of the trinuclear complexes, time-resolved emission spectra and temperature dependence of the emission decay of [Ru(dmb)2Re(Ph)]5+ were measured as a typical example. Fig. 5a shows the emission decays measured at various temperatures (243–333 K) after excitation at λex = 456 nm, where much weaker temperature dependence was observed compared to [Ru(dmb)3]2+ (Fig. 5b and S7 (ESI†)). It has been reported that emission decays of many Ru(II)30 trisdiimine and Re(I)31 diimine phosphine complexes are strongly dependent on temperature, becoming faster at higher temperature due to thermal interconversion from the lowest 3MLCT excited state to the triplet ligand field excited state (3LF), which causes rapid nonradiative decay (Fig. 6). Therefore, the formation process of the 3LF should be suppressed in the case of [Ru(dmb)2Re(Ph)]5+, as the extension of π* conjugation in the bridging ligand lowers the energy level of the 3MLCT excited state. Suppression of this nonradiative decay process might be a reason for the long emission lifetime. Fig. 7a shows emission spectra recorded at various times after excitation at 456 nm. The shape of these time-resolved spectra gradually changed, as faster decay was observed at shorter wavelengths (Fig. 7b). This might be caused by intramolecular energy transfer between the Ru and Re units.
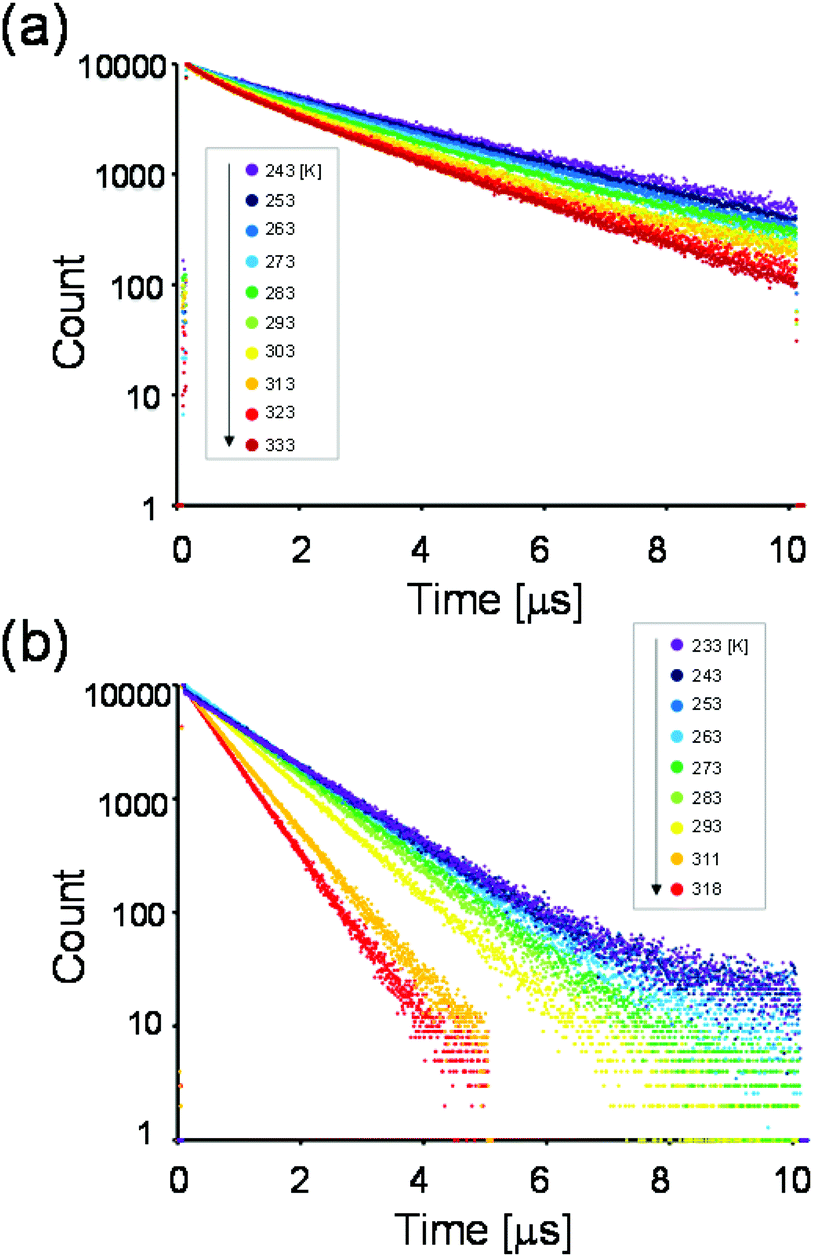 | ||
| Fig. 5 (a) Temperature dependence of the emission decay of [Ru(dmb)2Re(Ph)]5+. Decay curves were measured from 243 K (purple) to 333 K (dark red). Excitation wavelength was 456 nm and detection wavelength was 738 nm. (b) Temperature dependence of the emission decay of [Ru(dmb)3]2+. Decay curves were measured from 233 K (purple) to 318 K (red). Excitation wavelength was 456 nm and detection wavelength was 639 nm. The solvent was MeCN. | ||
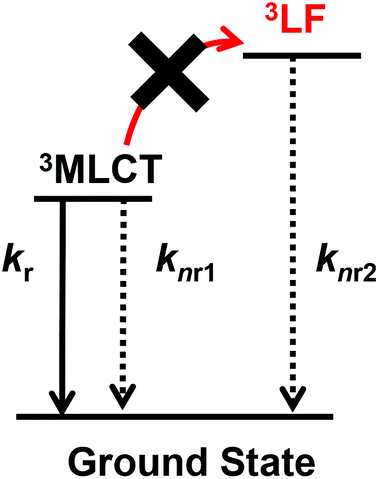 | ||
| Fig. 6 Jablonski diagram of [Ru(dmb)2Re(Ph)]5+. | ||
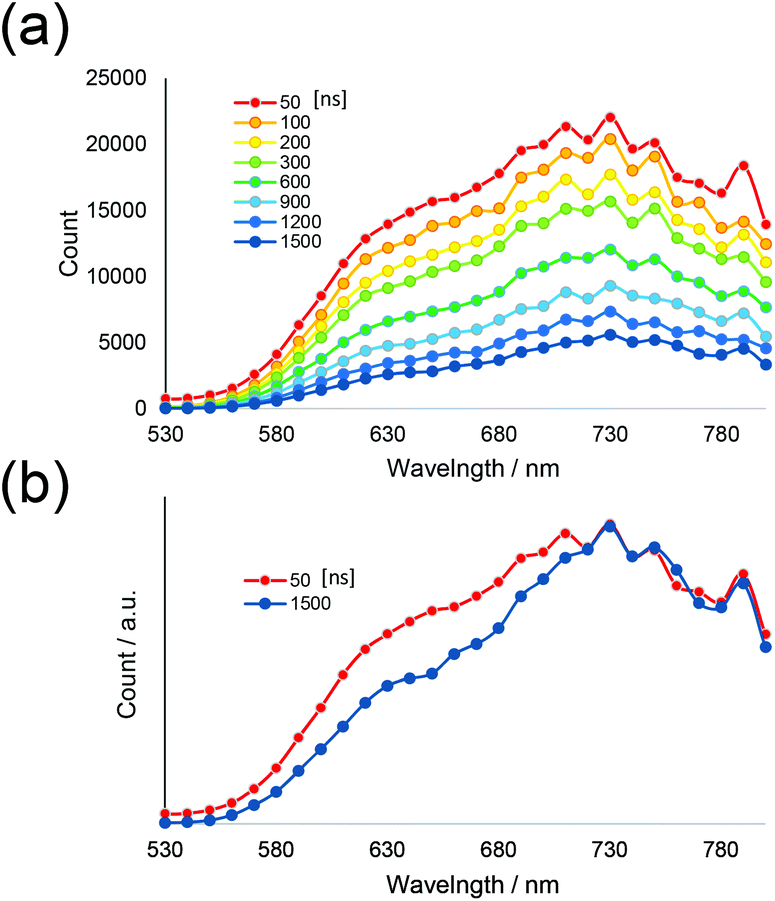 | ||
| Fig. 7 (a) Transient emission spectra of [Ru(dmb)2Re(Ph)]5+ (50–1500 ns after excitation) recorded by single-photon counting methods and (b) normalised spectra at 730 nm (50 ns (red) and 1500 ns (blue) after excitation). Excitation wavelength was 444 nm. The solvent was MeCN. | ||
Since the trinuclear complex [Ru(dmb)2Re(Ph)]5+ has both photosensitiser (Ru) and catalyst (Re) units for CO2 reduction, it was reasonably expected that this complex could act as a photocatalyst for CO2 reduction. A mixture of DMF and triethanolamine (5:1 v/v) containing 0.05 mM [Ru(dmb)2Re(Ph)]5+ and 0.1 M 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) was irradiated at λex = 600 nm for 22 h. Selective formation of CO was observed and the turnover number (TONCO) reached 115. This TONCO was higher than that found using a Ru(II)–Re(I) dinuclear complex with a N^N–CH2–CH2–N^N bridging ligand (N^N = 4′-methyl-bpy) (TONCO = 50) under the same reaction conditions, which has been reported to be one of the most efficient supramolecular photocatalysts for the reduction of CO2 using higher-energy light such as 480 nm,10 because the absorption at λ = 600 nm is stronger. Under these reaction conditions, most Ru(II) moieties should be mainly excited (Fig. 3a). After a reductive quenching process of the excited state of the Ru(II) unit by BIH, intramolecular electron transfer should proceed from the reduced Ru(II) unit to the Re(I) unit, followed by reduction of CO2 on the reduced Re(I) unit.
Conclusions
We successfully synthesised various trinuclear complexes from photofunctional complexes possessing vinyl or bromo groups as building blocks in a single step using the Mizoroki–Heck reaction. The obtained trinuclear complexes displayed strong absorption over a wide range of visible light and a long emission lifetime. [Ru(dmb)2Re(Ph)]5+ functioned as a photocatalyst for CO2 reduction, even by irradiation at 600 nm.Experimental
General procedures
1H NMR spectra were recorded using a JEOL AL400 (400 MHz) or an AL300 (300 MHz) instrument with the analyte dissolved in acetone-d6, MeCN-d3, or CDCl3. 31P NMR spectra were recorded using a JEOL ECX400 (400 MHz) instrument with the analyte dissolved in acetone-d6. IR spectra were recorded using a JASCO FT/IR-610 spectrometer with a resolution of 1 cm−1 and with the analyte dissolved in dichloromethane. ESI mass spectrometry (MS) was performed using a Shimadzu LC-MS-2010A system, with MeCN as the mobile phase. ESI time-of-flight MS was performed using a Waters LCT Premier instrument, with MeCN as the mobile phase. Size Exclusion Chromatography (SEC) was performed using a pair of Shodex PROTEIN KW-402J columns (300 mm long, 8.0 mm i.d.) with a KW-LG guard-column (50 mm long, 6.0 mm i.d.), a JASCO 880-51 degasser, an 880-PU pump, a MD-2010 Plus UV-vis photodiode-array detector, and a Rheodyne 7125 injector. The column temperature was maintained at 40 °C using a JASCO 860-CO oven. The eluent was a 1:1 (v/v) mixture of methanol and MeCN containing 0.5 M CH3COONH4, and the flow rate was 0.2 mL min−1. Separation of the trinuclear complexes was achieved using SEC with a pair of Shodex PROTEIN KW-2002.5 columns (300 mm long, 20.0 mm i.d.) with a KW-LG guard column (50 mm long, 8.0 mm i.d.) and a recycling preparative HPLC apparatus with a JASCO 870-UV detector. The eluent was a 1:1 (v/v) mixture of methanol and MeCN containing 0.15 M CH3COONH4, and the flow rate was 6.0 mL min−1. UV-vis absorption spectra were recorded using a JASCO V-670 instrument.
Emission measurements
Each compound was dissolved in MeCN and degassed using the freeze–pump–thaw method prior to measurement. Emission spectra were recorded at 25 °C using either a JASCO FP-8600 spectrofluorometer or a Hamamatsu C9920-02 system. The absolute emission quantum yields were evaluated using the Hamamatsu C9920-02 system equipped with an integrating sphere and a multichannel photodetector (PMA-12). Emission lifetimes were obtained using a HORIBA TemPro fluorescence lifetime system with an emission monochromator. The excitation light source was a NanoLED-560 pulse lamp and the instrumental response time was less than 0.1 ns. All emission decays were fitted by single or double exponential functions within appropriate χ2 (0.96–1.24). Transient emission spectra were recorded by the point-to-point measurement method using a Horiba FluoroCube time-correlated single-photon counting system. Sensitivity of the system at each detection wavelength was calibrated using correction coefficients determined from the difference between [Ru(4,4′-dimethyl-2-2′-bipyridine)3]2+ spectra recorded with the JASCO FP-8600 spectrofluorometer and the HORIBA TemPro fluorescence lifetime system.Photocatalytic reactions
A 4 mL aliquot of a solution in an 11 mL-cubic-quartz cell (with a light passage length of 1 cm) was irradiated with 600 nm monochromic light obtained from a Xe lamp with a band-pass filter (FWHM = 10 nm). The incident light intensity was 4.3 × 10−8 einstein per s. The gaseous reaction products (CO and H2) were analysed using a GC-TCD instrument (GL Science GC323). HCOOH was analysed using a capillary electrophoresis system (Otsuka Electronics Co., CAPI-3300I).Materials
N,N-Dimethylformamide (DMF) was dried over the molecular sieve 4 Å and distilled under a decreased pressure (10–20 mmHg). TEOA was distilled under a decreased pressure (<1 mmHg). The DMF and TEOA were maintained under Ar until they were used. 1,3-Dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH),11 4-methyl-4′-vinyl-2-2′-bipyridine (vbpy),32 4,4′-divinyl-2-2′-bipyridine (dvbpy),33 Ru(N^N)2Cl2 (N^N = bpy or dmb),34[Ru(CC,bpy)]2+,35[Ru(CC,dmb)]2+,35[Ir(CC,ppy)]+,36 and 4′-bromo-2,2′-bipyridine,37–39 Re(CO)5Br40 were prepared according to the reported methods with some modifications. All of the other reagents were of reagent grade quality and were used without further purification.
Synthesis
[Re(Brbpy)(CO)2(PEt3)2](PF6) ([Re(Br,Et)](PF6)). Re(PEt3)2(CO)3Br (100.0 mg, 0.1705 mmol) and silver triflate (48.5 mg, 0.189 mmol) were dissolved in THF (8 mL) and refluxed under Ar for 3 h. Precipitated AgBr was filtered with Celite. A yellow-green oil was collected after the solvent was removed under vacuum. This oil and 4-bromo-2,2′-bipyridine (Brbpy, 50.2 mg, 0.212 mmol) were dissolved in toluene (15 mL) and heated at 90 °C under Ar overnight. Brbpy (39.2 mg, 0.166 mmol) was added to the reaction mixture and heated overnight. After a second addition of Brbpy (55.6 mg, 0.235 mmol), the solution was further heated for 1 d. The solvent was evaporated to give an orange-red residue, which was purified by flash column chromatography on silica gel (collected as the second fraction using 100
:0–100:5 (v/v) CH2Cl2/MeOH as the eluent). The obtained orange-red solid was dissolved in MeOH to which a saturated methanol solution of NH4PF6 and a small amount of water were added. The solution was gradually evaporated until the orange-red solid precipitated. The solid was washed with water and Et2O and dried at 60 °C under vacuum. Yield: 112.8 mg (77.0%, over 2 steps). ESI-MS (in MeCN) m/z: 713 ([M − PF6]+). FT-IR (in CH2Cl2) νCO/cm−1: 1934, 1864. 1H NMR (297.60 MHz, acetone-d6): δ/ppm, 9.32 (d, J = 5.4 Hz, 1H, Brbpy-6), 9.14 (d, J = 6.0 Hz, 2H, Brbpy-6′), 9.02 (d, J = 2.2 Hz, 1H, Brbpy-3′), 8.90 (dd, J = 1.4, 8.2 Hz, 1H, Brbpy-3), 8.38 (dd, J = 6.9, 8.2 Hz, 1H, Brbpy-4), 8.03 (dd, J = 2.2, 6.0 Hz, 1H, Brbpy-5′), 7.89 (ddd, J = 1.4, 5.4, 6.9 Hz, 1H, Brbpy-5), 1.59–1.52 (m, 12H, P(CH2CH3)3), 0.92–0.83 (m, 18H, P(CH2CH3)3). Elemental Anal. Calcd for C24H37BrF6N2O2P3Re: C, 33.57; H, 4.34; N, 3.26. Found: C, 33.94; H, 4.18; N, 3.30.
Re(P(OEt)3)2(CO)3Br. Re(CO)5Br (1.17 g, 2.89 mmol) and P(OEt)3 (1.13 g, 6.82 mmol) were dissolved in toluene (100 mL), and the solution was refluxed under Ar for 28 h. After the solvent was evaporated, the residue was separated by column chromatography on alumina (collected as a first fraction, using 100
:0–0:100 (v/v) CH2Cl2/hexane as the eluents). The obtained white solids were dried under vacuum. Yield: 1.37 g (69.8%). FT-IR (in CH2Cl2) νCO/cm−1: 2069, 2048, 1970, 1920. 1H NMR (297.60 MHz, acetone-d6): δ/ppm, 4.19–4.10 (m, 12H, P(OCH2CH3)3), 1.31 (t, J = 6.9 Hz, 18H, P(OCH2CH3)3).
[Re(vbpy)(CO)2(P(OEt)3)2](PF6) ([Re(C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) C,OEt)](PF6)).
Re(P(OEt)3)2(CO)3Br (210.0 mg, 0.3077 mmol) and silver triflate (68.65 mg, 0.3433 mmol) were dissolved in THF (20 mL), and the solution was refluxed under Ar for 4 h. Precipitated AgBr was filtered by Celite. A brown oil was collected after the solvent was evaporated. Toluene (25 mL) containing the oil and 4-methyl-4′-vinyl-2,2′-bipyridine (vbpy, 72.3 mg, 0.368 mmol) was heated at 90 °C under Ar for 21.5 h. Evaporation of the solvent gave a brown residue, which was separated by flash column chromatography on silica gel (collected as a first yellow fraction, using 100:0–100:2 (v/v) CH2Cl2/MeOH as the eluent). The purification method was same as that for [Re(Br,Et)]+. Yield: 145.8 mg (51.7%). ESI-MS (in MeCN) m/z: 771 ([M − PF6]+). FT-IR (in CH2Cl2) νCO/cm−1: 1954, 1879. 1H NMR (297.60 MHz, acetone-d6): δ/ppm, 8.95 (d, J = 5.7 Hz, 1H, vbpy-6′), 8.87 (d, J = 5.9 Hz, 1H, vbpy-6), 8.78 (s, 1H, vbpy-3′), 8.68 (s, 1H, vbpy-3), 7.80 (d, J = 5.9 Hz, 1H, vbpy-5), 7.61 (d, J = 5.7 Hz, 1H, vbpy-5′), 6.97 (dd, J = 17.6, 10.9 Hz, 1H, vbpy–CHCH2), 6.44 (d, J = 17.6 Hz, 1H, vbpy–CHCH2 (trans)), 5.76 (d, J = 10.9 Hz, 1H, vbpy–CHCH2 (cis)), 3.83 (m, 12H, P(O–CH2–CH3)3), 2.60 (s, 3H, vbpy-CH3), 1.00 (t, J = 6.9 Hz, 18H, P(O–CH2–CH3)3). Elemental Anal. Calcd for C27H42F6N2O8P3Re: C, 35.41; H, 4.62; N, 3.06. Found: C, 35.64; H, 4.40; N, 3.18.
C,OEt)](PF6)).
Re(P(OEt)3)2(CO)3Br (210.0 mg, 0.3077 mmol) and silver triflate (68.65 mg, 0.3433 mmol) were dissolved in THF (20 mL), and the solution was refluxed under Ar for 4 h. Precipitated AgBr was filtered by Celite. A brown oil was collected after the solvent was evaporated. Toluene (25 mL) containing the oil and 4-methyl-4′-vinyl-2,2′-bipyridine (vbpy, 72.3 mg, 0.368 mmol) was heated at 90 °C under Ar for 21.5 h. Evaporation of the solvent gave a brown residue, which was separated by flash column chromatography on silica gel (collected as a first yellow fraction, using 100:0–100:2 (v/v) CH2Cl2/MeOH as the eluent). The purification method was same as that for [Re(Br,Et)]+. Yield: 145.8 mg (51.7%). ESI-MS (in MeCN) m/z: 771 ([M − PF6]+). FT-IR (in CH2Cl2) νCO/cm−1: 1954, 1879. 1H NMR (297.60 MHz, acetone-d6): δ/ppm, 8.95 (d, J = 5.7 Hz, 1H, vbpy-6′), 8.87 (d, J = 5.9 Hz, 1H, vbpy-6), 8.78 (s, 1H, vbpy-3′), 8.68 (s, 1H, vbpy-3), 7.80 (d, J = 5.9 Hz, 1H, vbpy-5), 7.61 (d, J = 5.7 Hz, 1H, vbpy-5′), 6.97 (dd, J = 17.6, 10.9 Hz, 1H, vbpy–CHCH2), 6.44 (d, J = 17.6 Hz, 1H, vbpy–CHCH2 (trans)), 5.76 (d, J = 10.9 Hz, 1H, vbpy–CHCH2 (cis)), 3.83 (m, 12H, P(O–CH2–CH3)3), 2.60 (s, 3H, vbpy-CH3), 1.00 (t, J = 6.9 Hz, 18H, P(O–CH2–CH3)3). Elemental Anal. Calcd for C27H42F6N2O8P3Re: C, 35.41; H, 4.62; N, 3.06. Found: C, 35.64; H, 4.40; N, 3.18.
Re(PPh3)2(CO)3Br. A toluene solution (50 mL) containing Re(CO)5Br (303 mg, 0.747 mmol) and PPh3 (457 mg, 1.74 mmol) was refluxed under N2 for 2 days. After cooling to room temperature, the white precipitate was collected and washed with toluene and hexane. Yield: 435 mg (66.7%). FT-IR (in CH2Cl2) νCO/cm−1: 2051, 1948, 1910. Elemental Anal. Calcd for C39H30BrO3P2Re: C, 53.55; H, 3.46. Found: C, 53.67; H, 3.66.
[Re(Br2bpy)(CO)2(PPh3)2](PF6) ([Re(Br2,Ph)](PF6)). Re(PPh3)2(CO)3Br (298.6 mg, 0.3414 mmol) and silver triflate (105.0 mg, 0.4087 mmol) were dissolved in CH2Cl2 (30 mL) and the solution was refluxed under N2 for 17 h. Precipitated AgBr was filtered by Celite. After evaporating the solvent, the obtained white residue and 4,4′-dibromo-2,2′-bipyridine (Br2bpy, 129.4 mg, 0.4120 mmol) were dissolved in toluene (30 mL, degassed using N2), and the solution was heated at 90 °C under N2 for 16 h. The orange precipitate was filtered off, washed with toluene and hexane, and purified by flash column chromatography on silica gel (collected as a first orange fraction, using 100
:0–100:5 (v/v) CH2Cl2/MeOH as the eluents). The purification method was same as that for [Re(Br,Et)]+. Yield: 321.7 mg (76.9%). ESI-MS (in MeCN) m/z: 1081 ([M − PF6]+). FT-IR (in CH2Cl2) νCO/cm−1: 1942, 1874. 1H NMR (297.60 MHz, acetone-d6): δ/ppm, 8.15 (d, J = 1.8 Hz, 2H, bpy-3,3′), 7.65 (d, J = 6.0 Hz, 2H, bpy-6,6′), 7.16–7.30 (m, 30H, 2PPh3), 6.84 (dd, J = 1.8, 6.0 Hz, 2H, bpy-5,5′). Elemental Anal. Calcd for C48H36Br2F6N2O2P3Re: C, 47.03; H, 2.96; N, 2.29. Found: C, 46.95; H, 3.03; N, 2.15.
[Re(dvbpy)(CO)2(PPh3)2](PF6) ([Re(CC2,Ph)](PF6)) was synthesized by a method similar to that for the synthesis of [Re(Br2,Ph)]+ using dvbpy instead of Br2bpy: yield: 56.4%. ESI-MS (in MeCN) m/z: 975 ([M − PF6]+). FT-IR (in CH2Cl2) νCO/cm−1: 1937, 1867. 1H NMR (297.60 MHz, acetone-d6): δ/ppm, 8.49 (d, J = 1.6 Hz, 2H, bpy-3,3′), 7.98 (d, J = 5.8 Hz, 2H, bpy-6,6′), 7.16–7.30 (m, 30H, 2PPh3), 7.03 (dd, J = 1.6, 5.8 Hz, 2H, bpy-5,5′), 6.81 (dd, J = 10.6, 17.4 Hz, 2H, –CHCH2), 6.27 (d, J = 10.6 Hz, 2H, –CHCH2 (cis)), 5.73 (d, J = 17.4 Hz, 2H, –CHCH2 (trans)). 31P NMR (400 MHz, acetone-d6): δ/ppm, 21.9 (s, 2P, PPh3), −143.6 (7, 1P, PF6−). HRMS (ESI-TOF) m/z: [M − PF6−]+ Calcd for C52H42N2O2P2Re 975.2283; Found 975.2284.
Re{P(p-FC6H4)3}2(CO)3Br. Re(CO)5Br (104 mg, 0.255 mmol) and P(p-FC6H4)3 (165 mg, 0.522 mmol) were dissolved in toluene (20 mL), and the solution was refluxed under N2 for 23 h. Evaporation of the solvent gave a yellow oil which was separated by column chromatography on silica gel (eluent: CH2Cl2). The obtained white solids were recrystallized with toluene–hexane, and dried under vacuum. Yield: 155 mg (61.8%). FT-IR (in CH2Cl2) νCO/cm−1: 2057, 1955, 1911. 1H NMR (399.65 MHz, acetone-d6): δ/ppm, 7.69–7.75 (m, 18H, m-Ph), 7.26 (dd, J = 8.8, 8.8 Hz, 18H, o-Ph). Elemental Anal. Calcd for C39H24BrF6O3P2Re: C, 47.67; H, 2.46. Found: C, 47.84; H, 2.70.
[Re(Br2bpy)(CO)2{P(p-FC6H4)3}2](PF6) ([Re(Br2,FPh)](PF6)). Re{P(p-FC6H4)3}2(CO)3Br (310 mg, 0.315 mmol) and silver triflate (92.5 mg, 0.359 mmol) were dissolved in THF (30 mL), and the solution was refluxed under Ar for 3 h. Precipitated AgBr was filtered by Celite. White solids were collected after the solvent was evaporated. The toluene solution (30 mL) containing the obtained residue and 4,4′-dibromo-2,2′-bipyridine (Br2bpy, 119 mg, 0.380 mmol) was heated at 90 °C under N2 for 16 h. After the solvent was evaporated, the obtained orange solids were purified by flash column chromatography on silica gel (collected as the first orange fraction, using 100
:0–100:5 (v/v) CH2Cl2/MeOH as the eluents). The purification method was the same as that for [Re(Br,Et)]+. Yield: 333 mg (79.2%). ESI-MS (in MeCN) m/z: 1189 ([Re(Br2bpy)(CO)2{P(p-FC6H4)3}2]+). FT-IR (in CH2Cl2) νCO/cm−1: 1943, 1874. 1H NMR (297.60 MHz, acetone-d6): δ/ppm, 8.72 (d, J = 1.8 Hz, 2H, Br2bpy-3,3′), 8.13 (d, J = 6.0 Hz, 2H, Br2bpy-6,6′), 7.46–7.38 (m, 14H, bpy-5,5′, m-Ph), 7.14 (dd, J = 8.7, 8.7 Hz, 12 H, o-Ph). Elemental Anal. Calcd for C48H36Br2F6N2O2P3Re: C, 47.03; H, 2.96; N, 2.29. Found: C, 46.95; H, 3.03; N, 2.15.
[Re(dvbpy)(CO)2{P(p-FC6H4)3}2](PF6) ([Re(CC2,FPh)](PF6)) was synthesized by a method similar to that for the synthesis of [Re(Br2,FPh)]+ using dvbpy instead of Br2bpy: yield: 50.8%. ESI-MS (in MeCN) m/z: 1083 ([M − PF6]+). FT-IR (in CH2Cl2) νCO/cm−1: 1939, 1869. 1H NMR (297.60 MHz, acetone-d6): δ/ppm, 8.73 (d, J = 4.4 Hz, 1H, bpy-6), 8.62 (s, 1H, bpy-3), 8.53 (s, 1H, bpy-3′), 8.13 (d, J = 6.2 Hz, 1H, bpy-6′), 7.43–6.79 (m, 28H, 6 × P(p-FC6H4)3, bpy-5,5′, 2 × –CHCH2), 6.29 (d, J = 17.7 Hz, 2H, 2 × –CHCH2(cis)), 5.75 (d, J = 11.2 Hz, 1H, –CHCH2(trans)), 6.29 (d, J = 11.2 Hz, 1H, –CHCH2(trans)). 31P NMR (400 MHz, acetone-d6): δ/ppm, 20.1 (s, 2P, P(p-FC6H4)3), −143.6 (7, 1P, PF6−). HRMS (ESI-TOF) m/z: [M − PF6−]+ Calcd for C52H36F6N2O2P2Re 1083.1718; Found: 1083.1689.
[Re(OEt)Re(Et)](PF6)2. [Re(Br,Et)](PF6) (20 mg, 0.023 mmol), [Re(C
C,OEt)](PF6) (21 mg, 0.023 mmol), Pd(OAc)2 (1.0 mg, 0.0045 mmol), PPh3 (2.4 mg, 0.0092 mmol) and AcONa (11.5 mg, 0.140 mmol) were dissolved in MeCN degassed using Ar (4 mL). The solution was refluxed at 75 °C under Ar for 14 h. An MeCN solution (4 mL) containing Pd(OAc)2 (1.0 mg, 0.0045 mmol) and PPh3 (2.4 mg, 0.0092 mmol) was added to the reaction mixture and refluxed for 20 h. The solvent was removed under vacuum to give an orange-red solid. The coupling reagents were removed by flash column chromatography on silica gel (100:0–100:2 CH2Cl2/MeOH as the eluents). The target complex was isolated using ion-exchange chromatography (SP Sephadex C-25; the eluent was a 1:1 v/v mixture of MeCN and water containing NH4PF6 (0–4 mM)). The solvent was evaporated until the solid precipitated. The solid was washed with water and Et2O and dried at 60 °C under vacuum. Yield: 27.7 mg (70.2%). 1H NMR (400 MHz, acetone-d6): δ/ppm, 9.30 (d, J = 5.7 Hz, 1H, γpy-6), 9.28 (d, J = 5.8 Hz, 1H, βpy-6), 9.10 (d, J = 5.8 Hz, 1H, δpy-6), 9.01 (s, 1H, γpy-3), 8.95 (s, 1H, βpy-3), 8.94 (s, 1H, δpy-3), 8.87 (d, J = 7.1 Hz, αpy-6), 8.67 (s, 1H, αpy-3), 8.37 (dd, J = 7.1, 8.0 Hz, 1H, δpy-4), 8.07 (m, 2H, –CHCH–), 8.03 (d, J = 5.8 Hz, 1H, βpy-5), 8.00 (d, J = 5.7 Hz, 1H, γpy-5), 7.87 (dd, J = 5.8, 7.1 Hz, 1H, δpy-5), 7.68 (d, J = 7.1 Hz, 1H, αpy-5), 3.92–3.86 (m, 12H, P(OCH2CH3)3), 2.67 (s, 3H, vbpy–CH3), 1.60–1.53 (m, 12H, P(CH2CH3)3), 1.04 (t, J = 7.3 Hz, 18H, P(OCH2CH3)3), 0.94–0.84 (m, 18H, P(CH2CH3)3). 31P NMR (400 MHz, acetone-d6): δ/ppm, 117.0 (s, 2P, P(OEt)3), −2.06 (s, 2P, PEt3), −143.5 (7, 2P, PF6−). FT-IR (in CH2Cl2) νCO/cm−1: 1954, 1930, 1880, 1860. ESI-MS (in MeCN) m/z: 702 ([M − 2PF6−]2+). Elemental Anal. Calcd for C51H78F12N4O10P6Re2: C, 36.17; H, 4.64; N, 3.31. Found: C, 36.05; H, 4.30; N, 3.33. HRMS (ESI-TOF) m/z: [M − 2PF6−]2+ Calcd for C51H78N4O10P4Re2 701.1879; Found: 701.1848. 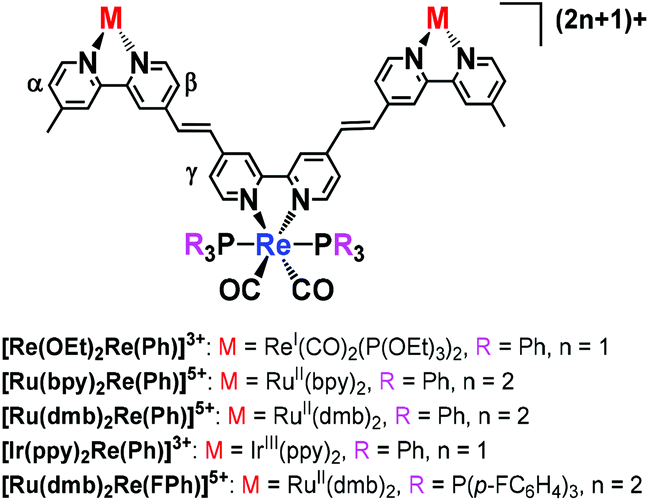
[Re(OEt)2Re(Ph)](PF6)3. [Re(Br2,Ph)](PF6) (10 mg, 0.0081 mmol), [Re(C
C,OEt)](PF6) (15 mg, 0.016 mmol), Pd(OAc)2 (0.37 mg, 0.0016 mmol), PPh3 (0.85 mg, 0.0032 mmol) and AcONa (3.3 mg, 0.041 mmol) were dissolved in MeCN degassed by N2 (2 mL). The solution was refluxed at around 75 °C under N2 for 2 days in dim light. An MeCN solution (2 mL) containing Pd(OAc)2 (0.37 mg, 0.0016 mmol) and PPh3 (0.85 mg, 0.0032 mmol) was added and refluxed 2 days again. Evaporation of the solvent gave brown solids, which were purified twice by ion-exchange chromatography (SP Sephadex C-25 and CM Sephadex C-25; the eluent was a 1:1 v/v mixture of MeCN and water containing NH4PF6 (0–8 mM)). The solvent was evaporated until solids were precipitated. The solid was washed with water and Et2O, and then dried at 60 °C under vacuum. Yield: 7.4 mg (31%). 1H NMR (400 MHz, acetone-d6): δ/ppm, 9.11 (d, J = 5.8 Hz, 2H, βpy-6), 8.97 (d, J = 5.2 Hz, 2H, αpy-6), 8.92 (s, 2H, βpy-3), 8.71 (s, 2H, αpy-3), 8.57 (s, 2H, γpy-3), 8.13 (d, J = 5.2 Hz, 2H, γpy-6), 8.03 (d, J = 5.8 Hz, 1H, βpy-5), 7.93 (d, J = 5.2 Hz, 2H, γpy-5), 7.72 (d, J = 5.2 Hz, 2H, αpy-5), 7.42–7.28 (m, 34H, –CHCH–, 2PPh3), 3.95–3.88 (m, 24H, 4P(OCH2CH3)3), 2.74 (s, 6H, αpy-CH3), 1.08 (t, J = 7.1 Hz, 36H, 4P(OCH2CH3)3). 31P NMR (400 MHz, acetone-d6): δ/ppm, 117.0 (s, 4P, –P(OEt)3), 21.8 (s, 2P, –PPh3), −143.4 (7, 3P, PF6−). FT-IR (in CH2Cl2) νCO/cm−1: 1954, 1940, 1878(br). ESI-MS (in MeCN) m/z: 820 ([M − 3PF6−]3+), 1303 ([M − 2PF6−]2+). HRMS (ESI-TOF) m/z: [M − 3PF6−]5+ Calcd for C102H118N6O18P6Re3 819.8528; Found: 819.8522.
[Ru(bpy)2Re(Ph)](PF6)5. [Re(Br2,Ph)](PF6) (15 mg, 0.012 mmol), [Ru(C
C,bpy)](PF6)2 (25 mg, 0.028 mmol), Pd(OAc)2 (0.52 mg, 0.0023 mmol), PPh3 (1.5 mg, 0.0057 mmol) and AcONa (1.5 mg, 0.018 mmol) were dissolved in MeCN degassed using N2 (10 mL). The solution was refluxed at 75 °C under N2 for 17 h in dim light. An MeCN solution degassed using N2 (10 mL) containing Pd(OAc)2 (0.52 mg, 0.0023 mmol) and PPh3 (1.3 mg, 0.0050 mmol) was added to the reaction mixture and refluxed for 1 d. Pd(OAc)2 (1.0 mg, 0.0045 mmol) and PPh3 (2.4 mg, 0.0092 mmol) were again added to the solution and the mixture was refluxed for 1 d. Evaporation of the solvent gave a red-black solid, which was purified by size exclusion chromatography. The red portion of the eluted solution was evaporated, dissolved in CH2Cl2 and washed twice with water containing NH4PF6. The red solid was obtained from the organic phase and dissolved in MeOH. A saturated methanol solution of NH4PF6 and a small amount of water were added to the dissolved solid. Evaporation of methanol gave a red solid, which was then washed with water and Et2O and dried at 60 °C under vacuum. Yield: 15 mg (42%). 1H NMR (400 MHz, acetone-d6): δ/ppm, 8.98 (s, 2H), 8.85–8.83 (m, 8H), 8.76 (s, 2H), 8.51 (s, 2H), 8.23–8.20 (m, 10H), 8.12–8.07 (m, 10H), 7.89 (d, J = 5.8 Hz, 2H), 7.84 (s, 4H), 7.73 (d, J = 5.8 Hz, 8H), 7.63–7.59 (m, 8H), 7.45 (d, J = 5.4 Hz, 2H), 7.32 (m, 30H, 2PPh3), 7.22 (d, J = 5.8 Hz, 2H), 2.65 (s, 6H, αpy-CH3). 31P NMR (400 MHz, acetone-d6): δ/ppm, 21.8 (s, 2P, –PPh3), −143.5 (7, 5P, PF6−). FT-IR (in CH2Cl2) νCO/cm−1: 1938, 1870. ESI-MS (in MeCN) m/z: 426 ([M − 5PF6−]5+), 571 ([M − 4PF6−]4+), 810 ([M − 3PF6−)]3+). Elemental Anal. Calcd for C114H90F30N14O2P7ReRu2: C, 47.82; H, 3.17; N, 6.85. Found C, 47.61; H, 3.22; N, 6.58. HRMS (ESI-TOF) m/z: [M − 5PF6−]5+ Calcd for C114H90N14O2P2ReRu2 427.8907; Found: 427.8919, [M − 4PF6−]4+ Calcd for C114H90F6N14O2P3ReRu2 571.1044; Found: 571.1010, [M − 3PF6−]3+ Calcd for C114H90F12N14O2P4ReRu2 809.7939; Found: 809.7933.
[Ru(dmb)2Re(Ph)](PF6)5. [Re(Br2,Ph)](PF6) (30 mg, 0.024 mmol), [Ru(C
C,dmb)](PF6)2 (47 mg, 0.049 mmol), Pd(OAc)2 (5.5 mg, 0.024 mmol), PPh3 (13 mg, 0.049 mmol) and AcONa (12 mg, 0.146 mmol) were dissolved in MeCN degassed using N2 (4 mL). The solution was refluxed at around 75 °C under N2 for 1 d in dim light. An MeCN solution degassed using N2 (4 mL) containing Pd(OAc)2 (5.5 mg, 0.024 mmol) and PPh3 (13 mg, 0.049 mmol) was added and then the solution was refluxed for 1 d. The solvent was removed under vacuum giving red-black solids. The solids were separated by size exclusion chromatography, and the red portion of the eluted solution was evaporated. The residue was dissolved in CH2Cl2 and washed twice with water containing NH4PF6. After evaporation, the obtained red solids were dissolved in MeOH, and a saturated methanol solution of NH4PF6 and a small amount of water were added to the solution. Evaporation of MeOH gave red solids, which were washed with water and Et2O, and dried at 60 °C under vacuum. Yield: 23 mg (32%). 1H NMR (300 MHz, acetone-d6): δ/ppm, 8.96 (s, 2H, βpy-3), 8.74 (s, 2H, αpy-3), 8.69 (s, 8H, dmb-3,3′), 8.51 (s, 2H, γpy-3), 8.10 (d, J = 6.2 Hz, 2H, γpy-6), 8.08 (d, J = 7.3 Hz, 2H, βpy-6), 7.98 (d, J = 5.8 Hz, 2H, γpy-5), 7.85–7.81 (m, 10H, αpy-6, dmb-6,6′), 7.72 (d, J = 5.6 Hz, 2H, βpy-5), 7.45–7.22 (m, 40H, −6PPh3, dmb-5,5′, –CHCH–), 7.23 (d, J = 6.2 Hz, 2H, αpy-5), 2.65 (s, 6H, αpy-CH3), 2.56 (s, 24H, dmb-CH3). 31P NMR (400 MHz, acetone-d6): δ/ppm, 21.7 (s, 2P, –PPh3), −143.5 (7, 5P, PF6−). FT-IR (in CH2Cl2) νCO/cm−1: 1939, 1870. ESI-MS (in MeCN) m/z: 450 ([M − 5PF6−]5+), 599 ([M − 4PF6−]4+), 847 ([M − 3PF6−]3+). HRMS (ESI-TOF) m/z: [M − 5PF6−]5+ Calcd for C122H106N14O2P2ReRu2 450.3158; Found: 450.3158, [M − 4PF6−]4+ Calcd for C122H106F6N14O2P3ReRu2 599.1357; Found: 599.1362, [M − 3PF6−]3+ Calcd for C122H106F12N14O2P4ReRu2 847.1691; Found: 847.1655.
[Ir(ppy)2Re(Ph)](PF6)3. [Re(Br2,Ph)](PF6) (11 mg, 9.2 μmol), [Ir(C
C,ppy)](PF6) (18 mg, 22 μmol), Pd(OAc)2 (1.1 mg, 4.9 μmol), PPh3 (2.8 mg, 11 μmol), and AcONa (1.6 mg, 19 μmol) were dissolved in MeCN degassed using N2 (10 mL). The solution was refluxed at around 75 °C under N2 for 1 d in dim light. An MeCN solution degassed using N2 (10 mL) containing Pd(OAc)2 (0.64 mg, 2.9 μmol) and PPh3 (1.2 mg, 4.6 μmol) was added and the reaction solution was refluxed for 1 d. An MeCN solution degassed using N2 (10 mL) containing Pd(OAc)2 (0.50 mg, 2.2 μmol) and PPh3 (1.2 mg, 4.6 μmol) was added again and the solution was refluxed for 1 d. Evaporation of the solvent gave brown solids, which were separated by size exclusion chromatography. The orange-brown portion of the eluted solution was evaporated, and the residue was dissolved in CH2Cl2 and washed twice with water containing NH4PF6. After evaporation of the organic phase, the orange solids were dissolved in methanol, and a saturated methanol of NH4PF6 and a small amount of water were added to the solution. After evaporation of methanol, the obtained orange solids were collected, washed with water and Et2O, and dried at 60 °C under vacuum. Yield: 2.0 mg (8.0%). 1H NMR (400 MHz, acetone-d6): δ/ppm, 9.01 (s, 2H, βpy-3), 8.78 (s, 2H, αpy-3), 8.56 (s, 2H, γpy-3), 8.25 (m, 6H), 8.09 (dd, J = 5.7, 6.2 Hz, 2H), 7.97–7.84 (m, 8H), 7.57 (d, J = 6.0 Hz, 2H), 7.32–7.16 (m, 38H), 7.07–6.93 (m, 10H), 6.36 (dd, J = 7.3, 7.4 Hz, 2H), 2.68 (s, 6H, αpy-CH3). 31P NMR (400 MHz, acetone-d6): δ/ppm, 21.8 (s, 2P, –PPh3), −143.5 (7, 3P, PF6−). FT-IR (in CH2Cl2) νCO/cm−1: 1939, 1870. ESI-MS (in MeCN) m/z: 771 ([M − 3PF6−]3+), 1229 ([M − 2PF6−]4+). HRMS (ESI-TOF) m/z: [M − 3PF6−]5+ Calcd for C118H90Ir2N10O2P2Re 770.8518; Found: 770.8478.
[Ru(dmb)2Re(FPh)](PF6)5 was synthesized from [Ru(CC,dmb)]2+ by a method similar to that for [Ru(dmb)2Re(Ph)]5+ using [Re(Br2,FPh)]+ instead of [Re(Br2,Ph)]+. Yield: 25%. 1H NMR (400 MHz, acetone-d6): δ/ppm, 8.94 (s, 2H, βpy-3′), 8.71 (s, 2H, αpy-3), 8.66 (s, 8H, dmb-3,3′), 8.56 (s, 2H, γpy-3), 8.18 (d, J = 6.0 Hz, 2H, γpy-6), 8.08 (d, J = 6.0 Hz, 2H, βpy-6′), 7.96 (d, J = 5.6 Hz, 2H, γpy-5), 7.86–7.82 (m, 12H, dmb-6,6′, –CHCH–), 7.69 (d, J = 5.6 Hz, 2H, αpy-6), 7.46–7.38 (m, 24H, αpy-5, βpy-5′, dmb-5,5′, m-Ph), 7.13–7.09 (m, 12 H, o-Ph), 2.63 (s, 3H, αpy-CH3), 2.56 (s, 24H, dmb-CH3). 31P NMR (400 MHz, acetone-d6): δ/ppm, 19.9 (s, 2P, –PPh3), −143.6 (7, 5P, PF6−). FT-IR (in CH2Cl2) νCO/cm−1: 1942, 1873. ESI-MS (in MeCN) m/z: 472 ([M − 5PF6−]5+), 626 ([M − 4PF6−]4+), 883 ([M − 3PF6−]3+). HRMS (ESI-TOF) m/z: [M − 5PF6−]5+ Calcd for C122H100F6N14O2P2ReRu2 471.9047; Found: 471.9044, [M − 4PF6−]4+ Calcd for C122H100F12N14O2P3ReRu2 626.1226; Found: 626.1216, [M − 3PF6−]3+ Calcd for C122H100F18N14O2P4ReRu2 883.1502; Found: 883.1474.
Notes and references
- R. Amadelli, R. Argazzi, C. A. Bignozzi and F. Scandola, J. Am. Chem. Soc., 1990, 112, 7099 CrossRef CAS.
- I.-W. Hwang, D. M. Ko, T. K. Ahn, Z. S. Yoon, D. Kim, X. Peng, N. Aratani and A. Osuka, J. Phys. Chem. B, 2005, 109, 8643 CrossRef CAS PubMed.
- N. Kaveevivitchai, R. Chitta, R. Zong, M. El Ojaimi and R. P. Thummel, J. Am. Chem. Soc., 2012, 134, 10721 CrossRef CAS PubMed.
- F. Li, Y. Jiang, B. Zhang, F. Huang, Y. Gao and L. Sun, Angew. Chem., Int. Ed., 2012, 51, 2417 CrossRef CAS PubMed.
- H. Ozawa and K. Sakai, Chem. Commun., 2011, 47, 2227 RSC.
- S. M. Arachchige, J. R. Brown, E. Chang, A. Jain, D. F. Zigler, K. Rangan and K. J. Brewer, Inorg. Chem., 2009, 48, 1989 CrossRef CAS PubMed.
- T. Stoll, M. Gennari, J. Fortage, C. E. Castillo, M. Rebarz, M. Sliwa, O. Poizat, F. Odobel, A. Deronzier and M.-N. Collomb, Angew. Chem., Int. Ed., 2014, 53, 1654 CrossRef CAS PubMed.
- K. Koike, S. Naito, S. Sato, Y. Tamaki and O. Ishitani, J. Photochem. Photobiol., A, 2009, 207, 109 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673 CrossRef CAS PubMed.
- Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Faraday Discuss., 2012, 155, 115 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22 CrossRef CAS PubMed.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326 CrossRef CAS PubMed.
- E. Kato, H. Takeda, K. Koike, K. Ohkubo and O. Ishitani, Chem. Sci., 2015, 6, 3003 RSC.
- V. Balzani, S. Campagna, G. Denti, A. Juris, S. Serroni and M. Venturi, Acc. Chem. Res., 1998, 31, 26 CrossRef CAS.
- R. Maity, H. Koppetz, A. Hepp and F. E. Hahn, J. Am. Chem. Soc., 2013, 135, 4966 CrossRef CAS PubMed.
- L. Cassidy, S. Horn, L. Cleary, Y. Halpin, W. R. Browne and J. G. Vos, Dalton Trans., 2009, 3923 RSC.
- P. J. Connors, D. Tzalis, A. L. Dunnick and Y. Tor, Inorg. Chem., 1998, 37, 1121 CrossRef CAS.
- D. Tzalis and Y. Tor, Chem. Commun., 1996, 1043 RSC.
- S. Goeb, A. De Nicola and R. Ziessel, J. Org. Chem., 2005, 70, 6802 CrossRef CAS PubMed.
- K. J. Arm and J. A. G. Williams, Dalton Trans., 2006, 2172 RSC.
- V. L. Whittle and J. A. G. Williams, Dalton Trans., 2009, 3929 RSC.
- S. Welter, N. Salluce, P. Belser, M. Groeneveld and L. De Cola, Coord. Chem. Rev., 2005, 249, 1360 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed.
- R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS.
- Rigorously excluding air contamination in the reaction vessel dramatically decreased the yield of [Re(OEt)Re(Et)]2+. However, adding too much air to the reaction vessel also decreased the yield. Air contamination that occurred when the cap of the vessel was opened to allow the reagents to be added supplied a suitable amount of air for [Re(OEt)Re(Et)]2+ to be produced. The role of air in the reaction is currently being investigated in our laboratory.
- G. F. Strouse, J. R. Schoonover, R. Duesing, S. Boyde, W. E. Jones and T. J. Meyer, Inorg. Chem., 1995, 34, 473 CrossRef CAS.
- Various decomposition products, including several kinds of mononuclear, dinuclear, and other kinds of trinuclear complexes, were observed.
- Z.-Y. Bian, H. Wang, W.-F. Fu, L. Li and A.-Z. Ding, Polyhedron, 2012, 32, 78 CrossRef CAS PubMed.
- S. Liu and K. S. Schanze, Chem. Commun., 2004, 1510 RSC.
- G. H. Allen, R. P. White, D. P. Rillema and T. J. Meyer, J. Am. Chem. Soc., 1984, 106, 2613 CrossRef CAS.
- K. Koike, N. Okoshi, H. Hori, K. Takeuchi, O. Ishitani, H. Tsubaki, I. P. Clark, M. W. George, F. P. A. John and J. J. Turner, J. Am. Chem. Soc., 2002, 124, 11448 CrossRef CAS PubMed.
- T. F. Guarr and F. C. Anson, J. Phys. Chem., 1987, 91, 4037 CrossRef CAS.
- J. Font, P. de March, F. Busque, E. Casas, M. Benitez, L. Teruel and H. Garcia, J. Mater. Chem., 2007, 17, 2336 RSC.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334 CrossRef CAS.
- T. F. Guarr and F. C. Anson, J. Phys. Chem., 1987, 91, 4037 CrossRef CAS.
- S. Metz and S. Bernhard, Chem. Commun., 2010, 46, 7551 RSC.
- A. Baron, C. Herrero, A. Quaranta, M. F. Charlot, W. Leibl, B. Vauzeilles and A. Aukauloo, Inorg. Chem., 2012, 51, 5985 CrossRef CAS PubMed.
- E. Hasegawa, T. Seida, N. Chiba, T. Takahashi and H. Ikeda, J. Org. Chem., 2005, 70, 9632 CrossRef CAS PubMed.
- X.-Q. Zhu, M.-T. Zhang, A. Yu, C. H. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501 CrossRef CAS PubMed.
- C. Kutal, M. A. Weber, G. Ferraudi and D. Geiger, Organometallics, 1985, 4, 2161 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: UV-vis absorption, emission spectra and emission decay curves of trinuclear complexes obtained, SEC charts of reaction solutions, temperature dependence of emission lifetime and electrochemical property of [Ru(dmb)2Re(Ph)]5+. See DOI: 10.1039/c5dt01717c |
| ‡ Present address: Department of Computer Science, School of Computer Science, Tokyo University of Technology, 1404-1, Katakura-cho, Hachio-ji-shi, Tokyo 192-0982, Japan. |
| This journal is © The Royal Society of Chemistry 2015 |