
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis and extraction studies of a new class of conformationally constrained (N,N,N′,N′-tetraalkyl)7-oxabicyclo[2.2.1]heptane-2,3-dicarboxamides†
Shikha
Sharma
a,
Surajit
Panja
b,
Arunasis
Bhattachariya
c,
Prem S.
Dhami
b,
Preetam M.
Gandhi
b and
Sunil K.
Ghosh
*a
aBio-Organic Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India. E-mail: ghsunil@barc.gov.in; Fax: +91-22-25505151; Tel: +91-22-25595012
bFuel Reprocessing Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
cRadiochemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
First published on 2nd June 2015
Abstract
A new class of conformationally constrained 7-oxabicyclo[2.2.1]heptane-2,3-dicarboxamides (OBDA) of three secondary amines was synthesized, and their extraction behavior for trivalent and tetravalent actinides in HNO3 medium was studied. Amongst the diamides, N,N-bis-2-ethylhexyl substituted diamide showed the best results for actinide extraction. This diamide also exhibited a very low level of extraction for Sr(II) and Ru(III) which is desirable, thus providing higher selectivity for actinides. The stripping of extracted metal ions was achieved using 0.1 M oxalic acid for Pu(IV) or pH ∼ 2 solution for Am(III). Third phase formation was not observed for the OBDA ligand even for a higher concentration of Eu up to 5 g L−1 with retention of good DEu. The tridentate nature of the OBDA ligand was ascertained by studying the IR and NMR spectra of the Eu(III) complex with the ligand. The OBDA showed the formation of a mixture of mono- and di-solvated species of Eu(III) as indicated by the slope analysis method and ESI-MS. Density functional theoretical (DFT) study was carried out to determine the energy optimized structure of the free ligand and its Am3+ complex.
Introduction
Spent nuclear fuel reprocessing is becoming a necessity in the generation of nuclear energy. The spent fuel is dissolved in nitric acid and the combined recovery of U(VI) and Pu(IV) is carried out by solvent extraction (PUREX process) by a solution of 1.1 M tri-n-butylphosphate (TBP) in n-dodecane.1 The acidic raffinate thus produced is known as high-level liquid waste (HLLW), which contains significant amounts of trivalent lanthanides and minor actinides (Am, Cm and Np). In addition, it also contains a small amount of Pu and fission products (137Cs, 90Sr, 106Ru etc.). Although the concentration levels are small, these actinides are α-emitting radioisotopes and need to be separated from HLLW for its management and safe disposal. Thus, partitioning and transmutation (P&T) strategy is becoming vital for the management of HLLW.2 A plethora of complexing reagents (solvents) have been developed over the last three decades for actinide partitioning purposes. The malonamide family of extractants was developed during the 1980s for the extraction of actinides from PUREX raffinate (DIAMEX process). The most promising member of this family was the DIAMEX reference compound N,N′-dimethyl-N,N′-dibutyl malonamide (DMDBTDMA) I (Fig. 1).3 Several phosphorus based compounds viz. trialkylphosphine oxides (TRPO process),4a diisodecylphosphoric acid (DIDPA),4b carbamoyl organophosphorous compounds (TRUEX process),4c–eetc. have also been developed for the same purpose. But they are non-incinerable and do not comply with the requirements of the CHON principle of extractant design. | ||
| Fig. 1 Structures of important diamides. | ||
The multidentate diglycolamide (DGA) family of extractants were introduced in the early 1990s for the extraction of different metal ions.5 The DGA substance class has close resemblance to the malonamides but possesses an additional arm, an ether group positioned between two acetamide functionalities. Amongst the diglycolamides (DGAs) synthesized, N,N,N′,N′-tetraoctyldiglycolamide (TODGA) II,5b–d and N,N,N′,N′-tetra-2-ethylhexyldiglycolamide (TEHDGA)5e,fIII (Fig. 1) have been found to be the most important extractants for the separation of trivalent actinides and lanthanides from HLLW. The change from malonamide (bidentate ligand) to DGA (tridentate ligand) significantly increased the affinity for both trivalent actinides and lanthanides. The main disadvantage of these DGA based extractants is the extraction of Sr(II) ions from the HLLW solution along with Ln(III) and Ac(III) ions.5b,g–i,6a This leads to the requirement of a greater number of stages for scrubbing. The higher extraction of strontium and other elements5j by TODGA was attributed to the higher basicity (KH) of the TODGA/n-dodecane (KH = 4.1)5b as compared to TEHDGA (KH = 1.72).5k Therefore, the extraction behavior is strongly dependent on the nature of the alkyl groups attached. To optimize the selectivity of extraction/stripping in terms of diluent, phase modifier and reagent concentration, unsymmetrical DGAs have also been developed.6a They have also been pre-organized on various tripodal platforms6b and as bis(DGA).6c
It is known that a small structural change in the substituent at the central carbon in malonamides causes a noticeable change in their extraction abilities.3c Similarly, DGA ligands with structural modifications in their skeleton were also attempted.7a The thioether analogs of DGAs,7b thiadiglycolamide (TGDA) have been made and employed in the extraction. The other modifications include (1) the increase in chain length from one carbon to two carbons between the central ether oxygen (oxydipropionamide or ODPA)/sulfur atom (thiadipropionamide or TDPA) and the amide moieties,7b (2) the addition of substituents on the carbons between the central oxygen atom and the amide moieties on either one or both sides of the central oxygen,7a (3) the replacement of the central oxygen by a (substituted) nitrogen atom,7c (4) synthesis of pillar[5]arene and calix[4]arene based DGA molecules7d,e and (5) synthesis of rigidified diglycolamides on a tetrahydrofuran platform.7a The extraction study experiments showed that these modifications changed some properties of extraction or back-extraction but did not change them in a meaningful way to be superior than TODGA II or TEHDGA III.
So far, the successful tridentate ligands designed and developed mostly consist of linear diglycolamides. The increase in chain length from one carbon to two carbons between the central ether oxygen atom and the amide moieties in ODPA has been attempted but with poor extraction abilities.7b As mentioned above, the only reported diglycolamides on a rigidified tetrahydrofuran platform (as a mixture of cis and trans-isomers) have been reported by Iqbal et al.7a We were curious to develop tridentate diamide based ligands 1 based on conformationally constrained 7-oxabicyclo[2.2.1]heptane-2,3-dicarboxylic acid, where the ether–oxygen is positioned at the bridgehead and a chain of two carbon atoms lies between the ether oxygen and the amide moieties (Fig. 2). The rigid coordination sphere created by the three donor groups (ether–oxygen and two amide groups) is expected to enhance binding kinetics as the coordination atoms are appropriately oriented for binding to a metal. The co-extraction of some fission product like Ru and Sr is a major issue with the currently used DGA classes of ligands. We were therefore curious to investigate the concept of the rigidity effect of donor groups on an oxabicyclo[2.2.1]heptane skeleton for the selective extraction of lanthanides and actinides. To the best of our knowledge, the syntheses of such simple oxy-diamides have not been attempted to study the effect of such structural modifications on extraction behavior. Herein, we describe the synthesis of three conformationally constrained bridged 7-oxabicyclo[2.2.1]heptane-2,3-dicarboxamides 1a–c (OBDA 1a–c) and their extraction properties for 241Am(III), (152+154)Eu(III), 239Pu(IV), 233U(VI) from the nitric acid medium using 15% isodecyl alcohol (IDA)/n-dodecane as a diluent mixture.
 | ||
| Fig. 2 Conformationally constrained diamides. | ||
Results and discussion
For the current study, three OBDAs 1a–c having different amine components have been synthesized from commercially available starting materials as shown in Scheme 1. The Diels–Alder reaction of furan and maleic anhydride gave the known exo adduct 2 in excellent yield, which was then hydrogenated using Pd/C as catalyst to give the known bridged tricyclic anhydride 3 in quantitative yield. Anhydride 3 was opened up with bis 2-ethylhexylamine to give the hemiamide 4 as an intermediate which was reacted in situ with another equiv. of bis 2-ethylhexylamine in the presence of diisopropylcarbodiimide (DIPC) and a catalytic amount of 4-dimethylaminopyridine (DMAP) to give the bridged OBDA 1a in excellent yield as a waxy solid. The other two dicarboxamides, OBDA 1b and OBDA 1c were similarly made in high yields from the anhydride 3 using dioctylamine and diisobutylamine, respectively (Scheme 1).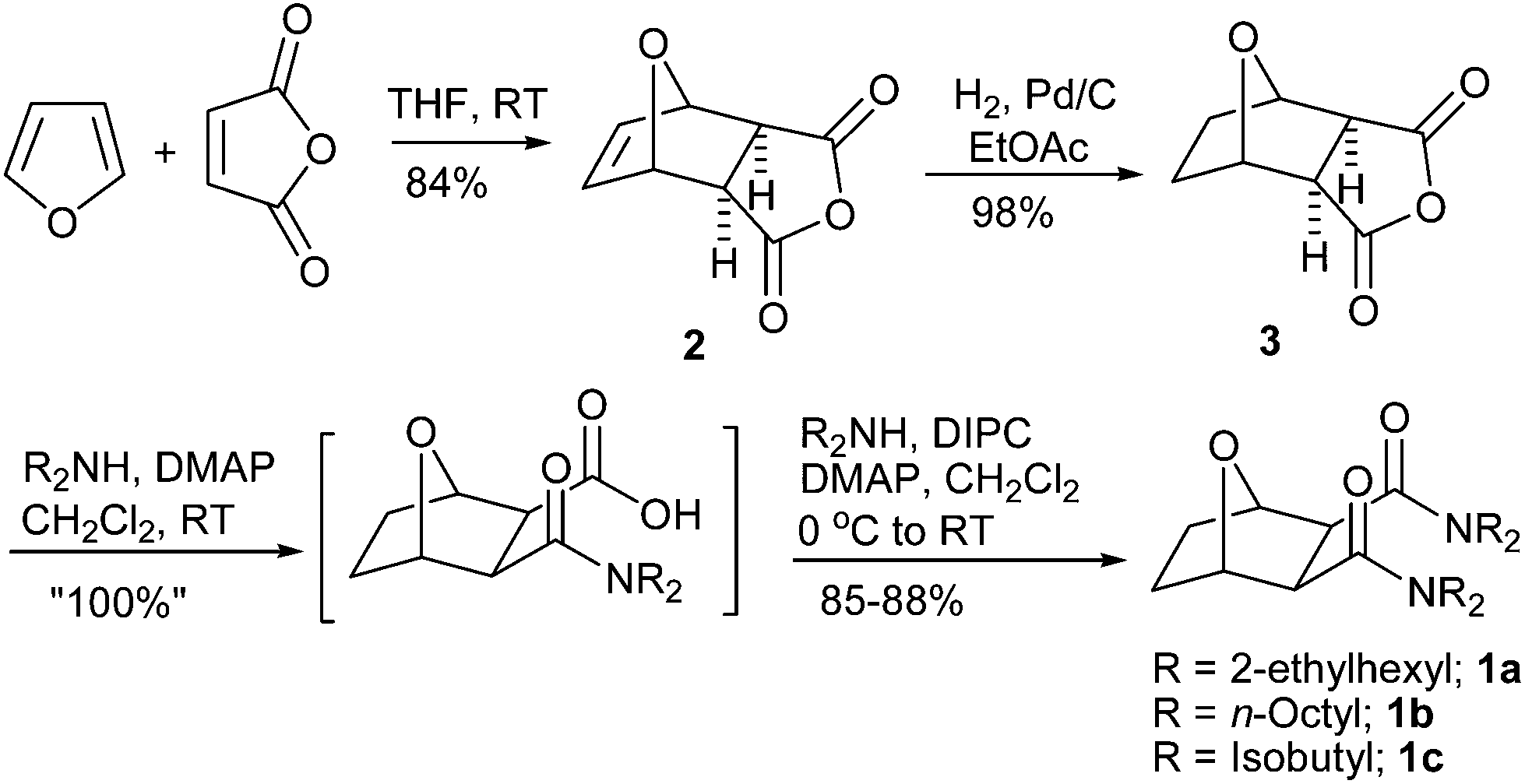 | ||
| Scheme 1 Synthesis of bridged 7-oxabicyclo[2.2.1]heptane-2,3-dicarboxamides. | ||
For extraction studies, IDA was used as a phase modifier to suppress the formation of the third phase by OBDA 1a in contact with 4 M HNO3 solution, and 15% was found to be enough to prevent that. The time of equilibration required for Am(III) and Pu(IV) by OBDAs was studied using 0.1 M OBDA 1a in 15% IDA/n-dodecane in the presence of 1 M HNO3. It was found that OBDA 1a showed very fast kinetics of equilibration (2 min) for both Am(III) and Pu(IV) (Fig. 3). The kinetics of extraction was found to be comparable to TEHDGA III under the same conditions. Therefore, in subsequent studies, 5 min equilibration time was maintained.
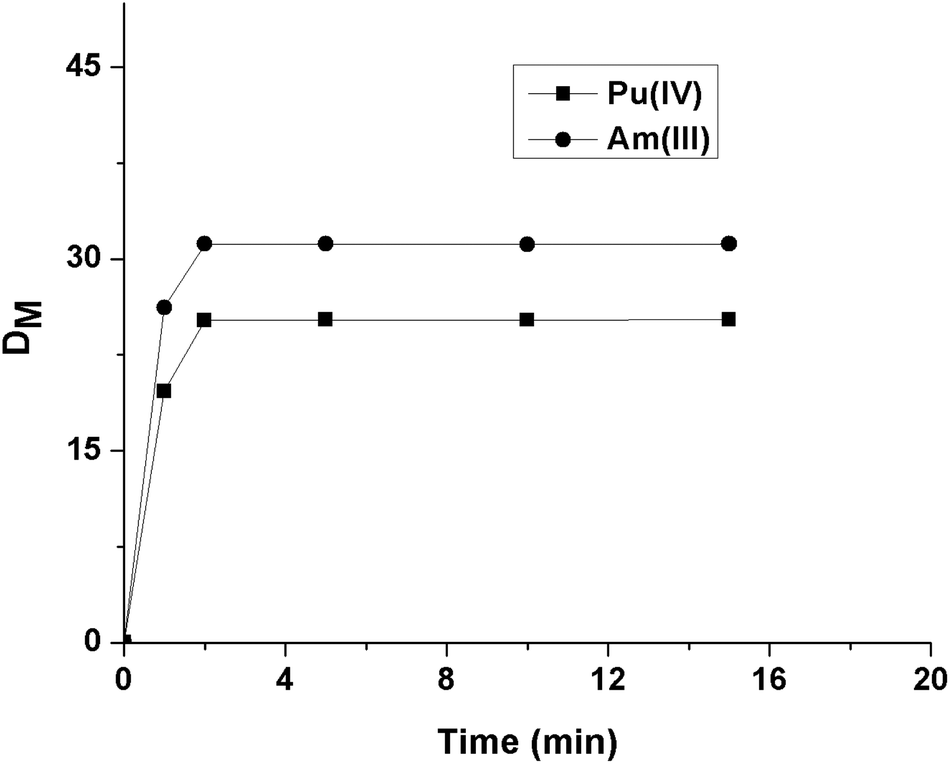 | ||
| Fig. 3 Kinetics of extraction of Pu(IV) and Am(III) using 0.1 M OBDA 1a in 15% IDA/n-dodecane from 1 M HNO3. | ||
The DAm value for OBDA 1a (Fig. 4) at 3 M HNO3 was found to be 52.3. For comparison purposes, the extraction of Am(III) was also studied with OBDA 1b and OBDA 1c at 3 M HNO3. The DAm value was slightly less for OBDA 1b (42 at 3 M HNO3). The solubility of OBDA 1c was poor in 15% IDA/n-dodecane. Hence, a solution of OBDA 1c in 30% IDA/n-dodecane was studied which also showed a DAm value of 40 at 3 M HNO3. Further studies were therefore performed with OBDA 1a only.
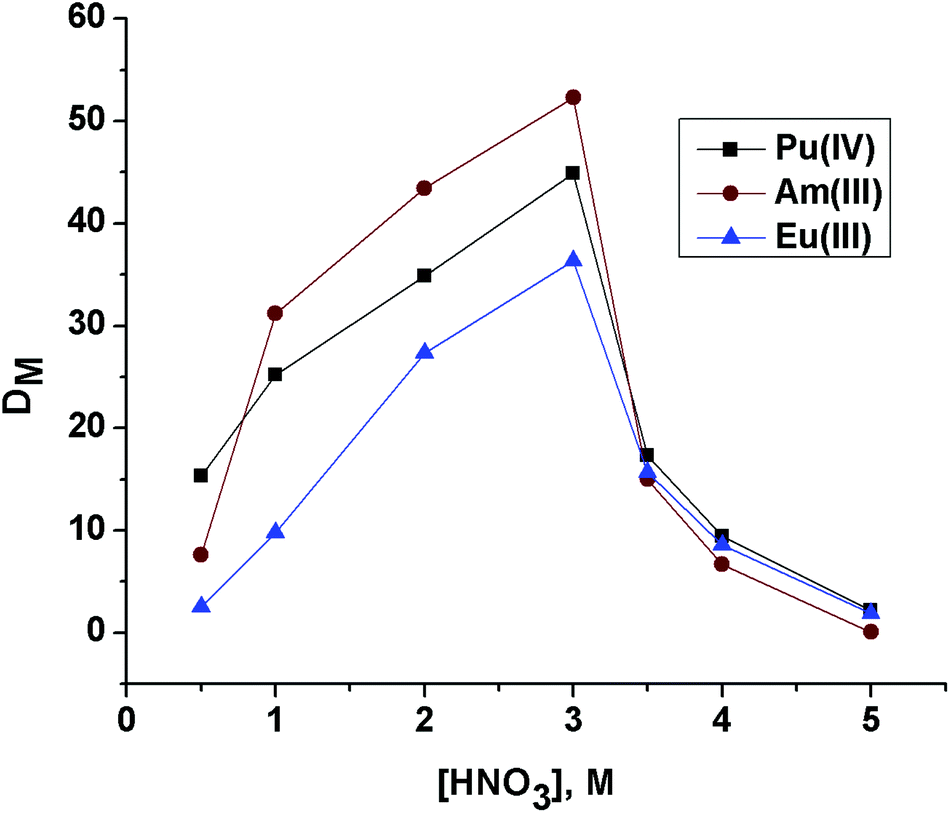 | ||
| Fig. 4 Effect of feed nitric acid concentration on the extraction of Pu(IV), Am(III) and Eu(III) using 0.1 M OBDA 1a in 15% IDA/n-dodecane. | ||
The effect of feed nitric acid concentration on the distribution ratio of Pu(IV), Am(III) and Eu(III) for 0.1 M OBDA 1a in 15% IDA/n-dodecane was investigated to explore the feasibility of using this ligand for the separation of actinides and lanthanides (Fig. 4). OBDA 1a showed an interesting trend in the distribution ratio of Pu(IV), Am(III) and Eu(III). In the case of OBDA 1a, an initial increasing trend of the distribution ratios for Pu(IV), Am(III) and Eu(III) was observed on increasing the feed nitric acid concentration. Beyond 3 M HNO3, the distribution ratios rapidly decreased. The results obtained for Am(III) were compared with the results obtained from TEHDGA III to understand the effect of conformational restrictions of the ligand structure on the extraction performance (Fig. 5). In the case of TEHDGA III, the distribution ratio continuously increased with increasing HNO3 concentration up to 6 M concentration. Between 3–4 M HNO3 (the commonly used acidity of nuclear waste), the DM values for OBDA 1a are comparable with TEHDGA III. Like TEHDGA III, OBDA 1a is a neutral extractant. Hence, the extraction mechanism takes place via nitrate ion assisted complex formation (eqn (1)). The subscript ‘org’ refers to species in the organic phase and the absence of subscript refers to those present in the aqueous phase:
| Mx+ + xNO3− + yOBDA 1aorg ⇄ [M(NO3)x(OBDA 1a)y]org | (1) |
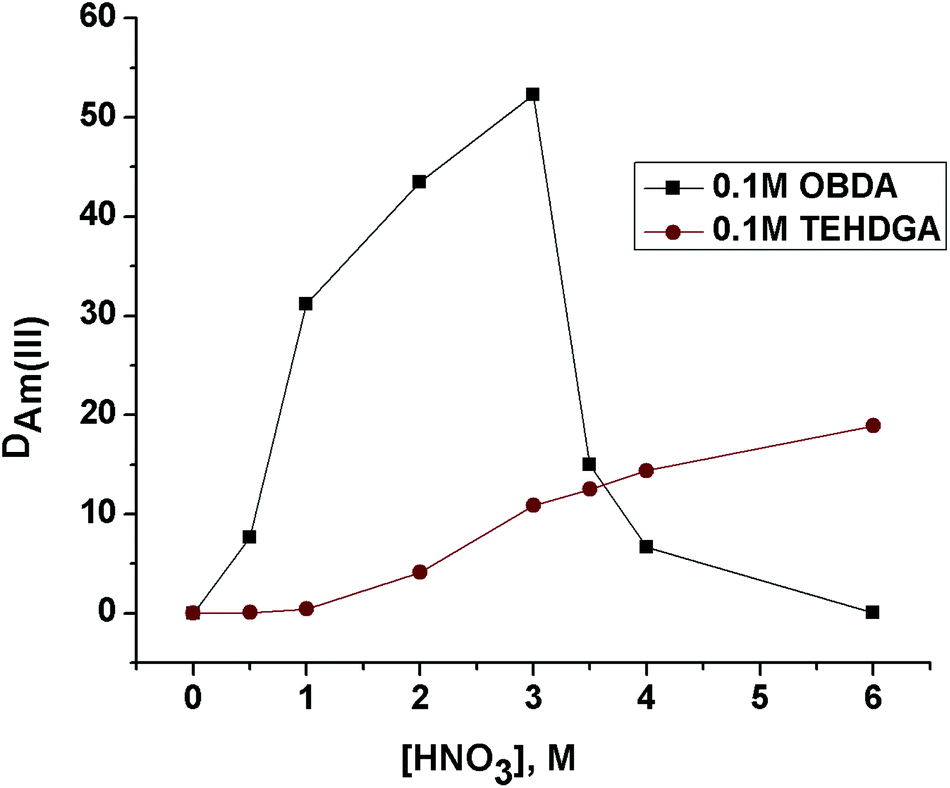 | ||
| Fig. 5 Comparative extraction of Am(III) using 0.1 M OBDA 1a and 0.1 M TEHDGA III in 15% IDA/n-dodecane at different feed nitric acid concentrations. | ||
So, increasing nitric acid concentration leads to increased formation of the neutral metal–nitrate complex leading to a higher DM value. This explains the trend observed for TEHDGA III and the increasing trend of DM for OBDA 1a on the increasing nitric acid concentration. But the decreasing trend in the distribution ratio beyond 3 M HNO3 for OBDA 1a needed more investigation. One possible reason for this abnormal behavior of OBDA 1a could be its stability at higher nitric acid concentration. This provoked us to study the stability behavior of OBDA 1a at different nitric acid concentrations. OBDA was found to be sensitive to hydrolytic degradation. In lower acid concentration, the degradation rate was found to be slow. In 1 M HNO3, the OBDA 1a was observed to be stable up to about 15 min after which degradation started and complete hydrolysis was observed in 2.5 h. In 3 M HNO3, the OBDA 1a solution in n-dodecane–IDA mixture is stable for a very short period of time. The hydrolysis pattern of OBDA 1a showed that after 5 min of contact with 3 M HNO3, its degradation was initiated followed by rapid degradation thereafter (Table 1, ESI†).8 The complete degradation of OBDA 1a took about 30 min resulting in the formation of bis-2-ethylhexylamine and the corresponding dicarboxylic acid, a product further independently confirmed by hydrolyzing the anhydride 3. It was therefore necessary to find out the ligand species responsible for the extraction of Pu(IV) and Am(III). Independent studies using either bis-2-ethylhexylamine or di-carboxylic acid of anhydride 3 or a mixture of both did not show any extraction of Pu(IV) and Am(III). This clearly proved that OBDA 1a is responsible for the extraction of Pu(IV) and Am(III). The OBDA molecules that are complexed to Pu(IV) or Am(III) did not undergo hydrolysis. This has been augmented by stripping studies of the OBDA 1a complexed to Pu(IV) or Am(III) with a higher concentration of nitric acid (6 M HNO3) where extracted metal ions are not stripped into the aqueous phase. So the combination of fast kinetics for complexation and good DM values for Pu(IV) and Am(III) at 3–4 M acidity (normally used in HLLW) prompted us to carry out further extraction studies using OBDA 1a.
To determine the stoichiometry of the extracted metal–ligand species for Pu(IV), Am(III) and Eu(III), the concentration of OBDA 1a was varied at a fixed aqueous acidity (1 M HNO3). The plot of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) DMvs. log[OBDA 1a] for Pu(IV), Am(III) and Eu(III) gave straight lines with varying slopes (Fig. 6). Pu(IV) was found to form a mono-solvated species with OBDA 1a whereas Am(III) and Eu(III) formed a mixture of mono-solvated and di-solvated species. The DM values for Pu, Am and Eu were studied at different nitrate ion concentrations at a fixed aqueous acidity (1 M HNO3). The plot of logDMvs. log[NO3−] showed a straight line with a slope of ∼4 for Pu(IV) and ∼3 for Am(III) or Eu(III) (Fig. 7) indicating that four nitrate ions are involved in the extraction of Pu(IV) while three nitrate ions are involved for Am(III) and Eu(III).
DMvs. log[OBDA 1a] for Pu(IV), Am(III) and Eu(III) gave straight lines with varying slopes (Fig. 6). Pu(IV) was found to form a mono-solvated species with OBDA 1a whereas Am(III) and Eu(III) formed a mixture of mono-solvated and di-solvated species. The DM values for Pu, Am and Eu were studied at different nitrate ion concentrations at a fixed aqueous acidity (1 M HNO3). The plot of logDMvs. log[NO3−] showed a straight line with a slope of ∼4 for Pu(IV) and ∼3 for Am(III) or Eu(III) (Fig. 7) indicating that four nitrate ions are involved in the extraction of Pu(IV) while three nitrate ions are involved for Am(III) and Eu(III).
 | ||
| Fig. 6 Plot of logDMvs. log[OBDA 1a] for (a) Am (b) Eu and (c) Pu. | ||
 | ||
| Fig. 7 Plot of logDMvs. logNO3− for (a) Am (b) Eu and (c) Pu. | ||
To determine the nature of the bonding between metal and ligand, the complex of Eu(NO3)3·5H2O with OBDA 1c9 was studied by IR and 1H NMR spectroscopy. IR spectra of the obtained complex10 (Fig. 8) indicates a strong binding of metal at the amide carbonyl group of the ligand. The ν C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O for OBDA 1c appeared at 1655 and 1632 cm−1 whereas for the metal–ligand complex it appeared at a lower wave number (1593 cm−1). This lowering in ν CO was attributed to the coordination of the metal at the carbonyl group of the ligand. The participation of the ether–oxygen of OBDA in binding with the metal in the metal ligand complex has been corroborated from the 1H NMR spectra (Fig. 9). A significant downfield shift of CH–O–CH resonance was observed after complexation with Eu [δ 4.86 (2 H, s, CHOCH) for free OBDA 1cvs. 5.96 (2 H, s, broad, CHOCH) for the complexed Eu]. NMR and IR studies confirmed the participation of amide carbonyls and ether–oxygen in metal ligand complex formation. The OBDA ligands thus function as tridentate ligands like DGA ligands. It will, therefore, be of interest to understand the structure and coordination modes in its complex using a density functional theoretical (DFT) study. ESI-MS spectra provided further insight on the metal ligand complex formation. The complex obtained was a mixture of two species mono- and di-solvated. The peaks at m/z 720.7 and 841.7 correspond to [(OBDA 1c)Eu(NO3)2(H2O)2]+ and [(OBDA 1c)Eu(NO3)3(H2O)4Na]+, respectively, while peaks at m/z 517.5 and 1096.7 correspond to [(OBDA 1c)2Eu(NO3)]2+ and [(OBDA 1c)2Eu(NO3)2]+, respectively (Fig. 1, ESI†).
O for OBDA 1c appeared at 1655 and 1632 cm−1 whereas for the metal–ligand complex it appeared at a lower wave number (1593 cm−1). This lowering in ν CO was attributed to the coordination of the metal at the carbonyl group of the ligand. The participation of the ether–oxygen of OBDA in binding with the metal in the metal ligand complex has been corroborated from the 1H NMR spectra (Fig. 9). A significant downfield shift of CH–O–CH resonance was observed after complexation with Eu [δ 4.86 (2 H, s, CHOCH) for free OBDA 1cvs. 5.96 (2 H, s, broad, CHOCH) for the complexed Eu]. NMR and IR studies confirmed the participation of amide carbonyls and ether–oxygen in metal ligand complex formation. The OBDA ligands thus function as tridentate ligands like DGA ligands. It will, therefore, be of interest to understand the structure and coordination modes in its complex using a density functional theoretical (DFT) study. ESI-MS spectra provided further insight on the metal ligand complex formation. The complex obtained was a mixture of two species mono- and di-solvated. The peaks at m/z 720.7 and 841.7 correspond to [(OBDA 1c)Eu(NO3)2(H2O)2]+ and [(OBDA 1c)Eu(NO3)3(H2O)4Na]+, respectively, while peaks at m/z 517.5 and 1096.7 correspond to [(OBDA 1c)2Eu(NO3)]2+ and [(OBDA 1c)2Eu(NO3)2]+, respectively (Fig. 1, ESI†).
 | ||
| Fig. 8 IR spectra of (a) OBDA 1c and (b) OBDA 1c complexed with Eu(NO3)3·5H2O. | ||
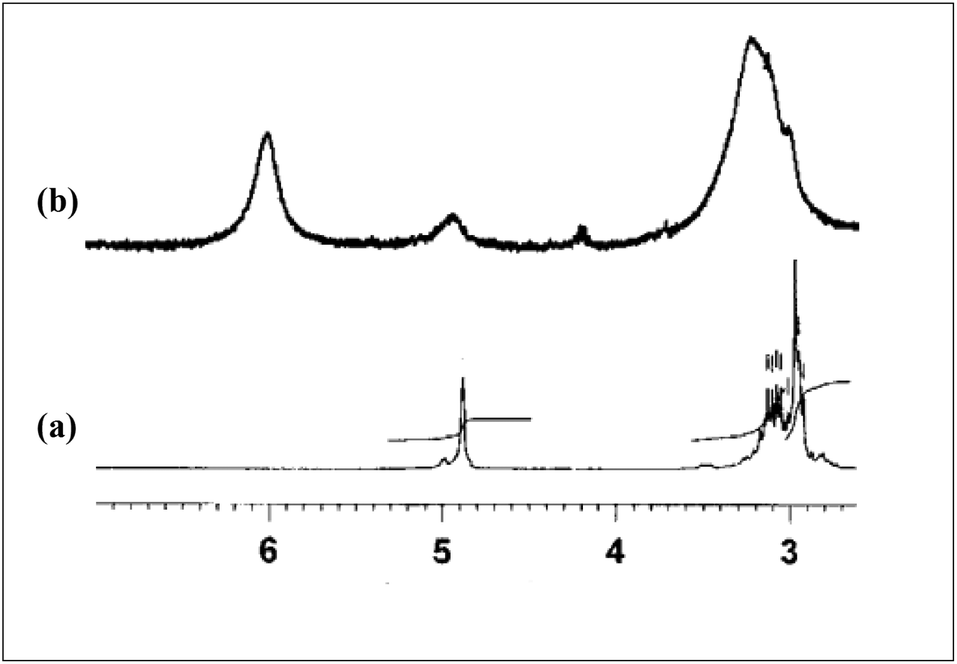 | ||
| Fig. 9 1H NMR spectra of the relevant portion (a) OBDA 1c and (b) OBDA 1c complexed with Eu(NO3)3·5H2O. | ||
The presence of macro concentrations of trivalent lanthanides in HLLW often leads to the formation of a third phase during ‘actinide partitioning’. To simulate the situation, the DM value of Eu(III) was studied with OBDA 1a at 3 M HNO3 with varying concentrations of Eu. No third phase formation was observed even up to 5 g L−1 of Eu. Also, significant DEu was found at higher concentrations (Table 2, ESI†).
Stripping of loaded metal ions from the organic phase is an important factor for the extractant to be considered for separation of actinides and lanthanides. It was possible for OBDA 1a loaded with Pu(IV) to be stripped by 0.1 M Oxalic acid (∼85% stripping in a single contact) whereas Am(III) was found to be stripped by pH 2 solution (∼90% stripping in a single contact).
The selectivity of OBDA 1a in reference to other elements present in HLLW was investigated in 3 M HNO3 and was found to be higher compared to TEHDGA III (Table 1). It was interesting to observe that DSr and DU values were low compared to those observed for TEHDGA III (DSr ∼ 0.65 and DU ∼ 2.1 for 0.1 M TEHDGA III/15% IDA)5e,k thus offer better purification of the trivalent actinides and lanthanides over the other constituents present in HLLW.
| Elements | Cs | Sr | Ru | Nb | Ce | U | Pu | Eu | Sb | Am |
|---|---|---|---|---|---|---|---|---|---|---|
| D M(OBDA) | <0.01 | 0.05 | 0.04 | <0.01 | 14.86 | 0.5 | 44.29 | 35.46 | <0.01 | 52.21 |
| D M(TEHDGA) | <0.01 | 0.65 | 0.05 | <0.01 | 13.17 | 2.1 | 34.16 | 9.15 | <0.01 | 11.15 |
DFT calculations were performed on the free OBDA with the di-methyl substituent on the amidic ‘N’ atoms in order to reduce the computational cost. The result shows that the ligand is most stable in the conformation, where two carbonyl oxygens of amide groups are in trans position (Fig. 10a). However, prior to the complexation with the metal ions, both the amidic oxygens should be in cis position. Two possible conformers with the amidic oxygens in the cis position were considered (Fig. 10b and 10c) and their relative energies were compared with respect to the most stable trans conformer. The results showed that the conformer ‘c’ (relative energy: 5.39 kcal mol−1) is more stable than the conformer ‘b’ (relative energy: 11.18 kcal mol−1). FTIR studies also showed the participation of the ethereal oxygen atom in the bonding with the metal ion, which is possible if the ligand forms complexes in the conformer ‘c’. The DFT study, therefore, substantiates the results of the FTIR studies. The slope analysis study indicated the presence of two OBDA molecules and three nitrate ions in the extracted species of Am3+. The guessed structure of the Am3+ complex was, therefore, prepared with two OBDA and three nitrate ions coordinated to the central Am3+ ion. The optimized geometry of the Am3+ complex is shown in Fig. 11. In this complex the amidic oxygens are closer to the central Am3+ ion with an ‘Am–O’ bond length of 2.51 ± 0.02 Å as compared to the ethereal oxygen atoms (2.70 ± 0.04 Å). The nitrate ions are, however, closer to the Am3+ ion as compared to the OBDA with an ‘Am–O’ distance of 2.403 ± 0.002 Å. In order to further understand the metal–ligand bonding the natural charges on the Am3+ ion and coordinating sites of the ligand molecules were calculated and the results are shown in Table 2. The electron density on the amidic oxygen atom increased from −0.60 e to −0.641 after complexation in spite of transfer of electron density to the Am3+ ion. This indicates more charge polarization in the ligand molecule after complexation due to the pulling of electron density by the amidic oxygen atoms from the adjacent carbon atom resulting in change in the electronic charge on the amidic carbon atom from −0.412 e to 0.714 e.
 | ||
| Fig. 10 Three different conformers of free OBDA ligand obtained by geometry optimization. | ||
 | ||
| Fig. 11 Energy optimized structure of the Am(III) complex of OBDA. | ||
| Q(Am) | Q(O)amide | Q(C)amide | Q(O)ether | |||
|---|---|---|---|---|---|---|
| In complex | In free ligand | In complex | In free ligand | In complex | In free ligand | In complex |
| 1.582 | −0.60 ± 0.01 | −0.641 ± 0.007 | −0.412 | 0.714 | −0.572 | −0.570 ± 0.001 |
Conclusion
A conformationally constrained class of diamides (OBDAs) has been found to be an excellent extractant for tri- and tetra-valent actinides as well as trivalent lanthanides in nitric acid medium. The synthesis of extractants is achieved from cheap commercially available materials in three steps in high yields and purity. OBDA 1a showed excellent extraction behavior with fast kinetics, and high loading capacity for lanthanides without third phase formation, very low extraction of fission products and easy stripping of the extracted metal ions. Very poor extraction of Sr(II) and Ru(III) by the ligand is advantageous because no scrubbing was needed before stripping. This class of molecules forms a mixture of mono-solvated and di-solvated species with Am and Eu as obtained by the slope analysis method and ESI-MS. OBDA 1a was found to be sensitive to hydrolytic degradation. This detrimental effect has been surpassed by fast kinetics of complexation and hydrolytic stability of the complexed OBDA as supported by good DM values at 3–4 M HNO3 and stripping studies with 6 M HNO3. DFT study indicates the participation of both carbonyl and ethereal oxygens in the bonding with metal ion, which is also supported by FTIR and NMR studies.Experimental
Synthesis of diamides OBDA 1a–c
CH).
General procedure 1
Extraction studies
Solutions of desired concentration of OBDA 1a–c were prepared in n-dodecane (with 15% iso-decanol as the phase modifier) and agitated with an equal volume of the aqueous phase (containing the requisite quantity of 239Pu/241Am tracer) in a rotary thermostated water bath for 5 minutes at 25.0 ± 0.1 °C. The two phases were then centrifuged and assayed by taking suitable aliquots from both the phases. The distribution ratio (DM) is defined as the ratio of concentration of metal ion in the organic phase to that in the aqueous phase. The valency of Pu during extraction was maintained as Pu(IV). For the preparation of Pu(IV) stock, NaNO2 was used as an oxidant in 1 M HNO3 solution. The oxidized Pu(IV) solution was subsequently extracted by 0.5 M 2-thenoyltrifluoroacetone (TTA) in xylene where Pu(IV) is quantitatively extracted. The loaded Pu was stripped by 7 M HNO3 and equilibrated 3 times with xylene to remove the dissolved TTA from the aqueous phase. The stripped Pu solution was used as stock for Pu(IV). For extraction studies of various fission products, HLLW solution generated from PUREX process was diluted in 3 M HNO3 and used as feed. HLLW originating from a research reactor fuel reprocessing plant from a particular batch having the composition of uranium (predominantly 238U) 7.51 g L−1, plutonium (predominantly 239Pu) 3.19 mg L−1, 137Cs 8.89 Ci L−1, 106Ru 7.99 Ci L−1, 144Ce 27.75 Ci L−1, 90Sr 4.0 Ci L−1, 125Sb 0.2 Ci L−1, 95Nb 14.32 mCi L−1 was used. In view of the high activity of HLLW, it was diluted by a factor of 100 to bring down the activity to a measurable level. Consequently, the activity of the radionuclide 152+154Eu became too low to determine and hence, had to be spiked. Owing to the pure β activity of 90Sr present in the diluted solution, 85+89Sr was spiked where a gamma emitter, namely, 85Sr, was used as a tracer. The acidity was finally adjusted to 3 M HNO3. All distribution studies were carried out in duplicate and the data were reproducible within an error limit of ±5%.Computational methods
Gas phase geometries of the free ligand (OBDA) and its Am3+ complex were optimized at the GGA level of density functional theory (DFT) by using Becke's exchange functional11a in conjunction with Perdew's correlation functional11b (BP86) with the generalized gradient approximation (GGA) where 60 electron core pseudopotentials (ECPs) along with the corresponding def-SV(P) basis set were selected for the Am3+ ion. All other lighter atoms were treated at the all electron (AE) level. All the calculations were performed using the TURBOMOLE program package.11c,dNotes and references
- J. L. Swanson, in Science and Technology of tributyl phosphate, ed. W. W. Schultz, L. L. Burger, J. D. Navratil and K. P. Bender, CRC Press Inc., Boca Raton, 1984, vol. III Search PubMed.
- (a) Actinide and Fission Product Partitioning and Transmutation: Status and Assessment report, OECD/NEA, Paris, 1999; (b) Potential Benefits and Impacts of Advanced Nuclear Fuel Cycles with Actinide Partitioning and Transmutation, in Nuclear Science, OECD/NEA, Paris, 2011 Search PubMed; (c) V. Anastasov, M. Betti, F. Boisson, F. Depisch, F. Houlbreque, R. Jeffree, I. Khamis, S. Lattemann, J. C. Miquel, S. Nisan and P. K. Tewari, Status of Minor Actinide Fuel Development, IAEA Nuclear Energy Series No. NF-T-4.6, 2009; (d) J. O. Denschlag, M. C. Duijvestijn, Th. Ethvignot, F. J. Hambsch, J. Katakura, Yu. V. Kibkalo, M. Lammer, T. Liu, V. M. Maslov, R. W. Mills, A. C. Wahl and S. V. Zhdanov, Fission Product Yield Data for the Transmutation of Minor Actinide Nuclear Waste, IAEA-TECDOCSTI/PUB/1286, 2008; (e) V. Smirnov, V. Sobolev, J. Somers, R. Srivenkatesan, A. Stanculescu, V. Subbotin, A. Surenkov, T. Suzuki, M. Szieberth, S. Taczanowski, J. Tommasi, I. Tretiakov, K. Tucek, J. Uhlir, D. Vidovic, H. Wider, S. Wang, Y. Wu, R. Zakirov, Q. Zeng and S. Zheng, Advanced Reactor Technology Options for Utilization and Transmutation of Actinides in Spent Nuclear Fuel, IAEA-TECDOC-1626, 2009.
- (a) L. B. Kumbahre, D. R. Prabhu, G.R. Mahajan, S. Sriram, V. K. Manchanda and L. P. Badheka, Nucl. Technol., 2002, 139, 253 Search PubMed; (b) V. K. Manchanda and P. N. Pathak, Sep. Purif. Technol., 2004, 35, 85 CrossRef CAS PubMed; (c) G. R. Mahajan, D. R. Prabhu, V. K. Manchanda and L. P. Badheka, Waste Manage., 1998, 18, 125 CrossRef CAS.
- (a) X. Liu, J. Liang and J. Xu, Solvent Extr. Ion Exch., 2004, 22, 163 CrossRef CAS; (b) S. Tachimori and H. Nakamura, J. Nucl. Sci. Technol., 1982, 19, 326 CrossRef; (c) E. P. Horwitz, D. G. Kalina, L. Kaplan, G. W. Mason and H. Diamond, Sep. Sci. Technol., 1982, 17, 1261 CrossRef CAS PubMed; (d) E. P. Horwitz and D. G. Kalina, Solvent Extr. Ion Exch., 1984, 2, 179 CrossRef CAS PubMed; (e) J. N. Mathur, M. S. Murli, P. R. Natarajan, L. P. Badheka, A. Banerji, A. Ramanujam, P. S. Dhami, V. Gopalakrishnan, R. K. Dhumwad and M. K. Rao, Waste Manage., 1993, 13, 317 CrossRef CAS.
- (a) Y. Sasaki, Y. Sugo, S. Suzuki and S. Tachimori, Solvent Extr. Ion Exch., 2001, 19, 91 CrossRef CAS; (b) S. A. Ansari, P. N. Pathak, V. K. Manchanda, M. Hussain, A. K. Prasad and V. S. Parmar, Solvent Extr. Ion Exch., 2005, 23, 463 CrossRef CAS; (c) G. Modolo, H. Asp, C. Schreinemachers and H. Vijgen, Solvent Extr. Ion Exch., 2007, 25, 703 CrossRef CAS PubMed; (d) S. J. Ansari, P. Pathak, P. K. Mohapatra and V. K. Manchanda, Chem. Rev., 2012, 112, 1751 CrossRef CAS PubMed; (e) S. Manohar, J. N. Sharma, B. V. Shah and P. K. Wattal, Nucl. Sci. Eng., 2007, 156, 96 CAS; (f) P. Deepika, K. N. Sabharwal, T. G. Srinivasan and P. R. Vasudeva Rao, Solvent Extr. Ion Exch., 2010, 28, 184 CrossRef CAS PubMed; (g) Z. X. Zhu, Y. Sasaki, H. Suzuki, S. Suzuki and T. Kimura, Anal. Chim. Acta, 2004, 527, 163 CrossRef CAS PubMed; (h) H. Suzuki, Y. Sasaki, Y. Sugo, A. Apichaibuol and T. Kimura, Radiochim. Acta, 2004, 92, 463 CrossRef CAS; (i) V. Kumar, A. Kumar, S. Mondal, J. N. Sharma, R. C. Hubli, P. K. Wattal and A. K. Suri, Sep. Purif. Technol., 2012, 98, 118 CrossRef CAS PubMed; (j) D. Magnusson, B. Christiansen, J. P. Glatz, R. Malmbeck, G. Modolo, D. Serrano-Purroy and C. Sorel, Solvent Extr. Ion Exch., 2009, 27, 26 CrossRef CAS PubMed; (k) J. N. Sharma, R. Ruhela, K. N. Harindaran, S. L. Mishra, S. K. Tangri and A. K. Suri, J. Radioanal. Nucl. Chem., 2008, 278, 173 CrossRef CAS PubMed.
- (a) J. Ravi, A. S. Suneesh, T. Prathibha, K. A. Venkatesan, M. P. Antony, T. G. Srinivasan and P. R. Vasudeva Rao, Solvent Extr. Ion Exch., 2011, 29, 86 CrossRef CAS PubMed; (b) D. Jańczewski, D. N. Reinhoudt, W. Verboom, C. Hill, C. Allignol and M. T. Duchesne, New J. Chem., 2008, 32, 490 RSC; (c) M. T. Murillo, A. G. Espartero, J. Sánchez-Quesada, J. de Mendoza and P. Prados, Solvent. Extr. Ion Exch., 2009, 27, 107 CrossRef CAS PubMed.
- (a) M. Iqbal, J. Huskens, W. Verboom, M. Sypula and G. Modolo, Supramol. Chem., 2010, 22, 827 CrossRef CAS PubMed; (b) Y. Sasaki, Y. Kitatsuji, Y. Sugo, Y. Tsubata, T. Kimura and Y. Morita, Solvent Extr. Res. Dev., 2012, 19, 51 CrossRef CAS; (c) Y. Sasaki, M. Ozawa, T. Kimura and K. Ohashi, Solvent Extr. Ion Exch., 2009, 27, 378 CrossRef CAS PubMed; (d) L. Wu, Y. Fang, Y. Jia, Y. Yang, J. Liao, N. Liu, X. Yang, W. Feng, J. Ming and L. Yuan, Dalton Trans., 2014, 43, 3835 RSC; (e) P. K. Mohapatra, A. Sengupta, M. Iqbal, J. Huskens, S. V. Godbole and W. Verboom, Dalton Trans., 2013, 42, 8558 RSC.
- To the best of our knowledge, this type of diamide hydrolysis in acid media is not reported. But, the amide hydrolysis by the carboxy-group of substituted maleamic acids is known. See: A. J. Kirby and P. W. Lancaster, J. Chem. Soc., Perkin Trans. 2, 1972, 1206 RSC.
- For this study OBDA 1c was chosen because of its relatively simpler 1H NMR spectra.
- Synthetic procedure for complex of Eu(NO3)3·5H2O with OBDA 1c: A solution of Eu(NO3)3·5H2O (85.6 mg, 0.2 mmol, 1 eq.) in a mixture of CH2Cl2/MeOH (90:10) was added to a solution of OBDA 1c (326 mg, 0.8 mmol, 4 eq.) in CH2Cl2. The reaction mixture was stirred at room temperature for 2 d. The resulting solution was evaporated and the residue was triturated with petrol several times to remove excess OBDA 1c. The residual product is the complex of Eu with OBDA 1c which was used for all the analyses.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822 CrossRef; (c) TURBOMOLE is a program package developed by the Quantum Chemistry Group at the University of Karlsruhe, Germany, 1988 Search PubMed; (d) R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt01691f |
| This journal is © The Royal Society of Chemistry 2015 |