
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The nitration pattern of energetic 3,6-diamino-1,2,4,5-tetrazine derivatives containing azole functional groups†
A.
Aizikovich
,
A.
Shlomovich
,
A.
Cohen
and
M.
Gozin
*
School of Chemistry, Faculty of Exact Sciences, Tel-Aviv University, Tel-Aviv 69978, Israel. E-mail: cogozin@gmail.com; Tel: +972-3-640-5878
First published on 25th June 2015
Abstract
One of the successful strategies for the design of promising new energetic materials is the incorporation of both fuel and oxidizer moieties into the same molecule. Therefore, during recent years, synthesis of various nitro-azole derivatives, as compounds with a more balanced oxygen content, has become very popular. In the framework of this effort, we studied nitration of N3,N6-bis(1H-tetrazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine (BTATz; 5) and its alkylated derivative N3,N6-bis(2-methyl-2H-tetrazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine 12, using a 15N-labeled nitration agent and monitoring and analyzing products of these reactions by 15N NMR. It was seen that the nitration of both compounds takes place only on the exocyclic (“bridging”) secondary amine groups. Possible tetranitro derivative isomers N,N′-(1,2,4,5-tetrazine-3,6-diyl)bis(N-(1-nitro-1H-tetrazol-5-yl)-nitramide) 6 and N,N′-(1,2,4,5-tetrazine-3,6-diyl)bis(N-(2-nitro-2H-tetrazol-5-yl)nitramide) 7, both of which have OB = 0% and calculated VODs of 9790 and 9903 m s−1, respectively, could not be observed in the reaction mixtures, during the in situ15N NMR monitoring of nitration of 5, using 15N-labeled nitrating agents. Following a similar strategy, a new analog of BTATz – N3,N6-Bis(1H-1,2,4-triazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine 15 was obtained and its nitration was studied. The reaction of 15 with a HNO3–Ac2O nitration mixture resulted in the formation of a new N3,N6-bis(3-nitro-1H-1,2,4-triazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine derivative 20 in a moderate yield. Structures and properties of 15 (in the form of its perchlorate salt, 16) and 20 were measured by FTIR, multinuclear NMR, MS, DSC and X-ray crystallography. It is important to note that compound 20 exhibits exothermic decomposition at 302 °C (DSC) and >353 N (sensitivity to friction), making it a highly-promising thermally-insensitive energetic material for further development.
Introduction
The history of the discovery and development of new energetic materials goes back to gunpowder, which was invented in Imperial China around the 9th century, in an attempt to create an immortality potion.1 This ground-breaking discovery led to the invention of fireworks and a variety of weapons in China and, later, throughout the world.2 Since the earliest findings, tremendous progress has been made in the rational design and preparation of novel energetic materials and compositions.3 Important properties of such materials, which are commonly taken into account by the designers are span heat of formation (HOF), density, melting and decomposition temperatures, sensitivity to friction and shock, velocity of detonation (VOD), synthetic complexity,4 as well as carbon content, nitrogen content and oxygen balance (OB).5Energetic materials typically contain both oxidizing and reducing functional groups in their molecular structure (or in the structure of their components). Under high temperature and pressure conditions, these materials would transform into more thermodynamically-stable products, including small molecules with low heats of formation, such as H2O, N2, CO, CO2, SO2 and metal oxides.6 The OB is a mathematical formula used to calculate the degree to which a given explosive or propellant could be oxidized. A “zero” OB value will be calculated when the chemical composition of the calculated energetic material will have the exact amount of oxygen atoms needed to convert all the carbon atoms to CO2, all hydrogen atoms to H2O, all sulphur atoms to SO2 and all metal atoms (if present in the material) to metal oxides. An energetic material would have a positive OB value if it contains more oxygen atoms than required for complete combustion and a negative OB value when the amount of oxygen atoms is insufficient for complete oxidation. The results of OB calculations were shown to have an excellent correlation with both sensitivity properties and the performance of energetic compounds and their formulations, which have a tendency to reach their best values as their OB values get closer to “zero”.7 When an energetic material has a negative OB value (an insufficient amount of oxygen for complete oxidation), it will typically exhibit an incomplete combustion, resulting in the formation of large amounts of toxic CO gas, smoke, soot and solid residues. Commonly, as the OB values for a certain explosive get lower, poorer performance for this explosive is observed, and the VOD and generated pressure for this explosive also become smaller.
In cases where an energetic material contains “too much oxygen” (has a positive OB value), the O2 produced during explosion absorbs a significant amount of energy, substantially reducing its explosive performance.8
There are several fascinating examples of energetic compounds possessing an OB value of 0%, such as the most potent chemical explosive known – octanitrocubane (ONC; R.E. factor = 2.38).9 Other examples include the recently prepared compound nitryl cyanide10 and the still synthetically-elusive nitrogen-rich compounds – 3,6-dinitro-1,2,4,5-tetrazine,11 2,4,6-trinitro-1,3,5-triazine,12 [1,2,3,4]-tetrazino-[5,6-e][1,2,3,4]-tetrazine-1,3,5,7-tetraoxide (TTTO),13 and (5-nitro-2H-tetrazol-2-yl)-methyl nitrate (Fig. 1).14
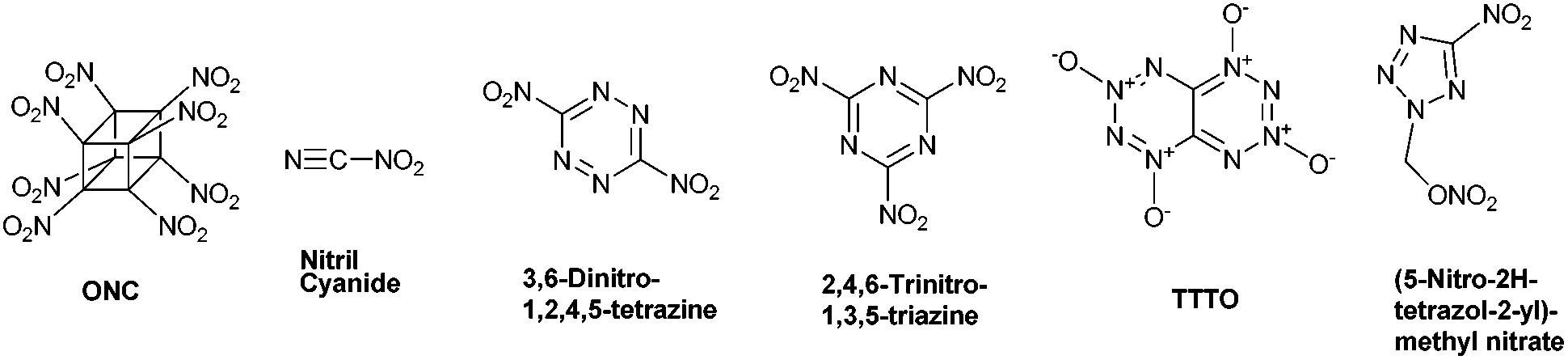 | ||
| Fig. 1 Structures of ONC, nitryl cyanide, 3,6-dinitro-1,2,4,5-tetrazine, 2,4,6-trinitro-1,3,5-triazine, TTTO and (5-nitro-2H-tetrazol-2-yl)-methyl nitrate. | ||
Due to their high density, highly positive enthalpy of formation, good detonation performances and excellent thermal stability in comparison with the conventional energetic materials, nitrogen-rich energetic compounds attract considerable scientific attention.15 The latter compounds were extensively explored during recent years for their great potential for both civilian and military applications such as in gas generators, low-signature propellants, as well as additives to pyrotechnics and explosives.16 Among popular building blocks used in the construction of nitrogen-rich energetic materials are tetrazole17 and tetrazine18 functional groups. However, the majority of nitrogen-rich organic compounds are oxygen-deficient. Since one of the most popular strategies for the design of promising new energetic materials is incorporating both fuel and oxidizer properties into a single molecule, one of the important challenges in this field is the preparation of new nitrogen-rich compounds with a low OB value. Therefore, in recent years, the synthesis and evaluation of various nitro-azole derivatives as compounds with a balanced oxygen content has become highly-popular (Fig. 2).19
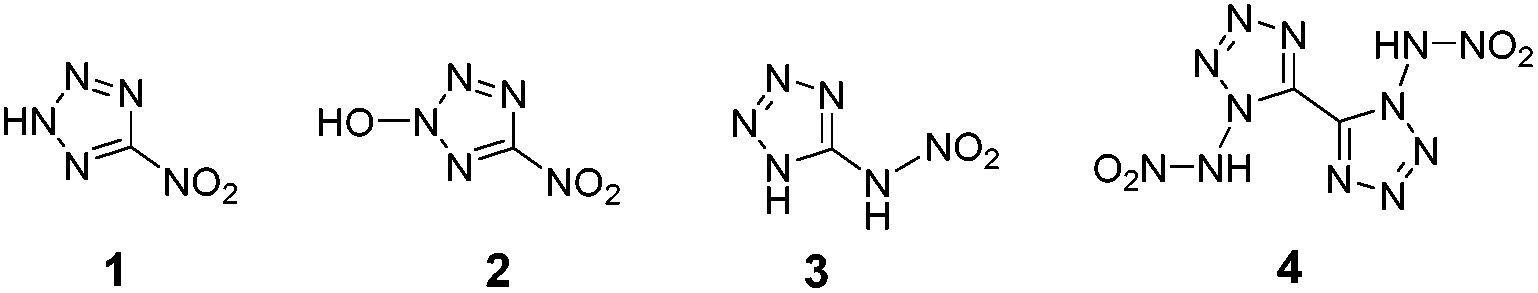 | ||
| Fig. 2 Structures of compounds 1 (OB = −7.0%), 2 (OB = +6.1%), 3 (OB = −12.3%) and 4 (OB = −3.1%). | ||
Results and discussion
In our perspective, as part of the framework of these efforts, one of the interesting challenges was exploring the synthesis of unreported tetra-nitro derivatives of N3,N6-bis(1H-tetrazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine (BTATz 5; OB = −64.5%)20 – isomers N,N′-(1,2,4,5-tetrazine-3,6-diyl)bis(N-(1-nitro-1H-tetrazol-5-yl)-nitramide) 6 and N,N′-(1,2,4,5-tetrazine-3,6-diyl)bis(N-(2-nitro-2H-tetrazol-5-yl)nitramide) 7, both of which have OB values of 0% and calculated VODs of 9790 and 9903 m s−1, respectively (Fig. 3, Table 1). Although it would seem that BTATz analogues exhibiting better OB values should be of significant interest, only a single example of such derivatives – 1,4-di-N-oxide 8 (OB = −45.7%) – was prepared by Chaves and coworkers.21a Unfortunately, compound 8 displayed decomposition at 134 °C (versus decomposition of BTATz at 318 °C), indicating that other derivatisation strategies should be investigated.21b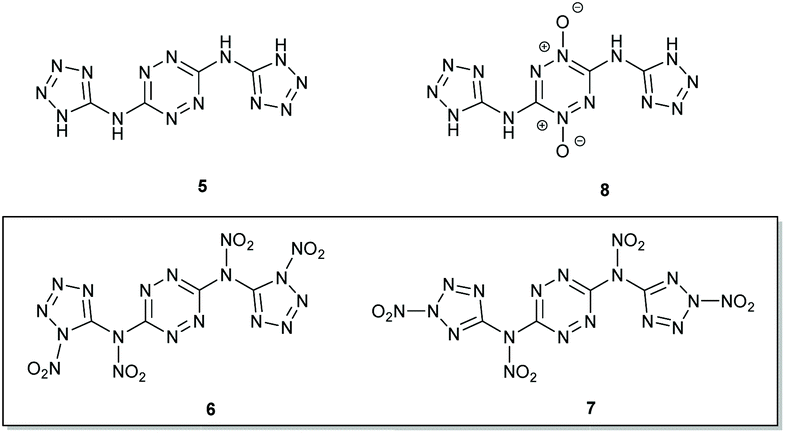 | ||
| Fig. 3 Structures of compounds 5, 6, 7 and 8. | ||
| D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | D–H⋯A/° |
|---|---|---|---|---|
| N1–H1⋯O4 | 0.83 | 2.20 | 2.9669 | 153.7 |
| N1–H1⋯O1 | 0.83 | 2.799 | 2.973 | 93.8 |
| N2–H2⋯O2 | 0.87 | 2.10 | 2.9401 | 162.1 |
| N6–H3⋯O1 | 0.80 | 2.06 | 2.8303 | 163 |
| N1–H1⋯N4 | 0.83 | 2.265 | 2.732 | 116 |
Approaches to prepare energetic molecules with improved OB values are frequently based on the conversion of NH groups in the starting materials into N–NO2 groups in the corresponding, more energetic, derivatives.22 Since the BTATz molecule has two pairs of NH groups in its structure, we decided to probe whether the nitration of this molecule would lead to the development of more potent energetic compounds (ultimately, to compounds 6 and 7). There are many methods for the conversion of amines to nitramines: using nitrating agents such as HNO3, mixtures of HNO3 and H2SO4, acetic anhydride and HNO3, nitrated silica gel and many others.23 Thus, our initial efforts were focused on the direct nitration of compound 5 under various reaction conditions. More specifically, we evaluated a series of nitrating conditions and temperature regimes which included the use of red fuming HNO3 and mixtures of either HNO3 and H2SO4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), HNO3 and acetic anhydride (1:1 v/v) or HNO3 and trifluoroacetic anhydride (1:1 v/v). However, all examined reaction conditions (and all examined temperature regimes) led to one of the two results: recovery of only the starting compound 5 at the end of the reaction or decomposition of 5.
:1 v/v), HNO3 and acetic anhydride (1:1 v/v) or HNO3 and trifluoroacetic anhydride (1:1 v/v). However, all examined reaction conditions (and all examined temperature regimes) led to one of the two results: recovery of only the starting compound 5 at the end of the reaction or decomposition of 5.
We further attempted to establish whether any nitration of compound 5 actually takes place, with the formed nitramines hydrolysing back to the starting material upon dilution of the purpose, we conducted in situ studies of the nitration of 5 by 15N NMR using Na15NO3 in concentrated H2SO4. A reference mixture of Na15NO3 in concentrated H2SO4 exhibited two 15N NMR signals at 383 ppm and 248 ppm, indicating the presence of 15NO3− and 15NO2+, respectively, and was consistent with previous reports.24 Subsequent addition of BTATz to this nitration mixture at 0 °C resulted in the appearance of a new 15N NMR signal at 336 ppm, which was assigned to the formation of a N–NO2 adduct. Although it was obvious that, under the explored conditions, no further nitration of 5 could be obtained, it was not clear which amine group in this compound underwent nitration (Fig. 4). Since the in situ15N NMR measurements did not help us to determine whether edocyclic 9/10 (routes A and B, Fig. 4) or exocyclic 11 (route C, Fig. 4) nitramines were formed, and since the resulting product could not be isolated for further analysis due to its instability, other approaches were required. Therefore, we synthesized a model compound – N3,N6-bis(2-methyl-2H-tetrazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine 12 – in which both endocyclic NH groups in the tetrazole rings were methylated (Fig. 5). The successful conversion of 5 to 12 was achieved by using dimethyl-sulfate in aqueous NaHCO3 at room temperature. A comparison of 1H and 13C NMR data obtained for 12 with published reports for 2-methyl-2H-tetrazole derivatives (versus 1-methyl-1H-tetrazole derivatives) strongly suggested that the methylation of 5 took place on the second nitrogen atom of the tetrazole ring.
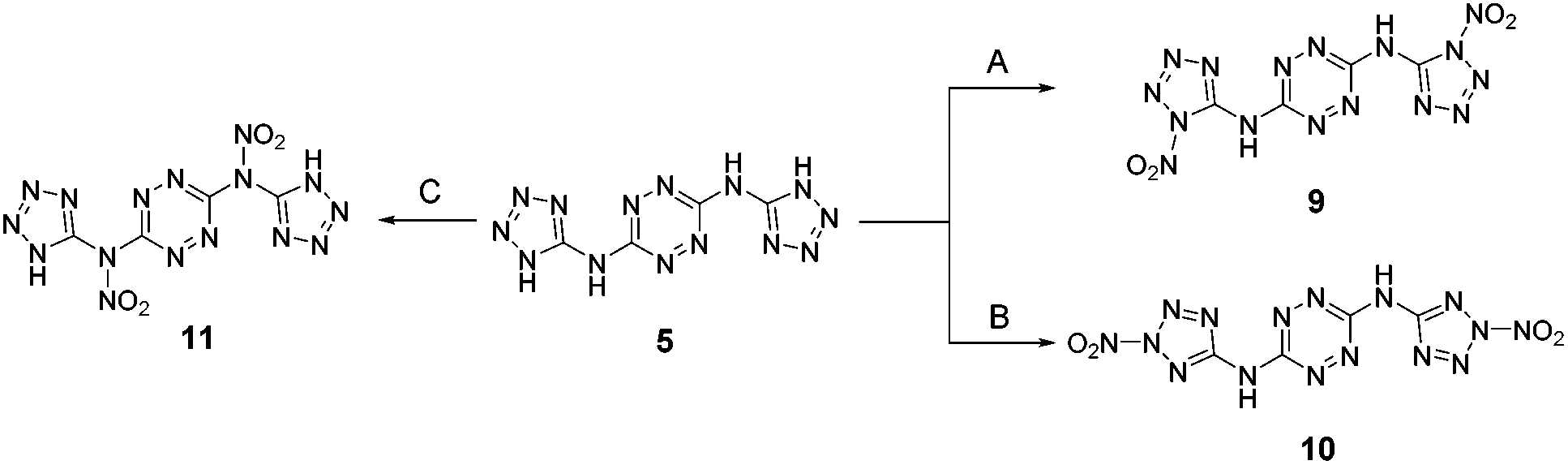 | ||
| Fig. 4 Possible nitrated derivatives of compound 5. Reaction conditions: Na15NO3, conc. H2SO4. | ||
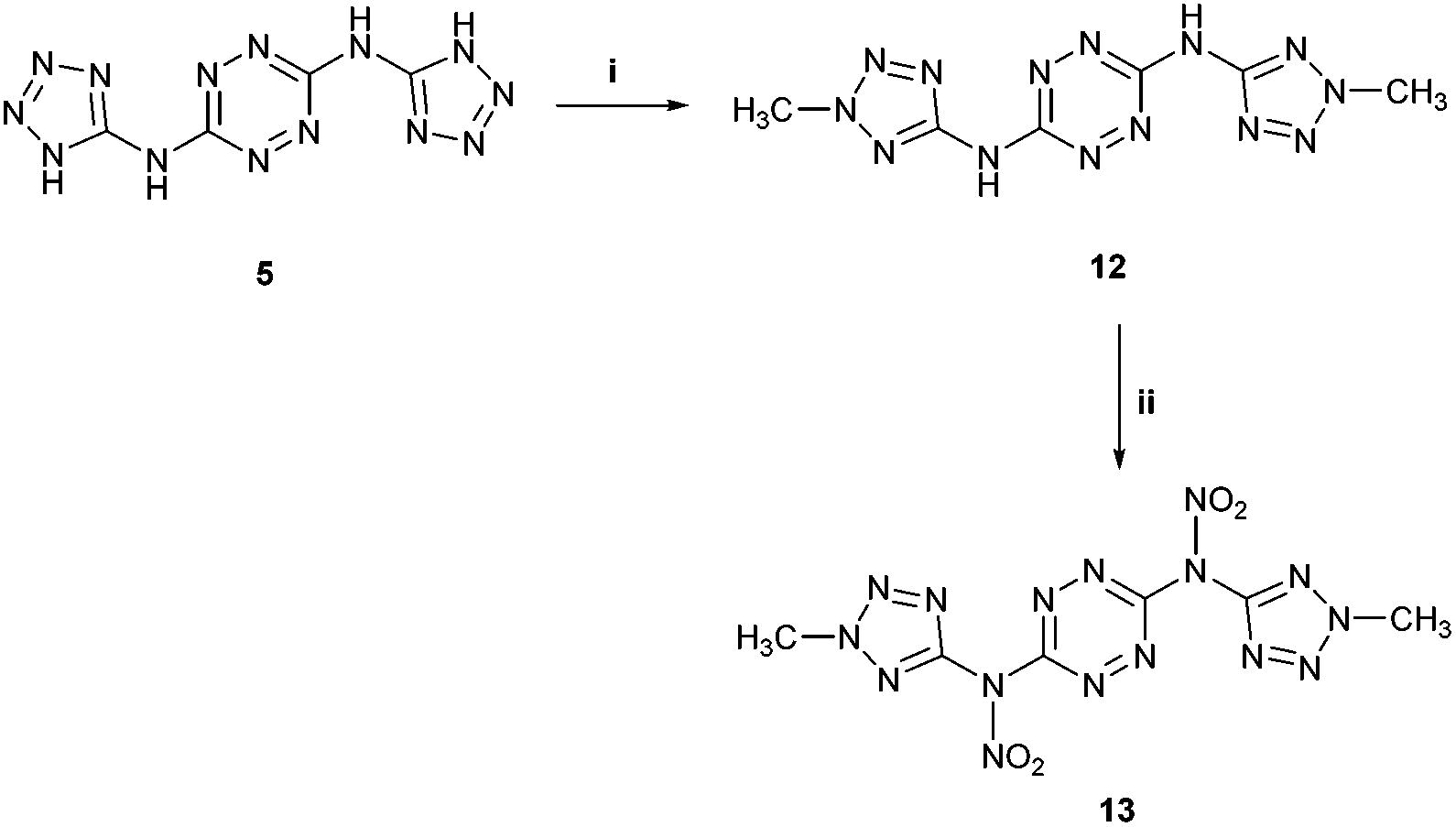 | ||
| Fig. 5 Synthesis of bis-N-methylated compound 12, with its subsequent nitration. Reaction conditions: (i) dimethylsulfate, NaHCO3, H2O, r.t. (ii) conc. HNO3, Ac2O. | ||
Further nitration of 12 with a mixture of concentrated HNO3 and acetic anhydride (1:2 v/v) resulted in the formation of the new compound N,N′-(1,2,4,5-tetrazine-3,6-diyl)-bis(N-(2-methyl-2H-tetrazol-5-yl)-nitramide 13 (Fig. 5). Unfortunately, nitramide 13 could not be fully characterized due to its very high sensitivity to impact and friction (primary explosive!). Also, based on 1H NMR analysis in a solution of DMSO-d6 or CD3CN, 13 underwent relatively quick hydrolysis back to the parent compound 12. 13C and 15N NMR studies in DMSO-d6 of the precipitate obtained in the nitration of 12 with a mixture of Na15NO3/HNO3 (prepared separately) and acetic anhydride also showed only the presence of the starting material 12. Yet, in situ15N NMR studies of the nitration of 12 with Na15NO3 in concentrated H2SO4 exhibited a new peak at 336 ppm (at the identical position of the nitramine's nitrate peak in compound 11), strongly supporting our hypothesis that both compounds 5 and 12 undergo nitration on their exocyclic NH groups.
After realizing that the nitration of 5 (and its bis-N-methyl-tetrazole analog 12) could not lead to the formation of stable nitramine products, we explored whether the nitration of the unreported 1,2,4-triazole analog of compound 5 – N3,N6-bis(1H-1,2,4-triazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine 15 would produce better results. The synthesis of compound 15 was achieved in an 81% yield, in a similar fashion to the synthesis of 5, by reacting 3,6-bis(3,5-dimethyl-1H-pyrazol-1-yl)-1,2,4,5-tetrazine (BPT, 14) with 1H-1,2,4-triazole-5-amine in sulfolane at 135 °C (Fig. 6). The corresponding, more soluble, energetic perchlorate salt 16 and nitrate salt 17 were prepared by treating 15 with HClO4 and HNO3, respectively (Fig. 6). The structure of 16 was confirmed by X-ray crystallography (Fig. 9). Subsequently, a direct nitration of compound 15 was explored under various reaction conditions, which included either HNO3, a mixture of H2SO4 and HNO3 (1:1; v/v), a mixture of CF3CO2H and HNO3 (1:1; v/v) or a mixture of NaNO3 and H2SO4. Under all tested temperature regimes, all aforementioned nitrating reagents led invariably to the formation of stable nitrate salt 17.
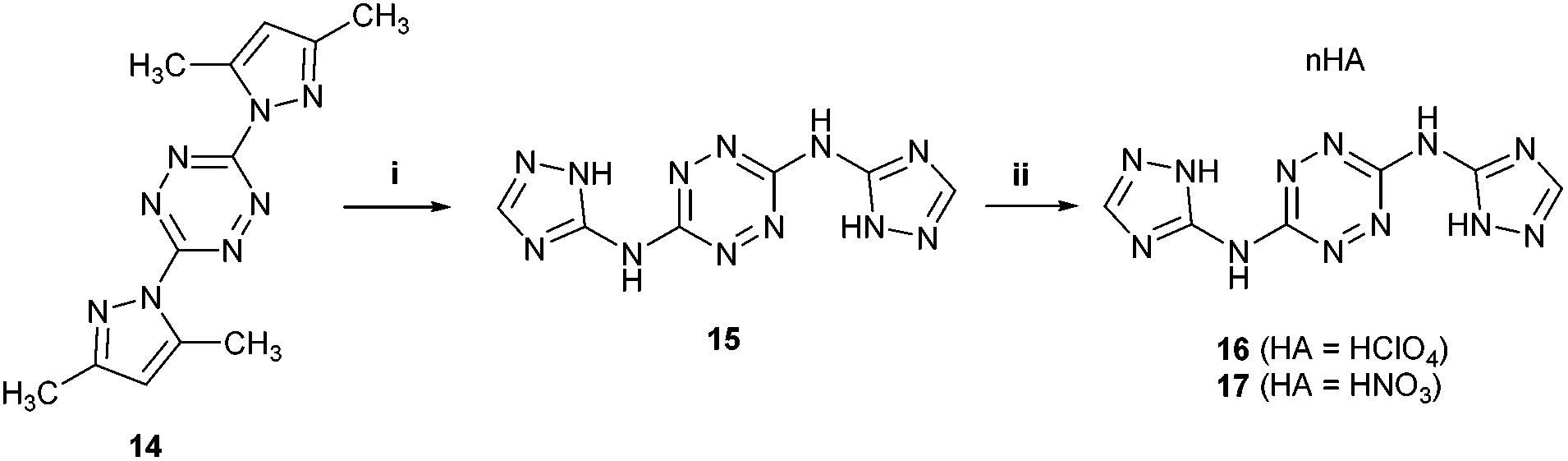 | ||
| Fig. 6 Preparation of compound 15 and its perchlorate salt 16 and nitrate salt 17. Reaction conditions: (i) 1H-1,2,4-triazole-5-amine, sulfolane, 135 °C; (ii) HClO4 or HNO3. | ||
In situ
15N NMR studies of the nitration of 15 with Na15NO3 in concentrated H2SO4 (at 0 °C) displayed a peak at 336 ppm (at the identical position of the nitramine's nitrate peak in compound 13), clearly indicating that the first nitration reaction takes place on the exocyclic NH groups of 15 and not on the CH or NH groups of its triazole rings (Fig. 7). This interesting observation demonstrates a similar nitration pattern for structurally related compounds 5, 12 and 15. We believe that similarly to 11 and 13, attempts for isolation of compound 18 result in a quick hydrolysis of the exocyclic nitramine group back to the original amine. In contrast to previous attempts, the nitration of 15 with a mixture of HNO3 and acetic anhydride (1:1; v/v) at 0 °C resulted in the formation of a new nitramine – N,N′-(1,2,4,5-tetrazine-3,6-diyl)bis(N-(3-nitro-1H-1,2,4-triazol-5-yl)-nitramide) 19, which was isolated by precipitation (Fig. 8). The latter compound could not be fully characterized due to its high sensitivity to impact and friction (primary explosive!). The solubility properties, sensitivity and thermal behaviour of 19 suggested its analogous structure to the exocyclic nitramine 13. Controlled gradual heating of approximately 0.2 mg of wet 19 in a glass capillary (suitable for melting point measurements) resulted via its explosion at 110–112 °C, shuttering the capillary.
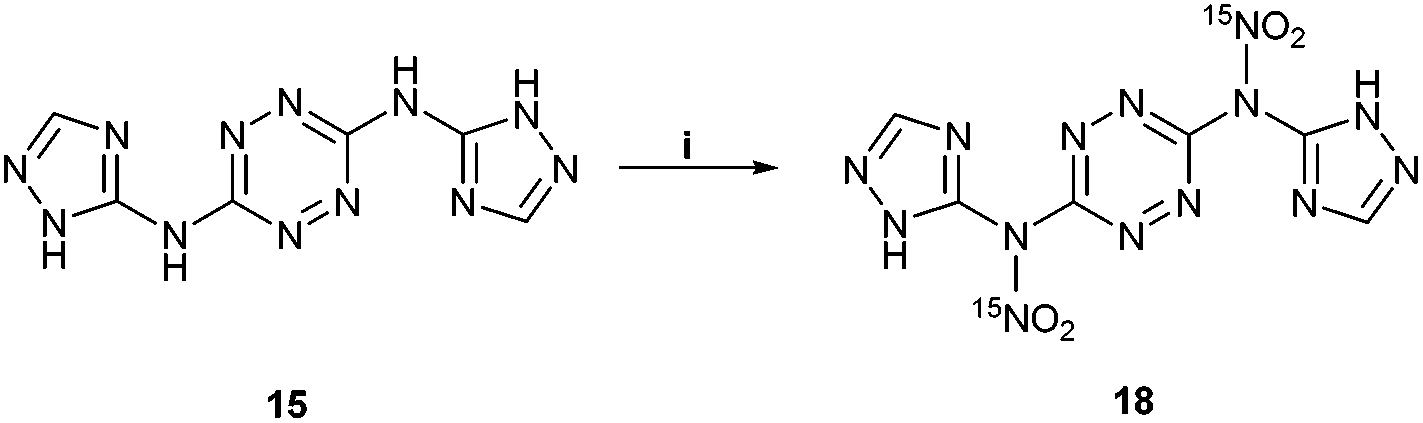 | ||
| Fig. 7 Formation of the proposed compound 18. Reaction conditions: (i) Na15NO3, H2SO4. | ||
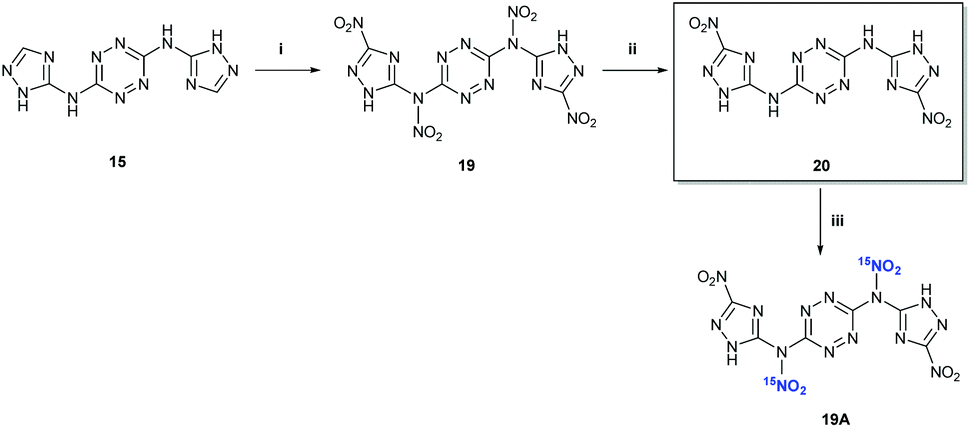 | ||
| Fig. 8 Synthesis of compounds 19, 20 and 19A. Reaction conditions: (i) conc. HNO3, Ac2O; (ii) CH3CN, 65 °C; (iii) Na15NO3/HNO3, Ac2O. | ||
15N NMR studies of the precipitate 19 (dissolved in DMSO-d6), obtained from nitration of 15 with a mixture of Na15NO3/HNO3 and acetic anhydride, showed the appearance of a new peak at 356.8 ppm (corresponding to C–15NO2 nitrogen), indicating the formation of a new derivative with nitrated triazole rings N3,N6-bis(3-nitro-1H-1,2,4-triazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine 20. No exocyclic nitramine derivatives (N–15NO2) of compound 15 were observed upon dissolution of 19 in DMSO-d6, most reasonably due to the hydrolysis of 19 into the hydrolytically stable 20. Also, the dissolution of 19 in hot CH3CN (with further heating at 65 °C for 30 min) immediately after its separation from the nitration reaction mixture, resulted in the formation of a new compound – N3,N6-bis(3-nitro-1H-1,2,4-triazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine 20 (Fig. 8). The structure of 20 was confirmed by X-ray crystallography (Fig. 10).
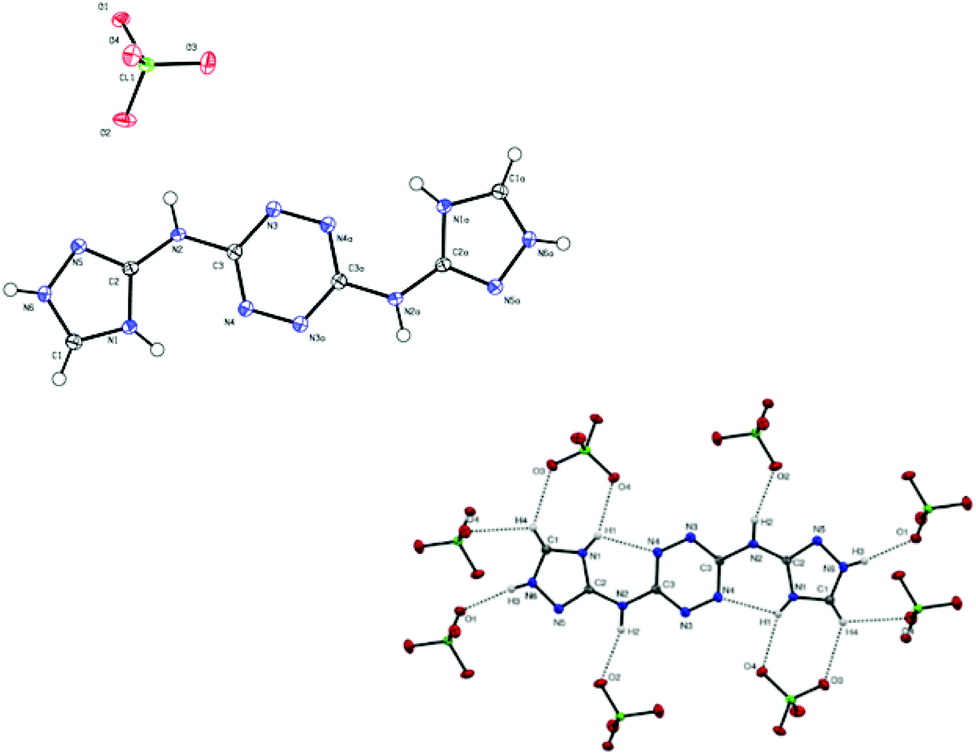 | ||
| Fig. 9 (top): Molecular structure of compound 16, (bottom): interactions between 16 and surrounding perchlorate anions. | ||
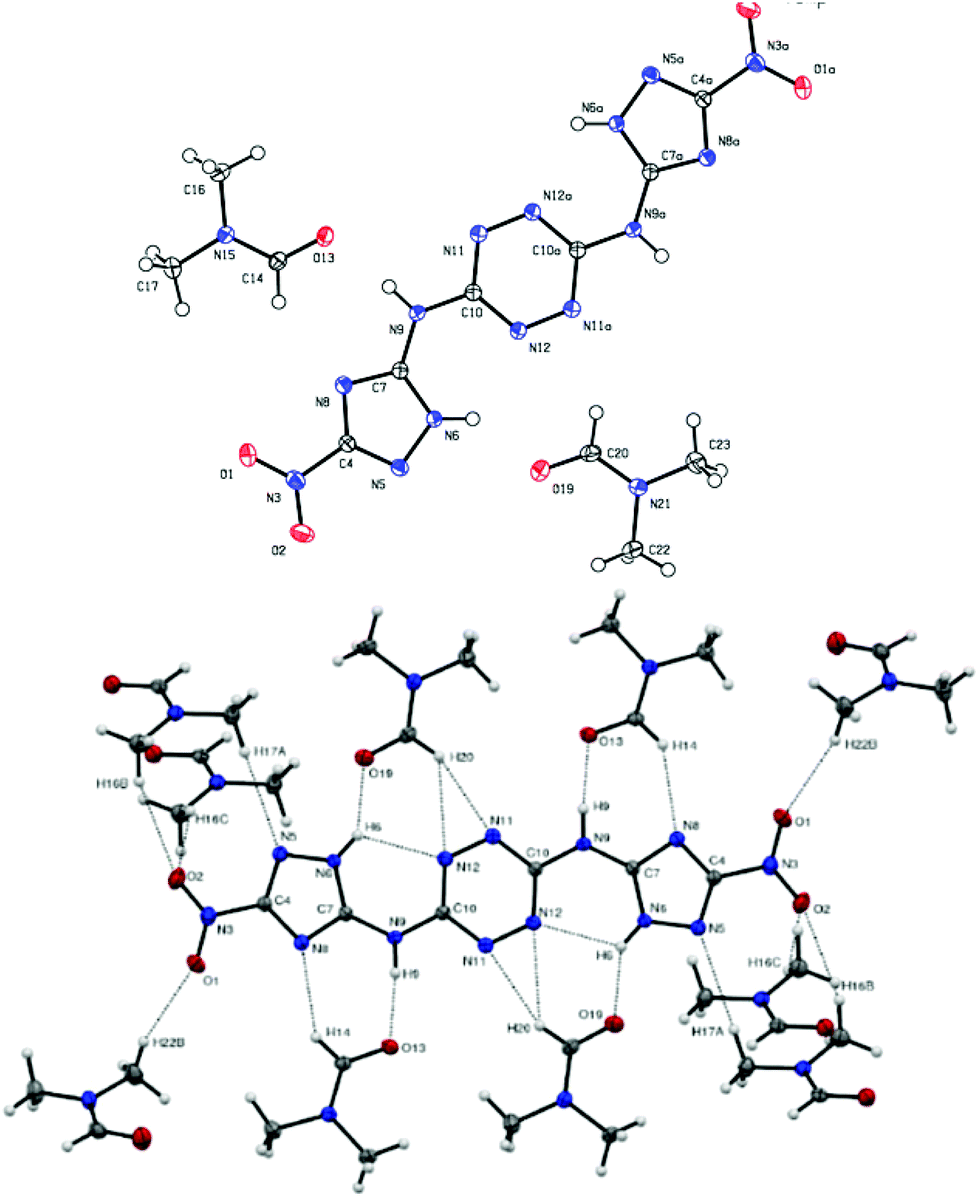 | ||
| Fig. 10 (top): Molecular structure of compound 20, (bottom): interactions between 20 and surrounding DMF molecules. | ||
In order to check whether the structure of 19 was correct, in situ15N NMR studies of the “back nitration” of 20 into exocyclic 15N nitramine 19A were conducted. In these experiments, a solid 20 was slowly added to a mixture of Na15NO3 in concentrated H2SO4 at 0 °C and, after 30 min, the reaction mixture was analysed by 15N NMR. A new peak at 336.3 ppm (corresponding to the exocyclic N–15NO2 nitrogen) appeared in the spectrum, indicating the formation of nitramine 19A (Fig. 8), which perfectly matches our previous observations and conclusions.
Crystal structures
X-ray measurements for single crystals of compounds 16 (perchlorate salt of compound 15; CCDC 1041768) and 20 (CCDC 1041769) were performed. The data for the crystals of 16 and 20 were collected using Mo Kα radiation (L = 0.71073 nm). An Oxford low-temperature device was used to keep the crystals at a constant temperature of 110 K during the entire data collecting period. Details of the X-ray data collection and structure refinements are summarized in the ESI.† A detailed examination of the crystal structures obtained for 16 and 20 showed no significant differences in bond lengths and torsion angles reported for structures of other compounds containing 1,2,4-triazole and 1,2,4,5-tetrazine functional groups.25 Both 16 and 20 molecules were found to be completely planar, due to an extensive delocalization of electrons in these molecules.Compound 16 was crystalized as solvent-free crystals with the monoclinic space group P2(1)/c and a cell volume of 757.49 Å3. A crystal unit cell of 16 contains eight molecules of nitrogen-rich cations and four perchlorate anions. The measured density for 16 was found to be 1.960 g cm−3. The nitrogen-rich cation is protonated at nitrogen atom N6 in both triazole rings. Fig. 9 shows interactions between hydrogen atoms in compound 16 and oxygen atoms in perchlorate anions (each nitrogen-rich cation interacts with eight perchlorate anions).
The hydrogen bonding parameters of these interactions are detailed in Table 1. The triazole moiety of 16 points towards nitrogen atom N6 and participates in an intramolecular hydrogen bond N1–H1⋯N4 with a D⋯A length of 2.732 Å and a D–H⋯A angle of 116°.
Compound 20 was crystalized as a monoclinic space group P2(1)/c and a cell volume of 1546.76 Å3 was calculated. The crystal unit cell of this compound contains eight molecules of 20 and ten molecules of DMF. The unit cell contains two additional disordered DMF molecules, which could not be reliably modelled by discrete atoms. Correspondingly, its contribution was subtracted from the diffraction pattern by the Squeeze technique, using the PLATON software.26 As a result, the density measured was 1.35 g cm−3, which is significantly lower than 1.87 g cm−3 – the calculated density for this compound.27Fig. 10 shows interactions between nitrogen and hydrogen atoms of compound 20 and molecules of DMF (each molecule of 20 interacts with 10 molecules of DMF). The bond length of the bridge corresponds to a C–N single bond (C10–N9 1.359 Å, N9–C7 1.370 Å). The triazole moiety points towards atom N6 and participates in an intramolecular hydrogen bond – N6–H6⋯N12 – which has a D⋯A length of 2.7781 Å and a D–H⋯A angle of 117° and an intermolecular hydrogen bond with the solvent (Table 2).
| D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | D–H⋯A/° |
|---|---|---|---|---|
| Symmetry codes: (i) 1 − x, −1/2 + y, 1/2 − z (ii) 1 + x, y, z (iii) −1 + x, y, z (iv) 1 − x, 1/2 + y, 1/2 − z (v) −1 + x, 1/2 − y, −1/2. | ||||
| N6–H6⋯N12 | 0.88 | 2.27 | 2.7781 | 117 |
| N6–H6⋯O19(i) | 0.88 | 1.94 | 2.6939 | 143 |
| N9–H9⋯O13(ii) | 0.88 | 1.86 | 2.7333 | 175 |
| C14–H14⋯N8(iii) | 0.95 | 2.43 | 3.2085 | 139 |
| C16–H16A⋯O13 | 0.98 | 2.46 | 2.8005 | 100 |
| C20–H20⋯N12(iv) | 0.95 | 2.58 | 3.2438 | 127 |
| C20–H20⋯N11(v) | 0.95 | 2.43 | 3.3178 | 155 = z |
Theoretical calculations, performance characteristics and thermal stabilities
Calculations were performed using the Gaussian 09 software.28 The geometric optimization of the structures and frequency analyses were carried out by using the B3LYP functional with the 6-311+G(d,p) basis set.29 The optimized structures of all materials correspond to, at least, a local energy minimum on the potential energy surface. In order to predict the energetic properties of new materials, the EXPLO5_v6.0130 software was used. This software utilizes an algorithm based on the Becker–Kistiakowsky–Wilson equation of state (BKW EOS) for gaseous detonation products. Thermal stability is a very important property of energetic materials. The onset temperatures for thermal decomposition of compounds 5, 12, 15, 16, 17 and 20 were determined using a differential scanning calorimeter (DSC). The results of these calculations and the experimental data are summarized in Table 3.| 5 | 6 | 7 | 9/10 | 11 | 12 | 13 | 15 | 16 | 17 | 18 | 19 | 20 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Nitrogen content. b Measured by DSC start of decomposition temperature. c Calculated density; (c*) density of dried powdered compounds was measured by gas pycnometry at 25 °C; (c**) density value that was obtained by a single-crystal X-ray diffraction. d Oxygen balance (for CaHbNcOdΩ = (a − 2b − 0.5d) × 1600/Mw). e Calculated enthalpy of formation. f Calculated energy of formation. g Calculated heat of detonation. h Calculated temperature of detonation i Calculated pressure of detonation. j Calculated volume of gaseous products. k Calculated velocity of detonation. | |||||||||||||
| Mw [g mol−1] | 248 | 428 | 428 | 338 | 338 | 276 | 366 | 246 | 447 | 372 | 336 | 426 | 336 |
| %Na [wt%] | 79 | 60 | 60 | 61.5 | 61.5 | 71 | 61 | 68 | 33.3 | 41.1 | 50 | 50 | 58 |
| T Dec [°C] | 309 | — | — | — | — | 306 | — | 350 | 246 | 167 | N/A | — | 302 |
| P [g cm−3] | 1.88 | 2.03 | 2.03 | 1.98 | 1.98 | 1.77 | 1.88 | 1.36* | 1.96** | 1.74* | 1.87 | 1.95 | 1.87 |
|
Ω
O2
[%] |
−64.6 | 0 | 0 | −23.6 | −23.6 | −92.6 | −48 | −97.5 | −25 | −43 | −47.6 | −18.7 | −47.6 |
| ΔHfe [kJ mol−1] |
918 | 1148 | 1334 | 113 | 964 | 1022 | 996 | 831 | −504 | −655 | 965 | 864 | 1032 |
| ΔU°ff [kJ kg−1] | 3789 | 2756 | 3190 | 3436 | 2933 | 3801 | 2808 | 3468 | −1056 | −1667 | 2952 | 2103 | 3151 |
| EXPLO5_v6.01 values: | |||||||||||||
| −ΔEU°g [kJ kg−1] |
4002 | 6048 | 6400 | 5492 | 5032 | 4185 | 5097 | 3728 | 2947 | 1921 | 5212 | 5315 | 5398 |
|
T
Eh [K] |
2905 | 4632 | 4856 | 4101 | 3872 | 2763 | 3597 | 2805 | 2630 | 1873 | 3782 | 4091 | 3891 |
| P C–J [kbar] | 301 | 443 | 456 | 415 | 394 | 265 | 333 | 124 | 251 | 161 | 329 | 390 | 339 |
| V Det . [m s−1] | 8809 | 9790 | 9903 | 9604 | 9429 | 8492 | 8932 | 6240 | 6711 | 6833 | 8830 | 9310 | 8903 |
| Gas vol.j [L kg−1] | 758 | 759 | 762 | 794 | 789 | 743 | 778 | 734 | 751 | 792 | 753 | 777 | 753 |
In contrast to compounds 15, 16 and 17, dinitro derivative 20 exhibited high thermal stability (Tdecomp. of 302 °C), high detonation velocity (VOD of 8903 m s−1) and high detonation pressure, performing comparably to BTATz 5. In addition, BAM measurements of dropping-hammer (7.67 Nm) and friction (>353 N) for compound 20 indicate a relatively high stability of this energetic material under mechanical stress (for comparison, the equivalent values for RDX are 7.5 Nm and 120 N, respectively).
Conclusions
A methodical study of nitration patterns was conducted on a series of 3,6-diamino-1,2,4,5-tetrazine derivatives, revealing the specificity of the nitration in this type of nitrogen-rich compounds, as well as the stability of the formed nitramines towards hydrolysis. The general motivation of this study was to evaluate whether energetic nitrogen-rich compounds, structurally-related to the N3,N6-bis(1H-tetrazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine (BTATz; 5), with improved oxygen balance characteristics, could be prepared. By using 15N-labeling methodology, we found that although nitramine derivatives of 5, as well as its dimethyl analogue N3,N6-bis(2-methyl-2H-tetrazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine (12), are formed in the reaction mixture, these nitramines could not be successfully isolated and quickly undergo hydrolysis back to the starting materials. Therefore, subsequent efforts were focused on more stable nitro derivatives of the newly synthesized compound N3,N6-bis(1H-1,2,4-triazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine (15). The nitration of 15 was studied by 15N NMR, showing a similar initial reactivity pattern of 5 and 12. Yet, due to the presence of 1,2,4-triazole functional groups in the structure of 15, a new C–NO2 derivative N3,N6-bis(3-nitro-1H-1,2,4-triazol-5-yl)-1,2,4,5-tetrazine-3,6-diamine (20) was obtained in a moderate yield. The structures of 16 (perchlorate salt of 15) and 20 (with 4 molecules of DMF) were determined by X-ray crystallography. Importantly, it was found that compound 20 exhibited high stability towards friction (>353 N) and very high thermal stability, decomposing at 302 °C (by DSC). The standard heats of formation for all compounds in this study were calculated on the B3LYP 6311G+d,p level of theory, showing highly-positive values of 831.5 kJ mol−1 and 1032.9 kJ mol−1 for 15 and 20, respectively. Detonation parameters for all compounds were calculated with the EXPLO5_v6.01 program. Theoretical density of 1.87 g cm−3 and velocity of detonation of 8903 m s−1 were calculated for 20, making it a highly promising new energetic compound.Experimental section
Caution! As certain triazole, tetrazole and tetrazine derivatives are unstable and can ignite or explode under the influence of impact, friction or heating, proper safety precautions should be taken when working with these materials. Lab personnel and equipment should be properly grounded and protective equipment including protective coat, Kevlar gloves, ear protection and face shield should be used.General information
All used chemical reagents and solvents were of analytical grade and were used as supplied, without further purification. 1H, 13C and 15N NMR spectra were recorded on a 400 MHz NMR spectrometer at 25 °C. The chemical shifts are given relative to the residual DMSO-d6 peaks or formamide (15N) as external standards and coupling constants are given in hertz (Hz). Infrared (IR) spectra were recorded on a FTIR spectrometer equipped with a diamond ATR unit. Mass spectra were recorded on a high-resolution ESI-Q-ToF machine. Elemental analyses (CHN) were performed at the service facility of the Hebrew University. Decomposition points were determined by differential scanning calorimetry (DSC). Measurements were performed at a heating rate of 5 °C min−1 in closed aluminium sample pans with a small hole in the lid under a nitrogen flow of 20 mL min−1 using an empty aluminium sample pan as a reference. Melting points were measured on a melting point apparatus in open glass capillaries. The impact sensitivities were tested according to STANAG 4489 using a BAM drop-hammer. The friction sensitivities were tested according to STANAG 4487 using a BAM friction tester. Experimental densities were obtained by pycnometry measurements at ambient temperature.General procedures
:20 v/v; 3 × 10 mL) and dried under vacuum to yield pure 12 (536 mg; 59%) as a red solid. DSC (5 °C min−1) 306 °C (decomp.). 1H NMR (400 MHz, DMSO-d6): δ 4.32 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 40.2, 159.0, 160.6. 13C DEPT135 (100 MHz, DMSO-d6): δ 40.2 (CH3). HRMS (ESI+): m/z = 277.1127 [M + H+]. Elemental analysis: calcd (%) for C6H8N14 C 26.09, H 2.96; found. C 26.29, H 2.86. FTIR (ATR): ν 3235 (w), 3018 (w), 2865 (w), 1588 (s), 1538 (s), 1428 (s), 1262 (m), 1197 (m), 1049 (s), 950 (s), 898 (w), 815 (w), 744 (m), 672 (m), 547 (s) cm−1.
Acknowledgements
The authors would like to acknowledge the financial support provided by the Tel Aviv University and Dr S. Lipstman, Dr C. Evangelisti, Dr D. Krepel, Dr H. Titi and S. Fishman for their valuable contribution.Notes and references
- http://chemistry.about.com/od/historyofchemistry/a/gunpowder.htm .
- http://kaleidoscope.cultural-china.com/en/136Kaleidoscope1.html .
- (a) L. E. Fried, M. R. Manaa, P. F. Pagoria and R. L. Simpson, Ann. Rev. Mater. Res., 2001, 31, 291–321 CrossRef CAS; (b) Q. Wu, W. Zhu and H. Xiao, J. Mater. Chem. A, 2014, 2, 13006–13015 RSC; (c) H. Martinez, Z. Zheng and W. R. Dolbier, J. Fluorine Chem., 2012, 143, 112 CrossRef CAS PubMed; (d) A. Dippold and T. Klapötke, J. Am. Chem. Soc., 2013, 135, 9931 CrossRef CAS PubMed; (e) V. Thottempudi, H. Gao and J. M. Shreeve, J. Am. Chem. Soc., 2011, 6464 CrossRef CAS PubMed.
- (a) P. F. Pagoria, G. S. Lee, A. R. Mitchell and R. D. Schmidt, Thermochim. Acta, 2002, 384, 187 CrossRef CAS; (b) A. K. Sikder and N. Sikder, J. Hazard. Mater., 2004, 112, 1 CrossRef CAS PubMed; (c) Z. Zeng, H. Gao, B. Twamley and J. M. Shreeve, J. Mater. Chem., 2007, 17, 3819 RSC; (d) G. Wang, H. Xiao, X. Ju and X. Gong, Propellants, Explos., Pyrotech., 2006, 31, 361 CrossRef CAS PubMed; (e) G. X. Wang, H. M. Xiao, X. J. Xu and X. H. Ju, Propellants, Explos., Pyrotech., 2006, 31, 102 CrossRef CAS PubMed; (f) J. Zhang and J. M. Shreeve, J. Am. Chem. Soc., 2014, 136, 4437 CrossRef CAS PubMed.
- (a) D. E. Chavez and M. A. Hiskey, J. Energy Mater., 1999, 17, 357–377 CrossRef CAS PubMed; (b) Q. Zhang, J. Zhang, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2013, 19, 11000 CrossRef CAS PubMed.
- (a) D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289 CrossRef CAS PubMed; (b) M. J. Rossi, J. C. Bottaro and D. F. McMillen, Int. J. Chem. Kinet., 1993, 25, 549 CrossRef CAS PubMed; (c) C. Darwich, T. M. Klapötke and C. M. Sabaté, Chem. – Eur. J., 2008, 14, 5756 CrossRef CAS PubMed.
- (a) Q. L. Yan and S. Zeman, Int. J. Quantum Chem., 2013, 113, 1049 CrossRef CAS PubMed; (b) M. Cartwright and J. Wilkinson, Propellants, Explos., Pyrotech., 2010, 35, 326 CrossRef CAS PubMed; (c) Q. Wu, W. Zhu and H. Xiao, J. Chem. Eng. Data, 2013, 58, 2748 CrossRef CAS.
- (a) M. Genetier, A. Osmont and G. Baudin, J. Phys.: Conf. Ser., 2014, 500, 192001 CrossRef; (b) M. J. Kamlet and H. G. Adolph, Propellants Explos., 1979, 4, 30 CrossRef CAS PubMed; (c) E. S. Shanley and G. A. Melhem, Process Saf. Prog., 1995, 14, 29 CrossRef CAS PubMed.
- (a) C. Zhang, Y. Shu, Y. Huang, X. Zhao and H. Dong, J. Phys. Chem. B, 2005, 109, 8978 CrossRef CAS PubMed; (b) B. Tan, X. Long and J. Li, Comput. Theor. Chem., 2012, 993, 66 CrossRef CAS PubMed; (c) M. X. Zhang, P. E. Eaton and R. Gilardi, Angew. Chem., Int. Ed., 2000, 39, 401 CrossRef CAS.
- M. Rahm, G. Bélanger-Chabot, R. Haiges and K. O. Christe, Angew. Chem., Int. Ed., 2014, 53, 6893 CrossRef CAS PubMed.
- T. Wei, W. Zhu, X. Zhang, Y. F. Li and H. Xiao, J. Phys. Chem. A, 2009, 113, 9404 CrossRef CAS PubMed.
- A. A. Korkin and R. J. Bartlett, J. Am. Chem. Soc., 1996, 118, 12244 CrossRef CAS.
- C. Steffen, K. Thomas, U. Huniar, A. Hellweg, O. Rubner and A. Schroer, J. Comput. Chem., 2010, 31, 2967 CAS.
- Y. Li, W. Liu and S. Pang, Molecules, 2012, 17, 5040–5049 CrossRef CAS PubMed.
- (a) T. M. Klapötke, J. Stierstorfer and A. U. Wallek, Chem. Mater., 2008, 20, 4519 CrossRef; (b) E. G. Gillan, Chem. Mater., 2000, 12, 3906 CrossRef CAS; (c) Y. H. Joo, B. Twamley, S. Garg and J. M. Shreeve, Angew. Chem., Int. Ed., 2008, 47, 6236 CrossRef CAS PubMed; (d) Y. Guo, H. Gao, B. Twamley and J. M. Shreeve, Adv. Mater., 2007, 19, 2884 CrossRef CAS PubMed; (e) G. Steinhauser and T. M. Klapötke, Angew. Chem., Int. Ed., 2008, 47, 3330 CrossRef CAS PubMed; (f) D. Chand, D. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2013, 1, 15383 RSC; (g) R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584 CrossRef CAS PubMed.
- (a) D. Srinivas, V. D. Ghule and K. Muralidharan, RSC Adv., 2014, 4, 7041 RSC; (b) G. H. Tao, B. Twamley and J. M. Shreeve, J. Mater. Chem., 2009, 19, 5850 RSC.
- (a) T. M. Klapötke and D. G. Piercey, Inorg. Chem., 2011, 50, 2732 CrossRef PubMed; (b) Y. H. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2010, 49, 7320 CrossRef CAS PubMed; (c) Y. H. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2009, 48, 564 CrossRef CAS PubMed.
- (a) H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., Int. Ed., 2004, 43, 1983 CrossRef CAS PubMed; (b) G. Fischer, G. Holl, T. M. Klapötke and J. Weigand, J. Thermochim. Acta, 2005, 437, 168 CrossRef CAS PubMed; (c) T. M. Klapötke and J. Stierstorfer, Helv. Chim. Acta, 2007, 90, 2132 CrossRef PubMed; (d) T. M. Klapötke, A. Preimesser and J. Stierstorfer, Z. Naturforsch., B: Chem. Sci., 2013, 68, 1310–1320 Search PubMed.
- (a) P. M. Cormick, M. Rovera and E. N. Durantini, J. Photochem. Photobiol. Chem., 2008, 194, 220 CrossRef PubMed; (b) M. Esseffar, E. Quintanilla, J. Z. Dvalos, J. L. M. Abboud and M. Yez, New J. Chem., 2002, 26, 1567 RSC; (c) V. D. Ghule, R. Sarangapani, P. M. Jadhav and S. P. Tewari, J. Mol. Model., 2011, 17, 1507 CrossRef CAS PubMed; (d) D. Srinivas, V. D. Ghule, S. P. Tewari and K. Muralidharan, Chem. – Eur. J., 2012, 18, 15031 CrossRef CAS PubMed; (e) H. Hao Wei, H. Gao and J. M. Shreeve, Chem. – Eur. J., 2014, 20(51), 16943–16952 CrossRef PubMed; (f) R. Haiges, G. Bélanger-Chabot, S. M. Kaplan and K. O. Christe, Dalton Trans., 2015, 44, 7586 RSC; (g) R. Haiges and K. O. Christe, Inorg. Chem., 2013, 52, 7249 CrossRef CAS PubMed.
- A. Saikia, R. Sivabalan, B. G. Polke, G. M. Gore, A. Singh, A. Subhananda Rao and A. K. Sikder, J. Hazard. Mater., 2009, 170, 306 CrossRef CAS PubMed.
- (a) D. E. Chavez, M. A. Hiskey and D. L. Naud, Propellants, Explos., Pyrotech., 2004, 29, 209 CrossRef CAS PubMed; (b) T. Wei, J. Wu, W. Zhu, C. Zhang and H. Xiao, J. Mol. Model., 2012, 18(8), 3467–3479 CrossRef CAS PubMed.
- M. Masuda, H. F. Mower, B. Pignatelli, I. Celan, M. D. Friesen, H. Nishino and H. Ohshima, Chem. Res. Toxicol., 2000, 13, 301 CrossRef CAS PubMed.
- (a) G. Olah, A. Orlinkov, A. B. Oxyzoglou and G. K. S. Prakash, J. Org. Chem., 1995, 60, 7348 CrossRef CAS; (b) J. G. Hoggett, R. B. Moodie, J. R. Penton and K. Schofield, Nitration and aromatic reactivity, 1971 Search PubMed.
- G. K. S. Prakash, L. Heiliger and G. A. Olah, Inorg. Chem., 1990, 29, 4965–4968 CrossRef CAS.
- (a) A. A. Dippold and T. M. Klapötke, Chem. – Eur. J., 2012, 18, 16742–16753 CrossRef CAS PubMed; (b) A. B. Sheremetev, N. V. Palysaeva, M. I. Struchkova and K. Y. Suponitsky, Mendeleev Commun., 2012, 22, 302–304 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- M. H. Keshavarz, J. Hazard. Mater., 2007, 145, 263–269 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., GAUSSIAN 09 (Revision D.01), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- M. Sućeska, Mater. Sci. Forum, 2004, 325, 465–466 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Contains copies of 1H and 13C NMR spectra for isolated compounds and 15N NMR spectra for 15N-labeled compounds formed in situ in the nitration experiments, and crystallographic data for compounds 16 and 20. CCDC 1041768 and 1041769. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01641j |
| This journal is © The Royal Society of Chemistry 2015 |