DOI:
10.1039/C5DT01633A
(Paper)
Dalton Trans., 2015,
44, 13119-13124
Alkoxy substituted halogenated closo-dodecaborates as anions for ionic liquids†
Received
30th April 2015
, Accepted 4th June 2015
First published on 24th June 2015
Abstract
Halogenated and alkoxylated closo-dodecaborates [B12X11OR]2− (X = Cl, Br; R = propyl, octyl, dodecyl) have been synthesized by halogenation of the known [B12H11OH]2− anion followed by alkylation in the superbasic medium DMSO/KOH. The obtained sodium salts were transformed by simple metathesis reactions in aqueous solution to the tetrabutylammonium ([NBu4]+) and 1-hexyl-3-methylimidazolium ([C6mim]+) salts. All compounds were fully characterized by heteronuclear NMR, IR and Raman spectroscopy, ESI mass spectrometry, and thermal analytical measurements. Selected anions were also structurally characterized as their [Ph4P]+ salts by single crystal X-ray diffraction. The [C6mim]+ salts are thermally stable up to more than 300 °C and show clear melting points. Surprisingly, the compound [C6mim]2[B12Cl11O-propyl] having the short propyl group bound to the boron cluster shows the lowest melting point (96 °C) of all the investigated compounds. Thus this compound is a rare member of the class of ionic liquids consisting of dianions.
Introduction
Halogenated boron cluster anions exhibit exceptional chemical and thermal stability. For instance the chlorinated closo-dodecaborate [B12Cl12]2− and 1-carba-closo-dodecaborate [HCB11Cl11]− anions have decomposition temperatures higher than 600 °C and 400 °C,1 respectively, and have been widely applied as weakly coordinating anions, for instance to stabilize reactive cations.2 These properties make these boron cluster anions potentially interesting for applications in thermally stable ionic liquids.3 Despite the still increasing importance of ionic liquids,4 the number of boron cluster-based ionic liquids is still limited. Salts containing carborates5 and stannaborates6 show quite low melting points. In contrast, the melting points of salts based on the dianionic perchlorinated dodecaborate [B12Cl12]2−,5e,7 which has been introduced by us as a weakly coordinating anion,8 are higher. Table 1 summarizes the melting points of selected low melting salts containing boron cluster anions.
Table 1 Melting points of selected low melting salts containing boron cluster anions
| Compound |
T
m/°C |
Ref. |
| [C2mim]2[B12Cl12] |
265 |
5e
|
| [C16mim]2[B12Cl12] |
105 |
5e
|
| [N2 2 2 4]2[B12Cl12] |
>300 |
7
|
| [N1 1 1 16]2[B12Cl12] |
104 |
7
|
| [C2mim][SnB11H11Et] |
106 |
6
|
| [C4mim][SnB11H11Bu] |
55 |
6
|
| [C2mim][HCB11H11] |
122 |
5a
|
| [C2mim][n-propyl-CB11H11] |
45 |
5a
|
| [C4mim][Et3NB12H11] |
128–130 |
10c
|
| [C4mim][dodecyl3NB12H11] |
25 |
10c
|
The very high price of the 1-carba-closo-dodecaborates prevents their broad application, while the parent closo-dodecaborate [B12H12]2− and its derivatives are relatively cheap and easily available.9 Therefore attempts have been made to develop ionic liquids based on trialkylammonio substituted single charged closo-dodecaborate anions [R3NB12H11]−.10 These anions led to salts of satisfying low melting points, but the chemical stability of these compounds is still in need of improvement, due to the presence of potentially reactive boron–hydrogen bonds. Very recently, we reported the chlorinated derivative [B12Cl11NMe3]−,11 which possesses improved chemical, thermal, and electrochemical stability. However, all attempts to introduce longer alkyl chains on the nitrogen atom have not been successful so far. To take advantage of the cheap price or simple synthesis, respectively, of the parent closo-dodecaborate cluster and the superior chemical and thermal properties of the perhalogenated derivatives we attempted to prepare low melting salts of the hitherto unknown halogenated [B12X11OR]2− anion (X = Cl, Br, R = alkyl). Herein the synthesis, full spectroscopic characterization, thermal and electrochemical properties, and crystal structures are reported.
Results and discussion
Synthesis of the halogenated and alkoxylated clusters [B12X11OR]2− (X = Cl, Br, R = alkyl)
We started from the known hydroxylated cluster [B12H11OH]2−, which was obtained by a literature procedure.12 Halogenation and O-alkylation gives the halogenated and alkoxylated clusters [B12X11OR]2−. In principle, these two steps can be performed in two different chronological orders. However, from the synthesis of [B12Cl11NMe3]− it is known that halogenation of an already N-alkylated cluster leads to two problems.11 The halogenation is sterically hindered and thus yields incompletely halogenated products and secondly the already present organic group may be partly halogenated as well. Therefore, we preferred to perform the halogenation first followed by a subsequent O-alkylation. The halogenation reactions were carried out in a boiling aqueous (for [B12Cl11OH]2−) or aqueous/methanolic (for [B12Br11OH]2−) solution and by using the elemental halides as halogenation reagents.1c,9 Due to the presence of small amounts of unreacted [B12H12]2− and dihydroxylated [B12H10]2−, small amounts of [B12X12]2− (X = Cl, Br) and [B12X10(OH)2]2− (X = Cl, Br) were obtained as impurities. The boron–oxygen bond is stable under these reaction conditions, thus only the hydrogen atoms are replaced by halides as required. The subsequent alkylation was performed in the superbasic medium KOH and DMSO.13Scheme 1 summarizes the complete reaction pathway.
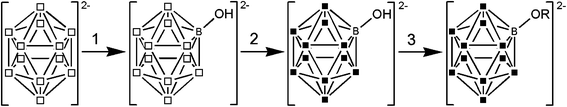 |
| | Scheme 1 Reaction pathway to [B12X11OR]2− (X = Cl, Br, R = alkyl). 1. H2SO4/H2O, ΔT; 2. Cl2 in H2O or Br2 in H2O/methanol, ΔT; 3. R–Br in KOH/DMSO, r.t. (□ = B–H, ■ = B–Cl/Br; R = alkyl). | |
Spectroscopic characterization of [B12X11OH]2− and [B12X11OR]2− (X = Cl, Br; R = alkyl)
All new compounds were fully characterized by heteronuclear NMR spectroscopy and by vibrational spectroscopy. The full set of data and all actual spectra are included in the ESI (section S.2†). The 11B NMR spectra of the hydroxylated cluster [B12H11OH]2− strongly depend on the solvent. In organic solvents and water (pH 7) the spectrum shows a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5:1 pattern. The oxygen-bonded boron atom B1 is observed at about 5 ppm, the five membered rings between −15 and −18 ppm and the antipodal boron atom B12 at −24 ppm. In strongly acidic solution the 11B NMR signal of the boron atom B1 is shifted to a higher field by about 5 ppm, while the chemical shifts of the other boron atoms are not affected. This is in agreement with protonation of the hydroxyl group in acidic solutions. After halogenation the spectrum changes from a 1:5:5:1 to a 1:10:1 pattern (chlorination or bromination does not lead to a difference), because the signals of the B2–6 and B7–11 atoms overlap. The boron atom B1 is significantly shifted to a higher field, while the antipodal boron atom B12 is shifted to a lower field. Subsequent alkylation does not change the 11B NMR pattern. Table 2 summarizes the 11B NMR chemical shift of the boron clusters.
:5:5:1 pattern. The oxygen-bonded boron atom B1 is observed at about 5 ppm, the five membered rings between −15 and −18 ppm and the antipodal boron atom B12 at −24 ppm. In strongly acidic solution the 11B NMR signal of the boron atom B1 is shifted to a higher field by about 5 ppm, while the chemical shifts of the other boron atoms are not affected. This is in agreement with protonation of the hydroxyl group in acidic solutions. After halogenation the spectrum changes from a 1:5:5:1 to a 1:10:1 pattern (chlorination or bromination does not lead to a difference), because the signals of the B2–6 and B7–11 atoms overlap. The boron atom B1 is significantly shifted to a higher field, while the antipodal boron atom B12 is shifted to a lower field. Subsequent alkylation does not change the 11B NMR pattern. Table 2 summarizes the 11B NMR chemical shift of the boron clusters.
Table 2
11B NMR shifts [ppm] of selected boron clusters in different solvents
| |
B1 |
B2–B7 |
B8–B11 |
B12 |
| B12H11OH (D2O, pH 7) |
5.4 |
−15.5 |
−18.0 |
−24.1 |
| B12H11OH (D2O, pH 1) |
0.7 |
−17.8 |
−19.1 |
−23.7 |
| B12H11OH (CD3CN) |
5.5 |
−15.7 |
−17.9 |
−24.0 |
| B12Cl11OH |
−7.3 |
−13.8 |
−13.8 |
−15.5 |
| B12Cl11O-R |
−7.5 |
−13.8 |
−13.8 |
−15.6 |
| B12Br11OH |
−4.2 |
−14.1 |
−14.1 |
−16.6 |
| B12Br11O-R |
−4.4 |
−14.4 |
−14.4 |
−16.7 |
The IR spectra of salts of the [B12X11OH]2− (X = H, Cl, Br) anions show the O–H vibration at about 3600 cm−1 (Fig. S16 and S19), which disappears after O-alkylation. In the alkoxy substituted derivatives the characteristic C–H vibration is present, whose intensity increases with the length of the alkyl chain. All IR- and Raman spectra are depicted in the ESI.† Negative ESI-mass spectra of all the compounds show the complete anions as the main signal. The isotopic patterns are in good compliance with the expected and simulated spectra. In addition, ion pairs were also detected in agreement with our previous reports.14
Thermal analysis
The thermal properties of the tetrabutylammonium and the 1-hexyl-3-methylimidazolium ([C6mim]+) salts of the new [B12X11OR]2− anions were investigated (see section S.2.10 in the ESI† for details). Thermogravimetric analyses (TGA) were carried out to investigate the decomposition reactions of the salts, and differential scanning calorimetry (DSC) measurements gave the melting points. The tetrabutylammonium salts have significantly lower decomposition temperatures than the [C6mim]+ salts and do not melt below 300 °C, therefore the following discussion is restricted to the [C6mim]+ salts. All thermo-analytical data referring to the [C6mim]+ salts are summarized in Table 3.
Table 3 Compilation of the thermo-analytical data for [C6mim]2[B12X11OR] (X = Cl, Br; R = propyl, octyl, dodecyl)
| Anion |
M
pa/°C |
Experimental mass lossb/% |
Theoretical mass lossc/% |
T
dd/°C |
|
The melting points were measured using Stuart SMP10 melting point apparatus. Values in brackets were obtained by differential scanning calorimetry.
Mass loss for the first decomposition step based on the TGA measurements.
Calculated for the loss of the alkyl chain of the anion.
Decomposition temperature
|
| [B12Cl11O-propyl]2− |
94–98 (96) |
5.15 |
4.72 |
350 |
| [B12Cl11O-octyl]2− |
138–143 (145) |
12.97 |
11.51 |
368 |
| [B12Cl11O-dodecyl]2− |
136–142 (144) |
16.08 |
16.29 |
355 |
| [B12Br11O-propyl]2− |
158–160 (161) |
3.95 |
3.07 |
335 |
| [B12Br11O-octyl]2− |
120–124 (126) |
8.89 |
7.69 |
349 |
| [B12Br11O-dodecyl]2− |
128–134 (139) |
7.46 |
11.08 |
240 |
The DSC measurements show only one endothermic peak in the heating curve and one exothermic peak in the cooling curve for most compounds. The melting points are in very good agreement with the melting points obtained by classical optical melting point determination. Also the heating and cooling curves obtained by DSC for [C6mim]2[B12Cl11O-propyl]2− are depicted in Fig. 1. There is no clear dependency of the melting points on either the length of the alkyl chain or the halogen atoms on the cluster. The salt [C6mim]2[B12Cl11O-propyl] shows the lowest melting point (96 °C) of all the investigated compounds and can be classified as an ionic liquid. Surprisingly, the smaller chloro substituted anion with the shortest alkyl chain leads to the lowest melting point among all the anions in this study. A similar behavior has also been observed for the carborane ionic liquids [C2mim][RCB11H11], which show an increase of the melting point, when the alkyl chain becomes longer than the propyl chain.5a
 |
| | Fig. 1 Heating and cooling curves for [C6mim]2[B12Cl11O-propyl] obtained by DSC. | |
While the perchlorinated [B12Cl12]2− anion decomposes above 600 °C,1c the thermal stability of the alkoxy-substituted anions is significantly reduced. In general the [NBu4]+ salts decompose at lower temperatures than the [C6mim]+ salts, which is exemplarily depicted for the [B12Cl11O-propyl]-anion in Fig. 2. All [C6mim]+ salts decompose above 335 °C with the exception of [C6mim]2[B12Br11O-dodecyl], which decomposes at 240 °C. The mass loss of the first decomposition step is in good agreement with splitting of the alkyl group and thus breaking the oxygen–carbon bond. Combined thermo-analytical and mass spectrometric measurements (Fig. S93, S97, and S101†) show typical fragments of alkyl groups and thus also confirm the assignment of the first decomposition step to the loss of the alkyl group. The subsequent decomposition steps at higher temperatures, which are probably related to the decomposition of the cation and at even higher temperature also to the boron cluster, cannot be assigned unambiguously.
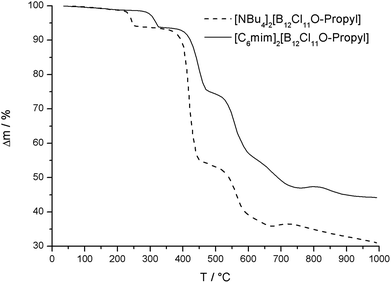 |
| | Fig. 2 Thermogravimetric analysis of [NBu4]2[B12Cl11O-propyl] and [C6mim]2[B12Cl11O-propyl]. | |
Crystal structures
All attempts to obtain crystals suitable for X-ray diffraction of the [NBu4]+ or [C6mim]+ salts were unsuccessful. However, by using the [PPh4]+ counter cation suitable crystals were easily grown from an acetonitrile/diethyl ether solution. The crystal structures of the hydroxo clusters [B12X11OH]2− (X = Cl, Br) and the alkylated clusters [B12X11OR]2− (X = Cl, Br; R = propyl, octyl) were determined (Table 4 and Fig. 3). In the halogenated hydroxo clusters [B12X11OH]2− (X = Cl, Br) the hydroxo group is disordered over two positions (see Fig. S111 and S112 in the ESI†) which is in contrast to the ordered structure of the non-halogenated derivative [B12H11OH]2−.12 Alkylation always leads to an ordered structure. The propyl substituted clusters are independent of the halogen substituent quite similar. In contrast, the structures containing the longer octyl chain vary depending on the halogen substituent on the boron cluster. All anions show more or less pronounced C–H⋯X (X = Cl, Br) hydrogen bonds.15 In all four structures the methylene group neighbored to the oxygen points to one halogen atom of the boron cluster in the ortho position. The hydrogen atoms of this methylene group show short intramolecular contacts to one or two halogen atoms. The H⋯Cl distances (273.9–291.1 pm) are shorter than the H⋯Br distances (294.2–309.5 pm). As a consequence, the B–O–C angles are smaller in the chloro derivatives (117–124°) than in the bromo derivatives (126–127°). In all structures the B–O bond is bent away from the chlorine atom, which forms the C–H⋯X contacts, indicating steric repulsion of the methylene group neighbored to the oxygen atom and the halogen atoms in the ortho position. The octyl chain in the structure of [B12Br11O-octyl]2− coils itself around the anion, while in the chlorinated cluster the octyl chain is pointing away from the anion. Several additional intramolecular C–H⋯Br contacts are present in the [B12Br11O-octyl]2− anion, nevertheless it remains unclear why the structures of the chloro and the bromo derivatives are so different. Additional intramolecular C–H⋯X contacts between anions and cations are present as well in all structures, which presumably significantly determine the packing and also the orientation in space of the alkyl chains.
 |
| | Fig. 3 Anions in the crystal structures of [PPh4]2[B12Cl11O-propyl], [PPh4]2[B12Cl11O-octyl], [PPh4]2[B12Br11O-octyl], and [PPh4]2[B12Br11O-propyl]. The Cl⋯H distances are given in pm. Ellipsoids are drawn at 50% probability and hydrogen atoms are drawn with arbitrary radii. | |
Table 4 Crystallographic parametersa
| |
[PPh4]2[B12Cl11OH] |
[PPh4]2[B12Br11OH] |
[PPh4]2[B12Cl11O-propyl] |
[PPh4]2[B12Cl11O-octyl] |
[PPh4]2[B12Br11O-propyl]·CH3CN |
[PPh4]2[B12Br11O-octyl]·OEt2 |
|
R
1 = ∑||Fo|−|Fc||/∑|Fo|, wR2 = (∑[w(Fo2 − Fc2)2]/∑[wFo4])1/2.
|
| Formula |
C48H40B12Cl11OP2 |
C48H40B12Br11OP2 |
C51H47B12Cl11OP2 |
C56H57B12Cl11OP2 |
C53H50B12Br11NOP2 |
C60H67B12Br11O2P2 |
| CCDC no. |
1057639
|
1057640
|
1057641
|
1057642
|
1057643
|
1057644
|
|
M
|
1214.41 |
1703.47 |
1257.50 |
1327.63 |
1787.61 |
1890.81 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
Triclinic |
Monoclinic |
Triclinic |
| Space group |
P21/n |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n |
P |
|
a/pm |
1495.69(4) |
1288.17(2) |
1127.145(18) |
1217.21(4) |
1180.24(4) |
1208.12(4) |
|
b/pm |
1015.60(3) |
1234.53(2) |
2847.14(5) |
1649.10(5) |
3885.26(14) |
1699.30(5) |
|
c/pm |
1809.74(5) |
1913.05(4) |
3005.22(5) |
1747.60(5) |
1396.73(4) |
1849.10(4) |
|
α/° |
90 |
90 |
113.9658(17) |
93.883(2) |
90 |
88.517(2) |
|
β/° |
100.432(2) |
108.9073(19) |
90.7492(14) |
103.004(3) |
91.786(2) |
77.871(2) |
|
γ/° |
90 |
90 |
94.0845(13) |
102.370(3) |
90 |
73.201(3) |
|
U/nm3 |
2.70359(12) |
2.87814(9) |
8.7810(3) |
3.31378(17) |
6.4017(4) |
3.55054(18) |
|
Z
|
2 |
2 |
6 |
2 |
4 |
2 |
|
μ(Mo-Kα)/mm−1 |
0.663 |
7.749 |
0.615 |
0.547 |
6.973 |
6.292 |
| No. of data collected |
15848 |
14897 |
91326 |
29031 |
43395 |
35549 |
| No. of unique data |
6304 |
6727 |
40498 |
12967 |
15102 |
16534 |
|
R
int
|
0.0235 |
0.0237 |
0.0284 |
0.0404 |
0.0729 |
0.0326 |
|
R
1, wR2 (I > 2σ(I))a |
0.0400, 0.0924 |
0.0284, 0.0523 |
0.0661, 0.1455 |
0.0589, 0.1537 |
0.0559, 0.1347 |
0.0364, 0.0648 |
|
R
1, wR2 (all data) |
0.0477, 0.0964 |
0.0437, 0.0570 |
0.0927, 0.1625 |
0.0701, 0.1605 |
0.0741, 0.1471 |
0.0671, 0.0734 |
Conclusion
Alkoxy substituted and perhalogenated closo-dodecaborates were synthesized for the first time and their spectroscopic, thermal and structural properties were determined. The alkoxy groups significantly lower the melting point of their salts compared to the [B12X12]2− anions. They are still quite resistant to oxidation as determined by cyclic voltammetry (see Fig. S110†) and their decomposition temperatures are still above 300 °C (Table 3), however, they are much lower than for the perhalogenated closo-dodecaborate [B12Cl12]2− and 1-carba-closo-dodecaborate [HCB11Cl11]− anions.1 The salt [C6mim]2[B12Cl11O-propyl] has a melting point of 96 °C and thus can be regarded as an ionic liquid. Interestingly the anion carrying the quite short propyl group leads to the salt with the lowest melting point. Ionic liquids containing doubly charged anions are rare,16,17 thus [C6mim]2[B12Cl11O-propyl] is a representative of this elusive class of compounds.
Experimental section
General remarks
All solvents and the following chemicals were used as purchased from the manufacturer: [nBu4N]Br (Aldrich, 99%), [C6mim]Cl (Aldrich, ≥97%), Et3N (Riedel-de Haen), acetonitrile (Fisher Scientific), DMSO (Roth, 99.5%), n-propyl bromide (Sigma-Aldrich, 99%), n-octyl bromide (Sigma-Aldrich, 99%), n-dodecyl bromide (Sigma-Aldrich, 97%), KOH (Grüssing, 85%), and NaOH (Roth, ≥99%). K2[B12H12] was prepared by a published procedure.9 [B12H11OH]2− was obtained from [B12H12]2− by a time and temperature controlled reaction with sulfuric acid.12 Synthetic and spectroscopic details for the preparation of [B12H11OH]2− (X = H, Cl, Br) anions are included in the ESI.†
IR spectra were measured on a Bruker ALPHA P FT-IR spectrometer equipped with a diamond ATR attachment. Raman spectra of samples in flame-sealed capillaries were recorded using a Bruker Equinox 55 FRA 106/S FT-Raman spectrometer equipped with a highly sensitive Ge detector and a Nd:YAG-Laser (1064 nm). NMR spectra were measured on a Bruker Avance 400 and a Bruker AvanceIII 600 spectrometer in 5 mm NMR tubes at room temperature. Air and moisture sensitive samples were measured in 5 mm NMR tubes equipped with a J. Young Teflon in a glass valve. Chemical shifts are given with respect to Me4Si (1H, 13C) and BF3·OEt2 (11B). Two dimensional NMR experiments were performed to assist the assignment of the spectra. Electrospray ionization (ESI) mass spectra were recorded using a Bruker micrOTOF instrument. Thermoanalytical measurements were done with a Mettler Toledo TGA/DSC 1 LSTAR System. The coupled thermogravimetric/mass spectrometric measurements (see ESI) were carried out with a Mettler Toledo TGA/DSC 1 LSTAR System connected to Pfeiffer Vacuum Thermostar Gas Analysis System and a Gas Controller GC 200. Melting points were measured using a Stuart SMP10 melting point apparatus.
Crystal structure determination
The [PPh4]+ salts used for diffraction experiments were synthesized by a simple metathesis reaction. 100 mg of the specific sodium salt were dissolved in 10 ml of water and a twofold molar excess of [PPh4]Br dissolved in 10 ml of water was added. After stirring for two hours the white precipitate was filtered off, washed with a small amount of cold water and dried under low pressure at 80 °C. Single crystals suitable for X-ray diffraction were obtained by slow diffusion of diethyl ether into a solution of the respective salt in acetonitrile. The single crystal X-ray diffraction studies were performed on an Oxford Diffraction Gemini E Ultra diffractometer equipped with a 2 K × 2 K EOS CCD area detector and a four-circle kappa goniometer using MoKα (0.71073 Å) radiation at 120 K. The crystals were mounted onto a cryo loop using fluorinated oil and frozen in the cold nitrogen stream of the goniometer. Details of the crystallographic data collection and the refinement parameters can be found in Table 4. The structures were solved by direct methods (SHELXS)18 using the program package OLEX2.19 Subsequent least-squares refinement on F2 (SHELXL)18 located the positions of the remaining atoms in the electron density maps. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions using a riding model. The data were corrected for absorption.
Synthesis of Na2[B12X11OR] (X = Cl, Br; R = propyl, octyl, dodecyl)
Here a general procedure is given. Yields and all spectroscopic data are included in the ESI.† 1.00 g (1.71 mmol in the case of the chloro cluster, 0.93 mmol in the case of the bromo cluster) of the specific sodium salts synthesized above was dissolved in 20 ml of DMSO. In the second step a tenfold molar excess of the alkyl bromide was added via a microliter syringe. After that the reaction was initiated by adding KOH (485 mg, 8.65 mmol in the case of the chloro cluster/264 mg, 4.70 mmol in the case of the bromo cluster). The colorless solutions remained colorless or turned pale yellow to yellow depending on the alkyl halide used. The mixtures were stirred at room temperature for two days. Subsequently the solutions were filtered and all volatiles were removed under low pressure and heating to 70 °C. The residue was washed with a small amount of isopropyl alcohol, filtered and dried at 120 °C.
Synthesis of [NBu4]2[B12X11OR] (X = Cl, Br; R = propyl, octyl, dodecyl)
Here a general procedure is given. Yields and all spectroscopic data are included in the ESI.† 100 mg of the specific sodium salt were dissolved in 10 ml of water. A twofold molar excess of tetrabutylammonium bromide was dissolved in 10 ml of water and the two solutions were combined and stirred for two hours. The white precipitate was filtered, washed with a small amount of cold water and dried under low pressure at 80 °C.
Synthesis of [C6mim]2[B12X11OR] (X = Cl, Br; R = propyl, octyl, dodecyl)
Here a general procedure is given. Yields and all spectroscopic data are included in the ESI.† 100 mg of the specific sodium salt were dissolved in 10 ml of water. A twofold molar excess of 1-hexyl-3-methylimidazolium chloride was dissolved in 10 ml of water and the two solutions were combined and stirred for two hours. The white precipitate was filtered, washed with a small amount of cold water and dried under low pressure at 80 °C.
Acknowledgements
We are grateful to Anke Helfer, Prof. Ulrich Scherf, Philipp Hagemann, Martina Henseleit, and Prof. Roland Görtz for the thermo-analytical measurements and to the crystallographic reviewer for providing helpful advice.
References
-
(a) C. A. Reed, Chem. N. Z., 2011, 75, 174–179 CAS;
(b) V. A. Popov, K. Y. Zhizhin, N. T. Kuznetsov, K. P. Staudhammer and V. M. Retivov, Adv. Mater. Res., 2009, 59, 96–100 CrossRef CAS;
(c) I. Tiritiris and T. Schleid, Z. Anorg. Allg. Chem., 2004, 630, 1555–1563 CrossRef CAS PubMed.
- See for review:
C. Knapp, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, 2013, vol. 1, pp. 651–679 Search PubMed.
- See for review:
A. Vöge and D. Gabel, in Boron Science, ed. N. S. Hosmane, CRC Press, Boca Raton, FL, 2012, pp. 807–825 Search PubMed.
-
(a) N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC;
(b) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
-
(a) A. S. Larsen, J. D. Holbrey, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 2000, 122, 7264–7272 CrossRef CAS;
(b) Y. Zhu, C. Ching, K. Carpenter, R. Xu, S. Selvaratnam, N. S. Hosmane and J. A. Maguire, Appl. Organomet. Chem., 2003, 17, 346–350 CrossRef CAS PubMed;
(c) Z. Yinghuai, K. Carpenter, C. C. Bun, S. Bahnmueller, C. P. Ke, V. S. Srid, L. W. Kee and M. F. Hawthorne, Angew. Chem., Int. Ed., 2003, 42, 3792–3795 (
Angew. Chem.
, 2003
, 115
, 3922–3925
) CrossRef PubMed;
(d) J. Dymon, R. Wibby, J. Kleingardner, J. M. Tanski, I. A. Guzei, J. D. Holbrey and A. S. Larsen, Dalton Trans., 2008, 2999–3006 RSC;
(e) M. Nieuwenhuyzen, K. R. Seddon, F. Teixidor, A. V. Puga and C. Viñas, Inorg. Chem., 2009, 48, 889–901 CrossRef CAS PubMed;
(f) N. Matsumi, M. Miyamoto and K. Aoi, J. Organomet. Chem., 2009, 694, 1612–1616 CrossRef CAS PubMed;
(g) S. A. Suarez, A. Foi, S. Eady, A. Larsen and F. Doctorovich, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, o417–o420 CAS;
(h) S. Liu, Z. Chen, Q. Zhang, S. Zhang, Z. Li, F. Shi, X. Ma and Y. Deng, Eur. J. Inorg. Chem., 2011, 1910–1920 CrossRef CAS PubMed.
- B. Ronig, I. Pantenburg and L. Wesemann, Eur. J. Inorg. Chem., 2002, 319–322 CrossRef CAS.
- N. Zhou, G. Zhao, K. Dong, J. Sun and H. Shao, RSC Adv., 2012, 2, 9830–9838 RSC.
- Selected references:
(a) C. Knapp and C. Schulz, Chem. Commun., 2009, 4991–4993 RSC;
(b) M. Kessler, C. Knapp, V. Sagawe, H. Scherer and R. Uzun, Inorg. Chem., 2010, 49, 5223–5230 CrossRef CAS PubMed;
(c) J. Derendorf, M. Keßler, C. Knapp, M. Rühle and C. Schulz, Dalton Trans., 2010, 39, 8671–8678 RSC;
(d) C. Bolli, J. Derendorf, M. Kessler, C. Knapp, H. Scherer, C. Schulz and J. Warneke, Angew. Chem., Int. Ed., 2010, 49, 3536–3538 (
Angew. Chem.
, 2010
, 122
, 3616–3619
) CrossRef CAS PubMed;
(e) M. Kessler, C. Knapp and A. Zogaj, Organometallics, 2011, 30, 3786–3792 CrossRef CAS.
- V. Geis, K. Guttsche, C. Knapp, H. Scherer and R. Uzun, Dalton Trans., 2009, 2687–2694 RSC.
-
(a) A. V. Agafonov, L. A. Butman, K. A. Solntsev, A. A. Vinokurov, N. A. Zhukova and N. T. Kuznetsov, Zh. Neorg. Khim., 1982, 27, 63–70 CAS;
(b) T. Peymann, E. Lork, M. Schmidt, H. Nöth and D. Gabel, Chem. Ber., 1997, 130, 795–799 CrossRef CAS PubMed;
(c) E. Justus, K. Rischka, J. F. Wishart, K. Werner and D. Gabel, Chem. – Eur. J., 2008, 14, 1918–1923 CrossRef CAS PubMed;
(d) E. Justus, A. Vöge and D. Gabel, Eur. J. Inorg. Chem., 2008, 5245–5250 CrossRef CAS PubMed.
- C. Bolli, J. Derendorf, C. Jenne, H. Scherrer, C. P. Sindlinger and B. Wegener, Chem. – Eur. J., 2014, 20, 13783–13792 CrossRef CAS PubMed.
- T. Peymann, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 2000, 39, 1163–1170 CrossRef CAS.
- T. Peymann, E. Lork and D. Gabel, Inorg. Chem., 1996, 35, 1355–1360 CrossRef CAS PubMed.
-
(a) D. Gabel, C. Knapp, T. Dülcks and J. Warneke, Phys. Chem. Chem. Phys., 2011, 13, 5712–5721 RSC;
(b) C. Jenne, M. Keßler and J. Warneke, Chem. – Eur. J., 2015, 21, 5887–5891 CrossRef CAS PubMed.
-
(a) C. B. Aakeroy, T. A. Evans, K. R. Seddon and I. Palinko, New J. Chem., 1999, 23, 145–152 RSC;
(b) J.-A. van den Berg and K. R. Seddon, Cryst. Growth Des., 2003, 3, 643–661 CrossRef CAS.
- Y. Yoshida, H. Tanaka, G. Saito, L. Ouahab, H. Yoshida and N. Sato, Inorg. Chem., 2009, 48, 9989–9991 CrossRef CAS PubMed.
- J. Pernak, M. Niemczak, F. Giszter, J. L. Shamshina, G. Gurau, O. A. Cojocaru, T. Praczyk, K. Marcinkowska and R. D. Rogers, ACS Sustainable Chem. Eng., 2014, 2, 2845–2851 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1057639–1057644. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01633a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence


