DOI:
10.1039/C5DT01620G
(Paper)
Dalton Trans., 2015,
44, 15334-15342
Porous frameworks constructed by non-covalent linking of substitution-inert metal complexes†
Received
30th April 2015
, Accepted 17th July 2015
First published on 21st July 2015
Abstract
The incorporation of active sites into metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) is an attractive way to functionalise these materials. However, the methodology to organise substitution-inert metal-based secondary building units (SBUs) with active sites into MOFs or PCPs via coordination driven self-assembly is severely limited. In this study, we successfully assembled substitution-inert paddle-wheel Rh(II) dimers to afford three novel porous frameworks, Rh2(ppeb)4(THF)2 (1-THF), Rh2(ppeb)4(3-pentanone)2 (1-PN) and Rh2(ppeb)4(1-adamantylamine)2 (1-AD) (ppeb = 4-[(perfluorophenyl)ethynyl]benzoate), by using non-covalent interactions. Multipoint arene–perfluoroarene (Ar–ArF) interactions, which allow the unidirectional face-to-face interaction mode of aromatic rings, were used to assemble the substitution-inert paddle-wheel Rh(II) dimers. The obtained frameworks were structurally characterisation by single crystal X-ray diffraction, and it is found that all structures exhibited a one-dimensional channel with active axial sites exposed to the pores. The porous properties of the obtained frameworks were also investigated by thermogravimetric analysis, gas adsorption and powder X-ray diffraction measurements. Moreover, the ligand substitution reaction at the active axial sites was examined at the crystalline state and the flexible structural transformation with the change of channel shapes and sizes was observed.
Introduction
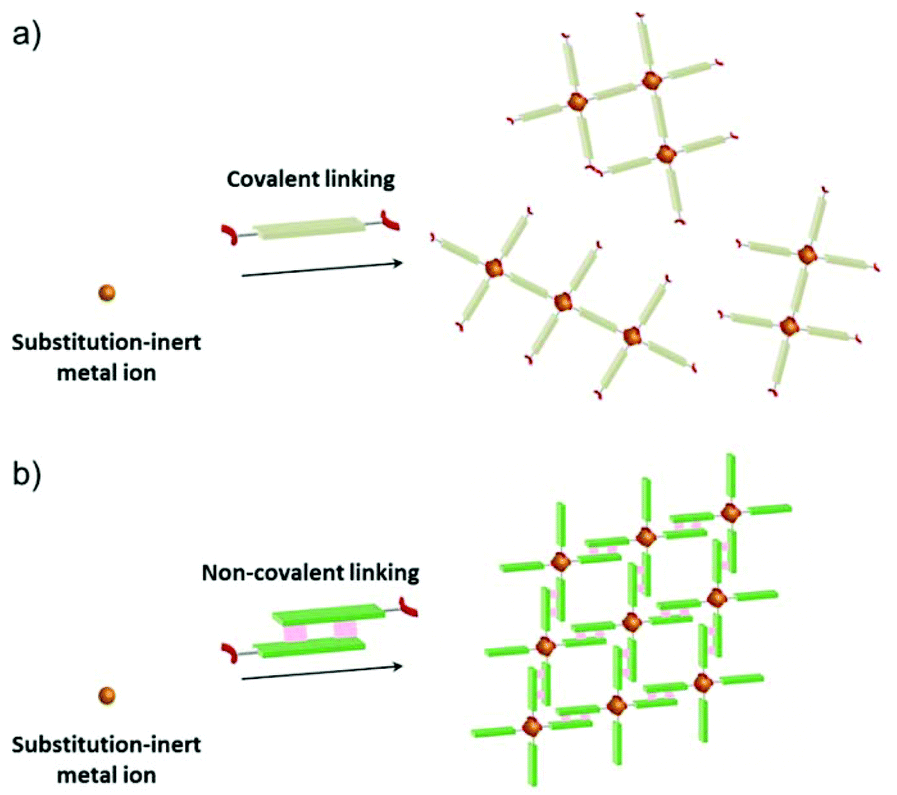
Metal–organic frameworks (MOFs) or porous coordination polymers (PCPs)1,2 are crystalline materials composed of inorganic building units and organic linkers and have attracted considerable attention as new types of porous materials. In particular, MOFs or PCPs with open metal sites are of interest because these sites can strongly interact with adsorbed molecules to afford specific functionalisation of the pore surfaces for adsorption/separation,3 heterogeneous catalysis,4 and sensing.5 One of the most straightforward strategies to construct such materials is to embed open metal sites on secondary building units (SBUs) of the framework.6 The Rh(II) paddle-wheel complex, which is composed of four substitution-inert carboxylate ligands and two substitution-labile axial ligands, would be an attractive SBU with D4h symmetry because the porous frameworks constructed are expected to show high stability arising from the substitution-inert carboxylate ligands and the high reactivity of the substitution-labile axial sites known as active centres for many catalytic reactions.7 However, it is hard to construct MOFs or PCPs by linking substitution-inert metal-based SBUs via covalent linkers (Scheme 1a) because the covalent linking of these SBUs often prevents the formation of single crystalline materials.8 Although there are many reports on single crystal structures of MOFs and PCPs consisting of substitution-labile SBUs with active sites (e.g., HKUST-1,6a MOF-74,6b and so on9), to the best of our knowledge, no single crystal structures of MOFs or PCPs constructed by substitution-inert SBUs with those sites have been reported so far.
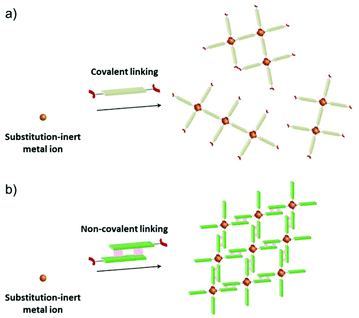 |
| | Scheme 1 Schematic illustration of the self-assembly of substitution-inert SBUs via (a) covalent linking and (b) non-covalent linking. | |
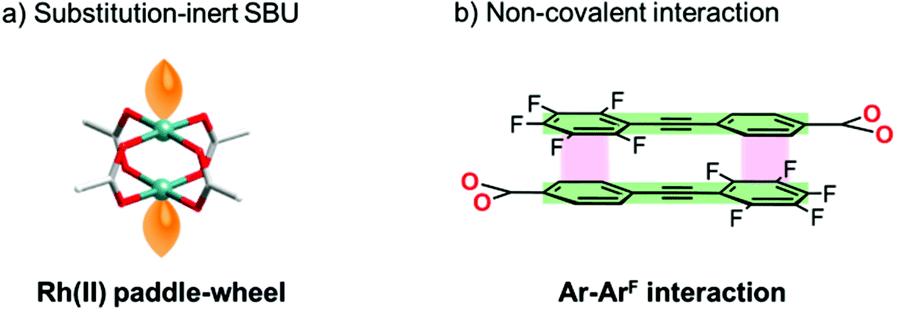
Here, we show an effective approach to construct a porous framework based on substitution-inert SBUs with active sites. Recently, many reports have proven that non-covalent interactions (e.g., hydrogen bonds,10 π–π interactions,11 combination of hydrogen bonds and π–π interactions,12 and van der Waals interactions13) can be promising tools for the construction of porous materials. In comparison with covalent linking, these interactions can work under ambient conditions to afford a comparable well-defined framework. A key to our success is the usage of this “non-covalent” interaction (Scheme 1b) for the self-assembly of Rh(II) paddle-wheel dimers (Scheme 2a). As a module for non-covalent linking, a monocarboxylate ligand with multipoint arene–perfluoroarene14 (Ar–ArF) interaction sites was designed (Scheme 2b). As we have previously reported this interaction enables the unidirectional face-to-face interaction mode of aromatic rings to work dominantly, and the controlled self-assembly of molecules can be achieved.15
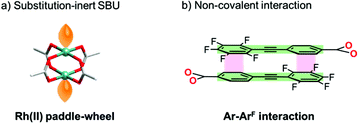 |
| | Scheme 2 Schematic illustration of (a) substitution inert SBU and (b) non-covalent interaction employed in this study. | |
Experimental
General methods
All solvents and reagents were of the highest quality available and used as received except for diethylamine and triethylamine. Diethylamine and triethylamine were dried by refluxing over KOH, distilled under argon, and degassed with a freeze-and-pump thaw. 4-[(Perfluorophenyl)ethynyl]benzoic acid (Hppeb) and Na4[Rh2(CO3)4] were prepared by using the literature methods.15,16 All syntheses were performed under an atmosphere of dry nitrogen or dry argon unless otherwise indicated. Elemental analysis was carried out on a J-SCIENCE LAB MICRO CORDER JM10 elemental analyser. The 1H NMR spectrum was acquired on a JEOL JNM-LA500 spectrometer, where chemical shifts in DMF-d7 were referenced to internal tetramethylsilane. The 19F NMR spectrum was acquired on a JEOL JNM-LA500 spectrometer, where chemical shifts in DMF-d7 were referenced to external trifluorotoluene. Thermogravimetric analyses (TGA) were carried out with a SHIMADZU DTG-60 under a nitrogen atmosphere. TGA profiles of 1-THF, 1-PN and 1-AD were measured at a heating rate of 20 K min−1. Fourier transformed infrared spectra were recorded on a Perkin-Elmer Spectrum 100 FT-IR spectrometer equipped with an ATR (Attenuated Total Reflectance) accessory.
Synthetic procedures
Synthesis of Rh2(ppeb)4(Et2O)2.
Na4[Rh2(CO3)4]·3H2O (59 mg, 0.10 mmol) and Hppeb (250 mg, 0.80 mmol) in 40 ml of dimethylacetamide/H2O mixed solvents were stirred for 8 h at 100 °C. After evaporation of the solvent, the residue was washed with water and dissolved in Et2O and subsequently purified by column chromatography using neutral alumina (Et2O as eluent). The yield of [Rh2(ppeb)4] was 44 mg (27% based on Na4[Rh2(CO3)4]·3H2O). 1H NMR (500 MHz, DMF-d7) δ ppm: 7.64 (d, J = 10.0 Hz, 2H), 7.98 (d, J = 10.0, 2H); 19F NMR (470.4 MHz, DMF-d7) δ ppm: −164.3 (dt, J = 4.7, 18.8 Hz, 2F), −154.5 (t, J = 18.8 Hz, 1F), −138.9 (dd, J = 4.7, 18.8 Hz, 2F); Anal. Calcd for Rh2(O2CC6H4C2C6F5)4(Et2O)2·2H2O: C, 49.96; H, 2.60; N, 0.00%. Found: C, 49.79; H, 2.60; N, 0.13%.
Syntheses of 1-THF, 1-PN, and 1-AD.
The three kinds of axial ligand substituted complexes were synthesised by the recrystallization of Rh2(ppeb)4(Et2O)2. Rh2(ppeb)4(THF)2 (1-THF) and Rh2(ppeb)4(3-pentanone)2 (1-PN) were obtained by the slow evaporation of the mixed solution of Et2O/THF and Et2O/3-pentanone containing Rh2(ppeb)4(Et2O)2, respectively. For Rh2(ppeb)4(1-adamantylamine)2 (1-AD), the recrystallisation was performed by layering the Et2O solution of 1-adamantylamine on the Et2O solution of Rh2(ppeb)4(Et2O)2. FTIR-ATR (cm−1): 441w, 466s, 522s, 583w, 646m, 699m, 761s, 774s, 850m, 861m, 884m, 965s, 988s, 1018w, 1042m, 1113m, 1141w, 1176w, 1286w, 1389s, 1496s, 1523m, 1549w, 1597m (COO−), 2227w (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 2876w, 2978w (1-THF). FTIR-ATR (cm−1): 411m, 465s, 522s, 582w, 645m, 699m, 725w, 761s, 774s, 813w, 849m, 859m, 964s, 989s, 1020m, 1113m, 1143w, 1178m, 1250w, 1288m, 1305m, 1391s, 1483m, 1496s, 1523m, 1548m, 1596m (COO−), 1677m, 1718w, 2227w (CC), 2980w (1-PN). FTIR-ATR (cm−1): 403w, 428w, 463s, 520s, 644w, 698m, 717w, 762s, 773s, 849m, 860m, 924m, 965s, 988s, 1017m, 1061w, 1094w, 1115m, 1141w, 1178w, 1290w, 1311w, 1385s, 1450w, 1496s, 1523m, 1547w, 1600m (COO−), 2228w (CC), 2847w, 2904w, 3275w (NH2), 3333w (NH2) (1-AD).
C), 2876w, 2978w (1-THF). FTIR-ATR (cm−1): 411m, 465s, 522s, 582w, 645m, 699m, 725w, 761s, 774s, 813w, 849m, 859m, 964s, 989s, 1020m, 1113m, 1143w, 1178m, 1250w, 1288m, 1305m, 1391s, 1483m, 1496s, 1523m, 1548m, 1596m (COO−), 1677m, 1718w, 2227w (CC), 2980w (1-PN). FTIR-ATR (cm−1): 403w, 428w, 463s, 520s, 644w, 698m, 717w, 762s, 773s, 849m, 860m, 924m, 965s, 988s, 1017m, 1061w, 1094w, 1115m, 1141w, 1178w, 1290w, 1311w, 1385s, 1450w, 1496s, 1523m, 1547w, 1600m (COO−), 2228w (CC), 2847w, 2904w, 3275w (NH2), 3333w (NH2) (1-AD).
X-ray crystallography
Crystals of 1-THF, 1-PN and 1-AD were mounted in a loop. Diffraction data at 123 K were measured on a RAXIS-RAPID Imaging Plate diffractometer equipped with confocal monochromated Mo-Kα radiation and data were processed using RAPID-AUTO (Rigaku). Structures were solved by direct methods and refined by full-matrix least squares techniques on F2 (SHELXL-97).17 All of the non-hydrogen atoms were anisotropically refined, while all of the hydrogen atoms (except for amine hydrogen atoms of 1-adamantylamine in 1-AD, which were found on a difference Fourier map and refined isotropically) were placed geometrically and refined with a riding model with Uiso constrained to be 1.2 times the Ueq of the carrier atom. For 1-THF, the diffused electron densities resulting from residual solvent molecules were removed from the data set using the SQUEEZE routine of PLATON and refined further using the data generated. CCDC reference numbers 1049445–1049447 (1-THF, 1-PN and 1-AD) (Table 1).
Table 1 Crystallographic data and structural refinements for 1-THF, 1-PN and 1-AD
| Compound |
1-THF
|
1-PN
|
1-AD
|
|
R
1 = ∑||Fo| − |Fc||/∑|Fo|.
wR2 = [∑(w(Fo2 − Fc2)2)/∑w(Fo2)2]1/2.
|
| Formula |
C68H32F20O10Rh2 |
C170H132F40O26Rh4 |
C92H70F20N2O11Rh2 |
| Formula weight |
1594.76 |
3762.40 |
1965.32 |
| Temperature (°C) |
−150 |
−150 |
−150 |
| Crystal system |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
C2/m (#12) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2) (#2) |
P (#2) |
|
a (Å) |
18.6449(7) |
15.1526(7) |
10.8625(8) |
|
b (Å) |
27.2267(14) |
15.3844(4) |
13.8827(10) |
|
c (Å) |
8.8932(4) |
19.5087(4) |
14.9217(11) |
|
α (°) |
90 |
99.0550(7) |
103.6458(15) |
|
β (°) |
107.4822(12) |
96.0045(7) |
93.1519(15) |
|
γ (°) |
90 |
112.8590(7) |
94.0312(16) |
|
V (Å3) |
4306.0(3) |
4069.14(17) |
2175.3(3) |
|
Z
|
2 |
1 |
1 |
|
D
c (g cm−3) |
1.230 |
1.535 |
1.500 |
|
μ (mm−1) |
0.471 |
0.514 |
0.483 |
|
F(000) |
1580 |
1900 |
994 |
R
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
0.0660 |
0.0554 |
0.0511 |
| wR2b |
0.1742 |
0.1875 |
0.1511 |
| Goodness-of-fit on F2 |
1.135 |
1.111 |
1.089 |
Crystal data for Rh2(ppeb)4(THF)2 (1-THF).
C68H32F20O10Rh2, Mr = 1594.76, monoclinic, space group C2/m, (#12), a = 18.6449(7), b = 27.2267(14), c = 8.8932(4) Å, β = 107.4822(12)°, V = 4306.0(3) Å3, Z = 2, T = 123(2) K, ρc = 1.230 g cm−3, μ(Mo-Kα) = 0.471 cm−1, 2θmax = 50.0°, λ(Mo-Kα) = 0.710747 Å, 16842 reflections measured, 3892 unique (Rint = 0.0312), 3549 (I > 2σ(I)) were used to refine 238 parameters, 6 restraints, wR2 = 0.1742 (I > 2σ(I)), R1 = 0.0660 (I > 2σ(I)), GOF = 1.135.
Crystal data for Rh2(ppeb)4(3-pentanone)2 (1-PN).
C170H132F40O26Rh4, Mr = 3762.40, triclinic, space group P, (#2), a = 15.1526(7), b = 15.3844(4), c = 19.5087(4) Å, α = 99.0550(7)°, β = 96.0045(7)°, γ = 112.8590(7)°, V = 4069.14(17) Å3, Z = 1, T = 123 (2) K, ρc = 1.535 g cm−3, μ(Mo-Kα) = 0.514 cm−1, 2θmax = 54.96°, λ(Mo-Kα) = 0.710747 Å, 40282 reflections measured, 18528 unique (Rint = 0.0223), 13311 (I > 2σ(I)) were used to refine 1081 parameters, 0 restraints, wR2 = 0.1875 (I > 2σ(I)), R1 = 0.0554 (I > 2σ(I)), GOF = 1.111.
Crystal data for Rh2(ppeb)4(1-adamantylamine)2 (1-AD).
C92H70F20N2O8Rh2, Mr = 1965.32, triclinic, space group P, (#2), a = 10.8625(8), b = 13.8827(10), c = 14.9217(11) Å, α = 103.6458(15)°, β = 93.1519(15)°, γ = 94.0312(16)°, V = 2175.3(3) Å3, Z = 1, T = 123 (2) K, ρc = 1.500 g cm−3, μ(Mo-Kα) = 0.483 cm−1, 2θmax = 54.94°, λ(Mo-Kα) = 0.710747 Å, 21333 reflections measured, 9889 unique (Rint = 0.031), 8336 (I > 2σ(I)) were used to refine 602 parameters, 6 restraints, wR2 = 0.1511 (I > 2σ(I)), R1 = 0.0511 (I > 2σ(I)), GOF = 1.089.
Results and discussion
Synthesis and characterization
Synthesis of the Rh(II) complex was performed by the reaction of Na4[Rh2(CO3)4]·3H2O with 8 eq. of Hppeb at 100 °C in DMA/H2O (3:1) for 8 h. A four substituted complex, Rh2(ppeb)4(Et2O)2, was obtained in 27% yield by subsequent column chromatography using neutral alumina (Et2O as the eluent). Note that the use of Na4[Rh2(CO3)4]·3H2O was crucial, and the reaction of Hppeb with carboxylate precursors, Rh(II) acetate or Rh(II) trifluoroacetate, did not afford the desired complex possibly due to the slow reaction rate of the di-substituted carboxylate complex.15 The slow evaporation of the THF/Et2O mixed solution of Rh2(ppeb)4(Et2O)2 afforded single crystals of Rh2(ppeb)4(THF)2 (1-THF) suitable for X-ray crystallography. 1-PN and 1-AD were also obtained by recrystallization of Rh2(ppeb)4(Et2O)2. The recrystallisation of Rh2(ppeb)4(Et2O)2 from slow evaporation of the mixed solution of Et2O/3-pentanone afforded Rh2(ppeb)4(3-pentanone)2 (1-PN). To obtain Rh2(ppeb)4(1-adamantylamine)2 (1-AD), recrystallisation was performed by layering Et2O solution of 1-adamantylamine on Et2O solution of Rh2(ppeb)4(Et2O)2. These three compounds were characterised by single-crystal X-ray diffraction and IR spectroscopy.
Crystal structures
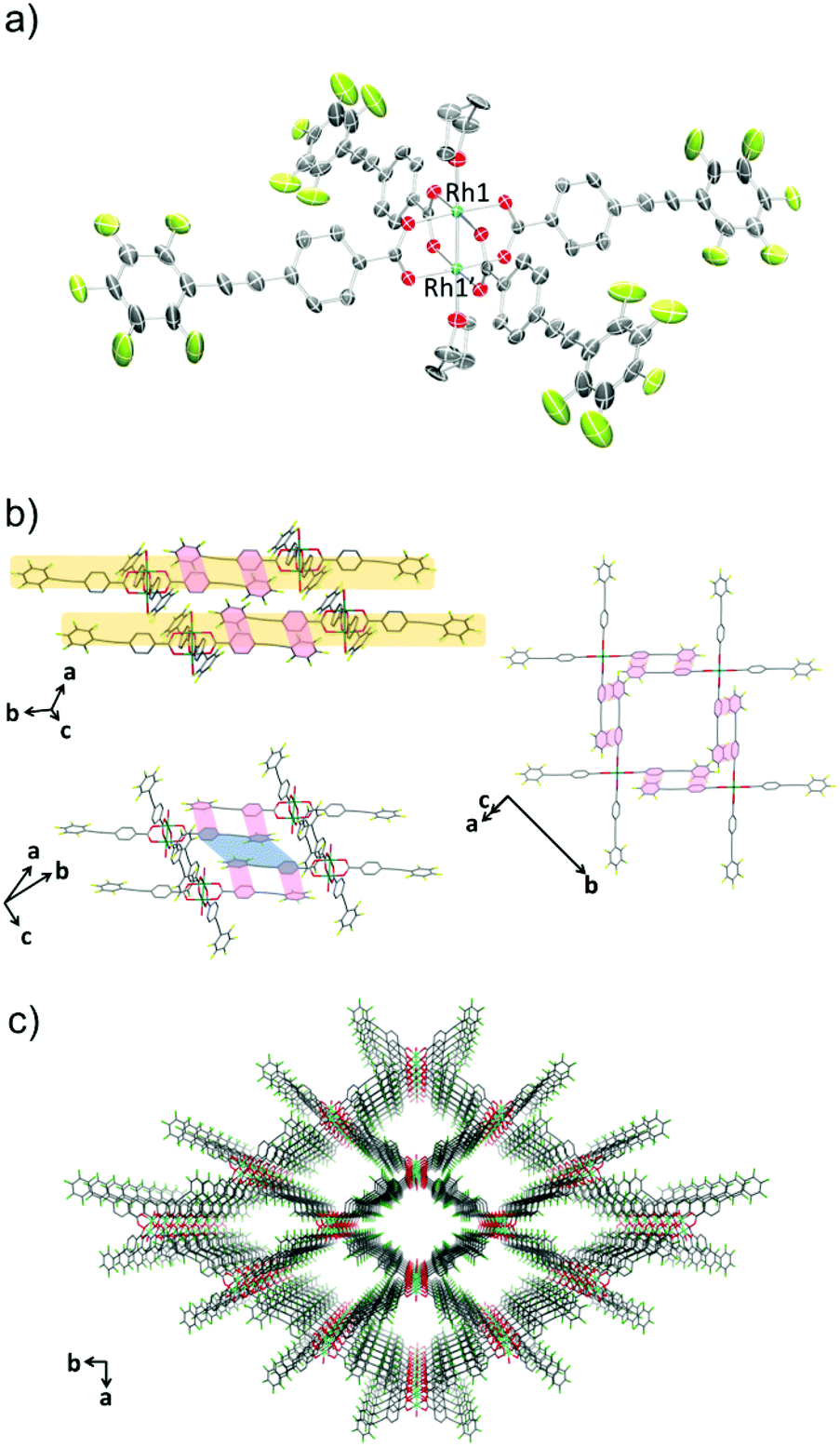
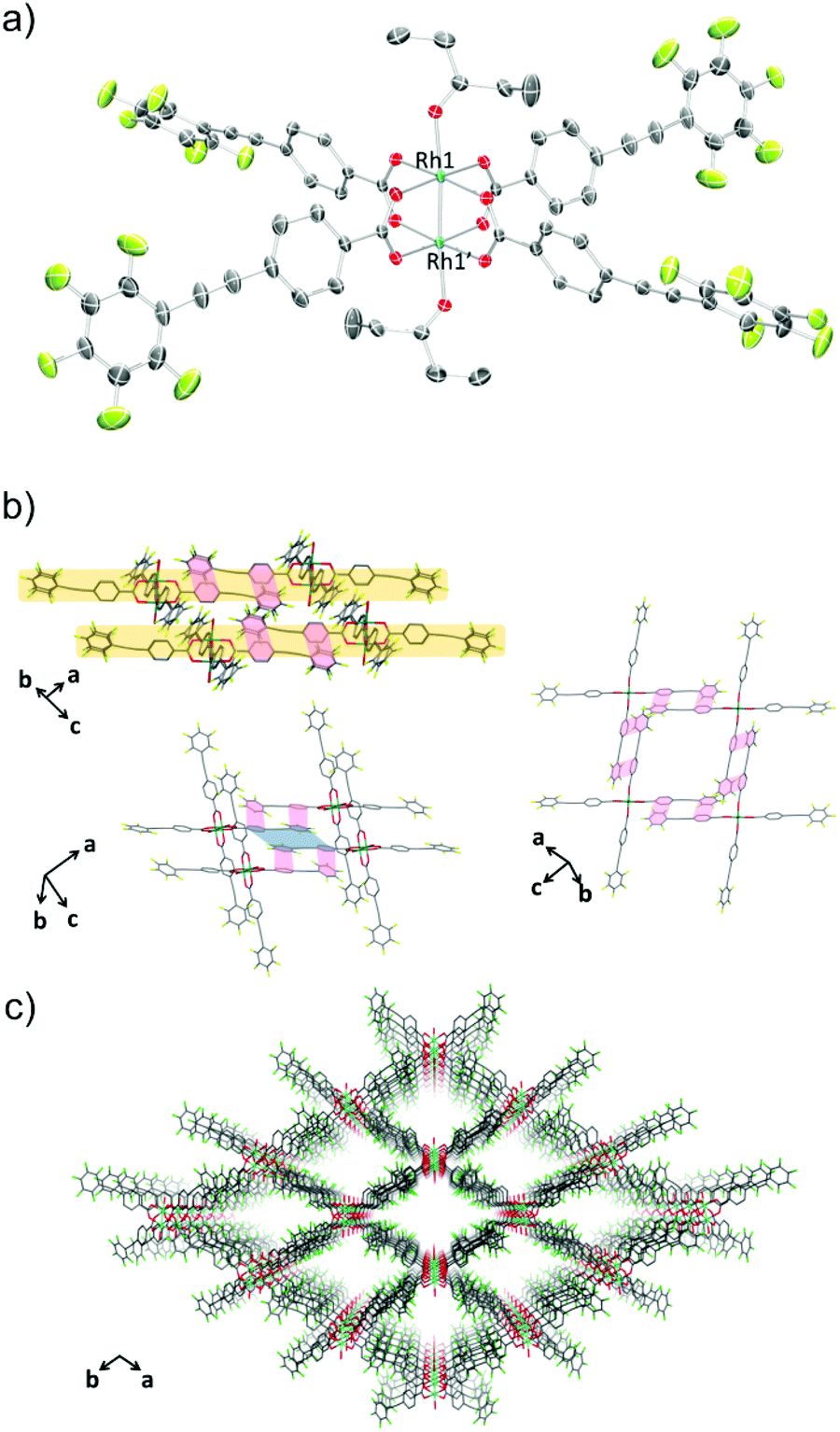
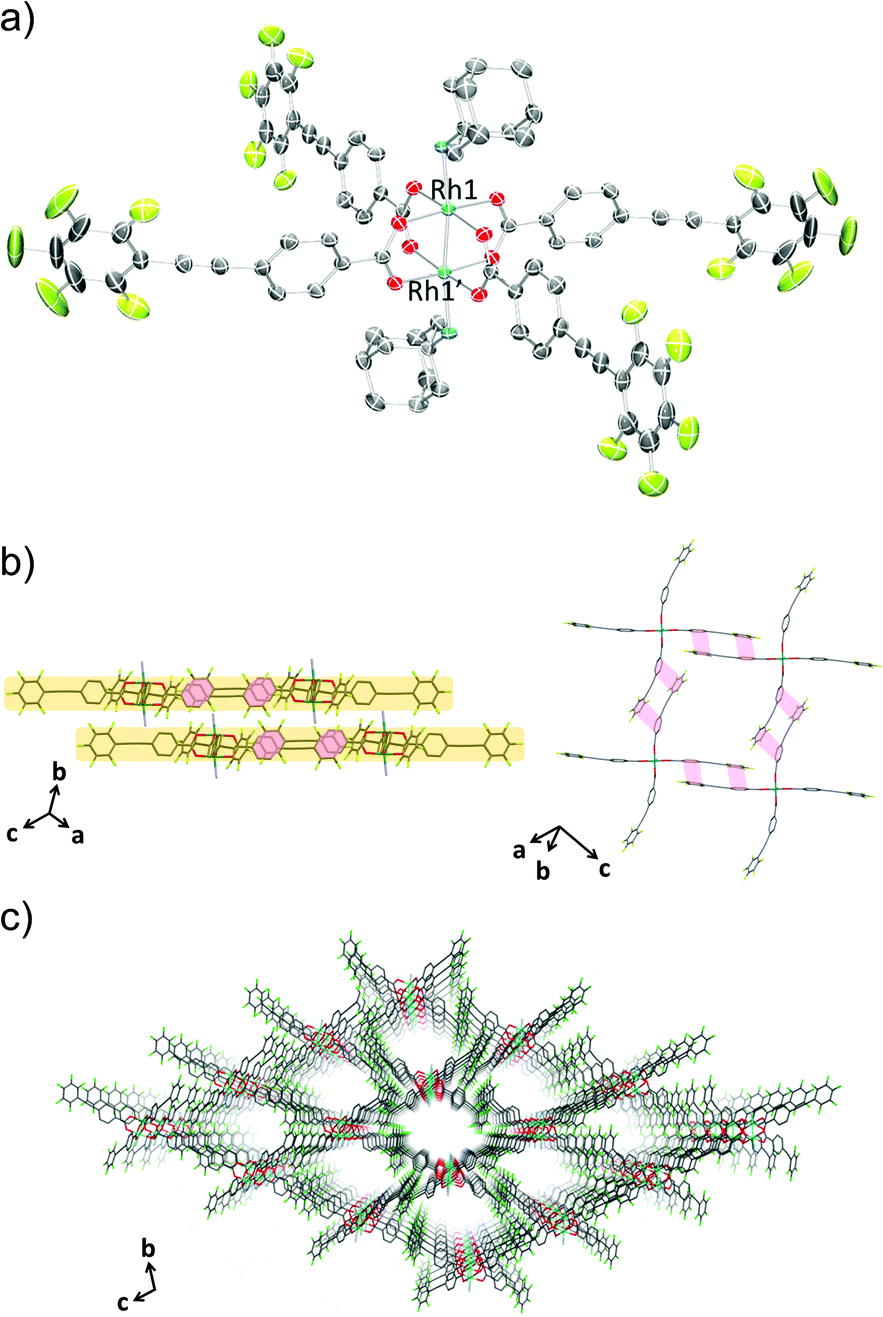
The crystal structure of 1-THF is comprised of a Rh(II) paddle-wheel with THF molecules coordinated at the axial sites (Fig. 1a). Four ppeb− ligands occupied the positions in the equatorial plane of each rhodium atom. The Rh1–Rh1′ distance was 2.3875(13) Å, which is in the range found for previously reported Rh(II) paddle-wheel dimers (2.316 to 2.486 Å).18 The angle between the phenylene and perfluorophenyl rings was estimated to be 18.4°.
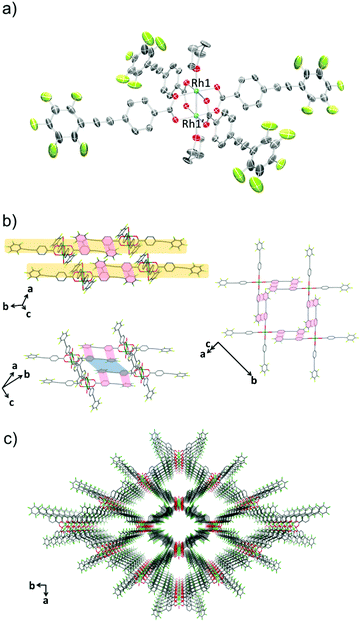 |
| | Fig. 1 (a) An ORTEP drawing of 1-THF (50% probability ellipsoids). Hydrogen atoms are omitted for clarity. (b) 2-D sheet structure of 1-THF. Ar–ArF interactions and π–π interactions are shown in red and blue lines respectively. Hydrogen atoms and carbon atoms of THF at the axial positions are omitted for clarity. (c) Crystal packing of 1-THF along the c axis. Hydrogen atoms, and carbon atoms of THF at the axial positions are omitted for clarity. O = red, C = grey, F = pale green, Rh = sea green. | |
In the crystal packing structure of 1-THF, the aforementioned multipoint Ar–ArF interaction played a key role in stabilising the molecular assembly. As shown in Fig. 1b, an infinite two-dimensional (2-D) square-grid sheet structure was formed via multipoint Ar–ArF interactions between the ligands. The mean interplanar separation between the phenylene and perfluorophenyl rings was 3.56(16) Å. These 2-D sheets were stacked through π–π interactions (denoted as blue line in Fig. 1b) to form a columnar structure of paddle-wheel units along the c axis. As a result, a porous structure with the channel entrance size19 of 13.8 × 12.1 Å2 was formed (Fig. 1c), and THF molecules were contained in the pores as guests. The recrystallisation of Rh2(ppeb)4(Et2O)2 from Et2O/3-pentanone afforded a similar crystal structure composed of a Rh(II) paddle-wheel complex bearing 3-pentanone at the axial sites, Rh2(ppeb)4(3-pentanone)2 (1-PN) (Fig. 2a). In the crystal structure of 1-PN, the mean interplanar separation between phenylene and perfluorophenyl rings was 3.49(12) Å, and the channel entrance size was estimated to be 12.9 × 10.5 Å2. It should be noted that the active sites of these complexes were exposed to the pores in both structures.
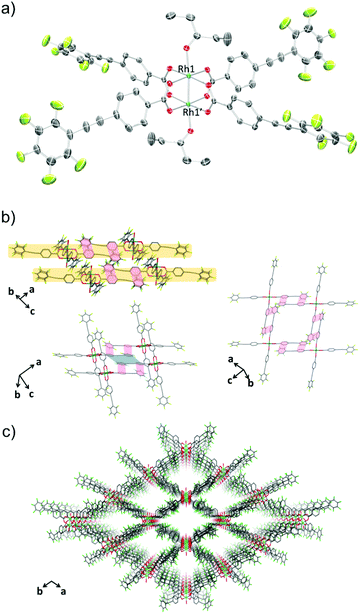 |
| | Fig. 2 (a) An ORTEP drawing of 1-PN (50% probability ellipsoids). Hydrogen atoms and crystal solvent molecules are omitted for clarity. (b) 2-D sheet structure of 1-PN. Ar–ArF interactions and π–π interactions are shown in red and blue lines respectively. Hydrogen atoms, carbon atoms of 3-pentanone at the axial positions and crystal solvent molecules observed in the channels are omitted for clarity. (c) Crystal packing of 1-PN along the c axis. Hydrogen atoms, and carbon atoms of 3-pentanone at the axial positions and crystal solvent molecules observed in the channels are omitted for clarity. O = red, C = grey, F = pale green, Rh = sea green. | |
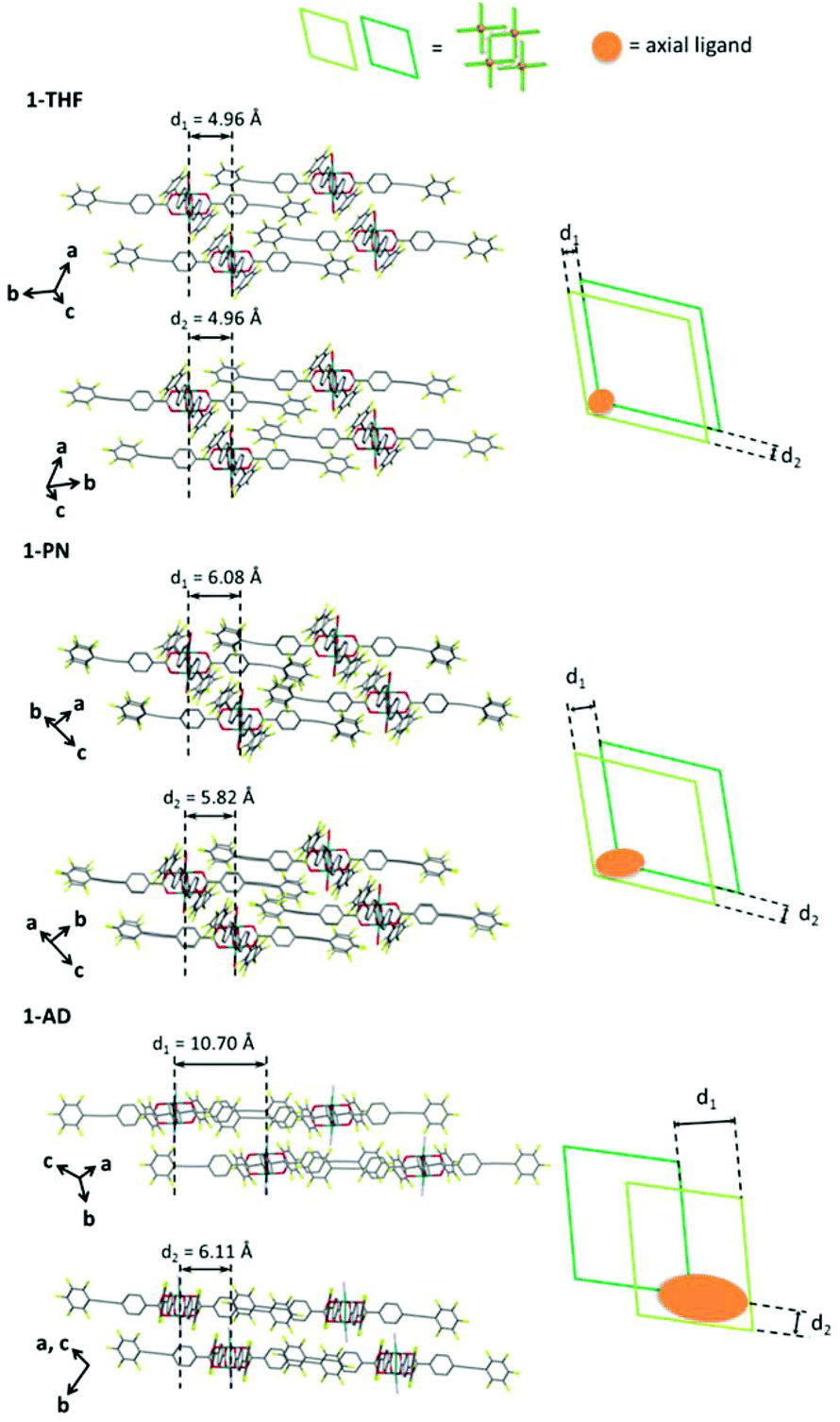
The reaction of Rh2(ppeb)4(Et2O)2 with 1-adamantylamine was subsequently performed to examine the adjustability of the framework to bulky molecules, and the targeted axial ligand substituted complex, Rh2(ppeb)4(1-adamantylamine)2 (1-AD, Fig. 3a), was obtained. The distance between two rhodium atoms in 1-AD was 2.4140(7) Å, which is comparable to that of 1-THF or 1-PN. For 1-AD, the angles between the phenylene and perfluorophenyl rings were 16.3° and 5.9°. In the crystal structure of 1-AD, a 2-D sheet structure stabilised by multipoint Ar–ArF interactions was also obtained (Fig. 3b). However, π–π interactions between the 2-D sheets were not observed because the bulkiness of the axial ligands prevented a close contact between sheets. Alternatively, the ppeb− ligands exhibited a conformation almost perpendicular to the 2-D sheets, and CF–π interactions20 between the ppeb− ligands formed inter-sheet stacking. As a result, a 1-D channel structure with an entrance size of 10.1 × 8.2 Å2 was obtained (Fig. 3c). The mean interplanar separation between the phenylene and perfluorophenyl rings was 3.50(16) Å. The axial ligands were also exposed to the pores despite the different stacking mode from 1-THF and 1-PN. The comparison between the assembling-patterns between these structures is shown in Fig. 4. The horizontal distances between the Rh dimeric nodes in neighbouring 2-D sheets (denoted as d1 and d2) were found to be d1 = d2 = 4.96 Å for 1-THF, d1 = 6.08 Å, d2 = 5.82 Å for 1-PN and d1 = 10.70 Å, d2 = 6.11 Å for 1-AD and were mainly different between the three structures, reflecting the bulkiness of the axial ligands. These crystallographic observations indicate that the 2-D sheet structures in these porous frameworks are well defined by the multipoint Ar–ArF interactions and that the interlayer stacking structures are controllable in response to the size and shape of the axial ligands.
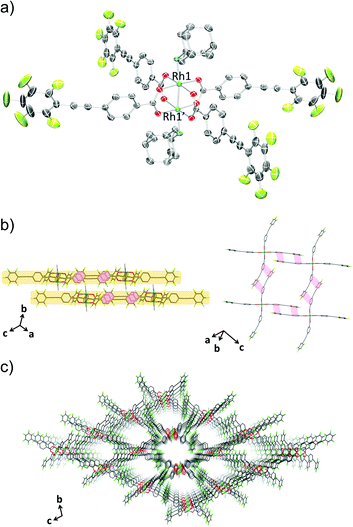 |
| | Fig. 3 (a) An ORTEP drawing of 1-AD (50% probability ellipsoids). Hydrogen atoms and crystal solvent molecules are omitted for clarity. (b) 2-D sheet structure of 1-AD. Ar–ArF interactions are shown in red lines. Hydrogen atoms, and carbon atoms of 1-adamantylamine at the axial positions and crystal solvent molecules observed in the channels are omitted for clarity. (c) Crystal packing of 1-AD along the a axis. Hydrogen atoms, and carbon atoms of 1-adamantylamine at the axial positions and crystal solvent molecules observed in the channels are omitted for clarity. O = red, C = grey, F = pale green, N = blue, Rh = sea green. | |
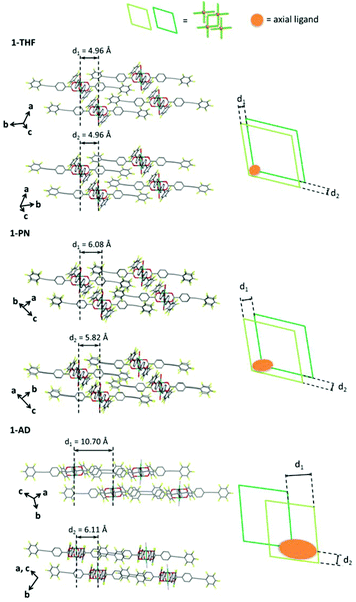 |
| | Fig. 4 Side view of the crystal packing (left) and the schematic illustration of the top view of the interlayer stacking structure (right) for 1-THF (top), 1-PN (middle) and 1-AD (bottom). In the schematic illustration, the axial ligands are represented as orange circles. | |
The effect of the axial ligands on the porous properties
The effect of the axial ligands on the porous properties of the frameworks was also investigated by several measurements. A TG curve of 1-AD exhibited no weight loss upon heating up to 284 °C (Fig. 5c), which indicated the strong coordination ability of the axial ligands to the metal centres. Upon further heating up to 600 °C, the degradation of the complex was observed. It should be noted that the removal of the guest molecules (Et2O) was not observed due to their highly volatile nature. The removal of the axial ligand was also not observed due to their strong coordination ability. Furthermore, the CO2 adsorption isotherms for 1-AD evacuated at 40 °C (Fig. 5f) and 120 °C (Fig. S1c in the ESI†) exhibited almost identical sorption profiles, suggesting the retention of the porous structure due to the “capping” of the active sites with the strongly coordinated axial ligands. In contrast, the TG curves of 1-THF and 1-PN indicated the release of the axial ligands in the temperature range of 100–170 °C with a weight loss of 5.30% (1-THF) and 5.82% (1-PN), respectively (Fig. 5a and b). The degradation of the complexes was observed in the temperature range of 327–600 °C. In the CO2 adsorption isotherms of 1-THF and 1-PN evacuated at 40 °C and 120 °C, the amount of adsorbed CO2 gradually increased as the pressure increased and showed hysteresis during the desorption process (Fig. 5d and e and Fig. S1a and b in the ESI†) and the adsorption amount is dependent on the temperature of pre-treatment, which may be attributed to the transformation of the interlayer stacking structure arising from the exchange or the loss of the axial ligands. These results clearly indicate the difference in the lability of the axial ligands between 1-AD and other two structures, 1-THF and 1-PN. Additionally, N2 adsorption measurements at 77 and 195 K were performed and almost no adsorption of N2 was observed for 1-THF and 1-PN (Fig. S2a and b, and Fig. S3a and b in the ESI†), suggesting the selective CO2 adsorption over N2 in these frameworks. In contrast to 1-THF and 1-PN, N2 was slightly adsorbed to 1-AD at 77 K (Fig. S2c in the ESI†), although no adsorption of N2 was observed at 195 K (Fig. S3c in the ESI†).
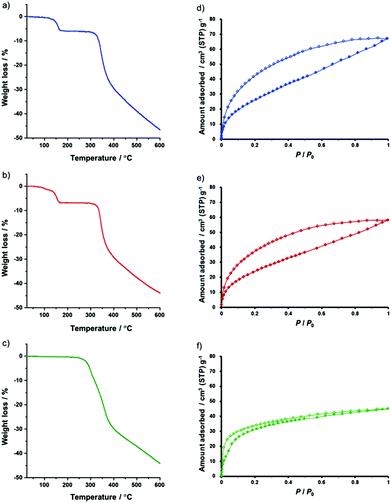 |
| | Fig. 5 TGA profiles of (a) 1-THF, (b) 1-PN and (c) 1-AD with a heating rate of 20 K min−1. CO2 sorption isotherms of (d) 1-THF, (e) 1-PN and (f) 1-AD at 195 K. Filled shapes: adsorption. Open shapes: desorption. The samples of 1-THF, 1-PN and 1-AD for the CO2 adsorption were evacuated at 40 °C before the measurements. | |
PXRD measurements of the samples after the removal of the guest molecules and the dissociation of the axial ligands were performed to investigate the structural change in these processes. The experiments were performed for 1-AD and 1-PN, which have the unremovable axial ligands and the removable ligands, respectively. Note that the PXRD patterns for the as-synthesized compounds were measured under the vapour of the guest molecules to prevent the loss of the guest molecules. In the case of 1-AD, the PXRD pattern changed after the evacuation at 40 °C (from A to B in Fig. S4 in the ESI†), suggesting the structural change attributed to the removal of the guest molecules. Subsequent evacuation of the sample at 120 °C gave the almost identical PXRD pattern (Fig. S4C in the ESI†), which indicates the strong coordination of axial ligands. These results are fully consistent with the result of TGA and CO2 adsorption measurements. In the case of 1-PN, the PXRD pattern changed after evacuation at 40 °C (from D to E in Fig. S5 in the ESI†) and the structural transformation accompanied by the removal of the guest molecules was observed. In contrast to 1-AD, the subsequent evacuation at 120 °C caused the significant change of the PXRD pattern (from E to F in Fig. S5 in the ESI†) and the structural transformation due to the dissociation of the axial ligands was indicated. At this stage, we could not determine the structure after the removal of the guest molecules or the axial ligands, and thus, the detailed discussion on the CO2 adsorption profiles was not allowed. However, the results of CO2 adsorption and PXRD measurements revealed that the crystallinity and porosity of the sample were maintained throughout the removal of the guest molecules and the dissociation of the axial ligands, and the framework structures transformed flexibly accompanied by these processes.
Additionally, the PXRD patterns before and after the CO2 adsorption were also examined. As shown in Fig. S6–9, in the ESI,† the samples of 1-PN and 1-AD did not show significant change in the PXRD pattern after the CO2 adsorption measurements regardless of the pre-treatment temperature. These results indicate that our frameworks recover their initial structures after the CO2 adsorption/desorption processes.
Flexible structural transformation during the ligand exchange reaction of the frameworks
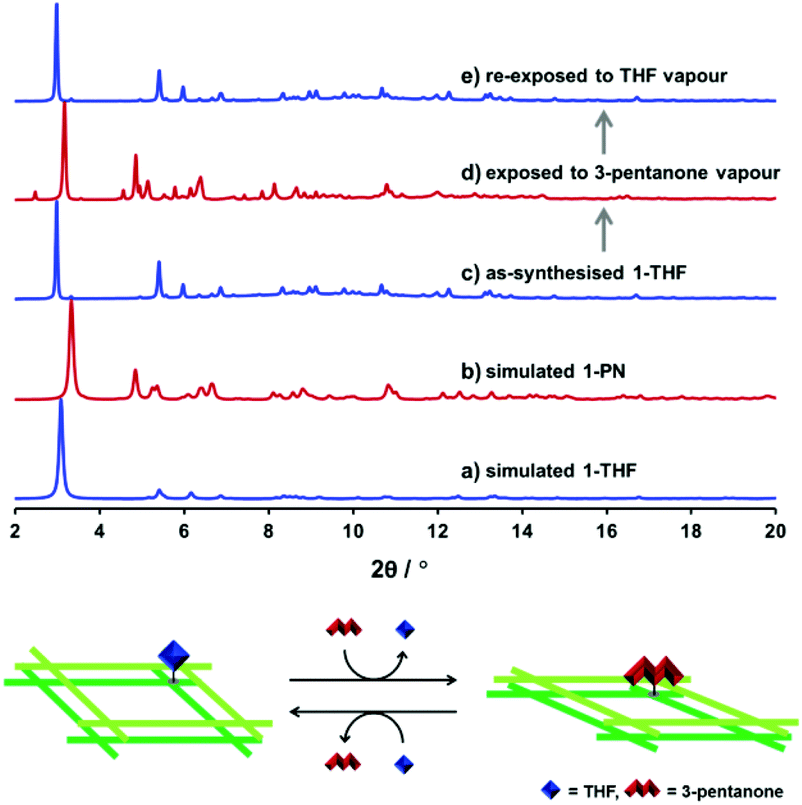
The structural flexibility of 1-THF and 1-PN and the reactivity of the open axial sites in these frameworks were examined by PXRD measurements. Initially, a crystalline sample of 1-THF was evacuated at room temperature for 1 h and exposed to 3-pentanone vapour for 1 h. PXRD patterns before and after the treatment suggested the complete structural transformation from 1-THF to 1-PN (Fig. 6c and d, blue and red lines). Subsequently, the obtained powder sample of 1-PN was evacuated at room temperature for 1 h and exposed to THF vapour for 1 h. The PXRD pattern of the sample treated with THF revealed the regeneration of 1-THF (Fig. 6e). These results indicate that the active sites of the SBUs in our framework effectively worked to control the structural flexibility of each phase.
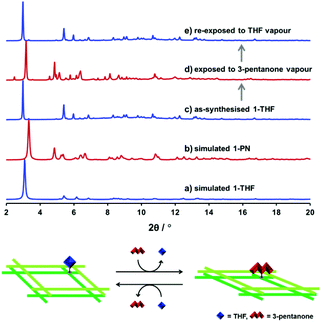 |
| | Fig. 6 Simulated XRD patterns of (a) 1-THF and (b) 1-PN and PXRD patterns of as-synthesised (c) 1-THF, (d) the sample of 1-THF exposed to 3-pentanone vapour, and (e) the sample re-exposed to THF vapour. Schematic illustration of the structural transformation between 1-THF and 1-PN induced by the reversible ligand substitution reaction (bottom) (λ = 0.79978 Å). | |
Conclusions
In conclusion, we demonstrated the construction and structural determination of porous frameworks composed of a substitution-inert complex with active sites. Three novel Rh(II) paddle-wheel complexes, Rh2(ppeb)4(X)2 (X = THF (1-THF), 3-pentanone (1-PN), and 1-adamantylamine (1-AD)), were successfully assembled to form porous frameworks with a solvent accessible columnar channel by utilizing the multipoint Ar–ArF interaction. The obtained supramolecular structures possessed active sites exposed to the channel, and the comparison of crystal packing of the three complexes revealed the structural difference induced by the size and the structure of the axial ligands. Furthermore, the porous properties of these frameworks were also examined by several experiments. Although the axial ligands of 1-AD did not dissociate upon heating and during the sorption process, the axial ligands of 1-THF and 1-PN were labile to the axial ligand substitution reaction, and the induced-fit structural transformation in response to the ligand substitution was observed in the crystalline state. We believe that our results presented here should be a key example to construct porous materials based on substitution-inert metal ions with active sites that can be applied for catalytic reactions and chemical sensors.
Acknowledgements
This work was supported by a Grant-in-Aid for Young Scientists (A) (No. 15H05480) (to M.K.), a Grant-in-Aid for Young Scientists (A) (No. 25708011) (to S.M.), a Grant-in-Aid for Challenging Exploratory Research (No. 26620160) (to S.M.), and a Grant-in-Aid for JSPS Fellows (No. K2S2605890) (to T.I.) from the Japan Society for the Promotion of Science. This work was also supported by a Grant-in-Aid for Scientific Research on Innovative Areas “AnApple” (No. 15H00889). The synchrotron radiation experiments were performed at the BL02B2 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2014B1428).
Notes and references
-
(a) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed;
(b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed;
(c) G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC;
(d) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC;
(e) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC;
(f) D. Zacher, O. Shekhah, C. Wöll and R. A. Fischer, Chem. Soc. Rev., 2009, 38, 1418–1429 RSC;
(g) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed;
(h) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed;
(i) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed;
(j) C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed;
(k) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974–986 CrossRef CAS PubMed.
-
(a) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed;
(b) B. D. Chandler, G. D. Enright, K. A. Udachin, S. Pawsey, J. A. Ripmeester, D. T. Cramb and G. K. H. Shimizu, Nat. Mater., 2008, 7, 229–235 CrossRef CAS PubMed;
(c) W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906–912 CrossRef CAS PubMed;
(d) J. Rabone, Y.-F. Yue, S. Y. Chong, K. C. Stylianou, J. Bacsa, D. Bradshaw, G. R. Darling, N. G. Berry, Y. Z. Khimyak, A. Y. Ganin, P. Wiper, J. B. Claridge and M. J. Rosseinsky, Science, 2010, 329, 1053–1057 CrossRef CAS PubMed;
(e) G. K. H. Shimizu, J. M. Taylor and S. Kim, Science, 2013, 341, 354–355 CrossRef CAS PubMed;
(f) H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
-
(a) E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed;
(b) S. Xiang, Z. Zhang, C. Zhao, K. Hong, X. Zhao, D. Ding, M. Xie, C. Wu, M. C. Das, R. Gill, K. M. Thomas and B. Chen, Nat. Commun., 2011, 2, 204 CrossRef PubMed;
(c) E. D. Bloch, M. R. Hudson, J. A. Mason, S. Chavan, V. Crocellà, J. D. Howe, K. Lee, A. L. Dzubak, W. L. Queen, J. M. Zadrozny, S. J. Geier, L.-C. Lin, L. Gagliardi, B. Smit, J. B. Neaton, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2014, 136, 10752–10761 CrossRef CAS PubMed;
(d) Y.-S. Bae, C. Y. Lee, K. C. Kim, O. K. Farha, P. Nickias, J. T. Hupp, S. T. Nguyen and R. Q. Snurr, Angew. Chem., Int. Ed., 2012, 51, 1857–1860 CrossRef CAS PubMed;
(e) Z. Guo, H. Wu, G. Srinivas, Y. Zhou, S. Xiang, Z. Chen, Y. Yang, W. Zhou, M. O'Keeffe and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 3178–3181 CrossRef CAS PubMed.
-
(a) C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS PubMed;
(b) Z. Zhang, Y. Chen, S. He, J. Zhang, X. Xu, Y. Yang, F. Nosheen, F. Saleem, W. He and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 12517–12521 CAS;
(c) W. Xuan, C. Ye, M. Zhang, Z. Chen and Y. Cui, Chem. Sci., 2013, 4, 3154–3159 RSC.
-
(a) B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693–1696 CrossRef CAS PubMed;
(b) N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dincă, J. Am. Chem. Soc., 2013, 135, 13326–13329 CrossRef CAS PubMed.
-
(a) S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS;
(b) N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
-
(a) P. Ceccherelli, M. Curini, M. C. Marcotullio and O. Rosati, Tetrahedron, 1991, 47, 7403–7408 CrossRef CAS;
(b) M. P. Doyle, Acc. Chem. Res., 1986, 19, 348–356 CrossRef CAS;
(c) M. P. Doyle, K. G. High, C. L. Neslonely, T. W. Clayton Jr. and J. Lin, Organometallics, 1991, 10, 1225–1226 CrossRef CAS;
(d) S. Tanaka, S. Masaoka, K. Yamauchi, M. Annaka and K. Sakai, Dalton Trans., 2010, 39, 11218–11226 RSC;
(e) D. K. Kumar, A. S. Filatov, M. Napier, J. Sun, E. V. Dikarev and M. A. Petrukhina, Inorg. Chem., 2012, 51, 4855–4861 CrossRef CAS PubMed;
(f) J. Hansen and H. M. L. Davies, Coord. Chem. Rev., 2008, 252, 545–555 CrossRef CAS PubMed;
(g) K. P. Kornecki, J. F. Briones, V. Boyarskikh, F. Fullilove, J. Autschbach, K. E. Schrote, K. M. Lancaster, H. M. L. Davies and J. F. Berry, Science, 2013, 342, 351–354 CrossRef CAS PubMed.
-
(a) G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed;
(b) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
-
(a) P. D. C. Dietzel, Y. Morita, R. Blom and H. Fjellvåg, Angew. Chem., Int. Ed., 2005, 44, 6354–6358 CrossRef CAS PubMed;
(b) B. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745–4749 CrossRef CAS PubMed;
(c) X.-S. Wang, S. Ma, P. M. Forster, D. Yuan, J. Eckert, J. J. López, B. J. Murphy, J. B. Parise and H.-C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7263–7266 CrossRef CAS PubMed;
(d) D. Yuan, D. Zhao, D. Sun and H.-C. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5357–5361 CrossRef CAS PubMed;
(e) M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876–16883 CrossRef PubMed;
(f) T. M. McDonald, D. M. D'Alessandro, R. Krishnac and J. R. Long, Chem. Sci., 2011, 2, 2022–2028 RSC;
(g) Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS PubMed;
(h) M. Pang, A. J. Cairns, Y. Liu, Y. Belmabkhout, H. C. Zeng and M. Eddaoudi, J. Am. Chem. Soc., 2012, 134, 13176–13179 CrossRef CAS PubMed.
-
(a) P. Li, Y. He, J. Guang, L. Weng, J. C.-G. Zhao, S. Xiang and B. Chen, J. Am. Chem. Soc., 2014, 136, 547–549 CrossRef CAS PubMed;
(b) W. Yang, A. Greenaway, X. Lin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, P. Hubberstey, S. Kitagawa, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 14457–14469 CrossRef CAS PubMed;
(c) M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed;
(d) S. A. Dalrymple and G. K. H. Shimizu, J. Am. Chem. Soc., 2007, 129, 12114–12116 CrossRef CAS PubMed;
(e) P. S. Nugent, V. L. Rhodus, T. Pham, K. Forrest, L. Wojtas, B. Space and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 10950–10953 CrossRef CAS PubMed;
(f) P. Li, Y. He, Y. Zhao, L. Weng, H. Wang, R. Krishna, H. Wu, W. Zhou, M. O'Keeffe, Y. Han and B. Chen, Angew. Chem., Int. Ed., 2015, 54, 574–577 CAS.
-
(a) M. J. Bojdys, M. E. Briggs, J. T. A. Jones, D. J. Adams, S. Y. Chong, M. Schmidtmann and A. I. Cooper, J. Am. Chem. Soc., 2011, 133, 16566–16571 CrossRef CAS PubMed;
(b) T. Kaczorowski, I. Justyniak, T. Lipińska, J. Lipkowski and J. Lewiński, J. Am. Chem. Soc., 2009, 131, 5393–5395 CrossRef CAS PubMed;
(c) M. Mastalerz, M. W. Schneider, I. M. Oppel and O. Presly, Angew. Chem., Int. Ed., 2011, 50, 1046–1051 CrossRef CAS PubMed;
(d) K. Sokołowski, W. Bury, I. Justyniak, D. Fairen-Jimenez, K. Sołtys, D. Prochowicz, S. Yang, M. Schröder and J. Lewiński, Angew. Chem., Int. Ed., 2013, 52, 13414–13418 CrossRef PubMed;
(e) G. Zhang, O. Presly, F. White, I. M. Oppel and M. Mastalerz, Angew. Chem., Int. Ed., 2014, 53, 1516–1520 CrossRef CAS PubMed;
(f) C. Redshaw, S. Jana, C. Shang, M. R. J. Elsegood, X. Lu and Z. X. Guo, Organometallics, 2010, 29, 6129–6132 CrossRef CAS.
-
(a) J. Lü, C. Perez-Krap, M. Suyetin, N. H. Alsmail, Y. Yan, S. Yang, W. Lewis, E. Bichoutskaia, C. C. Tang, A. J. Blake, R. Cao and M. Schröder, J. Am. Chem. Soc., 2014, 136, 12828–12831 CrossRef PubMed;
(b) W. Bury, I. Justyniak, D. Prochowicz, A. Rola-Noworyta and J. Lewiǹski, Inorg. Chem., 2012, 51, 7410–7414 CrossRef CAS PubMed;
(c) J. Lewiński, T. Kaczorowski, D. Prochowicz, T. Lipińska, I. Justyniak, Z. Kaszkur and J. Lipkowski, Angew. Chem., Int. Ed., 2010, 49, 7035–7039 CrossRef PubMed.
-
(a) H. Kim, Y. Kim, M. Yoon, S. Lim, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2010, 132, 12200–12202 CrossRef CAS PubMed;
(b) T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed;
(c) J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed;
(d) C. G. Bezzu, M. Helliwell, J. E. Warren, D. R. Allan and N. B. McKeown, Science, 2010, 327, 1627–1630 CrossRef CAS PubMed;
(e) T. Hasell, S. Y. Chong, M. Schmidtmann, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 7154–7157 CrossRef CAS PubMed;
(f) M. W. Schneider, I. M. Oppel, A. Griffin and M. Mastalerz, Angew. Chem., Int. Ed., 2013, 52, 3611–3615 CrossRef CAS PubMed.
-
(a) J. H. Williams, Acc. Chem. Res., 1993, 26, 593–598 CrossRef CAS;
(b) C. R. Patrick and G. S. Prosser, Nature, 1960, 187, 1021 CrossRef CAS PubMed.
- T. Itoh, M. Kondo, M. Kanaike and S. Masaoka, CrystEngComm, 2013, 15, 6122–6126 RSC.
- C. R. Willson and H. Taube, Inorg. Chem., 1975, 47, 533–535 Search PubMed.
-
G. M. Sheldrick, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
-
(a)
F. A. Cotton, C. A. Murillo and R. A. Walton, “Multiple Bonds Between Metal Atoms“, Springer Science and Business Media, New York, 3rd edn, 2005 Search PubMed;
(b) E. B. Royar and S. D. Robinson, Platinum Metals Rev., 1982, 26, 65–69 Search PubMed.
- The channel entrance size was estimated by considering the van der Waals radii of constituent atoms, and the atoms at axial ligands are omitted for the calculation.
-
(a) N. Hayashi, T. Mori and K. Matsumoto, Chem. Commun., 1998, 1905–1906 RSC;
(b) K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC;
(c) X. Xu, B. Pooi, H. Hirao and S. H. Hong, Angew. Chem., Int. Ed., 2014, 53, 1283–1287 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures of CO2 or N2 sorption isotherms, PXRD patterns. CCDC 1049445–1049447. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01620g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a