
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and X-ray structure analysis of cytotoxic heptacoordinate sulfonamide salan titanium(IV)-bis-chelates†
Tiankun
Zhao
a,
Martin
Grützke‡
a,
Kathrin H.
Götz
a,
Tetiana
Druzhenko
b and
Thomas
Huhn
*a
aFachbereich Chemie and Konstanz Research School Chemical Biology, Universität Konstanz, Universitätsstr. 10, D-78457 Konstanz, Germany. E-mail: thomas.huhn@uni-konstanz.de; Fax: +49-7531-884424; Tel: +49-7531-882283
bDepartment of Chemistry, Kiyv National Taras Shevchenko University, 64 Volodymyrska Street, Kyiv, 01601, Ukraine
First published on 20th August 2015
Abstract
A series of novel sulfonamide substituted heteroleptic salan titanium(IV)-bis-chelates complexed to 2,6-pyridinedicarboxylic acid were synthesized, structurally characterized and evaluated for their anticancer activity against two human carcinoma cell lines. All cytotoxic complexes showed complete inhibition of cell growth at active concentration, two complexes based on pyrrolidine and azepane substituted sulfonamides displayed IC50 values below 1.7 μM and are more cytotoxic than cisplatin in both tested cell lines. The azepane substituted complex [L3Ti(dipic)] exhibited excellent activity with an IC50 value of 0.5 ± 0.1 μM in Hela S3 and 1.0 ± 0.1 μM in Hep G2.
Introduction
Cisplatin with its good antitumor activity was approved as the first metal anticancer drug in the early 1970s.1 Since then it has been the leading drug in chemotherapy of different solid tumors such as genitourinary, colorectal and non-small cell lung cancers for more than three decades.2 However, platinum based drugs are usually associated with side effects and drug resistance.3 Consequently, there is a high demand for designing new non-platinum alternatives with improved pharmacological properties and better tolerability. Titanium complexes have been identified rather early as promising metal based anticancer drugs.4 Most of the titanium complexes reported as antitumor agents so far are derivatives of titanocene dichloride (Cp2TiCl2)5 and Budotitane [Ti(bzac)2(OEt)2] (Hbzac = 1-phenylbutane-1,3-dione),6 these complexes showed enhanced antitumor activity in various cell lines and reduced side effects compared to cisplatin. Unfortunately, their clinical trials are stagnant due to rapid hydrolysis in the range of minutes under physiological conditions.7 In comparison, diamino bis(phenolato) “salan” titanium(IV)-bis-alkoxides, a third class of anticancer active Ti(IV), proved to be less prone towards hydrolysis and highly active in different cell lines.8 Bioactivity and reactivity of salan-Ti(IV)-alkoxides are significantly influenced by the aromatic substitution pattern of the salan ligand. While halogen substitution improved cytotoxicity in favour of apoptosis8c increasing the steric bulk resulted in a loss of cytotoxicity.8b,e A polar nitro substituent resulted in decreased hydrolytic stability but had a beneficial influence on cytotoxicity, most probably by enhancing the solubility of the resulting complex in a biologically relevant medium.9 Replacement of the notorious labile alkoxides by the dianionic tridentate 2,6-pyridinedicarboxylic acid (dipic) let us recently developing an efficiently stabilized heptacoordinate salan Ti(IV)-bis-chelate.10 Even though, the reported complex is not redox-active and has a significantly increased aqueous stability in the range of several days, this complex is highly toxic in vitro and more importantly, has enhanced antitumor efficacy in an in vivo cervical cancer mouse model.8fExploring scope and limitations of the stability enhancing effect of the additional dipic-chelator11 we became interested in introducing the electron withdrawing sulfonamide motif which is known to be a privileged group in drug discovery.12 A large number of structurally novel sulfonamide derivatives were reported to show substantial antitumor activities in a variety of processes such as carbonic anhydrase inhibition,13 cell cycle arrest,14 disruption of microtubular assembly,15 and angiogenesis inhibition.16 Several combinations of sulfonamide with metal-based chemotherapeutic agents have been reported to show synergistic effects in the treatment of cancer processes.17 However, Ti(IV) complexes with non-coordinating and thus potentially bioavailable sulfonamide groups are not known.18 Herein, we present the synthesis and preliminary cytotoxicity study of several heptacoordinate Ti(IV)-salan dipic hetero-bis-chelates with non-coordinating sulfonamide groups.
Results and discussion
Complex synthesis
The synthetic route for the sulfonamide-based salan ligands is outlined in Scheme 1. Protection of salicylaldehyde (1) as anilide followed by chloro sulfonation and in situ deprotection during work up, afforded hitherto unknown sulfonylchloride 3 as common starting material in 81% overall yield.193 reacted smoothly with different cyclic secondary amines to give sulfonamide substituted salicylic aldehydes 4a–d. The corresponding salan ligands H2L1–4 were prepared by reductive amination with ethylendiamine and reductive alkylation using aqueous formaldehyde in analogy to a known procedure.8f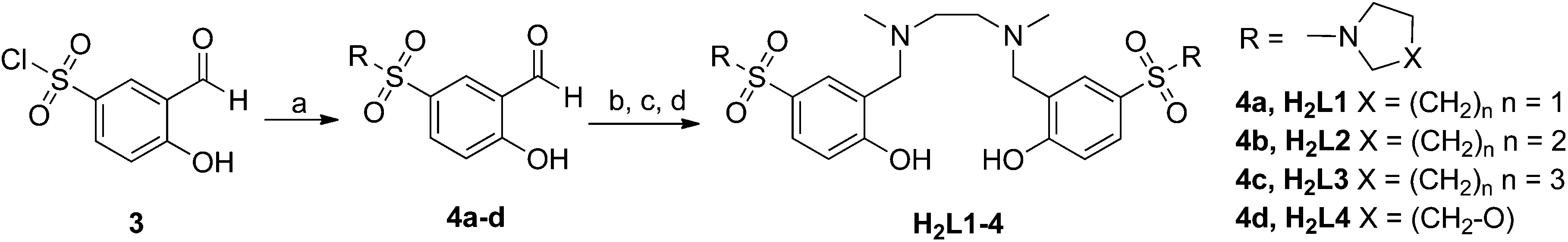 | ||
| Scheme 1 Synthesis of sulfonamide bearing salan ligands H2L1–4. Reagents and reaction conditions: (a) Amine, CH2Cl2, 0 °C to rt. (b) Ethylene diamine, MeOH, rt. (c) NaBH4, rt. CH2Cl2, MeOH, rt. (d) HCHO, AcOH, CH3CN, rt then NaBH4, 0 °C to rt. | ||
The synthesis of the sulfonamido-salan Ti(IV) complexes [L1–4Ti(dipic)] is summarized in Scheme 2.
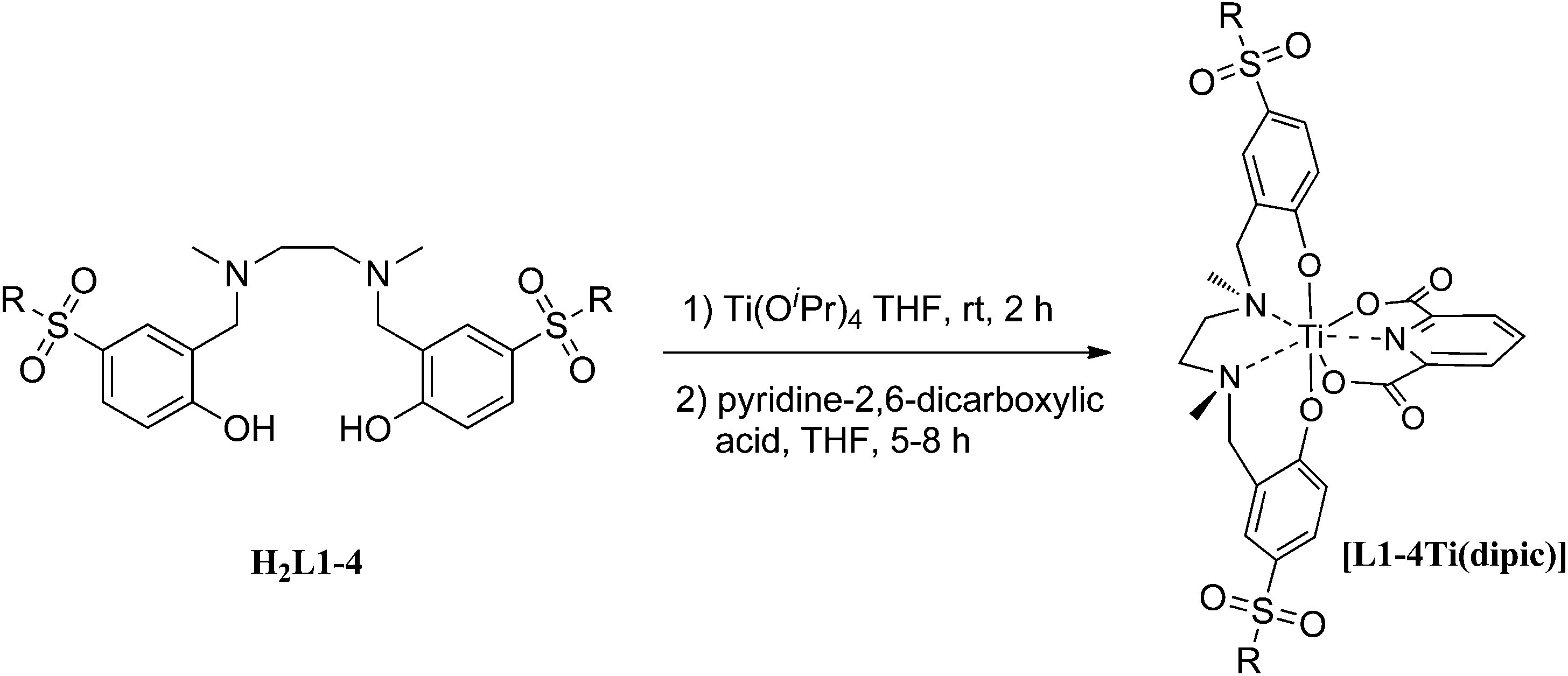 | ||
| Scheme 2 Synthesis of sulfonamido-salan Ti(IV) complexes [L1–4Ti(dipic)]. | ||
Metalation of salans H2L1–4 with titanium tetraisopropoxide in tetrahydrofuran resulted in the clean formation of the C2-symmetric cis-α-isomers in all cases as evident from the typical AB pattern of the diastereotopic benzylic protons (Ar–CH2–N) in the 1H NMR spectra recorded from the crude. [L1–4Ti(OiPr)2] were isolated in nearly quantitative yield as racemic mixtures of the Δ and Λ-isomers. In contrast to Ti(IV)-alkoxides of alkyl or halogen substituted salans which are dark orange to red colored and have their LMCT band accordingly in the UV-vis region, complexes [L1–4Ti(OiPr)2] are virtually colourless. The strongly electron withdrawing sulfonamide distinctly blue shifted the spectrum compared to alkyl or halogen substituted salans.8e In case of [L4Ti(OiPr)2] the isopropoxide was isolated and the λmax was determined to 308 nm. All sulfonamide substituted Ti(IV)-isopropoxides are sensitive in solution and decompose slowly over time. Consequently, they were directly converted to the corresponding dipic-derivatives.10 The ligand exchange reaction was initiated by the addition of THF to a mixture of crude [L1–4Ti(OiPr)2] and dipic to afford analytical pure [L1–4Ti(dipic)] in yields between 33–45% based on Ti(OiPr)4. The progress of the reaction was conveniently monitored by TLC. In contrast to previous experiments with non-sulfonamide bearing complexes10,11 the addition of less than 2 equiv. of dipic resulted in non-complete conversion even on prolonged reaction time. The excess dipic could be removed by carefully washing of a solution of [L1–4Ti(dipic)] in dichloromethane with dilute (1 mol L−1) sodium hydroxide solution. Complexes were then purified by column chromatography on silica gel and recrystallization.
Solid state structure and NMR studies
In the 1H-NMR spectra all complexes showed the characteristic signals for pseudo C2 symmetrical complexes, namely a coincidence of the signals of both halves of the salan backbone. As expected for complexes with electron withdrawing substituents, all signals are low-field shifted compared to the non-sulfonamide substituted dipic complex [L5Ti(dipic)]. For instance, the NMe-resonance of the dipic-bound protons in the sulfonamide bearing complexes appears around 0.1 ppm down-field shifted with respect to [L5Ti(dipic)]. The effect on the diastereotopic methylene protons is with 0.15 ppm slightly stronger. This demonstrates clearly that the sulfamido group of the salan ligand communicates via the Ti(IV) centre with the dipic ligand. The symmetry found in solution was confirmed in the solid by X-ray diffraction of single crystals of [L4Ti(dipic)] and [L5Ti(dipic)] (Fig. 1). Both crystallize in the monoclinic space group C2/c. In the asymmetric unit one molecule [L4Ti(dipic)] is accompanied by two molecules of acetone while [L5Ti(dipic)] crystalizes without additional solvent. In [L5Ti(dipic)] Ti(1), N(2) and C(12) are oriented on the two-fold axis which intersects the ethylendiamine bridge of the salan backbone between C(8) and C(8i). Consequently, the asymmetric unit contains only half a molecule. Selected bond length and angles are summarized in Table 1, crystallographic data are found in Table 4. With respect to the arrangement around the titanium centre, [L4Ti(dipic)] and [L5Ti(dipic)] compare extremely well with the other known solid state structures of Ti(IV)-salan dipicolinates.10,11 Both feature the pentagonal bipyramidal core structure of a heptacoordinate titanium centre with the phenolates in the apical position and the nitrogen atoms of the salan and the pyridine together with the carboxylates defining the equatorial plane. The bridging nitrogen atoms of the salan have the greatest deviation from the equatorial plane with 0.194(17) Å for N2 in [L4Ti(dipic)] and 0.284(8) Å for N1 in [L5Ti(dipic)]. The phenolate–Ti distance in [L5Ti(dipic)] (1.8672(11) Å) is comparable to that in [L4Ti(dipic)] (1.8586(15) Å and 1.8648(15) Å). In both complexes this distance is slightly elongated compared to the 1.84 Å found for the 2,4-dimethylsubstituted salan in ref. 10 indicating that the p-sulfonamido substituted [L4Ti(dipic)] as well as the unsubstituted homologue have a diminished donor strength compared with the 2,4-dimethylsubstituted complex. The MeN–Ti distance in both complexes is unaffected by the substitution in the vicinity of the salan and is with 2.3457(17) and 2.3757(18) for [L4Ti(dipic)] and 2.3695(14) Å for [L5Ti(dipic)] in the range of 2.350(2) Å and 2.384(2) of the previous characterized complex.10 Dipicolinic acid acts as bis-anionic tridentate coordinating to the titanium–salan fragment via two of the carboxylate oxygen atoms and its pyridine N atom. In [L4Ti(dipic)] both carboxylate groups of the dipic are pulled towards the Ti(IV) centre resulting in a twist out of plane with respect to the pyridine ring by 3.9(2)°. The Ti–carboxylate distances in [L4Ti(dipic)] differ with 2.0361(15) Å and 2.0551(14) Å quite strongly whereas the Ti–O distances in the previous characterized complex is more balanced (2.043(1) and 2.046(1) Å).10 In [L5Ti(dipic)] the Ti–carboxylate distance is with 2.0345(12) Å considerably shorter.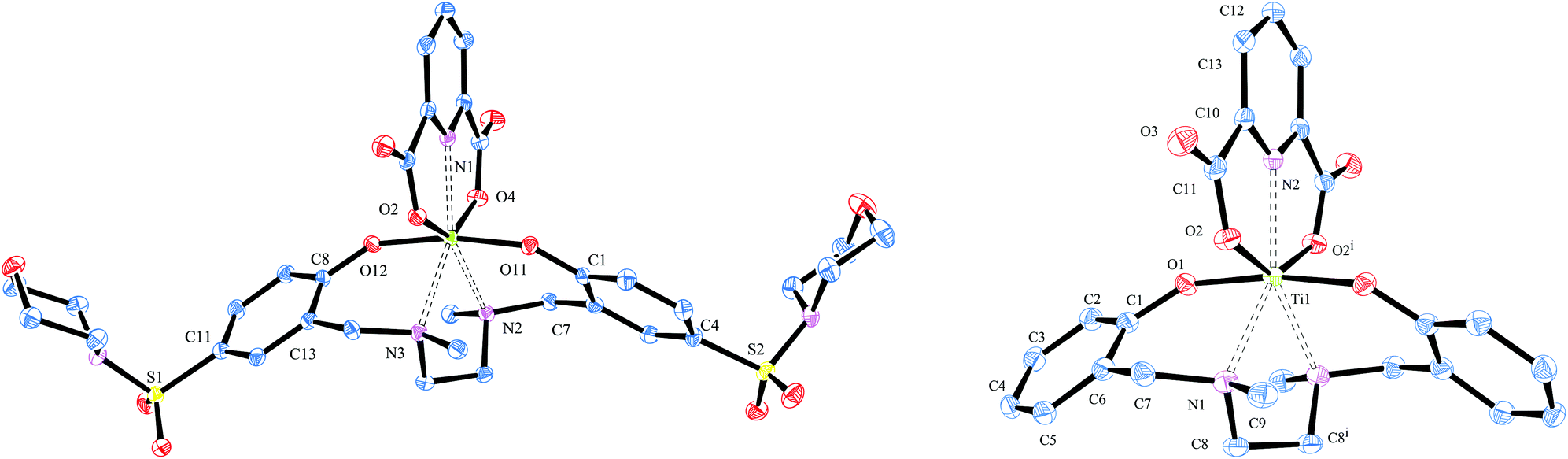 | ||
| Fig. 1 X-ray crystal structure of heptacoordinate [L4Ti(dipic)] (left) and [L5Ti(dipic)] (right). Both are exhibiting pseudo C2 symmetry and a distorted pentagonal-bipyramidal conformation of the ligands around the Ti(IV) center. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms and additional solvent molecules for [L4Ti(dipic)] are omitted for clarity. | ||
| [L4Ti(dipic)] | |||||
|---|---|---|---|---|---|
| Distances [Å] | Angles [°] | ||||
| Ti–O(11) | 1.8586(15) | O(11)–Ti–O(12) | 170.21(7) | O(11)–Ti–N(2) | 80.98(6) |
| Ti–O(12) | 1.8648(15) | O(11)–Ti–O(4) | 95.06(6) | O(12)–Ti–N(2) | 92.01(6) |
| Ti–O(4) | 2.0361(15) | O(12)–Ti–O(4) | 89.38(6) | O(4)–Ti–N(2) | 73.19(6) |
| Ti–O(2) | 2.0551(14) | O(11)–Ti–O(2) | 89.21(6) | O(2)–Ti–N(2) | 145.08(6) |
| Ti–N(1) | 2.1941(18) | O(12)–Ti–O(2) | 92.75(6) | N(1)–Ti–N(2) | 143.21(6) |
| Ti–N(2) | 2.3457(17) | O(4)–Ti–O(2) | 141.42(6) | O(11)–Ti–N(3) | 91.22(6) |
| Ti–N(3) | 2.3757(18) | O(11)–Ti–N(1) | 94.33(7) | O(12)–Ti–N(3) | 80.22(6) |
| O(12)–Ti–N(1) | 95.36(7) | O(4)–Ti–N(3) | 144.99(6) | ||
| O(4)–Ti–N(1) | 70.91(6) | O(2)–Ti–N(3) | 72.91(6) | ||
| O(2)–Ti–N(1) | 70.53(6) | N(1)–Ti–N(3) | 142.91(6) | ||
| N(2)–Ti–N(3) | 73.88(6) | ||||
| [L5Ti(dipic)] | |||||
|---|---|---|---|---|---|
| Distances [Å] | Angles [°] | ||||
| N(1)–Ti(1) | 2.3695(14) | O(1)#1–Ti(1)–O(1) | 171.17(7) | O(1)–Ti(1)–N(1) | 80.46(5) |
| N(2)–Ti(1) | 2.178(2) | O(1)#1–Ti(1)–O(2) | 89.00(5) | O(2)–Ti(1)–N(1) | 73.05(5) |
| O(1)–Ti(1) | 1.8672(11) | O(1)–Ti(1)–O(2) | 93.84(5) | O(2)#1–Ti(1)–N(1) | 143.95(5) |
| O(2)–Ti(1) | 2.0345(12) | O(2)–Ti(1)–O(2)#1 | 142.47(7) | N(2)–Ti(1)–N(1) | 143.46(3) |
| O(1)–Ti(1)–N(2) | 94.41(4) | O(1)–Ti(1)–N(1)#1 | 92.41(5) | ||
| O(2)–Ti(1)–N(2) | 71.23(3) | N(1)–Ti(1)–N(1)#1 | 73.08(7) | ||
| O(1)#1–Ti(1)–N(1) | 92.41(5) | ||||
Stability and hydrolysis
Stability tests were carried out using time resolved 1H NMR spectroscopy as previously reported.8b,e Briefly, to solutions of 15 μmol complexes [L1–4Ti(dipic)] in d8-THF a ∼1000 molar excess of D2O was added (t = 0) and 1H NMR spectra were recorded in certain time intervals. In case of [L5Ti(dipic)] the concentration was with 4.5 μmol much lower because of its very limited solubility in most solvents. The aim of the hydrolytic degradation study is to benchmark relative stability under controlled conditions which are certainly different from the hydrolysis in a biological environment. All complexes [L1–4Ti(dipic)] exhibit moderate hydrolytic stability with their half-lives ranging from 2–3.5 h (Table 2, Fig. S1 in the ESI†), and thus are more prone towards hydrolysis than is our previously reported Ti(IV)-bis-chelate with t1/2 ≫ 2 weeks under the same conditions10 but well within the range of other complexes recently tested to be bioactive.20 The non-sulfonamide bearing [L5Ti(dipic)] exhibited a much higher stability as evident from time resolved 1H NMR spectra (Fig. S9 of the ESI†). The reduced stability of sulfonamide bearing salan dipicolinates [L1–4Ti(dipic)] compared with the non-sulfonamide bearing [L5Ti(dipic)] can be rationalized by the electron withdrawing nature of the sulfonamide which renders the titanium more electrophilic and hence more ‘attractive’ towards the attack of OD− from the autoprotolysis of D2O. The lack of the ortho-substituent of the salan in complexes [L1–5Ti(dipic)] makes those complexes less efficiently shielded compared to the first hetero-bis-chelate from ref. 10 whose salan features a 2,4-dimethyl substitution pattern. In contrast to the oligonuclear hydrolysis products of bis(alkoxo) salan Ti(IV) complexes,8f,21 the hydrolysis of [L1–5Ti(dipic)] exclusively yields the corresponding salan ligands H2L1–5 as detected by NMR and HPLC (for a detailed one-to-one comparison see Fig. S1 and S2 in the ESI†).11Fig. 2 depicts a series of time resolved 1H NMR spectra recorded during the incubation of [L3Ti(dipic)] with D2O. After a total duration of 14 h only minute quantities of intact complex are detectable whereas the signals of the free ligands figure most prominently. For time resolved spectra recorded during the hydrolysis of the other complexes see Fig. S3–S5 of the ESI.†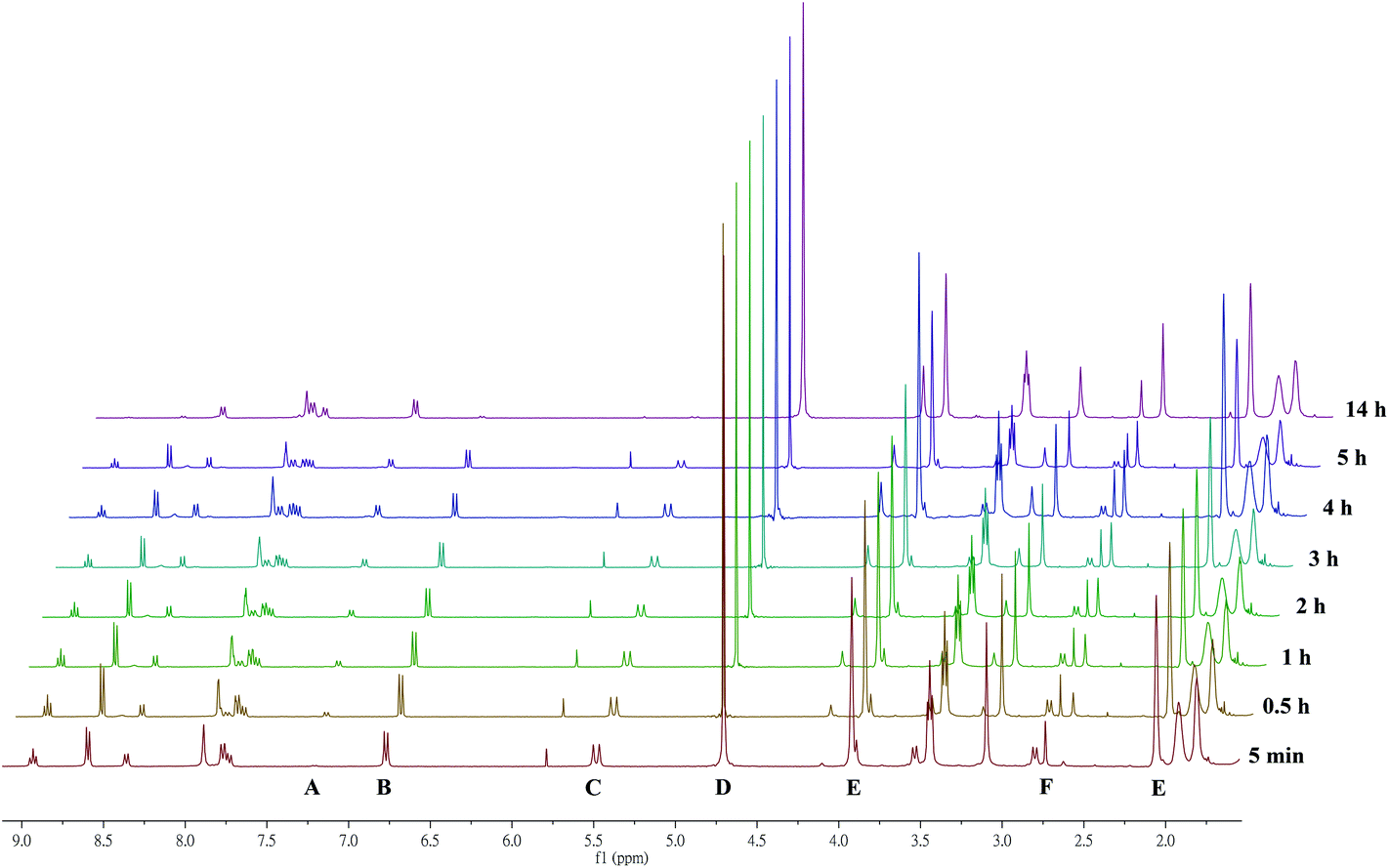 | ||
| Fig. 2 Example of a hydrolytic stability study of [L3Ti(dipic)] at 37 °C investigated by time resolved 1H NMR in aqueous (1000 equiv. D2O), d8-THF (0.4 mL) and 0.15 equiv. of 4-nitrotoluene as an internal reference. Spectra shown are from bottom to top measured at t = 5 min, 0.5 h, 1 h–5 h and 14 h after the addition of D2O. Signals marked are Har of free salan ligand H2L3 (A), Har of the [L3Ti(dipic)] (B), CarCH2 of [L3Ti(dipic)] (C), D2O (D), THF (E) and CH3 of 4-nitrotoluene (F). | ||
| Compound | t 1/2 (h) | Compound | t 1/2 (h) |
|---|---|---|---|
| a Detailed NMR-spectra are given in the ESI; error was estimated to be ±0.5 h. | |||
| [L1Ti(dipic)] | 2.0 | [L3Ti(dipic)] | 2.2 |
| [L2Ti(dipic)] | 3.5 | [L4Ti(dipic)] | 3.0 |
Biotest are run in DMEM medium which itself contains several inorganic salts, vitamines, amino acids and other constituents (see Experimental for details). To study the influence of these electrolytes on the stability of [L4Ti(dipic)] a series of time resolved 1H NMR with added medium was recorded. (cf. Fig. S6 in the ESI†). For this a solution of Dulbecco's Modified Eagle's Medium (dry formulation, Aldrich order no.: D5523-10X1L) with added sodium bicarbonate to make up for complete medium in D2O was prepared. This aqueous solution now containing all salts, amino acids, vitamins and glucose of the original cell medium was added as before to a solution of [L4Ti(dipic)] in d8-THF and NMR spectra measured immediately after. These measurements showed a slightly increased speed of hydrolysis which is well within the error margins (30 min) of those experiments (see Fig. S7 in the ESI†).
Next we investigated if the DMSO used for preparing stock solutions of complexes for the bio tests might interfere with the complex stability. Recently, McGowan et al. reported on the isolation of a biologically non active DMSO-coordinated titanium oxo species which forms upon the addition of the solvent to a 1,3-β-diketonato titanium(IV) complex during formulation of the drug for in vitro testing.22 As visible from time resolved 1H NMR spectra of [L4Ti(dipic)] recorded in d6-DMSO during 24 h at 37 °C (Fig. S8 of ESI†), no degradation or ligand exchange is detectable. Obviously, the heptacoordinate complex is efficiently stabilized by its two chelating ligands and withstands a potential ligand exchange against DMSO under these conditions.
Cytotoxicity assay
All compounds were tested for their cytotoxicity in the human cervix carcinoma cell line HeLa S3 and the human hepatocarcinoma cell line Hep G2 in a AlamarBlue-based cytotoxicity assay.23 IC50 values are given as mean values from three independent experiments each done in four replicates (further details of the assay are to be found in the Experimental section). The IC50 values of the synthesized compounds are summarized in Table 3. It was observed that compounds [L1–3Ti(dipic)] exhibited good to excellent bioactivity and reached maximum inhibition, that is a cell viability of 0% (for IC50-charts see Fig. S12 and S13 in the ESI†). Two of the most active complexes [L2Ti(dipic)] and [L3Ti(dipic)], with IC50 values below or close to 1 μmolar, are even more cytotoxic than cisplatin. Even though, the morpholinosulfonyl derivative [L4Ti(dipic)] showed the anticipated enhanced aqueous solubility, its bioactivity against Hela S3 is greatly reduced when compared with [L1–3Ti(dipic)] and even completely vanished against Hep G2 cells.| Compound reference | [L4Ti(dipic)] | [L5Ti(dipic)] |
|---|---|---|
| Chemical formula | C33H39N5O12S2Ti·2(C3H6O) | C25H25N3O6Ti |
| Formula Mass | 925.86 | 511.38 |
| Crystal system | Monoclinic | Monoclinic |
| a/Å | 38.9176(16) | 16.7824(13) |
| b/Å | 10.0754(5) | 14.9183(9) |
| c/Å | 21.8768(8) | 8.6437(7) |
| α/° | 90.00 | 90.00 |
| β/° | 93.133(3) | 91.497(6) |
| γ/° | 90.00 | 90.00 |
| Unit cell volume/Å3 | 8565.3(6) | 2163.3(3) |
| Temperature/K | 100(2) | 100(2) |
| Space group | C2/c | C2/c |
| No. of formula units per unit cell, Z | 8 | 4 |
| Absorption coefficient, μ/mm−1 | 0.370 | 0.447 |
| No. of reflections measured | 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 758 758 |
14886 |
| No. of independent reflections | 9115 | 2181 |
| R int | 0.1041 | 0.1342 |
| Final R1 values (I > 2σ(I)) | 0.0436 | 0.0332 |
| Final wR(F2) values (I > 2σ(I)) | 0.0835 | 0.0893 |
| Final R1 values (all data) | 0.0689 | 0.0449 |
| Final wR(F2) values (all data) | 0.0901 | 0.0916 |
| Goodness of fit on F2 | 1.022 | 1.043 |
The ligands H2L1–4 and dipic were investigated for their cytotoxicity as well to answer the question if the measured cytotoxicity might be an effect of liberated ligand. Dose–response curves for the ligands H2L1–5 were recorded in a concentration range comparable to that of the complexes. One obvious difference observed is the lower solubility of the ligand H2L1–4 compared to the corresponding complexes. While those were soluble close to the millimolar range during bio assays, the ligands showed limited solubility with the exception of H2L4. As a result, the ligands dose–response plots did not follow a sigmoidal trend (a comparison of complex and ligand toxicity is given in Table S11 of the ESI†). Instead, these curves show a shallow slope and monotonously approach towards some cytotoxicity. At the 100 μmolar regime bioactivity was detected albeit the maximum cell viability never reached below 30%. Raising the concentration even further leads to severe solubility problems. Only at this concentration regime where precipitation already was manifest highest cell viability was reached. Precipitation of H2L1–3 was detected during the assay when concentrations were higher than 100 μmolar by microscopic control of the cell assay. This precipitation might account for the found cytotoxicity of the ligands at highly elevated concentrations. In contrast, complexes [L1–3Ti(dipic)] showed potent cytotoxicity already at concentration in the μmolar regime with an inhibition rate of greater than 90%. The ligands H2L1–4 and dipic, however, showed a less pronounced cytotoxicity in this concentration regime. In contrast to the behaviour of the ligands, complexes [L1–3Ti(dipic)] lead to a complete loss of viability already at two orders of magnitude lower concentrations.
We have determined the half-lives of [L1–4Ti(dipic)] in the region of 2–3 h. Upon hydrolysis only ligands were found. The cytotoxicity of ligands does not follow a sigmoidal trend and reaches maximum inhibition only at concentrations higher than 100 μmolar. However, complexes show complete inhibition at much lower concentrations and have their IC50 values in the 1–3 μmolar regime. Based on the assumption that the ligand is not modified during incubation whereas the complexes decompose within a certain time, the found cytotoxicity can be rationalized by the following way: if cellular uptake of the complex would be rather slow, most of the complex would decompose to yield the ligand. As a result, the cytotoxicity profile of free ligand and complex should become very similar or even identical, which our data proves to be incorrect. Previous studies on salan type-Ti(IV) complexes suggested a serum protein-independent rapid cell penetrating mechanism,24 which recently was further corroborated by a high-resolution atomic absorption spectroscopy based study.25 The authors showed the rapid increase of the intracellular titanium concentration within hours after drug exposure for a wide selection of different Ti(IV)-species. For heptacoordinate hetero-bis-chelates recently a fast cellular uptake of the intact complex was proposed.11 Whether this obviously fast cellular uptake of [L1–3Ti(dipic)] results from the sulfonamide substitution by way of an active shuttling or if the solubility enhancing sulfonamide is making the complexes better accessible is debatable. Because the long-time stable [L5Ti(dipic)] showed a cytotoxicity in the low μmolar regime comparable to the other complexes, it does not necessarily has to rely on a fast uptake. What sets [L1–4Ti(dipic)] apart from complexes lacking the sulfonamide moiety are: (1) a greatly enhanced solubility and stability in DMSO facilitating the drug formulation, (2) a much enhanced aqueous solubility preventing precipitation of active drug from the media. Admittedly one has to note that the sulfonamide substitution reduces the long term stability of complexes in aqueous media. However, we could show that this does not necessarily limit the usefulness of those complexes because their cytotoxicity against Hela S3 and Hep G2 cells compares very well with other Ti(IV) based systems.
Conclusions
In summary, we have synthesized sulfonamide functionalised Ti(IV)-salan dipic bis-chelates with the sulfonamide being not part of the titanium's coordination sphere. Their structures were elucidated by means of NMR and by single crystal X-ray diffraction of [L4Ti(dipic)], and [L5Ti(dipic)]. Preliminary in vitro biological evaluation against two human cell lines revealed that [L2Ti(dipic)] and [L3Ti(dipic)] have cytotoxicity in the sub μmolar range and are 5–7 times more cytotoxic than cisplatin. The contribution of the sulfonamide group to this observation might be twofold: (1) complexes [L1–4Ti(dipic)] are notably better soluble in DMSO than the non-sulfonamide bearing homologue. This results in a better distribution in media and hence a possible better bioavailability in the cell based assy. (2) A possible role of the sulfonamido group during the process of cellular uptake cannot be ruled out and demands further biological studies. These novel sulfonamide Ti(IV)-bis-chelate complexes have highly potent anticancer activities. Further studies on the explanation of structure–activity relation are currently underway.Experimental section
All reagents, metal catalysts and solvents were obtained from commercial sources and were purified before use by standard methods.26 All experiments requiring dry atmosphere were carried out under nitrogen atmosphere using standard Schlenk technique. Silica gel 60 (40–63 μm) for flash chromatography was purchased from Macherey & Nagel (Düren). NMR spectra were measured on Bruker Avance III 400 and Bruker Avance DRX 600 spectrometers. Structure assignments were done based on 2D-NMR (COSY, HMBC, HSQC) experiments. 1H NMR chemical shifts are referenced with respect to the chemical shift of the residual protons present in the deuterated solvents (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm; d6-DMSO: δH = 2.50 ppm, δC = 39.52 ppm; d6-acetone: δH = 2.05 ppm, δC = 29.84, 206.26 ppm). UV-vis spectra were recorded on a Varian Cary 50 spectrophotometer and a Perkin Elmer Lambda 18 (200–600 nm) in dilute (∼10−5 M) THF solutions. IR spectra were recorded on a Perkin Elmer Spectrum 100 FTIR (ATR) spectrometer. Melting points are not corrected and were measured with a Krüss-Meltingpointmeter KFP I N. High resolution mass spectra were recorded of 1 μM dilute samples in acetonitrile–acetone–water-solutions on a Bruker micrOTOF II mass spectrometer. Elemental analyses were carried out in the micro analytical lab of the University of Konstanz using an Elementar Vario EL CHN analyzer.X-ray crystallography
Suitable single crystals of [L4Ti(dipic)] and [L5Ti(dipic)] were grown by slow diffusion of hexane into a saturated solution of [L4Ti(dipic)] in acetone or of hexane into a saturated solution of [L5Ti(dipic)] in dichloromethane. Crystal were directly picked from solution and covered in an inert oil and immediately placed in the cold N2-stream of the Oxford Cryostream 700 with nitrogen as coolant gas. Data collection for X-ray structure-determination was performed at a STOE IPDS-II diffractometer equipped with a graphite monochromated radiation source (λ = 0.71073 Å), an image plate detection system. The selection, integration, and averaging procedure of the measured reflex intensities, the determination of the unit cell by a least-squares fit of the 2θ values, data reduction, LP correction, and the space group determination were performed using the X-Area software package delivered with the diffractometer. A semiempirical absorption correction method was performed after indexing of the crystal faces. The structure was solved by direct methods (SHELXS-97)27 and refined by standard Fourier techniques against F square with a full-matrix leastsquares algorithm using SHELXL-97 and the WinGX (1.80.05)28 software package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions and refined with a riding model. Graphical representations were prepared with ORTEP-III.29 Crystallographic data (excluding structure factors) of [L4Ti(dipic)] and [L5Ti(dipic)] have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 1031337 and 1410575.Hydrolysis study
Hydrolysis tests were carried out by time-resolved 1H-NMR spectroscopy under the following conditions: Ti complex (15 μmol) for [L1–L4Ti(dipic)], 4-nitrotoluene as internal standard (0.3 mg, 2.25 μmol, 0.15 equiv.), D2O (0.27 mL, 1000 equiv.), d8-THF (0.4 mL). In case of [L5Ti(dipic)] the amounts were adjusted because of its lower solubility: (4.5 μmol) complex, D2O (0.08 mL, 1000 equiv.), and d8-THF (0.5 mL). 1H NMR were measured at 37 °C at given intervals and samples stored in-between at 37 °C. Data gathered by monitoring the decrease in isolated signals of the titanium-bound salan backbone and increase in the free salan ligand. Integrals are normalized against the internal standard. Control measurements were done in the absence of 4-nitrotoluene and showed no significant difference in the hydrolysis rate and products being formed. The hydrolysis experiments using cell medium were done in a similar manner with the exception of the amount of added D2O. While keeping the volume ratio as before the amount of available D2O is slightly diminished because of the added salts, amino acids, vitamins and glucose of the DMEM medium. Since the constituents of the medium interfere with the above standard, we used 1,4-dinitrobenzene instead. For an exact content of the used dry medium refer to ref. 30.Cytotoxicity assay
Cytotoxicity was measured on HeLa S3 and Hep G2 cells using an AlamarBlue (Thermo Scientific) assay.31 Alamarblue was purchased from BioSource Europe. Cells were cultivated at 37 °C in humidified 5% CO2 atmosphere using Dulbecco's DMEM-media (Invitrogen) containing 10% fetal calf serum (Biochrome AG), 1% penicillin and 1% streptomycin (both GIBCO). Cells were split twice per week. Both cell lines were tested on mycoplasma infections using a mycoplasma detection kit (Roche Applied Science). The cells were seeded in 96-well plates (4000 HeLa S3 cells per well or 8000 Hep G2 cells per well) and allowed to attach for 24 h. The cells were then incubated with different concentrations of the reagent to be tested. Compounds to be tested were dissolved in a suitable amount of DMSO and different concentrations were prepared by serial dilution with DMSO. One part of each DMSO solution is then added to 99 parts of medium. Cells were then incubated for 48 h with 100 μl of above medium containing 1% DMSO and a certain concentration of compound. The medium was then replaced by 100 μL medium containing 10% AlamarBlue (BioSource Europe) and the cells were incubated for 90 min. The fluorescence at 590 nm was measured after excitation at 530 nm using a Synergy HT Microplate Reader (BioTek). Raw readout data from the assay was corrected for background fluorescens by an “on-plate” blind containing only medium, 1% DMSO and Alamarblue but no cells (0-value). The background corrected absolute read-outs were then expressed as relative values with regard to an “on-plate” 100% standard containing untreated cells in medium with 1% DMSO. All data was then fitted to a sigmoidal dose–response model with variable slope (4 parameter logistic nonlinear regression model) using Sigma plot 10.0.32 Upper and lower boundaries as well as the slope were allowed to refine freely. All experiments were repeated at least three times on three different days with each experiment done in four replicates on the same plate. Replicates are treated with equal statistical weight; error-bars representing SEM. IC50 values are given as means from independent experiments, error values of IC50 are based on standard deviation of independent experiments.Complex syntheses
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 8.32 (s, 1H, Har), 8.15 (d, J = 6.4 Hz, 1H, Har), 7.22 (d, J = 6.4 Hz, 1H, Har); 13C NMR (CDCl3, 101 MHz): δ 195.4 (CO), 166.6 (Car), 135.9 (Car), 134.9 (Car), 133.8 (Car), 120.0 (Car). HRMS (ESI-TOF) m/z Calcd for C7H6ClO4S [M + H]+: 220.9670. Found: 220.9672.
:1) to give 4a as a yellow solid (2.85 g, 11.16 mmol, 83%). M.p. 158–159 °C; IR absorptions (cm−1, ATR): 2981, 2878, 1642, 1617, 1469, 1320, 1191, 1088, 751; 1H NMR (CDCl3, 400 MHz): δ 11.37 (s, 1H, OH), 9.95 (s, 1H, HCO), 8.10 (d, J = 2.4 Hz, 1H, Har), 7.92 (dd, J = 2.4, 8.8 Hz, 1H, Har), 7.09 (d, J = 8.8 Hz, 1H, Har), 3.24–3.20 (m, 4H, NCH2), 1.78–1.76 (m, 4H, NCH2
O), 8.32 (s, 1H, Har), 8.15 (d, J = 6.4 Hz, 1H, Har), 7.22 (d, J = 6.4 Hz, 1H, Har); 13C NMR (CDCl3, 101 MHz): δ 195.4 (CO), 166.6 (Car), 135.9 (Car), 134.9 (Car), 133.8 (Car), 120.0 (Car). HRMS (ESI-TOF) m/z Calcd for C7H6ClO4S [M + H]+: 220.9670. Found: 220.9672.
:1) to give 4a as a yellow solid (2.85 g, 11.16 mmol, 83%). M.p. 158–159 °C; IR absorptions (cm−1, ATR): 2981, 2878, 1642, 1617, 1469, 1320, 1191, 1088, 751; 1H NMR (CDCl3, 400 MHz): δ 11.37 (s, 1H, OH), 9.95 (s, 1H, HCO), 8.10 (d, J = 2.4 Hz, 1H, Har), 7.92 (dd, J = 2.4, 8.8 Hz, 1H, Har), 7.09 (d, J = 8.8 Hz, 1H, Har), 3.24–3.20 (m, 4H, NCH2), 1.78–1.76 (m, 4H, NCH2 ); 13C NMR (CDCl3, 101 MHz): δ 196.1 (CO), 164.5 (Car), 135.4 (Car), 133.8 (Car), 129.0 (Car), 119.9 (Car), 118.9 (Car), 48.1 (NCH2), 25.3 (NCH2
); 13C NMR (CDCl3, 101 MHz): δ 196.1 (CO), 164.5 (Car), 135.4 (Car), 133.8 (Car), 129.0 (Car), 119.9 (Car), 118.9 (Car), 48.1 (NCH2), 25.3 (NCH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2); ESI-MS m/z (%): 256 (90) [(M + H)+]; Anal. Calcd for C11H13NO4S: C, 51.75; H, 5.13; N, 5.49. Found: C, 51.78; H, 5.28; N, 5.55.
:1) (3.18 g, 11.8 mmol, 87%). M.p. 168–170 °C; IR absorptions (cm−1, ATR): 2927, 1662, 1292, 1154, 925, 742; 1H NMR (CDCl3, 400 MHz): δ 11.40 (s, 1H, OH), 9.96 (s, 1H, HCO), 8.03 (d, J = 2.4 Hz, 1H, Har), 7.87 (dd, J = 2.4, 8.8 Hz, 1H, Har), 7.12 (d, J = 8.8 Hz, 1H, Har), 3.01 (t, J = 4.0 Hz, 4H, NCH2), 1.68–1.63 (m, 4H, NCH2
H2); ESI-MS m/z (%): 256 (90) [(M + H)+]; Anal. Calcd for C11H13NO4S: C, 51.75; H, 5.13; N, 5.49. Found: C, 51.78; H, 5.28; N, 5.55.
:1) (3.18 g, 11.8 mmol, 87%). M.p. 168–170 °C; IR absorptions (cm−1, ATR): 2927, 1662, 1292, 1154, 925, 742; 1H NMR (CDCl3, 400 MHz): δ 11.40 (s, 1H, OH), 9.96 (s, 1H, HCO), 8.03 (d, J = 2.4 Hz, 1H, Har), 7.87 (dd, J = 2.4, 8.8 Hz, 1H, Har), 7.12 (d, J = 8.8 Hz, 1H, Har), 3.01 (t, J = 4.0 Hz, 4H, NCH2), 1.68–1.63 (m, 4H, NCH2 ), 1.47–1.41 (m, 2H, NCH2CH2
), 1.47–1.41 (m, 2H, NCH2CH2 ); 13C NMR (CDCl3, 101 MHz): δ 196.0 (CO), 164.7 (Car), 135.6 (Car), 134.0 (Car), 128.4 (Car), 120.0(Car), 118.9 (Car), 47.1 (NH2), 25.3 (NCH2H2), 23.6 (NCH2CH2H2); ESI-MS m/z (%): 270 (100) [(M + H)+]; Anal. Calcd for C12H15NO4S: C, 53.52; H, 5.61; N, 5.20. Found: C, 53.61; H, 5.45; N, 5.34.
:1) (1.35 g, 4.7 mmol, 33%); M.p. 152–154 °C; IR absorptions (cm−1, ATR): 2915, 1661, 1321, 1086, 997, 875, 736; 1H NMR (CDCl3, 400 MHz): δ 11.37 (s, 1H, OH), 9.95 (s, 1H, HCO), 8.08 (d, J = 2.4 Hz, 1H, Har), 7.90 (dd, J = 2.4, 8.8 Hz, 1H, Har), 7.10 (d, J = 8.8 Hz, 1H, Har), 3.29 (t, J = 4.0 Hz, 4H, NCH2), 1.74–1.72 (m, 4H, NCH2
); 13C NMR (CDCl3, 101 MHz): δ 196.0 (CO), 164.7 (Car), 135.6 (Car), 134.0 (Car), 128.4 (Car), 120.0(Car), 118.9 (Car), 47.1 (NH2), 25.3 (NCH2H2), 23.6 (NCH2CH2H2); ESI-MS m/z (%): 270 (100) [(M + H)+]; Anal. Calcd for C12H15NO4S: C, 53.52; H, 5.61; N, 5.20. Found: C, 53.61; H, 5.45; N, 5.34.
:1) (1.35 g, 4.7 mmol, 33%); M.p. 152–154 °C; IR absorptions (cm−1, ATR): 2915, 1661, 1321, 1086, 997, 875, 736; 1H NMR (CDCl3, 400 MHz): δ 11.37 (s, 1H, OH), 9.95 (s, 1H, HCO), 8.08 (d, J = 2.4 Hz, 1H, Har), 7.90 (dd, J = 2.4, 8.8 Hz, 1H, Har), 7.10 (d, J = 8.8 Hz, 1H, Har), 3.29 (t, J = 4.0 Hz, 4H, NCH2), 1.74–1.72 (m, 4H, NCH2 ), 1.62–1.59 (m, 4H, NCH2CH2
), 1.62–1.59 (m, 4H, NCH2CH2 ); 13C NMR (CDCl3, 101 MHz): δ 196.0 (CO), 164.4 (Car), 135.0 (Car), 133.3 (Car), 131.7 (Car), 120.0 (Car), 119.0 (Car), 48.4 (NH2), 29.3 (NCH2H2), 27.1(NCH2CH2H2); HRMS (ESI-TOF) m/z Calcd for C13H18NO4S [M + H]+: 284.0951. Found: 284.0959.
:1) (5.37 g, 19.79 mmol, 88%); M.p. 187–188 °C; IR absorptions (cm−1, ATR): 2993, 1685, 1668, 1348, 1328, 1260, 1225, 1150, 1111, 941, 742; 1H NMR (d6-DMSO, 400 MHz): δ 11.85 (s, 1H, OH), 10.32 (s, 1H, HCO), 7.93 (d, J = 2.0 Hz, 1H, Har), 7.81 (dd, J = 2.0, 8.8 Hz, 1H, Har), 7.22 (d, J = 8.8 Hz, 1H, Har), 3.7 (t, J = 4.0 Hz, 4H, OCH2), 2.85 (t, J = 4.0 Hz, 4H, NCH2); 13C NMR (d6-DMSO, 101 MHz): δ 189.6 (CO), 164.2 (Car), 134.8 (Car), 128.7 (Car), 124.9(Car), 122.3(Car), 118.6 (Car), 65.2 (OCH2), 45.8 (NCH2); HRMS (ESI-TOF) m/z Calcd for C11H14NO5S [M + H]+: 272.0587. Found: 272.0578.
); 13C NMR (CDCl3, 101 MHz): δ 196.0 (CO), 164.4 (Car), 135.0 (Car), 133.3 (Car), 131.7 (Car), 120.0 (Car), 119.0 (Car), 48.4 (NH2), 29.3 (NCH2H2), 27.1(NCH2CH2H2); HRMS (ESI-TOF) m/z Calcd for C13H18NO4S [M + H]+: 284.0951. Found: 284.0959.
:1) (5.37 g, 19.79 mmol, 88%); M.p. 187–188 °C; IR absorptions (cm−1, ATR): 2993, 1685, 1668, 1348, 1328, 1260, 1225, 1150, 1111, 941, 742; 1H NMR (d6-DMSO, 400 MHz): δ 11.85 (s, 1H, OH), 10.32 (s, 1H, HCO), 7.93 (d, J = 2.0 Hz, 1H, Har), 7.81 (dd, J = 2.0, 8.8 Hz, 1H, Har), 7.22 (d, J = 8.8 Hz, 1H, Har), 3.7 (t, J = 4.0 Hz, 4H, OCH2), 2.85 (t, J = 4.0 Hz, 4H, NCH2); 13C NMR (d6-DMSO, 101 MHz): δ 189.6 (CO), 164.2 (Car), 134.8 (Car), 128.7 (Car), 124.9(Car), 122.3(Car), 118.6 (Car), 65.2 (OCH2), 45.8 (NCH2); HRMS (ESI-TOF) m/z Calcd for C11H14NO5S [M + H]+: 272.0587. Found: 272.0578.
 ); 13C NMR (CDCl3, 101 MHz): δ 162.0 (Car), 129.2 (Car), 128.6 (Car), 127.3 (Car), 121.7 (Car), 116.9 (Car), 61.3 (CarH2), 53.8 (NH2H2N), 48.0 (NH2), 41.6 (NH3), 25.3 (NCH2H2); ESI-MS m/z (%): 567 (95) [(M + H)+]; Anal. Calcd for C26H38N4O6S2: C, 55.10; H, 6.76; N, 9.89. Found: C, 54.64; H, 6.67; N, 9.81.
); 13C NMR (CDCl3, 101 MHz): δ 162.0 (Car), 129.2 (Car), 128.6 (Car), 127.3 (Car), 121.7 (Car), 116.9 (Car), 61.3 (CarH2), 53.8 (NH2H2N), 48.0 (NH2), 41.6 (NH3), 25.3 (NCH2H2); ESI-MS m/z (%): 567 (95) [(M + H)+]; Anal. Calcd for C26H38N4O6S2: C, 55.10; H, 6.76; N, 9.89. Found: C, 54.64; H, 6.67; N, 9.81.
 ), 1.49–1.43 (m, 4H, NCH2CH2
), 1.49–1.43 (m, 4H, NCH2CH2 ); 13C NMR (CDCl3, 101 MHz): δ 162.0 (Car), 129.5 (Car), 128.7 (Car), 126.7 (Car), 121.7 (Car), 116.9 (Car), 61.4 (CarH2), 53.9 (NH2H2N), 47.1 (NCH2), 41.7 (NCH3), 25.3 (NCH2H2), 23.7 (NCH2CH2H2); ESI-MS m/z (%): 595 (85) [(M + H)+]; Anal. Calcd for C28H42N4O6S2: C, 56.54; H, 7.12; N, 9.42. Found: C, 56.55; H, 6.88; N, 9.24.
); 13C NMR (CDCl3, 101 MHz): δ 162.0 (Car), 129.5 (Car), 128.7 (Car), 126.7 (Car), 121.7 (Car), 116.9 (Car), 61.4 (CarH2), 53.9 (NH2H2N), 47.1 (NCH2), 41.7 (NCH3), 25.3 (NCH2H2), 23.7 (NCH2CH2H2); ESI-MS m/z (%): 595 (85) [(M + H)+]; Anal. Calcd for C28H42N4O6S2: C, 56.54; H, 7.12; N, 9.42. Found: C, 56.55; H, 6.88; N, 9.24.
 ), 1.47–1.43 (m, 8H, NCH2CH2
), 1.47–1.43 (m, 8H, NCH2CH2 ); 13C NMR (d6-DMSO, 101 MHz): δ 160.6 (Car), 128.5 (Car), 128.4 (Car), 127.5 (Car), 124.1 (Car), 115.7 (Car), 56.7 (CarH2), 53.4 (NH2H2N), 47.6 (NH2), 41.2 (NH3), 28.4 (NCH2H2), 26.3 (NCH2CH2H2); HRMS (ESI-TOF) m/z Calcd for C30H47N4O6S2 [M + H]+: 623.2932. Found: 623.2912.
H2), 56.6 (OH2), 53.4 (NH2H2N), 45.9 (NH2), 41.2 (NH3); HRMS (ESI-TOF) m/z Calcd for C26H39N4O8S2 [M + H]+: 599.2204. Found: 599.2203.
); 13C NMR (d6-DMSO, 101 MHz): δ 160.6 (Car), 128.5 (Car), 128.4 (Car), 127.5 (Car), 124.1 (Car), 115.7 (Car), 56.7 (CarH2), 53.4 (NH2H2N), 47.6 (NH2), 41.2 (NH3), 28.4 (NCH2H2), 26.3 (NCH2CH2H2); HRMS (ESI-TOF) m/z Calcd for C30H47N4O6S2 [M + H]+: 623.2932. Found: 623.2912.
H2), 56.6 (OH2), 53.4 (NH2H2N), 45.9 (NH2), 41.2 (NH3); HRMS (ESI-TOF) m/z Calcd for C26H39N4O8S2 [M + H]+: 599.2204. Found: 599.2203.
 (CH3)2), 4.60 (d, J = 14.4 Hz, 2H, CarCH2), 3.74 (dt, J = 4.6 Hz, 8H, O
(CH3)2), 4.60 (d, J = 14.4 Hz, 2H, CarCH2), 3.74 (dt, J = 4.6 Hz, 8H, O CH2N), 3.27 (d, J = 14.1 Hz, 2H, CarCH2), 2.98 (dt, J = 4.6 Hz, 8H, OCH2
CH2N), 3.27 (d, J = 14.1 Hz, 2H, CarCH2), 2.98 (dt, J = 4.6 Hz, 8H, OCH2 N), 2.90 (d, J = 9.4 Hz, 2H, N
N), 2.90 (d, J = 9.4 Hz, 2H, N CH2N), 2.46 (s, 6H, NCH3), 1.94 (d, J = 9.4 Hz, 2H, N
CH2N), 2.46 (s, 6H, NCH3), 1.94 (d, J = 9.4 Hz, 2H, N CH2N), 1.25 (d, J = 6.5 Hz, 12H, OCH(
CH2N), 1.25 (d, J = 6.5 Hz, 12H, OCH( )2); 13C NMR (CDCl3, 101 MHz): δ 166.3 (Car), 130.2 (Car), 130.0 (Car), 124.4 (Car), 122.6 (Car), 118.3 (Car), 79.6 (OH(CH3)2), 66.3 (OH2CH2N), 51.9 (CarH2), 47.5 (NCH3), 46.2 (OCH2H2N), 26.0 (NH2H2N), 25.7 (OCH(H3)), 25.5 (OCH(H3)); UV/Vis (THF): λmax (ε) = 252 nm (8383 M−1 cm−1), 308 nm (10004 M−1 cm−1); Anal. Calcd for C32H50N4O10S2Ti: C, 50.39; H, 6.61; N, 7.35. Found: C, 49.90; H, 6.55; N, 7.38.
:1) to give [L1Ti(dipic)] as a yellow solid (90.4 mg, 0.12 mmol, 33%). M.p. 250–251 °C; IR absorptions (cm−1, ATR): 2933, 1644, 1592, 1475, 1298, 1111, 945, 908, 783; 1H NMR (CDCl3, 400 MHz): δ 8.34 (dd, J = 7.0, 8.0 Hz, 1H, HPyr), 8.24 (d, J = 7.0 Hz, 2H, HPyr), 7.58–7.54 (m, 4H, Har), 6.52 (d, J = 8.0 Hz, 2H, Har), 5.39 (d, J = 16.0 Hz, 2H, CarCH2), 3.37 (d, J = 16.0 Hz, 2H, CarCH2), 3.32 (d, J = 8.0 Hz, 2H, N
)2); 13C NMR (CDCl3, 101 MHz): δ 166.3 (Car), 130.2 (Car), 130.0 (Car), 124.4 (Car), 122.6 (Car), 118.3 (Car), 79.6 (OH(CH3)2), 66.3 (OH2CH2N), 51.9 (CarH2), 47.5 (NCH3), 46.2 (OCH2H2N), 26.0 (NH2H2N), 25.7 (OCH(H3)), 25.5 (OCH(H3)); UV/Vis (THF): λmax (ε) = 252 nm (8383 M−1 cm−1), 308 nm (10004 M−1 cm−1); Anal. Calcd for C32H50N4O10S2Ti: C, 50.39; H, 6.61; N, 7.35. Found: C, 49.90; H, 6.55; N, 7.38.
:1) to give [L1Ti(dipic)] as a yellow solid (90.4 mg, 0.12 mmol, 33%). M.p. 250–251 °C; IR absorptions (cm−1, ATR): 2933, 1644, 1592, 1475, 1298, 1111, 945, 908, 783; 1H NMR (CDCl3, 400 MHz): δ 8.34 (dd, J = 7.0, 8.0 Hz, 1H, HPyr), 8.24 (d, J = 7.0 Hz, 2H, HPyr), 7.58–7.54 (m, 4H, Har), 6.52 (d, J = 8.0 Hz, 2H, Har), 5.39 (d, J = 16.0 Hz, 2H, CarCH2), 3.37 (d, J = 16.0 Hz, 2H, CarCH2), 3.32 (d, J = 8.0 Hz, 2H, N CH2N), 3.19–3.16 (m, 8H, NCH2), 2.82 (s, 6H, NCH3), 2.33 (d, J = 8.0 Hz, 2H, NCH2
CH2N), 3.19–3.16 (m, 8H, NCH2), 2.82 (s, 6H, NCH3), 2.33 (d, J = 8.0 Hz, 2H, NCH2 N), 1.77–1.74 (m, 8H, NCH2
N), 1.77–1.74 (m, 8H, NCH2 ); 13C NMR (CDCl3, 101 MHz): δ 169.1 (CO), 162.7 (Car), 149.5 (Car), 144.2 (Car), 129.7 (Car), 129.6 (Car), 129.3 (Car), 128.2 (Car), 126.3 (Car), 117.1 (Car), 63.9 (CarCH2), 54.3 (NH2H2N), 48.0 (NH2), 47.7 (NCH3), 25.3 (NH2H2); UV/Vis (THF): λmax (ε) = 210 nm (33120 M−1 cm−1), 240 nm (28330 M−1 cm−1), 375 nm (10340 M−1 cm−1); Anal. Calcd for C33H39N5O10S2Ti: C, 50.97; H, 5.05; N, 9.01. Found: C, 51.16; H, 5.17; N, 8.92; HRMS (ESI-TOF) m/z Calcd for C33H40N5O10S2Ti [M + H]+: 778.1691. Found: 778.1699.
); 13C NMR (CDCl3, 101 MHz): δ 169.1 (CO), 162.7 (Car), 149.5 (Car), 144.2 (Car), 129.7 (Car), 129.6 (Car), 129.3 (Car), 128.2 (Car), 126.3 (Car), 117.1 (Car), 63.9 (CarCH2), 54.3 (NH2H2N), 48.0 (NH2), 47.7 (NCH3), 25.3 (NH2H2); UV/Vis (THF): λmax (ε) = 210 nm (33120 M−1 cm−1), 240 nm (28330 M−1 cm−1), 375 nm (10340 M−1 cm−1); Anal. Calcd for C33H39N5O10S2Ti: C, 50.97; H, 5.05; N, 9.01. Found: C, 51.16; H, 5.17; N, 8.92; HRMS (ESI-TOF) m/z Calcd for C33H40N5O10S2Ti [M + H]+: 778.1691. Found: 778.1699.
 CH2N), 2.96–2.88 (m, 8H, NCH2), 2.82 (s, 6H, NCH3), 2.33 (d, J = 8.0 Hz, 2H, NCH2
CH2N), 2.96–2.88 (m, 8H, NCH2), 2.82 (s, 6H, NCH3), 2.33 (d, J = 8.0 Hz, 2H, NCH2 N), 1.62–1.59 (m, 8H, NCH2
N), 1.62–1.59 (m, 8H, NCH2 ), 1.41–1.38 (m, 4H, NCH2CH2
), 1.41–1.38 (m, 4H, NCH2CH2 ); 13C NMR (CDCl3, 101 MHz): δ 169.0 (CO), 162.7 (Car), 149.5 (Car), 144.3 (Car), 129.6 (Car), 129.4 (Car), 128.9 (Car), 128.1 (Car), 126.3 (Car), 117.0 (Car), 63.9 (CarH2), 54.2 (NH2H2N), 47.7 (NCH2), 47.0 (NCH3), 25.3 (NCH2H2), 23.5 (NCH2CH2H2). λmax (ε) = 215 nm (29690 M−1 cm−1), 240 nm (23870 M−1 cm−1), 375 nm (8640 M−1 cm−1); Found: 806.1993; Anal. Calcd for C35H43N5O10S2Ti: C, 52.17; H, 5.38; N, 8.69. Found: C, 52.25; H, 5.42; N, 8.53; HRMS (ESI-TOF) m/z Calcd for C35H44N5O10S2Ti [M + H]+: 806.2004.
); 13C NMR (CDCl3, 101 MHz): δ 169.0 (CO), 162.7 (Car), 149.5 (Car), 144.3 (Car), 129.6 (Car), 129.4 (Car), 128.9 (Car), 128.1 (Car), 126.3 (Car), 117.0 (Car), 63.9 (CarH2), 54.2 (NH2H2N), 47.7 (NCH2), 47.0 (NCH3), 25.3 (NCH2H2), 23.5 (NCH2CH2H2). λmax (ε) = 215 nm (29690 M−1 cm−1), 240 nm (23870 M−1 cm−1), 375 nm (8640 M−1 cm−1); Found: 806.1993; Anal. Calcd for C35H43N5O10S2Ti: C, 52.17; H, 5.38; N, 8.69. Found: C, 52.25; H, 5.42; N, 8.53; HRMS (ESI-TOF) m/z Calcd for C35H44N5O10S2Ti [M + H]+: 806.2004.
 CH2N), 3.19 (t, J = 4.0 Hz, 8H, NCH2), 2.80 (s, 6H, NCH3), 2.31 (d, J = 8.0 Hz, 2H, NCH2
CH2N), 3.19 (t, J = 4.0 Hz, 8H, NCH2), 2.80 (s, 6H, NCH3), 2.31 (d, J = 8.0 Hz, 2H, NCH2 N), 1.71–1.65 (m, 8H, NCH2
N), 1.71–1.65 (m, 8H, NCH2 ) 1.57–1.55 (m, 8H, NCH2CH2
) 1.57–1.55 (m, 8H, NCH2CH2 ); 13C NMR (CDCl3, 101 MHz): δ 169.0 (CO), 162.4 (Car), 149.5 (Car), 144.2 (Car), 132.2 (Car), 129.0 (Car), 128.6 (Car), 128.1 (Car), 126.3 (Car), 117.0 (Car), 63.9 (CarCH2), 54.3 (NH2H2N), 48.2 (NCH2), 47.7 (NCH3), 29.2 (NCH2H2), 27.0 (NCH2CH2H2); UV/Vis (THF): λmax (ε) = 210 nm (18360 M−1 cm−1), 242 nm (10180 M−1 cm−1), 375 nm (4054 M−1 cm−1); Anal. Calcd for C37H47N5O10S2Ti: C, 53.30; H, 5.68; N, 8.40. Found: C, 53.43; H, 5.82; N, 8.30; HRMS (ESI-TOF) m/z Calcd for C37H48N5O10S2Ti [M + H]+: 834.2317. Found: 834.2279.
); 13C NMR (CDCl3, 101 MHz): δ 169.0 (CO), 162.4 (Car), 149.5 (Car), 144.2 (Car), 132.2 (Car), 129.0 (Car), 128.6 (Car), 128.1 (Car), 126.3 (Car), 117.0 (Car), 63.9 (CarCH2), 54.3 (NH2H2N), 48.2 (NCH2), 47.7 (NCH3), 29.2 (NCH2H2), 27.0 (NCH2CH2H2); UV/Vis (THF): λmax (ε) = 210 nm (18360 M−1 cm−1), 242 nm (10180 M−1 cm−1), 375 nm (4054 M−1 cm−1); Anal. Calcd for C37H47N5O10S2Ti: C, 53.30; H, 5.68; N, 8.40. Found: C, 53.43; H, 5.82; N, 8.30; HRMS (ESI-TOF) m/z Calcd for C37H48N5O10S2Ti [M + H]+: 834.2317. Found: 834.2279.
 CH2N), 2.89 (t, J = 4.0 Hz, 8H, NCH2), 2.86 (s, 6H, NCH3), 2.60 (d, J = 8.0 Hz, 2H, NCH2
CH2N), 2.89 (t, J = 4.0 Hz, 8H, NCH2), 2.86 (s, 6H, NCH3), 2.60 (d, J = 8.0 Hz, 2H, NCH2 N); 13C NMR (d6-acetone, 101 MHz): δ 169.1 (CO), 164.2 (Car), 150.1 (Car), 146.4 (Car), 131.0 (Car), 130.4 (Car), 129.7 (Car), 128.3 (Car), 127.1 (Car), 117.8 (Car), 66.7 (OCH2), 63.9 (CarCH2), 54.8 (NH2H2N), 47.9 (NCH3), 47.1 (OCH2H2N); UV/Vis (THF): λmax (ε) = 210 nm (27190 M−1 cm−1), 245 nm (15900 M−1 cm−1), 360 nm (5930 M−1 cm−1); Anal. Calcd for C33H39N5O12S2Ti: C, 48.95; H, 4.86; N, 8.65. Found: C, 48.72; H, 4.75; N, 8.81; HRMS (ESI-TOF) m/z Calcd for C33H40N5O12S2Ti [M + H]+: 810.1589. Found: 810.1608.
O), 159.6 (Car), 149.9 (Car), 143.5 (Car), 129.7 (Car), 129.0 (Car), 128.1 (Car), 128.1 (Car), 125.9 (Car), 121.7 (Car), 116.4 (Car), 64.2 (CarCH2), 54.0 (NH2H2N), 47.7 (NCH3); UV/Vis (THF): λmax (ε) = 215 nm (23240 M−1 cm−1), 380 nm (6350 M−1 cm−1); HRMS (ESI-TOF) m/z Calcd for C25H26N3O6Ti [M + H]+: 512.1301. Found: 512.1316.
N); 13C NMR (d6-acetone, 101 MHz): δ 169.1 (CO), 164.2 (Car), 150.1 (Car), 146.4 (Car), 131.0 (Car), 130.4 (Car), 129.7 (Car), 128.3 (Car), 127.1 (Car), 117.8 (Car), 66.7 (OCH2), 63.9 (CarCH2), 54.8 (NH2H2N), 47.9 (NCH3), 47.1 (OCH2H2N); UV/Vis (THF): λmax (ε) = 210 nm (27190 M−1 cm−1), 245 nm (15900 M−1 cm−1), 360 nm (5930 M−1 cm−1); Anal. Calcd for C33H39N5O12S2Ti: C, 48.95; H, 4.86; N, 8.65. Found: C, 48.72; H, 4.75; N, 8.81; HRMS (ESI-TOF) m/z Calcd for C33H40N5O12S2Ti [M + H]+: 810.1589. Found: 810.1608.
O), 159.6 (Car), 149.9 (Car), 143.5 (Car), 129.7 (Car), 129.0 (Car), 128.1 (Car), 128.1 (Car), 125.9 (Car), 121.7 (Car), 116.4 (Car), 64.2 (CarCH2), 54.0 (NH2H2N), 47.7 (NCH3); UV/Vis (THF): λmax (ε) = 215 nm (23240 M−1 cm−1), 380 nm (6350 M−1 cm−1); HRMS (ESI-TOF) m/z Calcd for C25H26N3O6Ti [M + H]+: 512.1301. Found: 512.1316.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
Financial and scientific support by the Konstanz Research School Chemical Biology (KoRS-CB) is gratefully acknowledged. T.Z. thanks the KoRS-CB for a personal scholarship. The authors like to thank Dipl. Chem. Malin Bein for her help with the biological assays.Notes and references
- B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- (a) H. B. Gray, E. I. Stiefel, J. S. Valentine and I. Bertini, Biological Inorganic Chemistry: Structure and Reactivity, University Science Books, 2007 Search PubMed; (b) W. Kaim, B. Schwederski and A. Klein, Bioinorganic Chemistry – Inorganic Elements in the Chemistry of Life: An Introduction and Guide, Wiley, 2013 Search PubMed.
- (a) A. A. Argyriou, P. Polychronopoulos, G. Iconomou, E. Chroni and H. P. Kalofonos, Cancer Treat. Rev., 2008, 34, 368–377 CrossRef CAS; (b) S. R. McWhinney, R. M. Goldberg and H. L. McLeod, Mol. Cancer Ther., 2009, 8, 10–16 CrossRef CAS; (c) X. Yao, K. Panichpisal, N. Kurtzman and K. Nugent, Am. J. Med. Sci., 2007, 334, 115–124 CrossRef PubMed.
- (a) H. Köpf and P. Köpf-Maier, Angew. Chem., Int. Ed., 1979, 18, 477–478 CrossRef PubMed; (b) P. Köpf-Maier, Eur. J. Clin. Pharmacol., 1994, 47, 1–16 CrossRef; (c) E. Meléndez, Crit. Rev. Oncol. Hematol., 2002, 42, 309–315 CrossRef; (d) F. Caruso and M. Rossi, Mini-Rev. Med. Chem., 2004, 4, 49–60 CrossRef CAS; (e) A. M. Valentine, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2011 Search PubMed; (f) K. M. Buettner and A. M. Valentine, Chem. Rev., 2012, 112, 1863–1881 CrossRef CAS.
- (a) K. O'Connor, C. Gill, M. Tacke, F. J. Rehmann, K. Strohfeldt, N. Sweeney, J. M. Fitzpatrick and R. W. Watson, Apoptosis, 2006, 11, 1205–1214 CrossRef PubMed; (b) P. M. Abeysinghe and M. M. Harding, Dalton Trans., 2007, 3474–3482 RSC.
- (a) F. Caruso, M. Rossi, J. Tanski, R. Sartori, R. Sariego, S. Moya, S. Diez, E. Navarrete, A. Cingolani, F. Marchetti and C. Pettinari, J. Med. Chem., 2000, 43, 3665–3670 CrossRef CAS; (b) T. Schilling, K. B. Keppler, M. E. Heim, G. Niebch, H. Dietzfelbinger, J. Rastetter and A. R. Hanauske, Invest. New Drugs, 1996, 13, 327–332 CrossRef CAS.
- (a) J. H. Toney and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 947–953 CrossRef CAS; (b) X. Chen and L. Zhou, J. Mol. Struct. (THEOCHEM), 2010, 940, 45–49 CrossRef CAS PubMed.
- (a) M. Shavit, D. Peri, C. M. Manna, J. S. Alexander and E. Y. Tshuva, J. Am. Chem. Soc., 2007, 129, 12098–12099 CrossRef CAS PubMed; (b) D. Peri, S. Meker, M. Shavit and E. Y. Tshuva, Chem. – Eur. J., 2009, 15, 2403–2415 CrossRef CAS PubMed; (c) T. A. Immel, M. Debiak, U. Groth, A. Bürkle and T. Huhn, ChemMedChem, 2009, 4, 738–741 CrossRef CAS PubMed; (d) C. M. Manna, O. Braitbard, E. Weiss, J. Hochman and E. Y. Tshuva, ChemMedChem, 2012, 7, 703–708 CrossRef CAS PubMed; (e) T. A. Immel, U. Groth and T. Huhn, Chem. – Eur. J., 2010, 16, 2775–2789 CrossRef CAS PubMed; (f) T. A. Immel, U. Groth, T. Huhn and P. Öhlschläger, PLoS One, 2011, 6, e17869 CAS.
- (a) H. Glasner and E. Y. Tshuva, J. Am. Chem. Soc., 2011, 133, 16812–16814 CrossRef CAS PubMed; (b) H. Glasner and E. Y. Tshuva, Inorg. Chem., 2014, 53, 3170–3176 CrossRef CAS PubMed.
- T. A. Immel, M. Grützke, A. K. Späte, U. Groth, P. Öhlschläger and T. Huhn, Chem. Commun., 2012, 48, 5790–5792 RSC.
- M. Grützke, T. Zhao, T. A. Immel and T. Huhn, Inorg. Chem., 2015, 54, 6697–6706 CrossRef PubMed.
- A. Scozzafava, T. Owa, A. Mastrolorenzo and C. T. Supuran, Curr. Med. Chem., 2003, 10, 925–953 CrossRef CAS.
- A. Casini, A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, Curr. Cancer Drug Targets, 2002, 2, 55–75 CrossRef CAS.
- K. Fukuoka, J. Usuda, Y. Iwamoto, H. Fukumoto, T. Nakamura, T. Yoneda, N. Narita, N. Saijo and K. Nishio, Invest. New Drugs, 2001, 19, 219–227 CrossRef CAS.
- L. Hu, Z.-r. Li, Y. Li, J. Qu, Y.-H. Ling, J.-d. Jiang and D. W. Boykin, J. Med. Chem., 2006, 49, 6273–6282 CrossRef CAS PubMed.
- K. L. Lobb, P. A. Hipskind, J. A. Aikins, E. Alvarez, Y.-Y. Cheung, E. L. Considine, A. De Dios, G. L. Durst, R. Ferritto, C. S. Grossman, D. D. Giera, B. A. Hollister, Z. Huang, P. W. Iversen, K. L. Law, T. Li, H.-S. Lin, B. Lopez, J. E. Lopez, L. M. M. Cabrejas, D. J. McCann, V. Molero, J. E. Reilly, M. E. Richett, C. Shih, B. Teicher, J. H. Wikel, W. T. White and M. M. Mader, J. Med. Chem., 2004, 47, 5367–5380 CrossRef CAS PubMed.
- (a) T. Owa, T. Okauchi, K. Yoshimatsu, N. H. Sugi, Y. Ozawa, T. Nagasu, N. Koyanagi, T. Okabe, K. Kitoh and H. Yoshino, Bioorg. Med. Chem. Lett., 2000, 10, 1223–1226 CrossRef CAS; (b) A. J. Salmon, M. L. Williams, A. Innocenti, D. Vullo, C. T. Supuran and S.-A. Poulsen, Bioorg. Med. Chem. Lett., 2007, 17, 5032–5035 CrossRef CAS PubMed; (c) V. del Solar, A. Quiñones-Lombraña, S. Cabrera, J. M. Padrón, C. Ríos-Luci, A. Alvarez-Valdés, C. Navarro-Ranninger and J. Alemán, J. Inorg. Biochem., 2013, 127, 128–140 CrossRef CAS PubMed; (d) H. Dou, W. Zhong, L. Yang, T. Wang, H. Yan and Y. Hou, Bioorg. Med. Chem., 2012, 20, 4693–4700 CrossRef CAS PubMed.
- (a) For recent examples of sulfonamide coordinated TiIV-complexes see: ; (b) S. Hamura, T. Oda, Y. Shimizu, K. Matsubara and H. Nagashima, J. Chem. Soc., Dalton Trans., 2002, 1521–1527 RSC; (c) C. Lensink, G. J. Gainsford and M. J. R. Brandsma, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, m519–m520 CAS; (d) C. Lensink, J. Organomet. Chem., 1998, 553, 387–392 CrossRef CAS; (e) S. Pritchett, D. H. Woodmansee, P. Gantzel and P. J. Walsh, J. Am. Chem. Soc., 1998, 120, 6423–6424 CrossRef CAS.
- A. Rescigno, F. Bruyneel, A. Padiglia, F. Sollai, A. Salis, J. Marchand-Brynaert and E. Sanjust, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 799–807 CrossRef CAS PubMed.
- S. L. Hancock, R. Gati, M. F. Mahon, E. Y. Tshuva and M. D. Jones, Dalton Trans., 2014, 43, 1380–1385 RSC.
- M. Miller and E. Y. Tshuva, Eur. J. Inorg. Chem., 2014, 2014, 1485–1491 CrossRef CAS PubMed.
- R. M. Lord, J. J. Mannion, A. J. Hebden, A. E. Nako, B. D. Crossley, M. W. McMullon, F. D. Janeway, R. M. Phillips and P. C. McGowan, ChemMedChem, 2014, 9, 1136–1139 CrossRef CAS PubMed.
- R. Hamid, Y. Rotshteyn, L. Rabadi, R. Parikh and P. Bullock, Toxicol. In Vitro, 2004, 18, 703–710 CrossRef CAS.
- (a) A. D. Tinoco, E. V. Eames and A. M. Valentine, J. Am. Chem. Soc., 2008, 130, 2262–2270 CrossRef CAS; (b) A. D. Tinoco, H. R. Thomas, C. D. Incarvito, A. Saghatelian and A. M. Valentine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5016–5021 CrossRef CAS PubMed.
- J. Schur, C. M. Manna, E. Y. Tshuva and I. Ott, Metallodrugs, 2014, 1, 1–9 CrossRef.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Elsevier Science, 5th edn, 2003 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- sigmaaldrich.com/catalog/product/sigma/d5523 last accessed; 2015.03.02.
- R. D. Fields and M. V. Lancaster, Am. Biotechnol. Lab., 1993, 11, 48–49 CAS.
- Systat Software, Inc., http://www.systat.com, 2006.
- J. Balsells, P. J. Carroll and P. J. Walsh, Inorg. Chem., 2001, 40, 5568–5574 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of all compounds, crystallographic data for [L4Ti(dipic)] and [L5Ti(dipic)], colored representations of the cell viability plots, selected stability tests in different media and supplementary figures. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01618e |
| ‡ Present address: MEET Battery Research Center, University of Münster, Corrensstr. 46, D 48149 Münster, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |