
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe conjugates of ferrocene-1,1′-diamine and amino acids. A novel synthetic approach and conformational analysis†
Monika
Kovačević
a,
Ivan
Kodrin
*b,
Mario
Cetina
c,
Ivana
Kmetič
a,
Teuta
Murati
a,
Mojca Čakić
Semenčić
a,
Sunčica
Roca
d and
Lidija
Barišić
*a
aDepartment of Chemistry and Biochemistry, Faculty of Food Technology and Biotechnology, University of Zagreb, Pierottijeva 6, Zagreb, Croatia. E-mail: lidija.barisic@pbf.hr
bDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, Zagreb, Croatia. E-mail: ikodrin@chem.pmf.hr
cDepartment of Applied Chemistry, Faculty of Textile Technology, University of Zagreb, Prilaz baruna Filipovića 28a, Zagreb, Croatia
dNMR Centre, Ruđer Bošković Institute, Bijenička cesta 54, Zagreb, Croatia
First published on 12th August 2015
Abstract
A novel synthetic approach toward a poorly explored bioorganometallic consisting of ferrocene-1,1′-diamine bearing structurally and chirally diverse amino acid sequences is reported. Until now, ferrocene-1,1′-diamine was suitable for accommodating only identical amino acid sequences at its N-termini, leading to the symmetrically disubstituted homochiral products stabilized through a 14-membered intramolecular hydrogen-bonded ring as is seen in antiparallel β-sheet peptides. The key step of the novel synthetic pathway is the transformation of Ac–Ala– –Fn–
–Fn– OH (5) (Fn = 1,1′-ferrocenylene) to orthogonally protected Ac–Ala–
OH (5) (Fn = 1,1′-ferrocenylene) to orthogonally protected Ac–Ala– –Fn–
–Fn– Boc (7). The spectroscopic analysis (IR, NMR, CD) of the novel compounds, corroborated with DFT studies, suggests the interesting feature of the ferrocene-1,1′-diamine scaffold. The same hydrogen-bonding pattern, i.e. a 14-membered hydrogen-bonded ring, was determined both in solution and in the solid state, thus making them promising, yet simple scaffolds capable of mimicking β-sheet peptides. In vitro screening of potential anticancer activity in Hep G2 human liver carcinoma cells and Hs 578 T human breast cancer cells revealed a cytotoxic pattern for novel compounds (150–500 μM) with significantly decreased cell proliferation.
Boc (7). The spectroscopic analysis (IR, NMR, CD) of the novel compounds, corroborated with DFT studies, suggests the interesting feature of the ferrocene-1,1′-diamine scaffold. The same hydrogen-bonding pattern, i.e. a 14-membered hydrogen-bonded ring, was determined both in solution and in the solid state, thus making them promising, yet simple scaffolds capable of mimicking β-sheet peptides. In vitro screening of potential anticancer activity in Hep G2 human liver carcinoma cells and Hs 578 T human breast cancer cells revealed a cytotoxic pattern for novel compounds (150–500 μM) with significantly decreased cell proliferation.
Introduction
Reverse turns are structural features of proteins and peptides in which a polypeptide chain changes its direction enabling a protein to fold back on itself.1 Besides the role in protein folding,2 turns are involved in molecular recognition processes mediated through interactions of side chains within the turns with various receptor domains.1,3 Depending on the hydrogen-bonded ring size (7-, 10- or 13-membered), turns are classified as γ-, β- and α-turns, respectively. The β-turns are the most abundant motifs in folded proteins.1 As essential components of β-hairpins (the fundamental elements of anti-parallel β-sheets), the β-turns are considered as nucleators or initiation sites that bring the two peptide strands in close proximity, enabling their interactions through hydrogen bonding.4 While a proper β-sheet-mediated protein folding is essential for normal biological function, the protein misfolding or folding in a fatal way might cause extracellular or intracellular aggregate formation involved in triggering diseases.5 The small model compounds containing molecular templates capable of inducing β-sheet structures in attached peptide strands have been very useful in studies of β-sheet folding and interactions.6 Very recently, Sanjayan et al. gave an overview of the selected synthetic turn mimetics and hairpin nucleators reported during the past 30 years, with an emphasis on their great potential in the fields of peptide based drugs and organic asymmetric synthesis.7On the basis of the previous results, 1,n-disubstituted ferrocenes are established as molecular templates for the design of turns and β-sheet-like structures as the almost free rotating cyclopentadiene (Cp) rings are separated by about 3.3 Å which is ideal for interstrand hydrogen bonding in their conjugates comprised of natural amino acids or peptides.8 If peptide groups directly attached to the ferrocene template are taken into account, then three major classes of bioconjugates 1–3 derived from ferrocene-1,1′-dicarboxylic acid [(Fcd), 1],9 1′-amino-ferrocene-1-carboxylic acid [(Fca), 2]10 and ferrocene-1,1′-diamine [(Fcda), 3]11 need to be considered (Fig. 1). The ferrocene scaffolds Fcd, Fca and Fcda were recognized as turn-inducers in peptides 1–3 owing to the presence of 10- (1), 12- (2) and 14-membered (3) intramolecularly hydrogen bonded (IHB) rings.
 | ||
| Fig. 1 The ferrocene-containing peptides 1–3. | ||
Ferrocenes 3 derived from Fcda are only poorly examined. While peptides 1 and 2 have been the subject of numerous papers, only one paper describing the two symmetrically disubstituted homochiral peptides Fn–(NH–AA–Boc)2 (3a, AA = L- or D-Ala), derived from Fn(NHBoc)2, was reported by Kraatz et al.11 In that research, ferrocene-1,1′-diamine acted as a turn-inducing scaffold, facilitating the formation of 14-membered rings between attached symmetrical peptide chains, very similar to those found in antiparallel β-sheet peptides. This feature has not been yet observed in derivatives 1 and 2. It seems that the ferrocene-1,1′-diamine framework is a desired structural requirement if we want to further explore and utilize the connection between ferrocene-based peptides and the turn-inducing scaffolds capable of mimicking the natural structure of an antiparallel β-sheet. Therefore, we were prompted to find a new synthetic route to conjugates 3, but now bearing structurally and chirally diverse amino acid sequences at its N-termini to check their affinity to preserve the conformational motif comprised of two ferrocene-involved 10-membered IHB rings. Acting as a simple model made of only two amino acids, 3 could be considered as the lowest homologues and a step toward the more complex structures mimicking anti-parallel β-sheets.
In this paper, a new synthetic approach toward unsymmetrically substituted ferrocenes 3 is discussed and employed for the first time to prepare orthogonally protected Ac–Ala–NH–Fn–NH–Ala–Boc (3b). In addition, acetamide-protected Fn–(NH–Ala–Ac)2 (3c) is also reported (Scheme 1). Since previously described Boc-protected 3a was stabilized through the two simultaneous interchain NHFn⋯OCBoc IHBs engaged in the 14-membered ring, our goal was to explore whether the replacement of one or both Boc-groups of 3a with sterically less demanding Ac-groups in 3b and 3c will affect the IHB pattern. Therefore, the nonpolar and nonbulky side chain of Ala was not expected to interfere with hydrogen bonding.12 A detailed conformational analysis using spectroscopic (IR, NMR, CD) and computational study together with X-ray crystal structure analysis was performed to clarify IHB patterns of the synthesized compounds and to discuss (di)similarities in solution and in the solid state. Today, a lot of endeavour is directed at creating novel metal-based therapeutics with less negative medical and physical side-effects. Therefore, the newly synthesized derivatives were subjected to biological evaluation to test their potential anticancer activity.
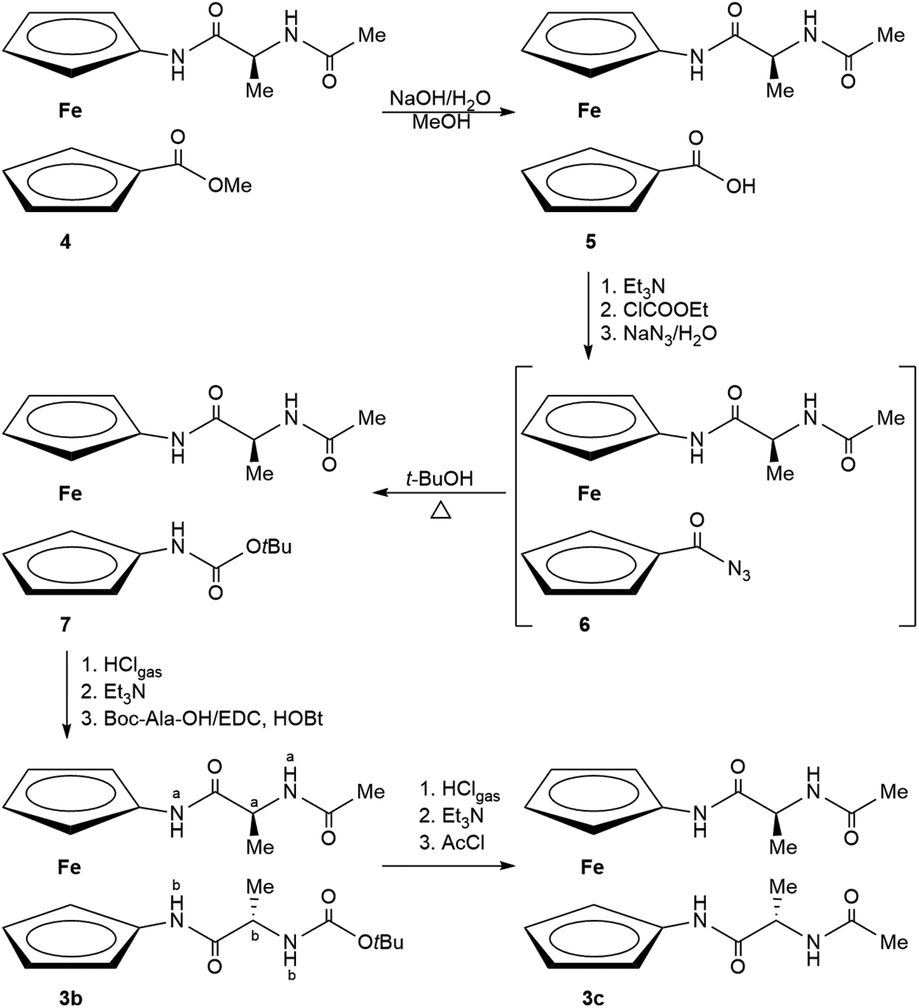 | ||
| Scheme 1 The synthesis of the goal compounds 3b and 3c (the atom numeration of 3b used for NMR analysis). | ||
Experimental
Materials and general methods
All reactions were carried out under an argon atmosphere. The CH2Cl2 used for synthesis and FTIR was dried (P2O5), distilled over CaH2, and stored over molecular sieves (4 Å). t-BuOH was dried with CaH2, distilled and stored over molecular sieves (4 Å). EDC (Acros Organics), HOBt (Aldrich), acetyl chloride (Aldrich) and alanine (Acros Organics) were used as received. The syntheses of Ac–Ala–Fca–OMe (4) had been previously described.13 The N-terminus of alanine was protected by using sodium hydroxide, aqueous dioxane and di-tert-butyldicarbonate to give Boc–Ala–OH. Products were purified by preparative thin layer chromatography on silica gel (Fluka Silica gel on TLC Al foils, silica gel matrix, with fluorescent indicator 254 nm, layer thickness 0.2 mm). Infrared spectra were recorded using CH2Cl2 solutions between NaCl windows by using a Bomem MB 100 mid FTIR spectrometer. (s) = strong, (m) = medium, (w) = weak. All 1H NMR spectra recorded at 600.133 MHz and 300 MHz and 13C NMR spectra recorded at 150.917 MHz and 75.432 MHz using a Bruker Avance spectrometer, were referenced to the residual solvent peak (CDCl3, 1H: 7.26 ppm, 13C: 77.16 ppm). In the case of the CDCl3/d6-DMSO mixture, the calibration was done using Me4Si. Double resonance experiments (COSY, NOESY and HMBC) were performed in order to assist in signal assignment. (s) = singlet, (d) = doublet, (q) = quartet, (dq) = doublet of quartet, (m) = multiplet. Unless otherwise noted, all spectra were recorded at 298 K. CD spectra were recorded using a Jasco-810 spectropolarimeter in CH2Cl2 and CH2Cl2/DMSO mixture. Molar ellipticity coefficients, [θ], are in degrees, the concentration, c, is given in mol L−1, and the path length, l, is given in cm, to give units for [θ] of deg cm2 dmol−1. NMR titrations were performed by adding 10 μL portions of d6-DMSO in NMR-tubes containing solutions of the examined peptides 3b and 3c in CDCl3 (c = 2.5 × 10−2 M). The spectra were recorded after each addition and d6-DMSO was added until no further changes in the chemical shifts of the NHs were observed. CD titrations were performed in a similar manner by stepwise addition of DMSO into cuvettes containing solutions of the examined peptides 3b and 3c in CH2Cl2 (c = 5 × 10−3 M). Mass spectra were recorded on a HPLC-MS system coupled with a triple–quadrupole mass spectrometer, operating in a positive ESI mode. High-resolution mass spectra were acquired using a 4800 MALDI TOF/TOF-MS Analyzer.Synthesis of Ac–Ala–NH–Fn–COOH (5)
A solution of 4 (500 mg, 1.34 mmol) in MeOH (3 ml) was heated at 80 °C in the presence of NaOH (53.7 mg, 1.34 mmol) and H2O (0.2 ml). After 1 h, the reaction mixture was evaporated to dryness. The residue was dissolved in 5% NaHCO3 and extracted with CH2Cl2 to remove the unreacted ester 4. The water layer was acidified with 20% HCl and extracted with EtOAc. The organic extracts were washed with a saturated solution of NaCl, dried with Na2SO4 and evaporated in vacuo. TLC purification (CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 1:1; Rf = 0.23) gave the desired product as an orange powder on removal of the solvent in vacuo (297.5 mg, 62%); mp 128–134 °C; IR (CH2Cl2): νmax/cm−1 = 3420 (w, NHfree), 3296 (m, NHassoc.), 3109–2800 (m, OH, COOH), 1710, 1697, 1653 (s, C
:EtOAc = 1:1; Rf = 0.23) gave the desired product as an orange powder on removal of the solvent in vacuo (297.5 mg, 62%); mp 128–134 °C; IR (CH2Cl2): νmax/cm−1 = 3420 (w, NHfree), 3296 (m, NHassoc.), 3109–2800 (m, OH, COOH), 1710, 1697, 1653 (s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OCOOH, CONH), 1576 (amide II); 1H NMR (600.133 MHz; CD3OD; Me4Si): δ (ppm) = 4.71 (s, 1H, HFn), 4.66 (s, 2H, HFn), 4.52–4.48 (m, 2H, HFn, CHAla), 4.29 (s, 1H, HFn), 4.27 (s, 1H, HFn), 4.01 (m, 2H, HFn), 2.05 (s, 3H, CH3Ac), 1.36 (d, J = 5.6 Hz, 3H, CH3Ala); 13C NMR, APT (150.917 MHz; CD3OD; Me4Si): δ (ppm) = 173.67 (COFn, COCOOH, COAc), 95.67, 79.51 (CqFn), 72.83, 72.38, 72.13, 67.04, 66.93, 64.46, 64.42 (CHFn), 50.98 (CHAla), 22.66 (CH3Ac), 18.03 (CH3Ala); ESI-MS (H2O:MeOH = 50:50): m/z = 381.1 [(M + Na)+].
OCOOH, CONH), 1576 (amide II); 1H NMR (600.133 MHz; CD3OD; Me4Si): δ (ppm) = 4.71 (s, 1H, HFn), 4.66 (s, 2H, HFn), 4.52–4.48 (m, 2H, HFn, CHAla), 4.29 (s, 1H, HFn), 4.27 (s, 1H, HFn), 4.01 (m, 2H, HFn), 2.05 (s, 3H, CH3Ac), 1.36 (d, J = 5.6 Hz, 3H, CH3Ala); 13C NMR, APT (150.917 MHz; CD3OD; Me4Si): δ (ppm) = 173.67 (COFn, COCOOH, COAc), 95.67, 79.51 (CqFn), 72.83, 72.38, 72.13, 67.04, 66.93, 64.46, 64.42 (CHFn), 50.98 (CHAla), 22.66 (CH3Ac), 18.03 (CH3Ala); ESI-MS (H2O:MeOH = 50:50): m/z = 381.1 [(M + Na)+].
Synthesis of Ac–Ala–NH–Fn–NHBoc (7)
Carbamate 7 was prepared starting from acid 5via unstable azide 6. Acid 5 (130 mg, 0.36 mmol) was suspended in water (0.1 ml) and sufficient acetone was added to complete the solution. After cooling to 0 °C, triethylamine (42 mg, 0.41 mmol) in acetone (0.65 ml) was added. While maintaining the temperature at 0 °C, a solution of ethyl chloroformate (49.2 mg, 0.45 mmol) in the same solvent (0.2 ml) was added and the mixture was stirred for 30 min at 0 °C. Thereafter, a solution of sodium azide (35.3 mg, 0.54 mmol) in water (0.1 ml) was added. The mixture was stirred for 1 h (0 °C) and acetone was removed in vacuo to leave the crude unstable azide 6, which was converted in situ to carbamate 7 by heating with dry t-BuOH (5 ml) at 65 °C for 5 h. The reaction mixture was evaporated to dryness and purified by preparative chromatography in EtOAc (Rf = 0.33) giving yellow powder of 7 (60 mg, 39%); mp 142–145 °C; IR (CH2Cl2): νmax/cm−1 = 3428 (s, NHfree), 3313 (w, NHassoc.), 1703 (s, COBoc), 1682, 1665 (s, COCONH), 1530, 1513 (amide II); 1H NMR (600.133 MHz; CDCl3; Me4Si): δ (ppm) = 8.30 (s, 1H, NHaFn), 6.58 (d, J = 7.5 Hz, 1H, NHAla), 6.23 (s, 1H, NHBoc), 4.61–4.53 (m, 3H, HFn, CHAla), 4.35–4.30 (m, 2H, HFn), 4.13–4.01 (m, 4H, HFn), 2.05 (s, 3H, CH3Ac), 1.50 [s, 9H, (CH3)3Boc], 1.44 (d, J = 6.9 Hz, 3H, CH3Ala); 13C NMR, APT (150.917 MHz; CDCl3; Me4Si): δ (ppm) = 171.20 (COFn), 170.54 (COAc), 80.54 (CqBoc), 65.93, 65.53, 65.47, 65.15, 62.95 (CHFn), 49.51 (CHAla), 28.55 [(CH3)3Boc], 23.37 (CH3Ac), 18.43 (CH3Ala); ESI-MS (H2O:MeOH = 50:50): m/z = 452.2 [(M + Na)+].
Synthesis of Ac–Ala–NH–Fn–NH–Ala–Boc (3b)
Carbamate 7 (500 mg, 1.16 mmol) was suspended in CH2Cl2 (3 ml) and HCl gas was bubbled through the suspension for 30 min at 0 °C. The obtained hydrochloride salt was suspended in CH2Cl2 and treated with NEt3 to give an unstable free base which was coupled with Boc–Ala–OH (440 mg, 2.33 mmol) using the standard EDC/HOBt method [EDC (533.6 mg, 2.78 mmol); HOBt (375.3 mg, 2.78 mmol)]. The reaction mixture was stirred at room temperature until complete consumption of the ferrocene starting material, as monitored by TLC. The standard work-up (washing with a saturated aqueous solution of NaHCO3, 10% aqueous solution of citric acid and brine, drying over Na2SO4 and evaporation in vacuo) followed by TLC purification of the crude products (EtOAc; Rf = 0.51) gave orange crystals of the desired product (408 mg, 70%); mp 141–143 °C; IR (CH2Cl2): νmax/cm−1 = 3439 (w, NHfree), 3310, 3253 (m, NHassoc.), 1684, 1665 (s, COCONH), 1571, 1506 (amide II); 1H NMR (600.133 MHz; CDCl3; Me4Si): δ (ppm) = 9.09 (s, 1H, NHaFn), 9.06 (s, 1H, NHbFn), 6.85 (d, J = 5.2 Hz, 1H, NH NHaAla), 5.36 (s, 2H, H-10, H-7), 5.21 (d, J = 6.3 Hz, 1H, NHbAla), 4.58 (dq, J = 8.0 Hz, 7.1 Hz, 1H, CHaAla), 4.24 (dq, J = 8.0 Hz, 7.1 Hz, 1H, CHbAla), 4.14 (s, 1H, H-2), 4.09 (s, 1H, H-5), 3.96 (s, 4H, H-3, H-4, H-8, H-9), 2.11 (s, 3H, CH3Ac), 1.46 [s, 9H, (CH3)3Boc], 1.39 (d, J = 6.9 Hz, 3H, CHa3Ala), 1.36 (d, J = 6.7 Hz, 3H, CHb3Ala); 13C NMR, APT (150.917 MHz; CDCl3; Me4Si): δ (ppm) = 171.94 (COaFn), 171.67 (CObFn), 171.45 (COAc), 156.80 (COBoc), 96.16 (C-1), 95.69 (C-6), 80.81 (CqBoc), 65.86 (C-8), 65.78 (C-9), 64.90 (C-3), 64.82 (C-4), 62.83 (C-7), 62.75 (C-10), 61.65 (C-2), 61.25 (C-5), 51.14 (CHbAla), 50.31 (CHaAla), 28.54 [(CH3)3Boc], 23.08 (CH3Ac), 17.84 (CHb3Ala), 17.45 (CHa3Ala); MALDI-HRMS m/z = 500.1726 (calculated for C23H32N4O5Fe = 500.1717).
Synthesis of Ac–Ala–NH–Fn–NH–Ala–Ac (3c)
The transformation of orthogonally protected peptide 3b (400 mg, 0.8 mmol) to Ac-analogue 3c was carried out by action of acetyl chloride (315 μl, 4.8 mmol): the free base, obtained via hydrochloride salt provided by deprotection of 3b in the above described manner, was cooled at 0 °C and acetyl chloride was added dropwise. After stirring in an ice bath for 15 min, TLC indicated that no residual starting material remained and the reaction mixture was poured into water and extracted with CH2Cl2. The combined organic phases were washed with brine, dried over Na2SO4 and evaporated to dryness in vacuo. The resulting crude product was purified by TLC on silica gel (n-BuOH:CH3COOH:H2O = 60:25:15; Rf = 0.64) to give an orange solid of 3c (325 mg, 92%); mp 112–114 °C; IR (CH2Cl2): νmax/cm−1 = 3438 (w, NHfree), 3306, 3252 (m, NHassoc.), 1686, 1678, 1665, 1650 (s, COCONH), 1570 (amide II); 1H NMR (300 MHz; CDCl3; Me4Si): δ (ppm) = 8.98 (s, 2H, NHFn), 6.19 (d, J = 5.2 Hz, 2H, NHAla), 5.23 (s, 2H, H-10, H-7), 4.50 (m, 2H, CHAla), 4.11 (s, 2H, H-2, H-5), 3.96 (s, 4H, H-3, H-4, H-8, H-9), 2.09 (s, 6H, CH3Ac), 1.41 (d, J = 6.9 Hz, 6H, CH3Ala); 13C NMR, APT (75.432 MHz; CDCl3; Me4Si): δ (ppm) = 171.40 (COFn), 171.37 (COAc), 95.56 (C-1, C-6), 65.79, 64.83 (C-3, C-4, C-8, C-9) 63.08, 61.82 (C-2, C-5, C-7, C-10), 50.35 (CHAla), 23.23 (CH3Ac), 17.63 (CH3Ala); MALDI-HRMS m/z = 442.1291 (calculated for C20H26N4O4Fe = 442.1298).
X-ray determination of 3b
The orange prismatic crystal with dimensions of 0.79 × 0.36 × 0.14 mm3 was obtained at room temperature by partial evaporation from a chloroform solution. The intensities were collected at 295 K on an Oxford Diffraction Xcalibur 2 diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å). The CrysAlisPro14 program was used for data collection and processing. The intensities were corrected for absorption using the multi-scan absorption correction method.14 The structure was solved by direct methods using SIR-200415 and refined by full-matrix least-squares calculations based on F2 using SHELXL-201316 integrated in the WinGX17 program package. Hydrogen atoms attached to the nitrogen atoms (N1–N4) have been found in the difference Fourier maps and their isotropic thermal parameters have been refined freely. Geometric restraint on the N–H distance was applied in the refinement. All other hydrogen atoms were included at calculated positions as riding atoms, with SHELXL-201316 defaults. The PLATON18 program was used for structure analysis and molecular and crystal structure drawing preparation. CCDC 1060677 contains the supplementary crystallographic data for this paper.Crystal data for 3b: C24H33Cl3FeN4O5, Mr = 619.74, orthorhombic space group P212121 (no. 19); a = 8.9475(4), b = 16.3934(11), c = 20.8167(17) Å; V = 3053.4(4) Å3; Z = 4; dx = 1.348 g cm−3; μ = 0.794 mm−1; RInt = 0.0344; x = −0.010(12); S = 0.981; R/wR = 0.0630/0.1550 for 356 parameters and 4443 reflections with I ≥ 2σ(I), R/wR = 0.1007/0.1803 for all 6637 independent reflections measured in the range 4.37°–θ –27.00°.
Computational details
Geometries of the selected compounds were analysed using MacroModel v10.319–21 using several different search methods with the OPLS_2005 force field.22 A ferrocene unit was frozen during optimization. Restrictions were imposed on the pseudo-torsion angle during conformational search, thus simulating rotation of two Cp rings. Conformers were then optimized using Gaussian09 (Revision D.01)23 at the B3LYP-D3/LanL2DZ level of theory using Grimme's dispersion with the original D3 damping function.24–26 The most stable conformers were reoptimized in chloroform at the B3LYP-D3/6-311+G(d,p) level of theory, using IEF-PCM for describing implicit solvent effects.27,28 Iron was modelled using the ECP set LanL2DZ. Molecules were visualized using Chem3D 201229 and GaussView 530 programs. The AIM2000 program was used for topological analysis of selected compounds.31,32 Hirshfeld surface analysis33,34 was performed using CrystalExplorer.35Biological evaluation
:1); Gibco, Paisley, UK) and Hs 578 T cells were cultivated in ATCC-formulated Dulbecco's modified Eagle's medium (DMEM, ATCC, USA) with supplemental bovine insulin (0.01 mg mL−1) (Sigma-Aldrich, St Louis, MO). Heat inactivated fetal bovine serum (Gibco, Paisley, UK) was added to make the complete growth medium for both cell lines in a final concentration of 10%. Cells were routinely cultured in 80 cm2 cell flasks (Nunc, Roskilde, Denmark) at 37 °C and under a humidified atmosphere of 5% CO2 in air. After reaching 70–90% confluence the cells were disaggregated using trypsin/EDTA (0.25% trypsin, 1 mM EDTA·4Na), counted and placed at the necessary density prior to sub-culture or seeding in wells for experimental needs.
Results and discussion
Synthesis of bioorganometallics 3b and 3c
The key intermediate 7, containing NH groups attached to both Cp rings, was obtained from Ac–Ala–Fca–OMe (4).13 In order to prevent racemisation of its chiral center, the saponification of the ester group of compound 4 was performed with an equimolar amount of a base under restricted conditions (1 h/80 °C). The resulting compound 5 was transformed to unstable azide 6 that was in situ subjected to a Curtius rearrangement by heating in t-BuOH to give orthogonally protected compound 7. In order to avoid the undesired conversion of the intermediate isocyanate group to the corresponding urea, t-BuOH was dried before use and the rearrangement temperature was limited to 65 °C. Otherwise, the temperature increment led to the formation of sym-urea derivative. Since TLC monitoring did not show any difference between the isocyanate intermediate and the azide precursor 6, the completion of the reaction was confirmed by the complete absence of the azide and isocyanate bands (2130 cm−1 and 2270 cm−1) in the IR spectrum of the crude reaction mixture. Boc-deprotection of 7 was conducted in acidic milieu, leaving the Ac protective group at the upper Cp intact. The obtained Ac–Ala–NH–Fn–NH2·HCl was processed with an excess of NEt3 to liberate the N-terminus, followed by coupling with activated Boc–Ala–OH to give orthogonally protected Ac–Ala–NH–Fn–NH–Ala–Boc (3b). Upon (i) Boc-deprotection and (ii) Ac-protection in the presence of acetyl chloride,37 Fn–(NH–Ala–Ac)2 (3c) was obtained (Scheme 1).Conformational analysis of 3b and 3c in solution
We herein presented a facile and efficient strategy for the preparation of novel bioorganometallics 3 containing peptide sequences of different structures and chirality attached at N–H functionalized Cp rings has been applied for the synthesis of their lowest homologues 3b and 3c. Considering that both carbamate carbonyl groups of their Kraatz's analogue 3a11 have been engaged in strong IHBs forming 14-membered rings in solution as well as in the solid state, we wanted to explore if the replacement of one or both bulky Boc groups with a Ac function will influence the IHB pattern, because of the decreasing steric hindrance. For this purpose, the spectral data of bioorganometallics 3b and 3c are compared to those related to the model (3a,1110,381110b and 1238) and reference compounds (8 and 9)10b (Fig. 2). The model compounds were chosen due to their structural and hydrogen-bonding abilities similar to those of the analysed peptides, while the reference compounds are characterized by their inability to form IHBs. | ||
| Fig. 2 The reference (8 and 9) and model compounds (10–12, 3a). | ||
Our endeavor to clarify the conformational properties of 3b and 3c began with IR analysis (Table 1). The two clearly distinct NH absorptions are indicative of a two-state equilibrium between non-bonded (>3400 cm−1) and hydrogen-bonded conformations (<3400 cm−1). The hydrogen bonding behaviour of the NH groups of 3b dominates this region of the spectra and is evidently independent of concentration (Fig. 3) and clearly intramolecular in nature [the ratio of the associated and free NH bands (1.8:1) did not change during the course of the experiment]. A closer inspection of Fig. 3 revealed that intensity of free NH bands of 3c remained unchanged upon dilution from 5 × 10−2 M to 1.25 × 10−2 M as opposed to the corresponding associated NH bands which were reduced up to 11%. Nevertheless, the proportion of hydrogen-bonded and free NH bands of 3c persisted unchanged (1.74:1) during further dilution. These data proved the predominance of the intramolecular HBs, but also the existence of the intermolecularly bonded NH groups of 3c.
 | ||
| Fig. 3 The NH stretching vibrations in concentration-dependent IR spectra of 3b and 3c in CH2Cl2. | ||
| Compound | ν NH | ν CO | δ | ||||
|---|---|---|---|---|---|---|---|
| Free | Assoc. | Amide I | NHaFn | NHbFn | NHBoc | NHAc | |
| a IR and NMR spectra were recorded in CH2Cl2 (c = 5 × 10−2 M). b No solution data available. | |||||||
| 3a | 9.00 | 5.11 | |||||
| 3b | 3439 | 3310 | 1684 | 9.06 | 9.02 | 5.21 | 6.85 |
| 3253 | 1665 | ||||||
| 3c | 3438 | 3306 | 1686 | 9.04 | 6.57 | ||
| 3252 | 1678 | ||||||
| 1665 | |||||||
| 1650 | |||||||
| 8 | 3436 | 1723 | 5.55 | ||||
| 9 | 3436 | 1684 | 6.49 | ||||
| 10 | 3425 | 3336 | 1697 | 5.55 | 6.83 | ||
| 11 | 3431 | 3328 | 1711 | 7.41 | 5.80 | ||
| 1687 | |||||||
| 12 | 3428 | 3328 | 1710 | 7.88 | 8.14 | 5.52 | |
| 1679 | |||||||
Although our previous studies of ferrocene peptides revealed the considerable interference of the bulky Boc group with hydrogen bonding,13,39 the similar ratios of hydrogen-bonded and non-bonded NH peak intensities in IR spectra of 3b and 3c exclude the steric hindrance of the Boc group. Taking into account that (i) hydrogen-bond-accepting urethane40 and the acetamide group10d appear at lower wavenumbers and (ii) urethane and the acetamide carbonyl groups of 3b and 3c are shifted to the lower region in comparison with non-bonded reference compounds 8 and 9, their engagement in hydrogen bonding is suggested. Thereby, non-bonded urethane carbonyl groups of the model compounds 11 and 12 absorb at higher wavenumbers.
The next step in the evaluation of the conformational preferences of the bioconjugates 3b and 3c in solution is interpretation of their NMR spectra. Indeed, all the amide proton resonances were recognized by analysis of coupling patterns and two-dimensional NMR spectra. [Due to the presence of the multiple resonances in the NMR spectra (Fig. S25 and S27†), one can assume that peptide 3c tends to partially aggregate in concentrated solution (50 mM),41 as it was observed by IR spectroscopy. Upon dilution of the NMR sample, these resonances underwent complete coalescence, (Fig. S26, S28 and S29†).] The dependence of the amide chemical shifts on the accessibility of the proton to the hydrogen-bond accepting site is well documented.42 The downfield chemical shifts (δ ≳ 7 ppm) in non-polar CDCl3 are generally considered to be assigned to the hydrogen-bonded NH protons. Herein, the resonances of NHFn at ∼9 ppm match very closely with those observed for the intramolecularly hydrogen-bonded amide proton of the model peptide 3a. Conversely, NHBoc and NHAc displayed the lowering of the chemical shift values compared to the amide group attached at the ferrocene core suggesting a lowered potential to experience IHBs. Furthermore, the correlation of NHBoc and NHAc chemical shifts of the examined peptides 3b and 3c with those observed for the non-bonded carbamate and acetamide groups of 8–12 does not support the possible hydrogen bond donating engagement of alanine moieties (Table 1).
In order to gain more insight into the conformational behaviour of these bioorganometallics, the concentration- and temperature-dependent NMR measurements were carried out. Since no significant concentration-dependent changes in chemical shifts of NHFn were seen (Δδ = 0.06–0.13 ppm), their participation in intramolecular HB is further supported (Fig. 4, Fig. S17 and S29†). The successive dilution did not considerably affect the chemical shift of non-bonded NHBoc proton of 3b (Δδ = 0.13 ppm), while NHAc protons of 3b and 3c experienced more appreciable upfield shifts (Δδ ≳ 0.4 ppm), which might account for their involvement to a lesser extent in intermolecular HB.
 | ||
| Fig. 4 Concentration dependent NH chemical shifts of peptides 3b and 3c [1H-NMR measurements were performed for a series of 6.25, 12.5, 25 and 50 mM solutions]. | ||
Amide proton temperature coefficients (Δδ/ΔT) are useful for the prediction of hydrogen bonding ability.43 Since ferrocene peptides are subjected to rapid decomposition in DMSO, their temperature dependences, i.e. exposure or shielding of NH groups to solvent, were measured in CDCl3.13,39,44 Low Δδ/ΔT values (−2.4 ± 0.5 ppb per K) correspond to both exposed and shielded amide protons of short peptides and thus are not very informative. The larger temperature dependencies are interpreted as indication of initially shielded NH groups that became exposed to the solvent upon dissociation of the self-associated aggregates or unfolding of ordered conformations at increased temperatures.45 Therefore, the low Δδ/ΔT value of the non-bonded NHBoc proton is attributed to its exposure to the solvent, while unfolding as well as dissociation lead to increased temperature dependences of initially shielded NHFn and NHAc, respectively (Fig. 5, Fig. S18 and S30†).
 | ||
| Fig. 5 Temperature dependent NH chemical shifts of peptides 3b and 3c (c = 2.5 × 10−2 M) in the temperature range of 298–328 K. | ||
Hydrophobic and hydrogen bonding interactions are of fundamental importance for the folding and misfolding behaviour of proteins.46 We decided, therefore, to investigate the hydrogen-bonding features of novel bioorganometallics by DMSO titration.47 DMSO is well-known to display a strong tendency to accept amide NH protons exposed to the solvent thereby causing the downfield shifts. If the amide protons are inaccessible to DMSO due to their engagement in HBs, no significant changes in chemical shifts will be observed. The conservation of high chemical shift values of NHFn protons upon titration with DMSO (Δδ < 0.2 ppm) indicates their participation in a strong IHB. In contrast, carbamate and acetamide NH protons experienced a significant modulation of chemical shifts (Δδ > 1.7 ppm), confirming the proposed non-hydrogen-bonded state for NHBoc as well as involvement of NHAc in a weak HB (Fig. 6, Fig. S19 and S31†).
 | ||
| Fig. 6 Solvent dependence of NH chemical shifts of 3b and 3c at varying concentrations of d6-DMSO in CDCl3 (c = 2.5 × 10−2 M, 298 K) to probe exposed vs. hydrogen-bonded amides. | ||
All these considerations on hydrogen-bonding behaviour of our peptides contribute to defining their conformational space based on interchain NHFn⋯OCBoc/Ac IHBs. The NOE contact between NHFn linked at one Cp ring and the tBu group belonging to the chain attached to another Cp ring supports the proposed interchain intramolecular hydrogen bonding engagement with the carbamate carbonyl group (Fig. 7).
 | ||
| Fig. 7 The interchain NOE connectivity between NHFn and tBu groups of 3b is depicted with arrows. | ||
It was shown that hydrogen bonding between podand chiral peptide chains of the previously reported ferrocenes 1–3a provokes chirality-organized structures detected by CD spectroscopy. The observed strong Cotton effects (Mθ ∼ 5000 deg cm2 dmol−1 for peptides 1 and Mθ ∼ 10000 deg cm2 dmol−1 for peptides 2) in the region of ferrocene-based transitions around 470 nm were ascribed to the highly organized chiral surrounding around the ferrocene unit.48 In addition, the earlier described L-Ala containing bioorganometallics 1, 2 and 3a displayed the positive Cotton effect attributed to the right-handed helicity. Considering that the Cotton effect reflects an average of the entire molecular population, the domination of P-helicity of herein studied peptides 3b and 3c is strongly supported, owing to their pronounced CD activity. DMSO, a potent hydrogen bond acceptor, is capable of disrupting the weak hydrogen bonds and thereby jeopardizes the conformational stability.10a,d,e The CD-data obtained upon treatment of peptides 3b and 3c with 20% of DMSO determined the conservation of ∼70% of the DMSO-free CD activity, certainly due to the existence of strong IHBs (Fig. 8).
 | ||
Fig. 8 The Cotton effects in chirality-organized ferrocene peptides [CH2Cl2 (c = 5 × 10−3 M), ( 3b, 3b,  3c)] and CH2Cl2 (c = 5 × 10−3 M) containing 20% of DMSO ( 3c)] and CH2Cl2 (c = 5 × 10−3 M) containing 20% of DMSO ( 3b, 3b,  3c)]. 3c)]. | ||
The peptides 3a–3c containing ferrocene-1,1′-diamine (Fcda) and Ala were shown to adopt a conformation defined by the 14-membered IHB-ring, resembling the hydrogen bonding in antiparallel β-sheet peptides. Since side-chain groups affect the backbone folding,49 our future work on ferrocenes 3 will include the replacement of one Ala unit with different amino acids to obtain a small library of Fcda-derived peptides. These simple model systems will enable us to determine the amino acids predisposed to get involved in the β-turn-mediated folding of peptides 3.50 Besides this, we will elongate the N-termini of the lowest homologues 3a–3c in order to establish models for extended β-sheet-like structures.
X-ray crystal structure analysis of 3b
An orange crystal of the ferrocene derivative 3b was grown at room temperature from chloroform solution and it crystallised in the chiral orthorhombic space group P212121 as a chloroform solvate. The absolute configuration of the stereogenic carbon atoms C12 and C20 of the L-Ala substituents is, as expected, S (Fig. 9).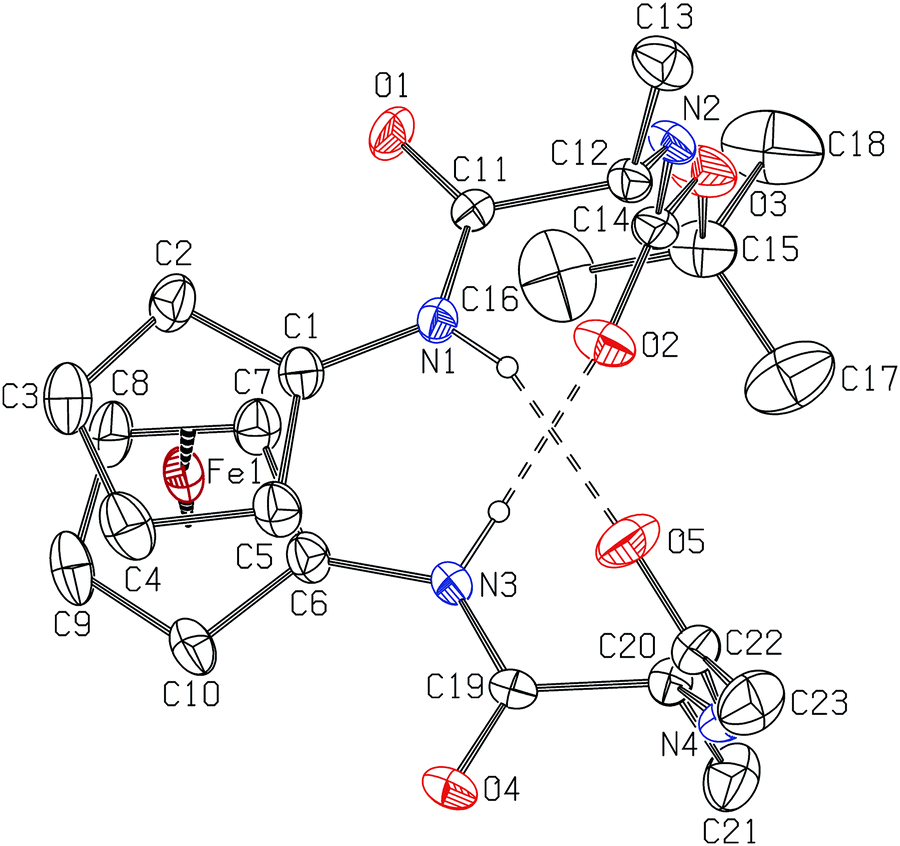 | ||
| Fig. 9 A molecular structure of 3b, with the atom numbering scheme, showing N–H⋯O intramolecular hydrogen bonds (indicated by dashed lines). Displacement ellipsoids for nonhydrogen atoms are drawn at the 20% probability level. Chloroform molecule and hydrogen atoms not included in intramolecular hydrogen bonds have been omitted for clarity. | ||
The bond lengths in the two peptide strands attached to the cyclopentadienyl (Cp) rings, as well as within the ferrocene core, present no unexpected features and are in good agreement with equivalent ones in 3a.11
The pseudo C1–Cg1–Cg2–C6 torsion angle of the ferrocene (Cg1 is centroid of the ring C1–C5 and Cg2 is centroid of the ring C6–C10) amounts to ca. +41°, i.e. a positive helical chirality (P) is observed. The value of this torsion angle also shows that a conformation of ferrocene is 1,2′, and that cyclopentadienyl (Cp) rings adopt a staggered conformation. The Cp rings are nearly coplanar to each other, with a tilt angle of 4.5(5)°.
The direction of the 1- and 1′-substituents of 3b is a consequence of intramolecular (IHB) N–H⋯O hydrogen bonds that link the juxtaposed strands. These IHBs are formed between amide N–H groups and carbonyl oxygen atoms (Fig. 9 and Table 2). The N1⋯O5 and N3⋯O2 hydrogen bonds form ten-membered rings of the R11(10) type,51 thus inducing a β-turn as in our previous published structure.52 In addition, the combination of these two IHBs forms a new ring of the R22(14) type. Because of the above mentioned IHBs, the non-hydrogen atoms of amide groups attached to Cp rings, N1/C11/O1, i.e. N3/C19/O4, are twisted for 17.7(8) and 11.5(9)° with respect to the mean planes of C1–C5 and C6–C10 ring atoms, respectively. In 3b, three intramolecular C–H⋯O hydrogen contacts are also observed (C2⋯O1, C16⋯O2 and C17⋯O2).
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|---|---|---|---|
| Symmetry codes:a −1/2 + x, 3/2 − y, 1 − z.b −1/2 + x, 1/2 − y, 1 − z.c Cg1 is centroid of the C6–C10 ring. | ||||
| N1–H⋯O5 | 0.86(5) | 2.06(5) | 2.859(7) | 156(4) |
| N3–H⋯O2 | 0.86(5) | 1.95(5) | 2.802(7) | 175(6) |
| C2–H⋯O1 | 0.93 | 2.58 | 2.976(8) | 106 |
| C16–H⋯O2 | 0.96 | 2.58 | 3.040(14) | 110 |
| C17–H⋯O2 | 0.96 | 2.33 | 2.975(13) | 124 |
| N2–H⋯O1 | 0.86(5) | 2.00(5) | 2.851(7) | 174(6)a |
| N4–H⋯O4 | 0.86(4) | 2.01(5) | 2.851(7) | 166(7)b |
| C24–H⋯Cg1c | 0.98 | 2.78 | 3.726(11) | 162 |
In the crystal, the molecules of 3b are self-assembled by two N–H⋯O hydrogen bonds, N2⋯O1 and N4⋯O4, both of them linking molecules into infinite zig-zag C(5) chains51 parallel to the a axis. The combination of these two chains generates sheets and leads to a (4,4) net53 (Fig. 10a). Chloroform molecules fill the gaps between the hydrogen-bonded molecules of 3b (Fig. 10b), and are linked to the ferrocene molecules by one C–H⋯π interaction (C24⋯Cg1, Table 2). A crystal packing diagram along the a axis reveals that hydrogen atoms of chloroform molecules point to the Cp rings of 3b, thus also participating in the formation of a two-dimensional network (Fig. 10b).
 | ||
| Fig. 10 A crystal packing diagram of 3b, viewed along the c axis (a) and the a axis (b), showing intramolecular and intermolecular N–H⋯O hydrogen bonds. Hydrogen bonds are indicated by dashed lines. | ||
Computational study of 3a, 3b and 3c
Our previously published papers about the conformational analysis of ferrocene containing peptides have resulted in close agreement between the experimental and DFT calculated data.13,48,54 Herein, we have decided to use the same approach to shed light on hydrogen bond patterns observed in solution as well as in the solid state of peptides 3a, 3b and 3c. The combination of a molecular and quantum mechanic approach has been used. The most stable conformers have been calculated with B3LYP-D3 functional and 6-311+G(d,p) basis sets. Iron was described with the LanL2DZ basis set. Chloroform was modelled using a polarizable continuum model (IEF-PCM).The generally accepted nomenclature of these and similar ferrocene containing compounds is displayed in Fig. 11. A pseudo-torsion angle ω is used to describe the rotation of two cyclopentadienyl rings. Depending on a relative value of the angle, the stereochemical descriptor 1,n′ is usually assigned with P- and M-label. Modified E/Z marks are used to unambiguously determine relative orientation of Cp–amide bonds. All of the marked hydrogen bonds in Fig. 12 were confirmed by Bader's AIM theory. Values of topological parameters of the bond critical points between the hydrogen-bond acceptor and the hydrogen atom [electron density ρ(r), Laplacian of the electron density ∇2ρ(r) and the energy density H(r)] were determined and compared with the Koch and Popelier criteria used to characterize hydrogen bonds.55,56 The results are presented in Table 3 and geometries of the most stable conformers are displayed in Fig. 12.
 | ||
| Fig. 11 Nomenclature of 1,n′-disubstituted ferrocenes with chiral substituents. (a) M/P isomers defined by a pseudo-torsion angle ω between two substituents; (b) E/Z isomers describing an orientation of Cp–amide bonds. | ||
 | ||
| Fig. 12 The most stable conformers of bioorganometallics 3a–c with selected hydrogen bonds. | ||
| Conformer | E rel | Stereochemical descriptors | ω pseudo-torsion angle | IHB pattern | NHFn⋯OCBoc/Ac (interchain) | NHFn⋯OCBoc/Ac (intrachain) | NHAla⋯OCAla (interchain) |
|---|---|---|---|---|---|---|---|
| 3a-1 | 0.00 | (P)-1,1′; E,E | 28.8 | A | 2.86 (2.86) | — | — |
| 3a-2 | 9.23 | (M)-1,5′; Z,Z | −59.0 | B | — | 3.03 (3.02) | 2.91 (2.91) |
| 3b-1 | 0.00 | (P)-1,1′; E,E | 28.3 | A | 2.86 (2.88) | — | — |
| 3b-1 | 7.34 | (M)-1,5′; Z,Z | −59.5 | B | — | 3.03 (2.99) | 2.92 (2.89) |
| 3c-1 | 0.00 | (P)-1,1′; E,E | 26.5 | A | 2.88 (2.88) | — | — |
| 3c-2 | 5.50 | (M)-1,5′; Z,Z | −60.3 | B | — | 2.98 (2.98) | 2.90 (2.90) |
In this study both newly synthesized derivatives 3b and 3c as well as Kraatz's analogue 3a11 were subjected to conformational analysis. The results were very similar for all the three ferrocene conjugates. The engagement of NHFn in hydrogen bonding almost exclusively predominated in all of the conformers. In the most stable conformers (3a-1, 3b-1 and 3c-1) the interchain NHFn⋯OCBoc/Ac hydrogen bonds between two opposite substituents were the only ones responsible for a folding of peptide chains. A 10-membered ring, also known as β-turn (IHB pattern A, Fig. 13) was formed. Consequently, methyl groups of each alanine pointed away from the place of interaction between substituents. Almost the same observation could be applied for the Boc and Ac groups, which also pointed away from the ferrocene. The most stable conformers of symmetrically substituted derivatives (3a-1 and 3c-1) adopted the C2 point group.
 | ||
| Fig. 13 The hydrogen bond patterns (A and B) observed in the most stable conformers of 3a, 3b and 3c in chloroform (PCM); R = Me, OtBu. Numerations of seven- and ten-membered rings are displayed. | ||
In comparison with these conformers, in which another potential hydrogen bond donor group, i.e. NHAla, was not engaged in the formation of any hydrogen bond, the second set of conformers (3a-2, 3b-2 and 3c-2) utilized all of the available NH groups in the formation of four hydrogen bonds. The observed IHB pattern B constituted two 7-membered rings (γ-turns) connected by intrachain NHFn⋯OCBoc/Ac hydrogen bonds. Additionally, two opposite substituents were connected by two interchain NHAla⋯OCAla hydrogen bonds engaged in the formation of 10-membered rings (β-turns). Again, conformers 3a-2 and 3c-2 with the same substituents adopt the C2 point group. The population of these conformers is more significant within 3b and 3c derivatives with sterically less demanding Ac group.
These results were compared with the experimental data. The calculated structure of the most stable conformer of 3b also adopted (P) helical chirality with a pseudo-torsion angle of ca. 28°, in comparison with 41° in an X-ray determined crystal structure. The CD experiments in solution also confirmed the domination of (P) helical forms. According to NMR experiments, the full engagement of NHFn groups in intramolecular hydrogen bonds, while NHBoc and NHAc groups are more prone to accomplish intermolecular hydrogen bonds, was also proven by the computational study.
Generally, hydrogen bonds can be related to a greater stabilization of conformers depending on specific IHB patterns. In our case, the NH groups of an alanine residue remained non-hydrogen bonded in the most populated conformers. This observation is in agreement with the X-ray determined structure of 3b. Furthermore, these groups are able to form additional intermolecular hydrogen bonds as it is observed in the crystal structure. Thus, we decided to investigate the effects of this interaction between two molecules of unsymmetrically substituted 3b as the most appropriate model for further analysis.
To gain better insight into close contacts we decided to employ Hirshfeld surface analysis. It summarizes the way in which one molecule interacts with neighbouring molecules. Fig. 14a shows the Hirshfeld surface mapped with dnorm values showing distances shorter than the sum of the van der Waals radii (red spots). Obviously, both substituents, the first protected with the Ac group (Fig. 14b), and the second protected with the Boc group (Fig. 14c) interact in the same way as neighbouring molecules by forming NH⋯O hydrogen bonds. To quantify these interactions we decided to calculate interaction energies between pairs of molecules cut out from the crystal structure and optimized in vacuo, therefore without PCM formalism used in conformational analysis.
 | ||
| Fig. 14 (a) Hirshfeld surface of 3b mapped with dnorm over the range −0.59 to 1.78 Å, areas with contact distances shorter than the sum of the van der Waals radii are coloured red (left – look from the side of the Ac protected substituent, right – look from the side of the Boc protected substituent); (b) optimized geometry of 3b pair of molecules connected through NH⋯O hydrogen bonds between Ac protected substituents (coloured yellow).; (c) optimized geometry of 3b pair of molecules connected through NH⋯O hydrogen bonds between Ac protected substituents (coloured red). | ||
First, we have compared the single molecule geometries of 3b (only heavy atoms) using experimental and both calculated structures (one optimized in CHCl3 and one in vacuo). After the superposition the calculated root-mean-square deviations (RMSD) were 0.35 Å and 0.33 Å, respectively. Obviously, there was no significant distortion of geometry upon optimization in vacuo and in CHCl3 modelled as polarizable continuum.
Second, we have compared each pair of molecules connected by intermolecular hydrogen bonds between substituents. As mentioned above, the unsymmetrically substituted 3b has two distinctive molecular pairs depending on whether the hydrogen bond occurred between Ac or Boc protected substituents of each molecule (Fig. 14b and c). After optimization and comparison with the crystal structure, the obtained RMSD values were 1.29 Å and 0.99 Å, respectively. The calculated interaction energies corrected by basis set superposition error (BSSE) suggest a favourable interaction between two molecules of each chosen molecular pair. The values were −58.9 kJ mol−1 and −62.1 kJ mol−1 (corrected by basis set superposition error, BSSE), respectively. Without BSSE correction the obtained values were −66.3 kJ mol−1 and −72.1 kJ mol−1. For comparison, the calculated interaction energy for two symmetrically substituted 3a molecules was −62.9 kJ mol−1 (with BSSE) and −73.9 kJ mol−1 (without BSSE). This could also be a good indication of similar packing of molecules in symmetrically substituted 3c for which, a single crystal, unfortunately, was not obtained.
Third, reorganization terms were also calculated as differences between the energies of each molecule based on the geometry adopted in the optimized dimer and the geometry of the most stable conformer. The corresponding values were less than 2 kJ mol−1, thus requiring very small reorganization of a single molecule geometry during crystal packing.
Fourth, the strength of interaction between a pair of molecules in the crystal structure of 3b was compared with the similar interaction between two peptide strands in one molecule. The interaction energy between strands (engaging two hydrogen bonds) was estimated as the energy difference of two optimized geometries, the “open” conformer obtained from 3b-1 by adjusting the pseudo-torsion angle to approximately 180°, in which two substituents were not able to interact, and the most stable conformer 3b-1, both optimized in vacuo. Considering all the approximations made in this approach, the calculated interaction energy amounts to −86 kJ mol−1 (not corrected by BSSE), i.e. for about 15 percent more than the interaction energy calculated between pair of 3b molecules.
By taking into account all of these observations, one might conclude that derivatives 3 are capable of preserving the same IHB pattern consisting of two 10-membered β-turns in solution as well as in the solid state, thus making them a promising, yet simple scaffolds capable of mimicking antiparallel β-sheet peptides.
Biological evaluation of bioconjugates 2a, 3b and 3c
The ferrocene conjugates 3b and 3c synthesised in this work and previously reported Ac–Ala–Fca–Ala–OMe (2a)10e were screened in vitro for their potential anticancer activity in Hep G2 human liver carcinoma cells and Hs 578 T human breast cancer cells. [Since the comparable conformational pattern based on two simultaneous IHBs was seen in solution and in the solid state of analogue 2a, we decided to test its biological activity as well.]The MTT assay, as an example of a widely applicable colorimetric endpoint assay, enables the indirect measurement of cytotoxicity as it involves the evaluation of mitochondrial dehydrogenase activity within the treatment period. In this method, the reduction of water-soluble tetrazolium salt MTT by metabolically active eukaryotic cells leads to precipitation of the colored formazans. It is assumed that dye reduction will be proportional to the number of viable cells in the exponential growth phase. Herein, the cells were treated with ferrocene and ferrocene conjugates 2a, 3b and 3c at a range of concentrations 50–500 μM and cell viability was determined after 72 h. The summarized results of cytotoxicity evaluation in Hep G2 and Hs 578 T cancer cell lines with the MTT bioassay are presented in Fig. 15. The IC50 values, presented in Table 4, were derived from the equations of related polynomial trend lines for each ferrocene conjugate.
 | ||
| Fig. 15 In vitro anti-proliferative effect of ferrocene and ferrocene conjugates 2a, 3b and 3c against Hep G2 human liver carcinoma cells (A) and Hs 578 T human breast cancer cells (B) obtained with the MTT assay after 72 h exposure. Data are presented as percentage of control: mean ± SEM of 3 experiments with 4 measurements within each experiment for each concentration. Statistical significance vs. control: ap < 0.001; bp < 0.005; cp < 0.01; dp < 0.025; ep < 0.05. | ||
| Compound | IC50 (μM) | |
|---|---|---|
| Hs 578 T | Hep G2 | |
| [n.d.] – non-determined IC value for the corresponding incubation period, out of applied concentration range. | ||
| Ferrocene | [n.d.] | [n.d.] |
| 3b | 280.77 | 259.33 |
| 3c | [n.d.] | [n.d.] |
| 2a | 116.49 | 89.87 |
Most of the metallodrugs currently tested and used in cancer treatment are based on platinum, in spite of negative medical and physical side-effects. For cisplatin, these include poor aqueous solubility, a high excretion rate from the body, loss of appetite (anorexia), development of drug resistance after continued drug dosage, high toxicity especially to the kidneys and bone marrow, and the most inconvenient, inability to distinguish between healthy and carcinoma cells.57 Therefore, many efforts are focused on the investigation of the novel metal-based therapeutics, i.e. ferrocene conjugates with similar antineoplastic activity and fewer side effects as an alternative for the platinum complex.
Ferrocene–acridine conjugates are pronounced among numerous ferrocene derivatives tested for antiproliferative purposes due to their highly cytotoxic activity.58 The most significant anticancer applications of ferrocene derivatives is referred to hydroxyferrocifens, obtained by the replacement of one phenyl ring of the active metabolite tamoxifen with a ferrocene moiety.59
The presented data (Fig. 15, Table 4) reveal a cytotoxic potential of ferrocene conjugates 2a, 3b and 3c against both cell lines. Ferrocene by itself had no significant effect on cell proliferation in concentrations up to 500 μM. However, a statistically significant reduction of the number of viable cells after treatment with 500 μM ferrocene (p < 0.001 for Hep G2 cells, p < 0.05 for Hs 578 T) was observed. Ferrocene conjugates 2a, 3b and 3c in concentrations of 150–500 μM significantly decreased cell proliferation (p < 0.001–p < 0.025) (Fig. 15) and the highest cytotoxicity is obtained by compound 2a in Hep G2 cells as well as in Hs 578 T cells with a slightly higher IC50 value (Table 4). For compound 3c, IC50 data were out of applied concentration range indicating low activity (Table 4). Liver carcinoma cells Hep G2 were generally more sensitive to the tested compounds, but the trend of cytotoxicity was the same in the both cell lines – from the weakest effect provoked by compound 3c to the most pronounced cell viability inhibition with compound 2a.
The obtained IC50 values for herein studied ferrocene-containing peptides 2a, 3b and 3c are still above IC50 for doxorubicin and cisplatin (literature data: 0.1–15 μM; e.g. for cisplatin in Hep G2 cells 4.7 ± 0.4 μM) determined in different human cancer cell lines.39,60,61 Nevertheless, compound 2a compared to others possesses the most prominent cytotoxic activity against human breast and liver cancer cells. Hence, the conformational space of the tested peptides, based on two simultaneous interchain hydrogen bonds [NHFn⋯OCBoc/Ac (3b, 3c) as well as NHFn⋯OCCOOMe and NHAla⋯OCAc (2a)], is not of decisive influence on their biological activity. Our previous work on ferrocene peptides revealed the contribution of lipophilicity to biological activity.39 Thus, the improved bioactivity of the bioorganometallics 2a and 3b is likely to be due to their increased lipophilicity (2a, Rf = 0.24; 3b, Rf = 0.51) in comparison with the more polar peptide 3c (Rf = 0.12). Therefore, the synthesis of similar compounds with profound biological activity is planned in our future work.
Conclusions
The employed synthetic route paves the way to bioorganometallics 3 containing ferrocene-1,1′-diamine substituted with peptide sequences of different structures and chirality that are expected to form intra- and intermolecular hydrogen bonds. A synthesis of orthogonally protected homochiral product Ac–Ala–NH–Fn–NH–Ala–Boc (3b) is reported for the first time. The detailed conformational analysis performed on novel compounds Ac–Ala–NH–Fn–NH–Ala–Boc (3b) and Fn–(NH–Ala–Ac)2 (3c) shows a great preference for the formation of 14-membered rings (also labelled as two simultaneous 10-membered β-turns) between two substituents, regardless of Boc and/or Ac protection groups. The same pattern persists in solution, as well as in the solid state where it is accompanied by favorable intermolecular hydrogen bonds in the formation of infinite zig-zag chains.Derivatives Ac–Ala–NH–Fn–NH–Ala–Boc (3b), Fn–(NH–Ala–Ac)2 (3c) and Ac–Ala–Fca–Ala–OMe (2a) decreased cell proliferation in Hep G2 and Hs 578 T cancer cell lines showing a cytotoxic ability in concentrations of 150–500 μM.
Whether these observations can be generalized to include other ferrocene-1,1′-diamine based derivatives remain to be seen. However, these results encourage us to extend our research on the synthesis of more complex systems. Together with the combination of spectroscopy, crystallography and computational analysis extended with biological evaluation tests it clearly establishes a protocol for extracting valuable information about systems capable of mimicking antiparallel β-sheet peptides.
With regard to the previously reported scaffolds Fcd and Fca, the herein employed ferrocene-1,1′-diamine (Fcda), as a constituent of yet unexplored group of peptidomimetics 3, shows a high potential to be considered as a stable and synthetically easily modifiable scaffold capable of preserving β-turns in solution as well as in the solid state, thus replicating the hydrogen bonding pattern of peptide β-sheets.
Acknowledgements
This research was supported by the Ministry of Science, Education and Sports of the Republic of Croatia (Grant Numbers 058-1191344-3122, 119-1193079-3069 and 0582184-2232), Croatian Science Foundation under the project 7444 and University of Zagreb (PP1.30). Computational resources were provided by the Croatian National Grid Infrastructure (http://www.cro-ngi.hr) at Zagreb University Computing Centre (SRCE).Notes and references
- G. D. Rose, L. M. Gierasch and J. A. Smith, Adv. Protein. Chem., 1985, 37, 1–109 CrossRef CAS PubMed.
- A. M. C. Marcelino and L. M. Gierasch, Biopolymers, 2008, 89, 380–391 CrossRef CAS PubMed.
- B. Eckhardt, W. Grosse, L.-O. Essen and A. Geyer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18336–18341 CrossRef CAS PubMed.
- (a) B. Odaert, F. Jean, C. Boutillon, E. Buisine, O. Melnyk, A. Tartar and G. Lippens, Protein Sci., 1999, 8, 2773–2783 CrossRef CAS PubMed; (b) A. A. Fuller, D. Du, F. Liu, J. E. Davoren, G. Bhabha, G. Kroon, D. A. Case, H. J. Dyson, E. T. Powers, P. Wipf, M. Gruebele and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11067–11072 CrossRef CAS PubMed.
- D. J. Selkoe, Nature, 2003, 426, 900–904 CrossRef CAS PubMed.
- (a) P. Chitnumsub, W. R. Fiori, H. A. Lashuel, H. Diaz and J. W. Kelly, Bioorg. Med. Chem., 1999, 7, 39–59 CrossRef CAS PubMed; (b) J. S. Nowick, E. M. Smith, J. W. Ziller and A. J. Shaka, Tetrahedron, 2002, 58, 727–739 CrossRef CAS; (c) O. Khakshoor and J. S. Nowick, Curr. Opin. Chem. Biol., 2008, 12, 722–729 CrossRef CAS PubMed; (d) J. S. Nowick, Acc. Chem. Res., 2008, 41, 1319–1330 CrossRef CAS PubMed; (e) P.-N. Cheng, J. D. Pham and J. S. Nowick, J. Am. Chem. Soc., 2013, 135, 5477–5492 CrossRef CAS PubMed.
- R. V. Nair, S. B. Baravkar, T. S. Ingole and G. J. Sanjayan, Chem. Commun., 2014, 50, 13874–13884 RSC.
- (a) S. Chowdhury, G. Schatte and H.-B. Kraatz, Angew. Chem., Int. Ed., 2008, 47, 7056–7059 CrossRef CAS PubMed; (b) A. Lataifeh, S. Beheshti and H.-B. Kraatz, Eur. J. Inorg. Chem., 2009, 3205–3218 CrossRef CAS; (c) T. Moriuchi and T. Hirao, J. Inclusion Phenom. Macrocyclic Chem., 2012, 74, 23–40 CrossRef CAS.
- (a) A. Nomoto, T. Moriuchi, S. Yamazaki, A. Ogawa and T. Hirao, Chem. Commun., 1998, 1963–1964 RSC; (b) T. Moriuchi, A. Nomoto, K. Yoshida, A. Ogawa and T. Hirao, J. Am. Chem. Soc., 2001, 123, 68–75 CrossRef CAS PubMed; (c) T. Moriuchi, T. Nagai and T. Hirao, Org. Lett., 2005, 7, 5265–5268 CrossRef CAS PubMed; (d) T. Moriuchi, T. Nagai and T. Hirao, Org. Lett., 2006, 8, 31–34 CrossRef CAS PubMed; (e) T. Moriuchi, A. Nomoto, K. Yoshida and T. Hirao, J. Organomet. Chem., 1999, 589, 50–58 CrossRef CAS; (f) T. Moriuchi, A. Nomoto, K. Yoshida and T. Hirao, Organometallics, 2001, 20, 1008–1013 CrossRef CAS; (g) T. Moriuchi and T. Hirao, Acc. Chem. Res., 2010, 43, 1040–1051 CrossRef CAS PubMed.
- (a) L. Barišić, M. Dropučić, V. Rapić, H. Pritzkow, S. I. Kirin and N. Metzler-Nolte, Chem. Commun., 2004, 2004–2005 RSC; (b) L. Barišić, M. Čakić, K. A. Mahmoud, Y.-n. Liu, H.-B. Kraatz, H. Pritzkow, S. I. Kirin, N. Metzler-Nolte and V. Rapić, Chem. – Eur. J., 2006, 12, 4965–4980 CrossRef PubMed; (c) L. Barišić, V. Rapić and N. Metzler-Nolte, Eur. J. Inorg. Chem., 2006, 4019–4021 CrossRef; (d) J. Lapić, D. Siebler, K. Heinze and V. Rapić, Eur. J. Inorg. Chem., 2007, 14, 2014–2024 CrossRef; (e) M. Čakić Semenčić, D. Siebler, K. Heinze and V. Rapić, Organometallics, 2009, 28, 2028–2037 CrossRef; (f) M. Čakić Semenčić, K. Heinze, C. Förster and V. Rapić, Eur. J. Inorg. Chem., 2010, 1089–1097 CrossRef.
- S. Chowdhury, K. A. Mahmoud, G. Schatte and H.-B. Kraatz, Org. Biomol. Chem., 2005, 3, 3018–3023 Search PubMed.
- M. G. Bomar, B. Song, P. Kibler, K. Kodukula and A. K. Galande, Org. Lett., 2011, 13, 5878–5881 CrossRef CAS PubMed.
- L. Barišić, M. Kovačević, M. Mamić, I. Kodrin, Z. Mihalić and V. Rapić, Eur. J. Inorg. Chem., 2012, 11, 1810–1822 CrossRef.
- Oxford Diffraction, Xcalibur CCD System. CrysAlisPro, Oxford Diffraction Ltd, Abingdon, England, 2013 Search PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- Maestro, version 9.7, Schrödinger, LLC, New York, NY, 2014 Search PubMed.
- MacroModel, version 10.3, Schrödinger, LLC, New York, NY, 2014 Search PubMed.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Henrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440–467 CrossRef CAS.
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1998, 37, 785–789 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- E. Cances and B. Mennucci, J. Math. Chem., 1998, 23, 309–326 CrossRef CAS.
- ChemBio3D Ultra 2012 v13, PerkinElmer Inc Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView 5.0, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- F. Biegler-König, J. Schömbohm and D. J. Bayles, J. Comput. Chem., 2001, 22, 545–559 CrossRef.
- F. Biegler-König and J. Schönbohm, J. Comput. Chem., 2002, 23, 1489–1494 CrossRef PubMed.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, Crystal Explorer (Version 3.1), University of Western Australia, 2012 Search PubMed.
- R. I. Freshney, Culture of Animal Cells – a Manual of Basic Technique, John Wiley & Sons, Inc., Hoboken, New Jersey, 5th edn, 2005 Search PubMed.
- M. Kovačević, V. Rapić, I. Lukač, K. Molčanov, I. Kodrin and L. Barišić, J. Mol. Struct., 2013, 1048, 349–356 CrossRef.
- S. Djaković, D. Siebler, M. Čakić Semenčić, K. Heinze and V. Rapić, Organometallics, 2008, 27, 1447–1453 CrossRef.
- M. Kovačević, K. Molčanov, K. Radošević, V. Gaurina Srček, S. Roca, A. Čače and L. Barišić, Molecules, 2014, 19, 12852–12880 CrossRef PubMed.
- (a) S. Ganesh and R. Jayakumar, Pept. Res., 2003, 59, 249–256 CrossRef; (b) V. S. Ananthanarayanan and T. S. Cameron, Int. J. Pept. Protein Res., 1988, 31, 399–411 CrossRef CAS PubMed; (c) L. Ning, W. De-Ning and Y. Sheng-Kang, Polymer, 1996, 37, 3045–3047 CrossRef.
- D. Lehnherr and R. R. Tykwinski, Org. Lett., 2007, 9, 4583–4586 CrossRef CAS PubMed.
- (a) S. H. Gellman, G. P. Dado, G.-B. Liang and B. R. Adams, J. Am. Chem. Soc., 1991, 113, 1164–1173 CrossRef CAS; (b) A. Pardi, G. Wagner and K. Wuthrich, Eur. J. Biochem., 1983, 137, 445–454 CrossRef CAS PubMed.
- (a) M. Llinás and M. P. Klein, J. Am. Chem. Soc., 1975, 97, 4731–4737 CrossRef; (b) H. Kessler, Angew. Chem., 1982, 94, 509–520 ( Angew. Chem., Int. Ed. Engl. , 1982 , 21 , 512–523 ) CrossRef CAS; (c) M. Iqbal and P. Balaram, Biopolymers, 1982, 21, 1427–1433 CrossRef CAS; (d) E. K. S. Vijayakumar and P. Balaram, Biopolymers, 1983, 22, 2133–2140 CrossRef CAS PubMed; (e) N. H. Andersen, J. W. Neidigh, S. M. Harris, G. M. Lee, Z. Liu and H. Tong, J. Am. Chem. Soc., 1997, 119, 8547–8561 CrossRef CAS; (f) N. J. Baxter and M. P. Williamson, J. Biomol. NMR, 1997, 9, 359–369 CrossRef CAS PubMed; (g) H.-J. Lee, H.-M. Park and K. B. Lee, Biophys. Chem., 2007, 125, 117–126 CrossRef CAS PubMed; (h) T. Cierpicki, I. Zhukov, R. A. Byrd and J. Otlewski, J. Magn. Reson., 2002, 157, 178–180 CrossRef CAS PubMed.
- (a) F. E. Appoh, T. C. Sutherland and H.-B. Kraatz, J. Organomet. Chem., 2004, 689, 4669–4677 CrossRef CAS; (b) J. Lapić, G. Pavlović, D. Siebler, K. Heinze and V. Rapić, Organometallics, 2008, 27, 726–735 CrossRef.
- E. S. Stevens, N. Sugawara, G. M. Bonora and C. Toniolo, J. Am. Chem. Soc., 1980, 102, 7048–7050 CrossRef CAS.
- (a) K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS PubMed; (b) A. W. Fitzpatrick, T. P. J. Knowles, C. A. Waudby, M. Vendruscolo and C. M. Dobson, PloS Comput. Biol., 2011, 7, e1002169 CrossRef CAS PubMed.
- (a) T. P. Curran, K. A. Marques and M. V. Silva, Org. Biomol. Chem., 2005, 3, 4134–4138 RSC; (b) S. Hanessian, G. Papeo, K. Fettis, E. Therrien and M. T. P. Viet, J. Org. Chem., 2004, 69, 4891–4899 CrossRef CAS PubMed; (c) S. K. Maji, D. Haldar, D. Bhattacharyya and A. Bannerjee, J. Mol. Struct., 2003, 646, 111–123 CrossRef CAS; (d) S. Vijayalakshmi, R. B. Rao, I. L. Karle and P. Balaram, Biopolymers, 2000, 53, 84–98 CrossRef CAS PubMed; (e) R. M. Jain, K. R. Rajashankar, S. Ramakumar and V. S. Chauhan, J. Am. Chem. Soc., 1997, 119, 3205–3211 CrossRef CAS; (f) R. S. Herrick, R. M. Jarret, T. P. Curran, D. R. Dragoli, M. B. Flaherty, S. E. Lindyberg, R. A. Slate and L. C. Thornton, Tetrahedron Lett., 1996, 37, 5289–5292 CrossRef CAS; (g) A. Mollica, M. Paglialunga Paradisi, D. Torino, S. Spisani and G. Lucente, Amino Acids, 2006, 30, 453–459 CrossRef CAS PubMed; (h) F. Sladojevich, A. Guarna and A. Trabocchi, Org. Biomol. Chem., 2010, 7, 916–924 RSC.
- V. Kovač, M. Čakić Semenčić, I. Kodrin, S. Roca and V. Rapić, Tetrahedron, 2013, 69, 10497–10506 CrossRef.
- (a) D. B. Dahl, Z. Bohannan, Q. Mo, M. Vannucci and J. Tsai, J. Mol. Biol., 2008, 378, 749–758 CrossRef CAS PubMed; (b) A. Q. Zhou, D. Caballero, C. S. O'Hern and L. Regan, Biophys. J., 2013, 105, 2403–2411 CrossRef CAS PubMed.
- J. S. Nowick and S. Insaf, J. Am. Chem. Soc., 1997, 119, 10903–10908 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- J. Lapić, S. Djaković, M. Cetina, K. Heinze and V. Rapić, Eur. J. Inorg. Chem., 2010, 106–114 CrossRef.
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460–1494 CrossRef.
- (a) J. Lapić, S. Djaković, I. Kodrin, Z. Mihalić, M. Cetina and V. Rapić, Eur. J. Org. Chem., 2010, 2512–2524 CrossRef; (b) S. Djaković, I. Kodrin, V. Smrečki, P. Novak, Z. Mihalić, D. Žiher, J. Lapić and V. Rapić, Tetrahedron, 2014, 70, 2330–2342 CrossRef; (c) M. Čakić-Semenčić, V. Kovač, I. Kodrin, L. Barišić and V. Rapić, Eur. J. Inorg. Chem., 2015, 112–123 CrossRef.
- U. Koch and P. L. A. Popelier, Phys. Chem., 1995, 99, 9747 CrossRef CAS.
- P. L. A. Popelier, J. Phys. Chem. A, 1998, 102, 1873 CrossRef CAS.
- J. C. Swarts, S. J. Vosoloo, S. J. Cronje, W. C. Du Plessis, C. E. Van Rensburg, E. Kreft and J. E. Van Lier, Anticancer Res., 2008, 28, 2781–2784 CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- G. Jaouen, S. Top, A. Vessières, G. Leclercq and M. J. McGlinchey, Curr. Med. Chem., 2004, 11, 2505–2017 CrossRef CAS PubMed.
- J. A. McAlpine, H. T. Lu, K. C. Wu, S. K. Knowles and J. A. Thomson, BMC Cancer, 2014, 14, 621–633 CrossRef PubMed.
- R. Cortés, M. Crespo, L. Davin, R. Martín, J. Quirante, D. Ruiz, R. Messeguer, C. Calvis, L. Baldomà and J. Badia, Eur. J. Med. Chem., 2012, 54, 557–566 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1060677. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01610j |
| This journal is © The Royal Society of Chemistry 2015 |