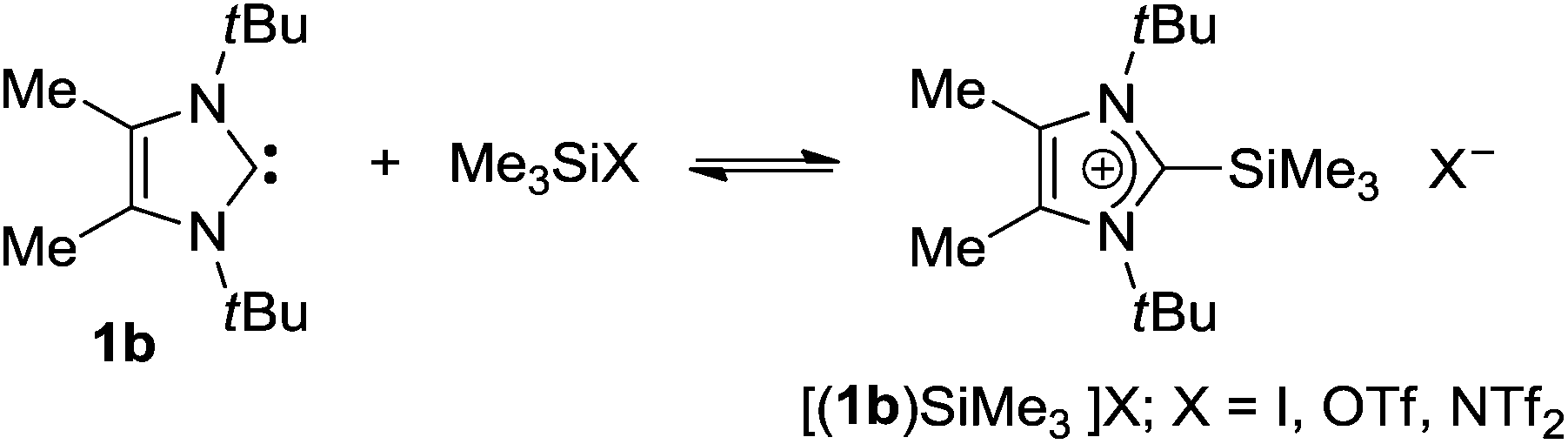DOI:
10.1039/C5DT01362C
(Paper)
Dalton Trans., 2015,
44, 9400-9408
Frustrated N-heterocyclic carbene–silylium ion Lewis pairs†
Received
10th April 2015
, Accepted 20th April 2015
First published on 20th April 2015
Abstract
The reaction of the N-heterocyclic carbene 1,3-di-tert-butyl-4,5-dimethylimidazolin-2-ylidene (1b) with trimethylsilyl iodide, triflate and triflimidate [Me3SiX, X = I, CF3SO3 (OTf), (CF3SO2)2N (NTf2)] by mixing the neat, liquid starting materials afforded the corresponding 2-(trimethylsilyl)imidazolium salts [(1b)SiMe3]X as highly reactive, white crystalline solids. Only the triflimidate (X = NTf2) proved to be stable in solution and could be characterized by means of NMR spectroscopy (in C6D5Br) and X-ray diffraction analysis, whereas dissociation into free 1b and Me3SiOTf was observed for the triflate system, in agreement with the trend derived by DFT calculations; the iodide was too insoluble for characterization. The compounds [(1b)SiMe3]X showed the reactivity expected for frustrated carbene–silylium pairs, and treatment with carbon dioxide, tert-butyl isocyanate and diphenylbutadiyne gave the 1,2-addition products [(1b)CO2SiMe3]X (X = I, OTf, NTf2), [(1b)C(NtBu)OSiMe3]OTf and [(1b)C(Ph)C(SiMe3)CCPh]OTf, respectively. Upon reaction with [AuCl(PPh3)], metal–chloride bond activation was observed, with formation of the cationic gold(I) complexes [(1b)Au(PPh3)]X (X = OTf, NTf2).
Introduction
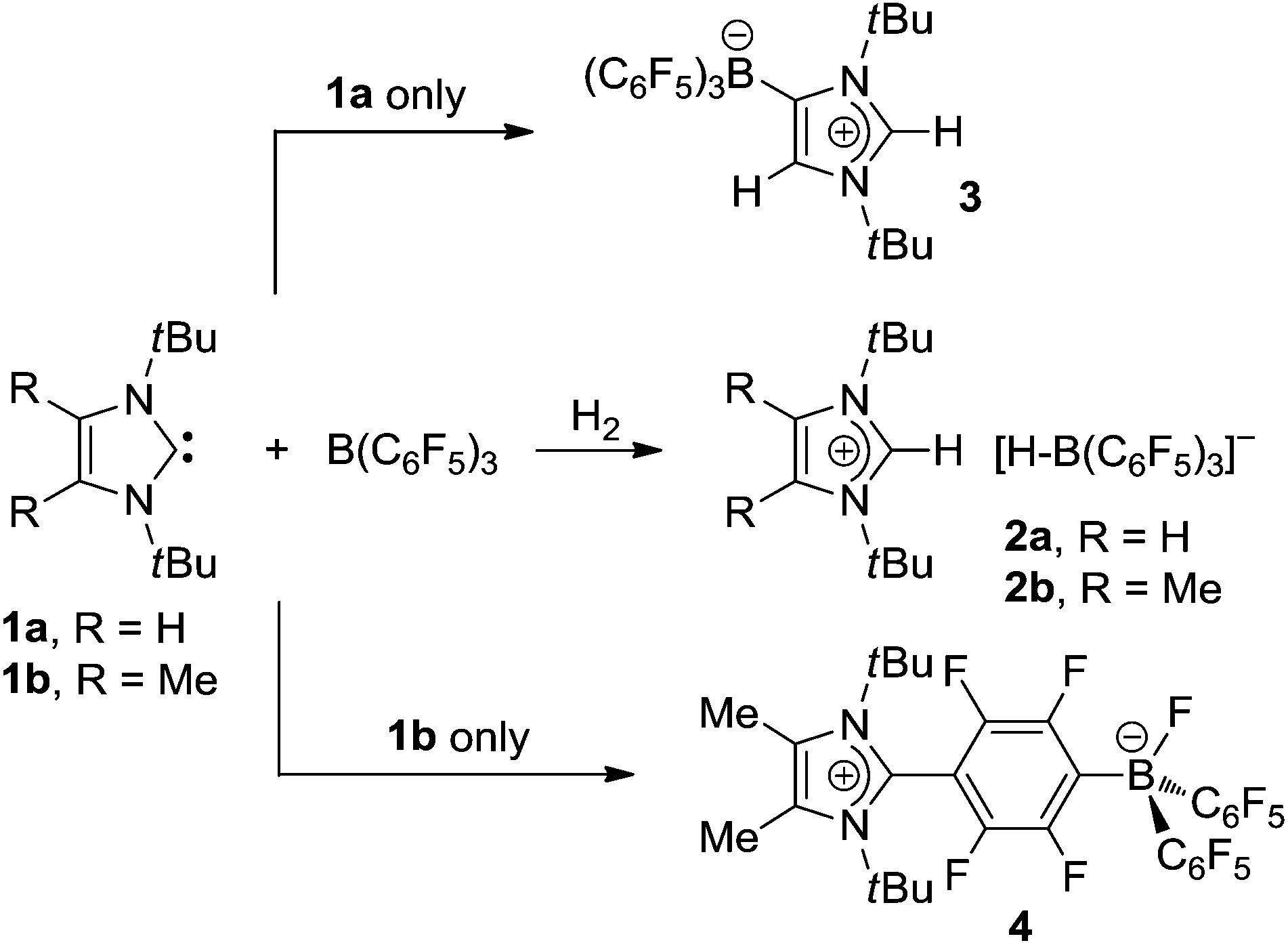
Frustrated Lewis pairs (FLPs), such as the well-studied phosphine–borane combination P(tBu)3/B(C6F5)3,1–5 represent a growing class of systems that are capable of activating small molecules, most notably dihydrogen, as a result of the unquenched and mutual reactivity of the Lewis base and acid.6 Accordingly, exposure of an equimolar solution of P(tBu)3 and B(C6F5)3 in toluene to an atmosphere of H2 swiftly affords the phosphonium borate [tBuPH][HB(C6F5)3] under ambient conditions.1,2 Extensive theoretical studies3–5 of heterolytic dihydrogen splitting with this system consistently indicate the importance of secondary noncovalent interactions for the formation of an encounter complex (or prepared Lewis pair) in which the donor and acceptor sites are suitably pre-organized for synergistic interaction with an incoming H2 molecule. A similar mechanistic proposal had been developed for the frustrated carbene–borane Lewis pair 1a/B(C6F5)3,7 which exhibits a particularly strong propensity for heterolytic dihydrogen cleavage;7,8 this can be ascribed to an enhanced cumulative acid–base strength and to the strongly exergonic formation of the imidazolium salt 2a (Scheme 1).9
 |
| | Scheme 1 Reactivity of frustrated carbene–borane Lewis pairs. | |
The high reactivity of this FLP was also exploited for the activation and fixation of numerous other small molecules,10i.e. ammonia,8 tetrahydrofuran,7 alkynes,11 white phosphorus,12 sulfur,13 carbon dioxide and nitrous oxide,14 and it was also used as a dehydrogenation reagent.15 In the absence of substrates, however, 1a/B(C6F5)3 does not become isolable as a weakly bound “normal” carbene–borane adduct, but slowly undergoes self-deactivation within 2 h at room temperature to form the significantly more stable “abnormal” adduct 3.7 Although initially unwanted, this observation triggered the development of anionic N-heterocyclic carbenes with a weakly coordinating anionic moiety (WCA-NHC), which can be formally derived from 3 by deprotonation.16,17 During our quest to prevent such deactivation reactions, numerous FLPs were studied with variation of the N-heterocyclic carbene (NHC),18,19 and also of the borane component.20 Thereby, carbene 1b proved particularly useful,21 since the presence of methyl groups in the 4,5-positions affords a sterically even more demanding carbene ligand, and self-deactivation of 1b/B(C6F5)3 is significantly slowed down in comparison with 1a/B(C6F5)3. Nevertheless, formation of the zwitterionic fluoroborate 4 by C–F activation was found to proceed within 4 d at room temperature (Scheme 1).14
As an alternative to boranes, more strongly Lewis acidic silylium ions have also been introduced to FLP chemistry,22 and it was shown that triarylsilylium salts of the type [Ar3Si][B(C6F5)4] react with bulky tertiary phosphines to afford frustrated Lewis pairs that are able to split dihydrogen irreversibly if an encounter complex with appropriate spatial requirements is formed.23 More recently, the thermally robust phosphine–silylium adduct [(tBu)3P-Si(iPr)3][B(C6F5)4] was reported to undergo H2 and D2 cleavage at 90 °C with formation of the silane (iPr)3SiH and the phosphonium borate [(tBu)3PH][B(C6F5)4].24 In view of these results, it was an obvious choice to aim at combining NHCs with silylium ions; however, for the present study, more readily available silylium sources such as the commercially available trimethylsilyl iodide, triflate and triflimidate [Me3SiX, X = I, CF3SO3 (OTf), (CF3SO2)2N (NTf2)] were chosen. In previous reports, the reactions of Me3SiI and Me3SiOTf with sterically less encumbered NHCs furnished the stable 2-(trimethylsilyl)imidazolium salts (“normal carbene–silylium adducts”) 5,256,26 and 7,27,28 whereas treatment of the more bulky carbene 1a with Me3SiOTf resulted in rapid formation of the 4-(trimethylsilyl)imidazolium triflate 8 (“abnormal carbene–silylium adduct”, Fig. 1).29 The latter reaction clearly indicates that the formal addition of Me3Si+ to the C2 carbene atom in 1a would afford a frustrated Lewis pair, which rearranges to the more stable abnormal adduct 8 in a similar fashion as described for 3 (vide supra). Therefore, the reactivity of 1b towards Me3SiX was investigated in the expectation that the presence of the 4,5-methyl substituents might render the corresponding carbene–silylium adducts [(1b)SiMe3]X (X = I, OTf, NTf2), more stable and even isolable. It should be noted that these studies are also highly relevant in view of the interest in NHC-mediated activation of tetravalent silicon compounds, since NHC–Si interactions have been hypothesized for the mechanistic course of various catalytic reactions such as cyanosilylation, CO2 reduction with silanes or polymerisation of silicon-containing compounds.30
 |
| | Fig. 1 Reported examples of (trimethylsilyl)imidazolium salts. | |
Results and discussion
Generation of carbene–silylium adducts
Our initial efforts to isolate the carbene–silylium adducts [(1b)SiMe3]X (X = I, OTf, NTf2) by mixing 1b with Me3SiX were ambiguous, and only the reaction with Me3SiNTf2 produced a precipitate of the triflimidate [(1b)SiMe3]NTf2 from hexane or toluene solution in satisfactory yield. Since the silyl reagents and also carbene 1b are liquid at room temperature, equimolar amounts of 1b and Me3SiX were mixed by blending in a Teflon vial under argon, resulting in immediate solidification to afford [(1b)SiMe3]I, [(1b)SiMe3]OTf and [(1b)SiMe3]NTf2 as colourless materials in quantitative yield and analytically pure form after careful washing with cold hexane in order to remove any trace of unreacted starting materials (Scheme 2). NMR spectroscopic characterisation in solution was hampered by the high reactivity of the compounds, which decompose rapidly in deuterated solvents such as CDCl3, CD2Cl2 and THF-d6 and are only sparingly soluble in toluene-d8. However, instructive NMR spectra could be recorded for the triflate and triflimidate salts in deutero-bromobenzene (C6D5Br), although the samples always showed contamination with the corresponding imidazolium salt, in which a hydrogen atom replaces the Me3Si group. The 13C NMR spectra reveal distinct differences, since the spectrum of [(1b)SiMe3]OTf shows a resonance at 210.5 ppm, indicating the presence of the free carbene 1b (δ = 210.2 ppm, in C6D5Br; δ = 212.2 ppm in C6D6).21 Accordingly, the 1H NMR spectrum exhibits the resonances for the free carbene at 1.55 (tBu) and 2.09 ppm (Me) and also for the methyl groups of Me3SiOTf at 0.20 ppm. The formation of only minor amounts of the expected adduct [(1b)SiMe3]OTf can be deduced from the observation of characteristic 13C NMR signals at 155.5 and 5.4 ppm, which can be assigned to the C2 and SiCH3 carbon atoms of the 2-(trimethylsilyl)imidazolium cation. Integration of the corresponding signals in the 1H NMR spectrum reveals more than 90% dissociation into the starting materials.
 |
| | Scheme 2 Reaction of 1b with trimethylsilyl iodide, triflate and triflimidate; mixing liquid 1b and liquid Me3SiX affords quantitatively solid [(1b)SiMe3]X. | |
In contrast, the NMR spectra of [(1b)SiMe3]NTf2 (in C6D5Br) indicate complete consumption of the free carbene 1b, and full conversion is indicated by strong 13C NMR signals at 156.0 (C2) and 5.3 ppm (SiCH3). The 1H NMR spectrum shows signals at 1.99, 1.35 and 0.23 ppm in a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18:9 ratio, attributable to the different methyl groups in [(1b)SiMe3]+, and the 29Si NMR spectrum gives rise to a signal at 4.5 ppm, which falls in the range observed for the related compounds 5 (−5.1 ppm),256 (4.5 ppm),26 and 7 (−1.0 ppm),27,28 and is significantly upfield from the chemical shift of silylating agents including Me3SiNTf2.31 It should be noted that the high reactivity of the systems 1b/Me3SiX has so far precluded further investigation of these putative equilibrium reactions, e.g. by temperature-dependent NMR studies.
:18:9 ratio, attributable to the different methyl groups in [(1b)SiMe3]+, and the 29Si NMR spectrum gives rise to a signal at 4.5 ppm, which falls in the range observed for the related compounds 5 (−5.1 ppm),256 (4.5 ppm),26 and 7 (−1.0 ppm),27,28 and is significantly upfield from the chemical shift of silylating agents including Me3SiNTf2.31 It should be noted that the high reactivity of the systems 1b/Me3SiX has so far precluded further investigation of these putative equilibrium reactions, e.g. by temperature-dependent NMR studies.
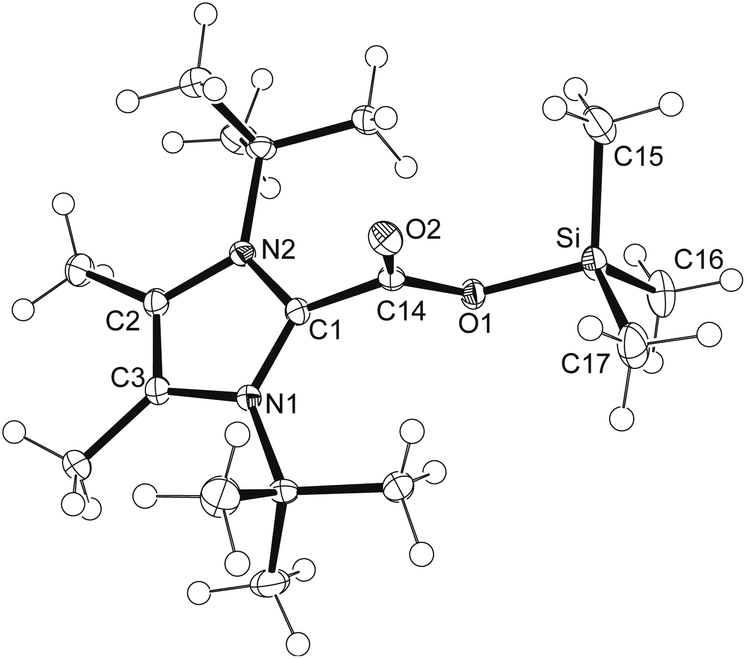
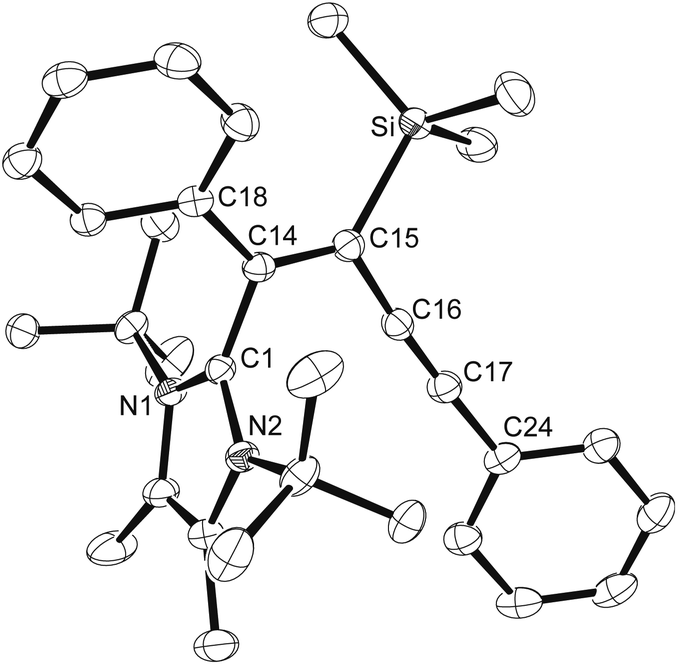
Single crystals of [(1b)SiMe3]NTf2 suitable for X-ray diffraction analysis were obtained as a hemisolvate from C6D5Br solution, and the molecular structure is shown in Fig. 2. Remarkably, the cation is strongly twisted and exhibits a long Si–C1 bond of 1.9671(13) Å, with the silicon atom lying 1.078(2) Å above the imidazole (C1–N1–C2–C3–N1) plane (r.m.s. deviation 0.02 Å).‡ The tBu groups are forced to the opposite side of the heterocycle with deviations of 0.554(2) Å (C4) and 0.473(2) Å (C10) of the tertiary carbon atoms from the plane. A similarly distorted structure is found for the same cation with the azidodiborate counterion [(C6F5)3BN3B(C6F5)3]−, whereas the sterically less congested compound 7 (Fig. 1) shows a shorter Si–C1 bond length of 1.9330(12) Å and a smaller displacement (0.232 Å) of the Si atom, indicating a smaller degree of “frustration”.32 For comparison, significantly shorter bonds are found for unperturbed 2-silylimidazolium cations of the type [NHC-SiH3]+ (1.920(2) Å)33 and [NHC-SiI3]+ (1.911(3) Å).34
 |
| | Fig. 2 ORTEP diagram of [(1b)SiMe3]NTf2 with thermal ellipsoids drawn at 50% probability. Selected bond lengths (Å) and angles (°): Si–C14 1.8655(15), Si–C15 1.8650(14), Si–C16 1.8681(14), Si–C1 1.9671(13), N1–C1 1.3653(16), N1–C2 1.3949(17); C15–Si–C14 113.36(7), C15–Si–C16 101.00(6), C14–Si–C16 109.96(7), C15–Si–C1 105.10(6), N1–C1–N2 106.35(11). Solvent is omitted. | |
Theoretical calculations of carbene–silylium adduct formation
To rationalize the reactivity of the carbene 1b towards the reagents Me3SiX (X = I, OTf, NTf2), the thermodynamics of adduct formation were calculated for the ion pairs [(1b)SiMe3]X and also for the corresponding adducts [(1c)SiMe3]X containing the sterically less encumbered carbene 1,3,4,5-tetramethylimidazolin-2-ylidene (1c). The anions were generally placed underneath the plane of the resulting 2-(trimethylsilyl)imidazolium salt, and the structures were freely refined. We employed the functionals M05-2X, M06-2X and B97-D, which were developed by Zhao and Truhlar35 and by Grimme36 to describe conveniently noncovalent and long-range dispersion interactions that can be expected to contribute significantly to the overall binding, in particular for the sterically congested adducts of carbene 1b. Table 1 summarizes the enthalpies (ΔH) and Gibbs free energies (ΔG) at 298 K for the formation of the adducts [(1b)SiMe3]X and [(1c)SiMe3]X, and Fig. 3 shows a bar diagram for the values derived with the B97-D functional. In addition, the gas-phase association energies were calculated for the reaction of the carbenes 1b and 1c with a “naked” [Me3Si]+ cation in the absence of any counterion (Table 1). Since the reverse reaction, the dissociation of [(1b)SiMe3]+ and [(1c)SiMe3]+ into the free carbenes and [Me3Si]+, can be defined as the trimethylsilylium affinity (TMSA = –ΔH),37 high TMSA values of ca. 75 kcal mol−1 (for 1b) and ca. 92 kcal mol−1 (for 1c) can be derived for both carbenes as expected for the combination of very strong nucleophiles and electrophiles. Nevertheless, the TMSA value of 1b is significantly smaller than that of 1c, which confirms the lower stability (by ca. 17 kcal mol−1) of the sterically overcrowded system. Notably, all calculations reproduce the highly distorted structure of the cation [(1b)SiMe3]+ found in the solid state (Fig. 2).†
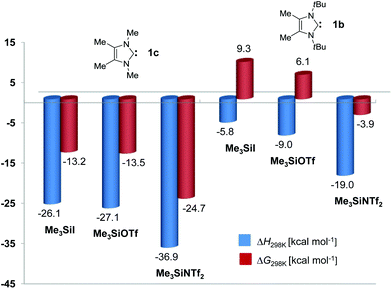 |
| | Fig. 3 Bar diagram showing the enthalpies (ΔH) and Gibbs free energies at 298 K (ΔG) for the reaction of 1b (right) and 1c (left) with Me3SiX (X = I, OTf, NTf2) at the B97D/6-311G(d,p) level of theory. | |
Table 1 Enthalpies (ΔH) and Gibbs free energies (ΔG) at 298 K in kcal mol−1 for the formation of carbene–silylium adductsa
| Compoundb |
M05-2X |
M06-2X |
B97-D |
| ΔH |
ΔG |
ΔH |
ΔG |
ΔH |
ΔG |
|
M05-2X, M06-2X, and B97-D/6-311G(d,p) enthalpies and Gibbs free energies at 298 K.
1b = 1,3-di-tert-butyl,4,5-dimethylimidazolin-2-ylidene; 1c = 1,3,4,5-tetramethylimidazolin-2-ylidene; if present, the anion X was placed underneath the plane of the resulting 2-trimethylimidazolium salt, and the structure was freely refined.
−ΔH = TMSA (trimethylsilylium affinity).37
|
| [(1b)SiMe3]I |
−0.2 |
14.6 |
−1.8 |
13.1 |
−5.8 |
9.3 |
| [(1b)SiMe3]OTf |
−0.3 |
13.8 |
−5.4 |
12.7 |
−9.0 |
6.1 |
| [(1b)SiMe3]NTf2 |
−11.2 |
4.1 |
−14.0 |
1.6 |
−19.0 |
−3.9 |
| [(1b)SiMe3]+ |
−73.7c |
−56.8 |
−75.4c |
−58.3 |
−77.5c |
−60.2 |
| [(1c)SiMe3]I |
−22.8 |
−10.2 |
−23.4 |
−10.3 |
−26.1 |
−13.2 |
| [(1c)SiMe3]OTf |
−24.0 |
−11.2 |
−26.2 |
−11.0 |
−27.1 |
−13.5 |
| [(1c)SiMe3]NTf2 |
−35.3 |
−22.4 |
−36.5 |
−22.2 |
−36.9 |
−24.7 |
| [(1c)SiMe3]+ |
−92.3c |
−77.2 |
−93.0c |
−77.5 |
−92.1c |
−77.1 |
The calculations for the ion pairs [(1b)SiMe3]X and [(1c)SiMe3]X reveal a marked influence of the counterion X, and in particular, the triflimidates (X = NTf2) of both systems are significantly more stable than the corresponding triflates and iodides; this reflects the effect of a highly delocalized counteranion,38 but might also indicate a favourable interaction with the 2-(trimethylsilyl)imidazolium cation. Since such interactions also play an important role in the solid state, we believe that the gas-phase calculations at hand will allow us to predict or substantiate trends in reactivity. All three functionals produce qualitatively similar results, with the B97-D consistently yielding lower energies. The following discussion will be restricted to the B97-D values, which are assembled in Fig. 3. For [(1c)SiMe3]X, the formation of all adducts is markedly exothermic and exergonic, which clearly suggests that frustrated Lewis pair behaviour cannot be expected for the combinations 1c/TMSX and that complete formation of ion pairs can be expected. In contrast, the adducts [(1b)SiMe3]X are significantly less stable, with their stability following the order [(1b)SiMe3]I (ΔH = −5.8 kcal mol−1) < [(1b)SiMe3]OTf (ΔH = −9.0 kcal mol−1) < [(1b)SiMe3]NTf2 (ΔH = −19.0 kcal mol−1). Although entropy effects should not be overestimated, it can be stated that the formation of the triflimidate was calculated to be slightly exergonic (ΔG = −3.9 kcal mol−1), whereas the triflate (ΔG = 6.1 kcal mol−1) and iodide (ΔG = 9.3 kcal mol−1) form endergonically. These theoretical results are in full agreement with the experimental observation that dissolution of the triflate in bromobenzene leads to predominant dissociation into 1b/Me3SiOTf, whereas the starting materials 1b/Me3SiNTf2 are not observed for the triflimidate; this allowed us to characterize the latter by means of NMR spectroscopy and X-ray diffraction analysis (vide supra).
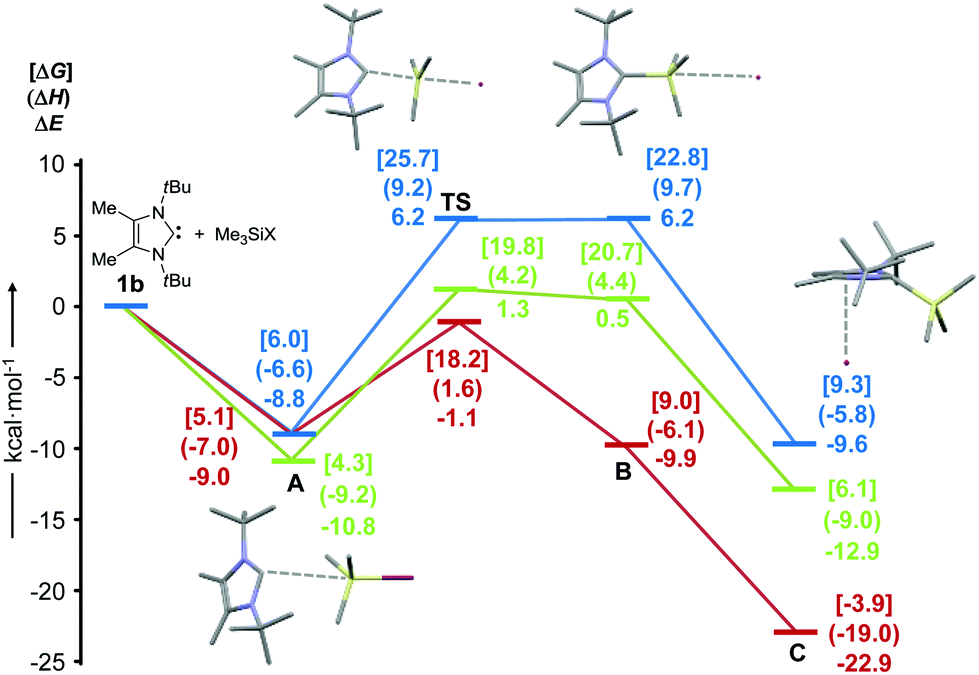
To establish a potential pathway for the formation of the 2-(trimethylsilyl)imidazolium salts [(1b)SiMe3]X, we assumed that the initial binding between the Lewis basic carbene 1b and the Lewis acidic Me3SiX reagent will occur according to the established model of n–σ* interaction,30,39 which leads to polarization of the adjacent Si–X bond by formation of a hypervalent silicon species. In the case of strong polarization, this may be followed by subsequent heterolytic Si–X bond cleavage and ionization. Since the same considerations are valid for the reverse reaction, we obtained suitable starting geometries based on the calculated structures of [(1b)SiMe3]X (C), which were modified by placing the counterion X opposite to the carbene ligand. Full refinement without any geometrical constraints afforded a second set of ionic minimum structures B with weak Si–X interactions, which are 15.8 (X = I), 13.4 (X = OTf) and 13.0 kcal mol−1 (X = NTf2) higher in energy than the global minima. For all three systems, we were then able to locate transition states (TS), which connect the ionic local minima B with covalent adducts of the type 1b/Me3SiX (A). The transition state structures exhibit hypercoordinate silicon atoms with distorted trigonal–bipyramidal geometries (Ccarbene–Si–I = 178.2°, Ccarbene–Si–O = 178.1° and Ccarbene–Si–N = 170.5°), in which the trigonal-planar [Me3Si]+ cation (C–Si–C angle sums = 360°) is flanked by loosely bound counterions and carbene ligands (Ccarbene–Si = 2.434 Å for X = I, Ccarbene–Si = 2.401 Å for X = OTf, Ccarbene–Si = 2.612 Å for X = NTf2). It is particularly noteworthy that very low barriers are found for the iodide and triflate systems (ΔΔE ≈ 0 kcal mol−1, X = I; ΔΔE = 0.8 kcal mol−1, X = OTf),40 which can be expected to rearrange readily to 1b/Me3SiX, whereas moderately higher barriers are found for both the formation and cleavage of [(1b)SiMe3]NTf2. The resulting adducts A are located −8.8 (X = I), −10.8 (X = OTf) and −9.0 kcal mol−1 (X = NTf2) below the energy of the free, isolated starting materials, but, surprisingly, they do not display any direct C–Si contacts between the carbene carbon and the silicon atoms, as indicated by very long Ccarbene–Si distances of 3.965 Å for X = I, 4.117 Å for X = OTf and 3.933 Å for (X = NTf2). Instead, the TMS groups display a series of short intermolecular C–H⋯C and C–H⋯N contacts in the range 2.53–2.78 Å, which probably contribute significantly to the overall binding energy.
It should be emphasized that the potential-energy profiles shown in Fig. 4 can be expected to be strongly affected by solvent effects, since a transition from covalent to ionic structures is undergone, which proceeds via strongly polarized transition states. Nevertheless, we believe that the gas-phase calculations at hand allow us to rationalise the different reactivities of the 1b/Me3SiX systems, with the most weakly coordinating triflimidate ion affording a persistent ionic compound [(1b)SiMe3]NTf2, whereas the corresponding iodide and triflate only form by mixing the neat starting materials, but are readily cleaved upon dissolution.
 |
| | Fig. 4 Potential-energy profile for the reaction of 1b with Me3SiX (X = I, blue; X = OTf, green; X = NTf2, red). Values correspond to ΔE = zero-point uncorrected B97-D/6-311G(d,p) electronic energies, ΔH = enthalpies at 298 K (round brackets, and ΔG = Gibbs free energies at 298 K (square brackets). Stick models are provided for the stationary points of the 1b/Me3SiI system. TS = transition state. | |
Reactivity of carbene–silylium Lewis pairs – addition reactions
The structural distortions determined experimentally and theoretically for the carbene–silylium adducts [(1b)SiMe3]X, together with their obvious lability, prompted us to investigate their behaviour as potential frustrated Lewis pairs. However, dihydrogen cleavage with the triflate and triflimidate systems proved unsuccessful under the conditions described for isolable phosphine–silylium adduct [(tBu)3P-Si(iPr)3][B(C6F5)4].24 In view of the general interest in the reduction of carbon dioxide by silanes, which can for instance be mediated by frustrated Lewis pairs,41 silylium ions,42 and N-heterocyclic carbenes,43 carbon dioxide fixation was studied by purging diethyl ether suspensions of [(1b)SiMe3]X (X = I, OTf, NTf2) with CO2. The adducts [9]X were isolated as voluminous white precipitates in excellent yields for X = I, OTf, whereas for X = NTf2, the reaction was accompanied by the formation of significant amounts of imidazolium triflimidate (Scheme 3). Although slow decomposition in chlorinated solvents was observed in a similar fashion as described for carbene–borane systems,14,44 satisfactory NMR spectra could be recorded in CD2Cl2, affording 13C NMR signals for the carboxylic group at ca. 158 ppm, similar to the value for the adducts 1b·CO2 (165.5 ppm)14 and 1b·CO2·B(C6F5)3 (157.7 ppm)14 and shifted by approximately 33 ppm to lower field compared to free CO2.45 Crystals of [9]OTf suitable for X-ray diffraction analysis were obtained from THF solution at −30 °C; the resulting molecular structure is shown in Fig. 5. The structural features of the CO2 moiety, with C–O bonds of 1.2083(16) and 1.3090(15) Å, are identical within error limits to the values found for the carbene–borane adduct 1b·CO2·B(C6F5)3, and the same perpendicular orientation (86.5°) to the imidazole plane is also observed.14
 |
| | Scheme 3 Addition reactions of [(1b)SiMe3]X with carbon dioxide, tert-butyl isocyanate and diphenylbutadiyne. | |
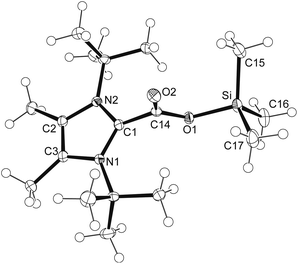 |
| | Fig. 5 ORTEP diagram of cation in [9]OTf with thermal ellipsoids at 50% probability. Selected bond lengths (Å) and angles (°): Si–O1 1.7399(10), Si–C16 1.8417(16), Si–C17 1.8469(15), Si–C15 1.8507(15), C1–C14 1.5129(17), N1–C1 1.3506(16), C14–O1 1.3090(15), C14–O2 1.2083(16); O1–Si–C16 101.30(6), O1–Si–C17 108.30(6), C14–O1–Si 123.60(9), N1–C1–C14 124.78(11), N1–C1–N2 109.79(11). | |
The fixation of the CO2 congener tert-butyl isocyanate was also studied, and its addition to a suspension of [(1b)SiMe3]OTf in diethyl ether afforded the adduct [10]OTf as a colourless solid in 79% yield (Scheme 3). The 1H NMR spectrum (in CD2Cl2) exhibits four singlets at 0.22, 1.35, 1.73 and 2.42 ppm in a 9:9:18:6 ratio, which can be assigned to the Me3Si and the three different types of methyl groups, respectively. Single crystals of [10]OTf were obtained from Et2O/CH2Cl2 solution at −30 °C, and the molecular structure was established by X-ray diffraction analysis (Fig. 6). The compound crystallizes with two independent cations and anions in the asymmetric unit with very similar structural parameters, which will be discussed for cation 1 only. The trimethylsilyl group is bound to the oxygen atom with Si–O = 1.7103(10) Å, which affords a shorter C1–C14 bond length of 1.5038(18) Å and a longer C14–O1 bond length of 1.3564(16) Å in comparison to neutral NHC-isocyanate adducts.46 The entire isocyanate moiety (Si–O1–C14–N3–C15) is planar and adopts a perpendicular orientation (89.4°) to the imidazole ring.
 |
| | Fig. 6 ORTEP diagram of one of the two independent cations in [10]OTf with thermal ellipsoids at 50% probability. Selected bond lengths (Å) and angles (°) in cation 1 [cation 2]: Si–O1 1.7103(10) [1.7106(10)], Si–C19 1.8444(17) [1.8486(17)], Si–C20 1.8498(15) [1.8432(18)], Si–C21 1.8509(17) [1.8484(17)], C1–C14 1.5038(18) [1.5019(18)], O1–C14 1.3564(16) [1.3577(16)], C14–N3 1.2555(18) [1.2537(18)], N3–C15 1.4897(16) [1.4873(16)]; O1–Si–C19 111.64(7) [101.71(6)], O1–Si–C20 110.01(7) [110.50(7)], C14–O1–Si 122.23(8) [120.91(8)], N1–C1–C14 124.79(12) [125.56(12)], C14–N3–C15 131.66(12) [131.82(12)]. | |
The reaction of frustrated amine–borane and phosphine–borane Lewis pairs with conjugated diynes has been reported to proceed either by 1,2- or 1,4-addition to afford 1,3-enyne or 1,2,3-butatriene derivatives, respectively.47–49 Treatment of diphenylbutadiyne with [(1b)SiMe3]OTf in chlorobenzene solution at room temperature gave the trans-1,2-addition product [11]OTf in 51% yield as a reddish brown solid. The 1H NMR spectrum shows three singlets at 0.42, 1.69 and 2.62 ppm in a 9:18:6 ratio for the methyl groups together with two sets of multiplets between 7.16 and 7.52 ppm for the phenyl rings. In the 13C NMR spectrum, the signals at 91.9, 106.7, 139.3 and 146.3 ppm can be assigned to a conjugated enyne moiety. 1,2-Addition of the carbene–silylium pair can be confirmed by X-ray diffraction analysis (Fig. 7), and the molecular structure exhibits the characteristic features of an enyne framework, i.e. C14–C15 = 1.3649(18) Å, C15–C16 = 1.4301(19) Å, C16–C17 = 1.208(2) Å.48,49 It should be noted that addition of amine–borane and phosphine–borane pairs was found to occur in a reverse manner, with the Lewis base, unlike the carbene in [11]OTf, attached to the inner carbon atom.47–49
 |
| | Fig. 7 ORTEP diagram of the cation 11 in [11]OTf with thermal ellipsoids at 50% probability. Selected bond lengths [Å] and angles: Si–C15 1.9211(14), C1–C14 1.4951(17), C14–C15 1.3649(18), C14–C18 1.4783(18), C15–C16 1.4301(19), C16–C17 1.208(2), C17–C24 1.4316(19); N1–C1–N2 108.66(11), C15–C14–C1 114.89(11), C17–C16–C15 174.63(15). | |
Reactivity of carbene–silylium Lewis pairs – activation of metal–halide bonds
The high reactivity of the carbene–silylium pairs prompted us to employ [(1b)SiMe3]OTf and [(1b)SiMe3]NTf2 as metal-free carbene-transfer reagents by taking advantage of the favourable formation of volatile trimethylsilyl halides.50 In view of the prominent role of cationic gold(I) complexes in homogeneous catalysis,51 the gold(I) chloride complex [AuCl(PPh3)] was chosen as model system. Treatment of this starting material with [(1b)SiMe3]OTf and [(1b)SiMe3]NTf2 in chlorobenzene solution furnished the complexes [(1b)Au(PPh3)]X, [12]X (X = OTf, NTf2), in high yield as colourless solids, which were isolated by precipitation with hexane and filtration (Scheme 4). NMR spectra were recorded in CD2Cl2 solution, revealing 31P NMR signals at ca. 38 ppm and 13C NMR signals at ca. 184 ppm with 2JCP = 125, which is in agreement with the data reported for other NHC-phosphine gold(I) complexes.52
 |
| | Scheme 4 Synthesis of cationic gold–carbene complexes. | |
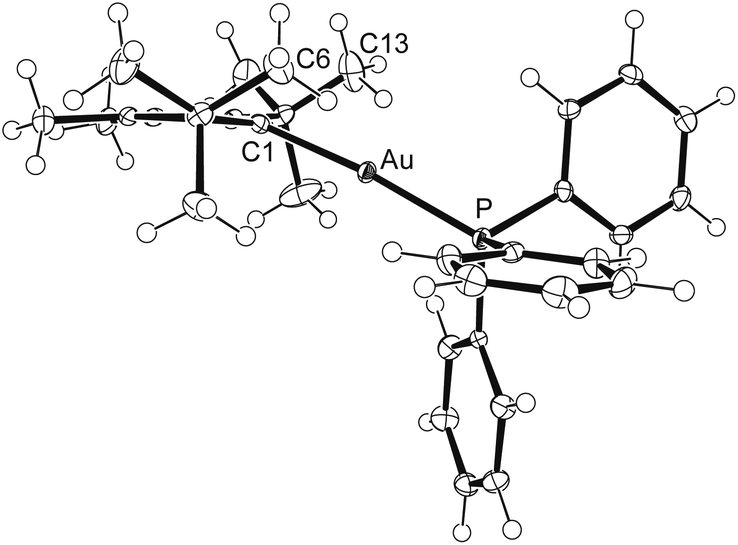
The molecular structures of both complexes were established by X-ray diffraction analyses, revealing almost identical structural parameters. Fig. 8 shows an ORTEP diagram of the cation in [12]NTf2, and further discussion will be confined to this compound. On first glance, the structural characteristics are unremarkable, with the cation displaying an almost linear C1–Au–P axis of 174.43(10)° and Au–C1 and Au–P distances of 2.063(3) and 2.2777(9) Å, which fall in the ranges found for related systems.16,17,52 For instance, gold–carbon and gold–phosphorus bond lengths of 2.044(4) Å and 2.275(1) Å were reported for the closely related complex [Au(1a)(PPh3)]PF6.53 However, closer inspection of the structures reveals distinct differences, since the carbene ligand in the latter complex shows the expected coplanar orientation with the C–Au–P axis, whereas pronounced twisting is observed for 12, with the gold atom being displaced by 0.753(6) Å from the imidazole plane, providing a remarkably large “pitch angle” of 20.5°.54 In addition, two conspicuously short Au⋯H contacts of ca. 2.4 Å are found, involving a methyl hydrogen atom of each tert-butyl group (at C6 and C13, Fig. 8). These are among the shortest gold–hydrogen bonds known,55 and their occurrence, together with the overall structural distortion, can be ascribed to hindered rotation of the tBu substituents due to steric interaction with the 4,5-methyl groups (buttressing effect).18,21,56 It is anticipated that coordination to other metal ions such as Ru(II), Rh(I) or Ir(I) might lead to C–H activation and formation of cyclometallated NHC complexes.57
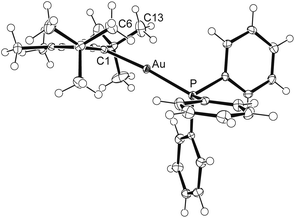 |
| | Fig. 8 ORTEP diagram of the cation in [12]NTf2 with thermal ellipsoids at 50% probability. Selected bond lengths [Å] and angles in [12]OTf/[12]NTf2: Au–C1 2.063(4)/2.068(3), Au–P 2.2777(10)/2.2777(9), C1–N1 1.353(5)/1.355(4), C2–N1 1.362(5)/1.360(4); C1–Au–P 176.52(11)/174.43(10), N1–C1–N2 106.6(3)/106.5(3), N1–C1–Au 125.5(3)/124.7(2), N2–C1–Au 126.8(3)/125.2(2). | |
DFT calculations were carried out to assess the energy difference between the twisted structure found in the solid state and a structure with a coplanar orientation of the imidazole plane towards the C1–Au–P axis. Therefore, the solid state structure of 12 in [12]NTf2 was used as starting coordinates and fully refined to afford the minimum structure B with similar structural characteristics, e.g. pitch angle = 20.1° (Fig. 9). Attempts to calculate an in-plane minimum structure failed; however, we were able locate a transition state (TS) with a pitch angle close to zero (1.4°), which connects B with a second, energetically slightly favoured (ΔΔE = 0.5 kcal mol−1) minimum structure A (pitch angle = 22.9°). This process represents the inversion of the carbene ligand at the donor carbon atom (analogous to the known inversion process of amines), which interconverts the two twisted structures A and B, and the small barrier of 2.9 kcal mol−1 indicates that the observation of this process cannot be monitored by NMR spectroscopy.
 |
| | Fig. 9 Potential-energy profile for the interconversion between the out-of-plane structures A and B, which proceeds via a coplanar transition state (TS). Values correspond to ΔE = zero-point uncorrected B97-D/6-311G(d,p) electronic energies, ΔH = enthalpies at 298 K (round brackets, and ΔG = Gibbs free energies at 298 K (square brackets). | |
Conclusions
It was demonstrated that the stability of trimethylsilylium adducts of the sterically demanding N-heterocyclic carbene 1b is strongly affected by the nature of the counterion and increases with decreasing nucleophilicity of the ion X or with increasing Lewis acidity of the silylating agent Me3SiX (X = I, OTF, NTf2).31,38 For all systems 1b/Me3SiX, the reactivity expected for frustrated Lewis pairs, such as CO2 fixation, was observed, but it is unclear whether the reactions occur by insertion into the C–Si bond of an isolable carbene–silylium adduct or proceed by stepwise addition of Lewis acid and base, and the reaction path is again likely to depend on the counterion X. Accordingly, the term “frustrated Lewis pair” has to be used with caution, since the carbene and the counterion compete for the Lewis acid component, the trimethylsilylium ion, which cannot be expected to be freely available at any time. This might also explain the inability of these systems to split dihydrogen. Alternatively, these carbene–silylium combinations can also be regarded as “tamed” silylium ions,58 which have found numerous applications in Lewis acid catalysis;59 and in view of numerous reports on NHC-mediated activation of silicon(IV) compounds,30 the potential use of carbene–silylium Lewis pairs for applications in catalysis can be envisaged. As demonstrated for the preparation of cationic NHC–gold(I) complexes, the systems 1b/Me3SiX promote metal–halide bond activation and allow for the simultaneous introduction of carbene 1b and a weakly coordinating counterion such as triflate or triflimidate, which might become a useful method for the preparation of coordinatively unsaturated and/or catalytically active transition metal complexes.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft through grant TA 189/9-1. E. T. is grateful to the Deutsche Bundesstiftung Umwelt (DBU) for a scholarship.
Notes and references
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- A. J. V. Marwitz, J. L. Dutton, L. G. Mercier and W. E. Piers, J. Am. Chem. Soc., 2011, 133, 10026–10029 CrossRef CAS PubMed.
- T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435–2438 CrossRef CAS PubMed.
- S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402–1405 CrossRef CAS PubMed.
- T. A. Rokob, I. Bakó, A. Stirling, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2013, 135, 4425–4437 CrossRef CAS PubMed.
-
(a)
Frustrated Lewis Pairs I, ed. G. Erker and D. W. Stephan, Springer-Verlag, Berlin, 2013 Search PubMed;
(b)
Frustrated Lewis Pairs II, ed. G. Erker and D. W. Stephan, Springer-Verlag, Berlin, 2013 Search PubMed;
(c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed;
(d) D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC;
(e) D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535 RSC;
(f) G. Erker, C. R. Chim., 2011, 14, 831–841 CrossRef CAS PubMed.
- D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428–7432 CrossRef CAS PubMed.
- P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433–7437 CrossRef CAS PubMed.
- T. A. Rokob, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2009, 131, 10701–10710 CrossRef CAS PubMed.
-
E. L. Kolychev, E. Theuergarten and M. Tamm, in Frustrated Lewis Pairs II, ed. G. Erker and D. W. Stephan, Springer-Verlag, Berlin, 2013, vol. 334, pp. 121–155 Search PubMed.
- M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6594–6607 CrossRef CAS.
- D. Holschumacher, T. Bannenberg, K. Ibrom, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2010, 39, 10590–10592 RSC.
- D. Holschumacher, C. G. Daniliuc, P. G. Jones and M. Tamm, Z. Naturforsch., B: Chem. Sci., 2011, 66, 371–377 CrossRef CAS PubMed.
- E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 43, 1651–1662 RSC.
- A. Jana, I. Objartel, H. W. Roesky and D. Stalke, Inorg. Chem., 2009, 48, 7645–7649 CrossRef CAS PubMed.
- S. Kronig, E. Theuergarten, C. G. Daniliuc, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2012, 51, 3240–3244 CrossRef CAS PubMed.
- E. L. Kolychev, S. Kronig, K. Brandhorst, M. Freytag, P. G. Jones and M. Tamm, J. Am. Chem. Soc., 2013, 135, 12448–12459 CrossRef CAS PubMed.
- S. Kronig, E. Theuergarten, D. Holschumacher, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2011, 50, 7344–7359 CrossRef CAS PubMed.
- D. Holschumacher, C. Taouss, T. Bannenberg, C. G. Hrib, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2009, 6927–6929 RSC.
- E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem. – Eur. J., 2012, 18, 16938–16946 CrossRef CAS PubMed.
- A. A. Grishina, S. M. Polyakova, R. A. Kunetskiy, I. Císařová and I. M. Lyapkalo, Chem. – Eur. J., 2011, 17, 96–100 CrossRef CAS PubMed.
-
(a) A. Schäfer, M. Reissmann, A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2011, 50, 12636–12638 CrossRef PubMed;
(b) A. Schäfer, M. Reißmann, A. Schäfer, M. Schmidtmann and T. Müller, Chem. – Eur. J., 2014, 20, 9381–9386 CrossRef PubMed.
- M. Reißmann, A. Schäfer, S. Jung and T. Müller, Organometallics, 2013, 32, 6736–6744 CrossRef.
- T. J. Herrington, B. J. Ward, L. R. Doyle, J. McDermott, A. J. P. White, J. P. Andrew, P. A. Hunt and A. E. Ashley, Chem. Commun., 2014, 50, 12753–12756 RSC.
- N. Kuhn, T. Kratz, D. Bläser and R. Boese, Chem. Ber., 1995, 128, 245–250 CrossRef CAS PubMed.
- J. J. Weigand, K.-O. Feldmann and F. D. Henne, J. Am. Chem. Soc., 2010, 132, 16321–16323 CrossRef CAS PubMed.
- D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 7264–7265 CrossRef CAS PubMed.
- F. D. Henne, E.-M. Schnöckelborg, K.-O. Feldmann, J. Grunenberg, R. Wolf and J. J. Weigand, Organometallics, 2013, 32, 6674–6680 CrossRef CAS.
- G. R. Whittell, E. I. Balmond, A. P. M. Robertson, P. M. Alasdair, S. K. Patra, M. F. Haddow and I. Manners, Eur. J. Inorg. Chem., 2010, 2010, 3967–3975 CrossRef PubMed.
- M. J. Fuchter, Chem. – Eur. J., 2010, 16, 12286–12294 CrossRef CAS PubMed.
- B. Mathieu and L. Ghosez, Tetrahedron, 2002, 58, 8219–8226 CrossRef CAS.
- [(1b)SiMe3][(C6F5)3BN3B(C6F5)3] was prepared by reaction of the FLP 1b/B(C6F5)3 with trimethylsilyl azide; 7 was prepared as previously described.27 The X-ray crystal structures of both compounds are presented in the ESI.†.
- Y. Xiong, S. Yao and M. Driess, Z. Naturforsch., B: Chem. Sci., 2013, 68b, 445–452 Search PubMed.
- A. C. Filippou, Y. N. Lebedev, O. Chernov, M. Straßmann and G. Schnakenburg, Angew. Chem., Int. Ed., 2013, 52, 6974–6978 CrossRef CAS PubMed.
-
(a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS;
(b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
-
(a) S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed;
(b) S. Grimme, WIREs Comput. Mol. Sci., 2011, 1, 211–228 CrossRef CAS PubMed.
- M. F. Ibad, P. Langer, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2011, 133, 21016–21027 CrossRef CAS PubMed.
- S. Antoniotti, V. Dalla and E. Duñach, Angew. Chem., Int. Ed., 2010, 49, 7860–7888 CrossRef CAS PubMed.
-
(a) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS PubMed;
(b) R. R. Holmes, Chem. Rev., 1996, 96, 927–950 CrossRef CAS PubMed;
(c) W. B. Jensen, Chem. Rev., 1978, 78, 1–22 CrossRef CAS.
- Furthermore, this indicates a very shallow potential energy surface (PES), which might be held responsible for the fact that the transition state for the triflate (green line) has a lower Gibbs free energy than the second ionic minimum structure B. Since all thermodynamic data are computed by means of statistical thermodynamics, and vibrations are treated as harmonic oscillators, especially for low frequency modes, large errors can be expected, see: P. Y. Ayala and H. B. Schlegel, J. Chem. Phys., 1998, 108, 2314–2325 CrossRef CAS PubMed.
- A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed.
- A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2012, 51, 2981–2984 CrossRef PubMed.
-
(a) S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS PubMed;
(b) S. N. Riduan, J. Y. Ying and Y. Zhang, ChemCatChem, 2013, 5, 1490–1496 CrossRef CAS PubMed.
- A. Winkler, M. Freytag, P. G. Jones and M. Tamm, J. Organomet. Chem., 2015, 775, 164–168 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, J. M. Alexander, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- M. Temprado, S. Majumdar, X. Cai, B. Captain and C. D. Hoff, Struct. Chem., 2013, 24, 2059–2068 CrossRef CAS PubMed.
- T. Voss, T. Mahdi, E. Otten, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 2367–2378 CrossRef CAS.
- C. M. Mömming, G. Kehr, B. Wibbeling, R. Fröhlich, B. Schirmer, S. Grimme and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 2414–2417 CrossRef PubMed.
- P. Feldhaus, B. Schirmer, B. Wibbeling, C. G. Daniliuc, R. Fröhlich, S. Grimme, G. Kehr and G. Erker, Dalton Trans., 2012, 41, 9135–9142 RSC.
- T. Böttcher, B. S. Bassil, L. Zhechkov, T. Heine and G.-V. Röschenthaler, Chem. Sci., 2012, 4, 77–83 RSC.
-
(a) M. Jia and M. Bandini, ACS Catal., 2015, 1638–1652 CrossRef CAS;
(b) A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864–876 CrossRef CAS PubMed;
(c) B. Alcaide and P. Almendros, Acc. Chem. Res., 2014, 47, 939–952 CrossRef CAS PubMed;
(d) Y.-M. Wang, A. D. Lackner and F. D. Toste, Acc. Chem. Res., 2014, 47, 889–901 CrossRef CAS PubMed;
(e) W. E. Brenzovich, Angew. Chem., Int. Ed., 2012, 51, 8933–8935 CrossRef CAS PubMed;
(f) L.-P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129–3139 RSC;
(g) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232–5241 CrossRef CAS PubMed;
(h) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776–1782 RSC;
(i) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed;
(j) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed;
(k) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed.
- S. Gaillard, P. Nun, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 5402–5408 CrossRef CAS.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 5625–5635 CrossRef CAS PubMed.
-
(a) P. L. Arnold and S. T. Liddle, Chem. Commun., 2006, 3959–3971 RSC;
(b) O. Kühl, Coord. Chem. Rev., 2009, 253, 2481–2492 CrossRef PubMed.
- H. Schmidbaur, H. G. Raubenheimer and L. Dobrzańska, Chem. Soc. Rev., 2014, 43, 345–380 RSC.
- R. W. Alder, P. R. Allen and S. J. Williams, J. Chem. Soc., Chem. Commun., 1995, 1267–1268 RSC.
-
(a) N. Bramananthan, E. Mas-Marzá, F. E. Fernandéz, C. E. Ellul, M. F. Mahon and M. K. Whittlesey, Eur. J. Inorg. Chem., 2012, 2213–2219 CrossRef CAS PubMed;
(b) S. Caddick, F. Cloke, N. Geoffrey, P. B. Hitchcock, K. de Lewis and K. Alexandra, Angew. Chem., Int. Ed., 2004, 43, 5824–5827 CrossRef CAS PubMed;
(c) N. M. Scott, R. Dorta, E. D. Stevens, A. Correa, L. Cavallo and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 3516–3526 CrossRef CAS PubMed;
(d) O. V. Zenkina, E. C. Keske, R. Wang and C. M. Crudden, Organometallics, 2011, 30, 6423–6432 CrossRef CAS;
(e) G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2011, 30, 5487–5492 CrossRef CAS;
(f) O. Rivada-Wheelaghan, M. Roselló-Merino, J. Díez, C. Maya, J. López-Serrano and S. Conejero, Organometallics, 2014, 33, 5944–5947 CrossRef CAS.
- A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2012, 51, 4526–4528 CrossRef CAS PubMed.
- H. F. T. Klare, F. T. Hendrik and M. Oestreich, Dalton Trans., 2010, 39, 9176–9184 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section including the synthesis and characterization of all compounds; details of the X-ray crystal structure determination and electronic structure calculations. CCDC 1052442–1052450. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01362c |
| ‡ A search of the Cambridge Database [C. R. Groom and F. H. Allen, Angew. Chem. Int. Ed., 2014, 53, 662–671] for the fragment {SiMe3 bonded to three-coordinate carbon} gave 2393 hits with 4604 individual fragments. The C–Si bond lengths ranged from 1.627 to 2.051 Å, with an average of 1.876 Å; our value would rank as tenth longest in this list. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence