DOI:
10.1039/C5DT01284H
(Paper)
Dalton Trans., 2015,
44, 13156-13162
Novel helical assembly of a Pt(II) phenylbipyridine complex directed by metal–metal interaction and aggregation-induced circularly polarized emission†
Received
3rd April 2015
, Accepted 10th June 2015
First published on 10th June 2015
Abstract
Pt(II) phenylbipyridine complexes possessing bis(phenylisoxazolyl)phenylacetylene ligands self-assembled to form stacked aggregates via Pt–Pt, π–π stacking, and dipole–dipole interactions. The assembled structures were influenced by the solvent properties. Non-helical assemblies found in chloroform displayed metal–metal-to-ligand charge transfer absorption and emission, whereas helical assemblies formed in toluene showed aggregation-induced enhancement of emission and aggregation-induced circularly polarized luminescence. The rates of the association and dissociation of the assemblies were significantly reduced in toluene, and the non-helical structures formed in chloroform were surprisingly memorized.
Introduction
Self-assembly of functional molecules offers a powerful tool for the development of discrete supramolecular organizations that display fascinating characteristics, such as optical, magnetic, mesogenic, and macroscopic properties.1 During self-assembly, reversible intermolecular interactions bind the molecular components together to define the direction and dimension of the target molecular organizations. Thus, self-assembly of molecular components in a desired manner can enable the generation of tailored functional organizations with the required properties. A variety of intermolecular interactions have been applied to the self-assembly. Reversible metallophilic interactions of d8- and d10-metals, including Au–Au,2 Pd–Pd,3 and Pt–Pt4 interactions, have received significant attention due to the development of polymeric linear metal arrays with unique photochemical properties and mesoscopic behaviors.5 Particularly, self-assemblies of square-planar Pt(II) complexes have displayed excellent phosphorescent properties. Although many self-assemblies of cationic Pt(II) complexes through Pt–Pt interactions have been reported,4 reports on the self-assembly of uncharged Pt(II) complexes in a common organic solution have been limited.6
We have reported that tris(phenylisoxazolyl)benzenes assemble to form helical assemblies, displaying unique photonic properties.7 During the course of the studies, the intermolecular dipole–dipole interaction of the isoxazolyl moieties has been crucial for developing the supramolecular helical assemblies. We envisioned developing a dimensionally organized platinum array via self-assembly of the neutral platinum complex with the aid of the intermolecular dipole–dipole interactions. In this study, new Pt(II)phenylbipyridine complexes possessing the bis(phenylisoxazolyl)phenylacetylene ligands S- and R-1, showing unusual aggregation-induced enhancement of emission (AIEE) and strong aggregation-induced circularly polarized luminescence (AICPL) are described (Scheme 1).
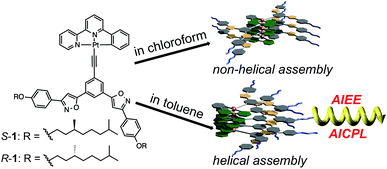 |
| | Scheme 1 Structure of molecules S- and R-1 and schematic representation of their self-assemblies in chloroform and in toluene. | |
Results and discussion
Synthesis
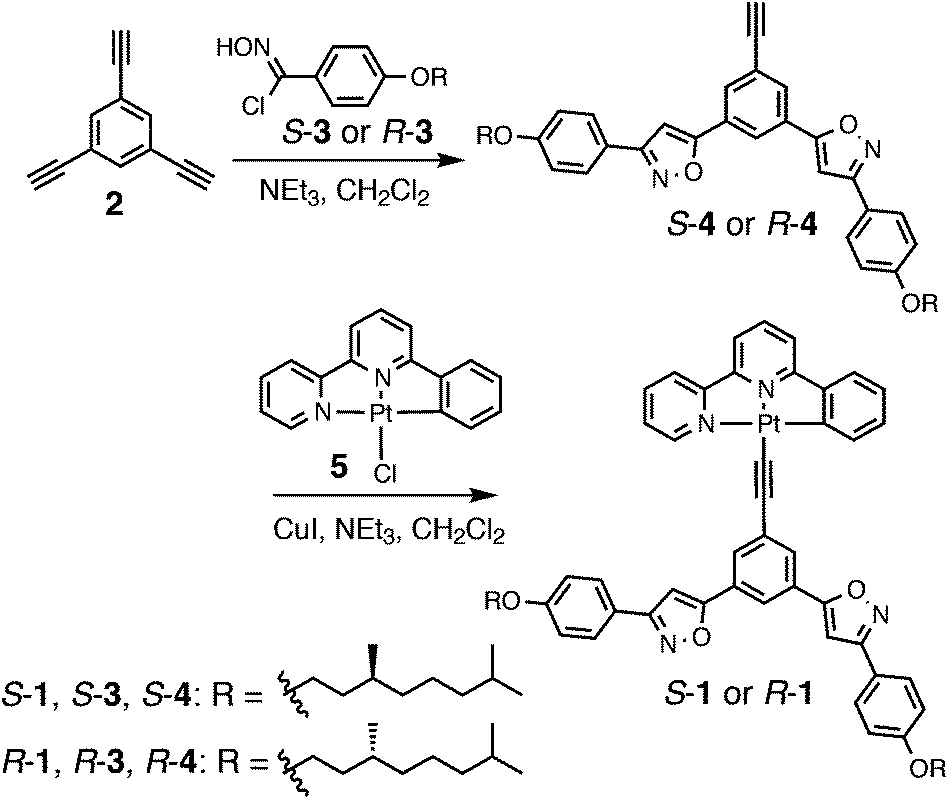
The synthesis of Pt(II) phenylbipyridine complexes S- and R-1 is shown in Scheme 2. Bis(phenylisoxazolyl)phenylacetylene ligands S- and R-4 were synthesized via 1,3-dipolar cycloaddition of 1,3,5-triethynylbenzene 2 with the p-alkoxyphenylnitrile oxides prepared in situ from the corresponding hydroximinoyl chlorides S- and R-3 by the treatment of triethylamine. Ligands S- and R-4 were introduced to the platinum center of 5 through a ligand exchange reaction catalyzed by copper(I) ion to afford the desired products S- and R-1.
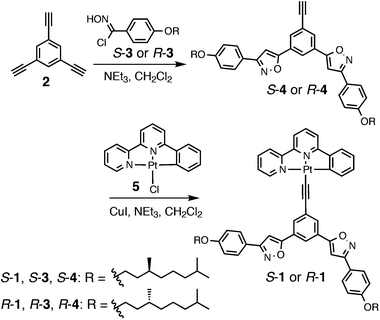 |
| | Scheme 2 Synthesis of Pt(II) phenylbipyridine complexes S- and R-1. | |
Self-assembly in chloroform
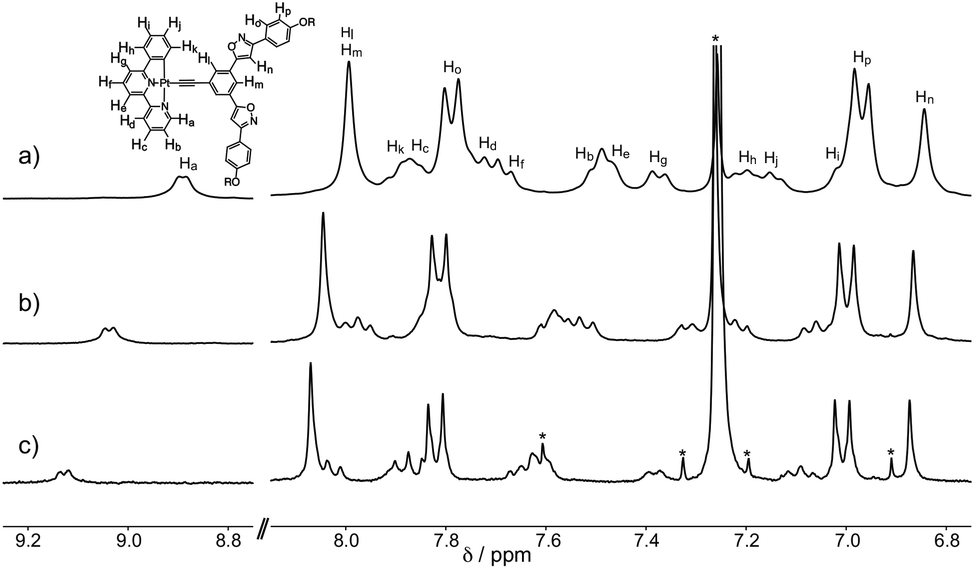
The self-assembling behavior of S-1 in chloroform-d1 was studied using 1H NMR spectroscopy. The 1H NMR signals of S-1 were concentration dependent (Fig. 1 and S2†). Aromatic protons Ha–Hp shifted upfield by increasing the concentration from 1.0 to 26.0 mmol L−1, indicating that S-1 formed stacked assemblies in which the aromatic protons are placed in the shielding regions produced by the aromatic rings of a neighboring S-1. A plot of the chemical shift changes of the protons versus the concentrations of S-1 produced hyperbolic curves. Non-linear curve-fitting analysis by applying the isodesmic model produced the estimated assembly-induced shifts (Δδ = –0.57, −0.20, −0.04, and −0.08 ppm for Ha, Hl, Hn, and Hp, respectively) and the association constant (KE = 93 ± 8 L mol−1). The phenylbipyridyl protons displayed larger upfield shifts than those of the other aromatic rings (Table S1†). These findings implied that S-1 stacked as piles along with the platinum center. The association constant of S-1 is larger than that of tris{(4-decyloxyphenyl)isoxazolyl}benzene (KE = 3.7 ± 0.3 L mol−1).7d Pt–Pt interactions accompanied by the π–π stacking and the dipole–dipole interactions drove the assembly of S-1.
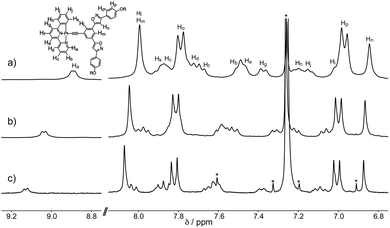 |
| | Fig. 1 Concentration-dependent 1H NMR spectra of S-1 at 298 K in chloroform-d1. The concentrations of S-1 are (a) 21.6, (b) 5.6, and (c) 1.0 mmol L−1. *indicates solvent peaks. | |
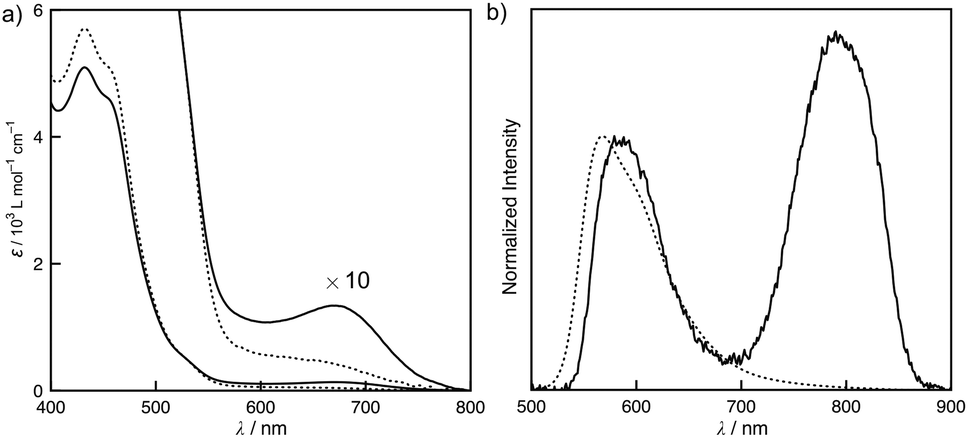
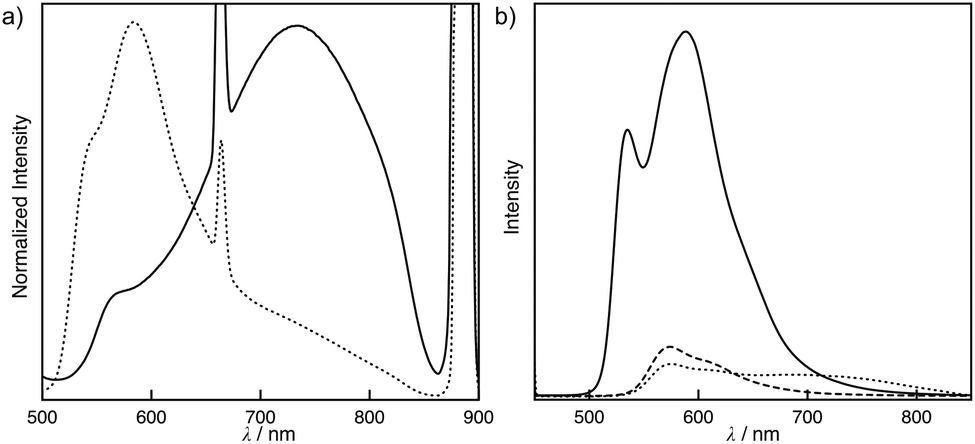
The absorption and emission spectra of S-1 provide insight into the intermolecular interactions throughout the self-assembly. Fig. 2 shows the concentration-dependent absorption and emission spectra of S-1 in chloroform. At a low concentration, the monomer absorption bands appeared at 431 and 456 nm, which primarily consist of HOMO → LUMO and HOMO−2 → LUMO excitations given by the TD-DFT calculation (Fig. S4†). The former transition was assigned to the mixed metal–ligand-to-ligand charge transfer (MLLCT) band. When a solution of S-1 was concentrated ten times, a new broad absorption band was obtained at 675 nm, which is recognized as a metal–metal-to-ligand charge transfer (MMLCT) band of the Pt–Pt bonds (Fig. 2a).4,6 In the diluted solution of S-1, a triplet MLCT emission of S-1 was only observed at 568 nm (Fig. 2b). After concentrating the solution, a 3MMLCT emission of the Pt–Pt bonds appeared at 790 nm.4,6 The concentration-dependent MMLCT absorption and emission reveal that the metallophilic interactions of the platinum centers obviously direct the stacked self-assembly of S-1. Our previous studies have shown that tris(phenylisoxazolyl)benzene-based stacked assemblies equilibrate between the P- and M-helical structures, which are biased by the presence of chiral side chains in cyclohexane.7c,d,g In contrast, S-1 was Circular Dichroism (CD) silent, even in the assembled state (Fig. S6†). Competitive solvation of chloroform most likely resulted in a weak association constant of 93 L mol−1, reflecting the limited sizes of the assemblies formed in solution. A high degree of polymerization for the assemblies of S-1 is required to adopt the helical organization, resulting in tight steric communication between the chiral side chains. The small sizes of the assemblies retain certain flexibility in molecular motions. The quantum yields (ϕ) of the phosphorescence decreased (ϕ = 0.027 at [S-1] = 5 × 10−4 mol L−1 and 0.003 at [S-1] = 5 × 10−3 mol L−1) after the assembly.
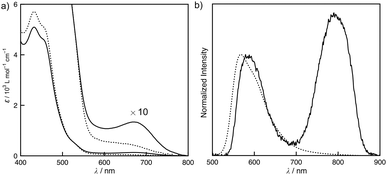 |
| | Fig. 2 Concentration-dependent (a) UV-vis absorption and (b) emission spectra of S-1 in chloroform at 25 °C. The concentrations of the solutions are 0.49 (dotted line) and 5.17 (solid line) mmol L−1. λex = 444 nm. | |
Helical self-assembly in toluene
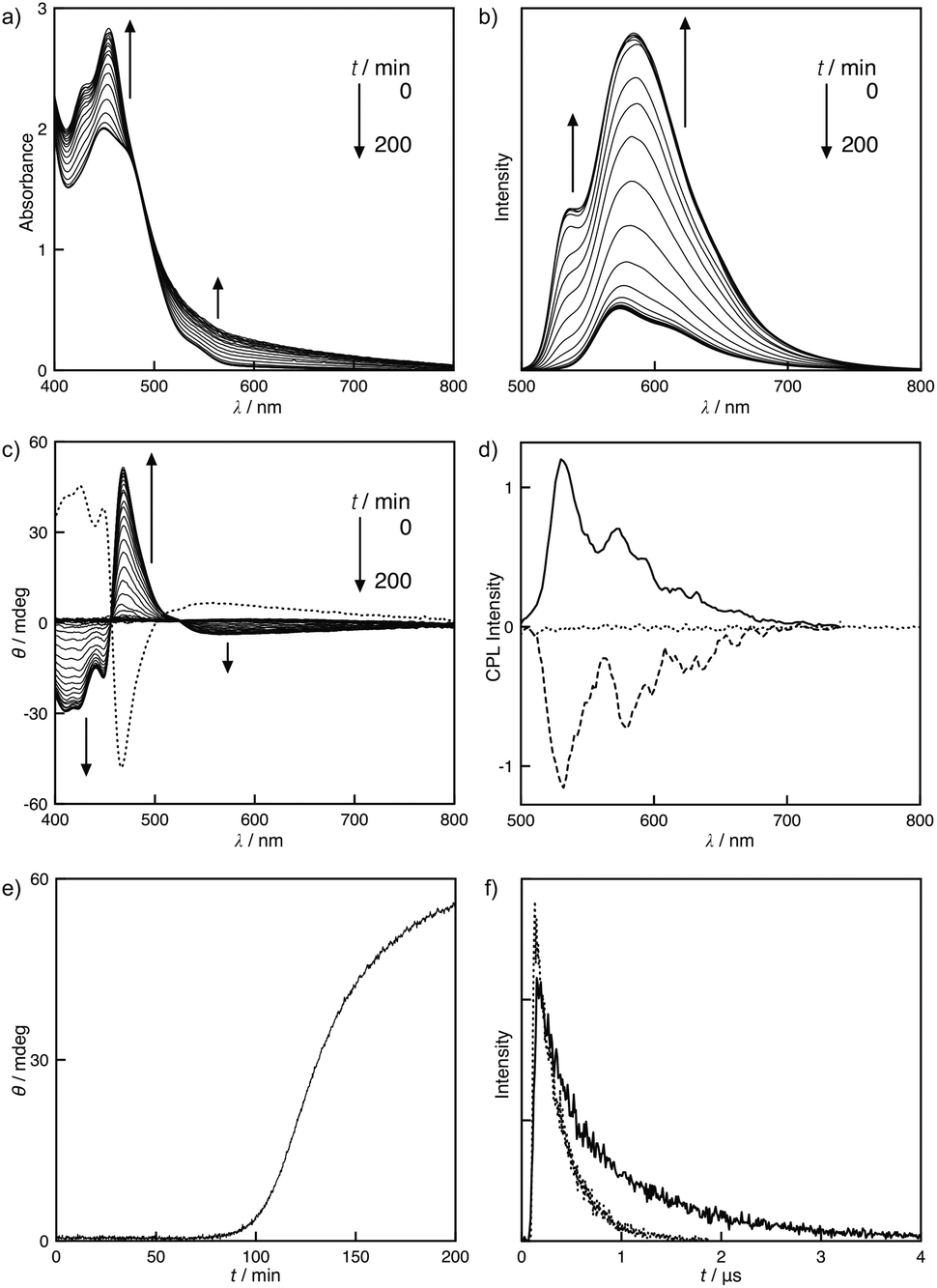
The solvent system directly influenced the molecular assembly of S-1. A toluene solution of S-1 led to the unique self-assembling behavior. Fig. 3a and 3b display the MLLCT absorption band at 449 nm and the 3MLCT emission band at 573 nm at 50 °C, which are assignable to the monomeric form of S-1. After cooling the toluene solution to 25 °C, an unusual time dependence was observed. New bands at 429 and 454 nm gradually emerged, implying that the self-assembling process was slow on a human time scale (Fig. 3a). The Pt–Pt bond formation was supported by the broad MMLCT absorption, which appeared at approximately 600 nm during the self-assembly. Accordingly, the polymeric platinum array was directed by the Pt–Pt interaction as well as the π–π stacking and the dipole–dipole interactions in toluene. To determine the size of the assemblies in toluene, dynamic light scattering (DLS) measurements were performed. The large assemblies were formed with a hydrodynamic radius of 1.3 ± 0.3 μm in toluene, while the sizable assemblies were not detected in chloroform (Fig. S8†). This slow assembling process was monitored using CD and emission spectra. When the solution was cooled to 25 °C, the CD was silent. After a brief period, the positive Cotton effects were strongly enhanced at 480 nm, resulting in a dissymmetry factor gabs of 0.002, whereas the negative Cotton effects were observed on the MMLCT bands (Fig. 3c). The CD spectra of S- and R-1 displayed a mirror image relationship, suggesting that S- and R-1 formed helical assemblies, and the helical sense was directed by the chiral side chains. Plots of ellipticity θ versus time resulted in a sigmoidal response, implying that the assembly process was auto-catalytic (Fig. 3e). The time-dependent emission changes were also observed on the assembly. The emission was found at 573 nm when the solution was cooled to 25 °C. New bands at 535 and 584 nm emerged after the solution was allowed to sit for a certain amount of time (Fig. 3b).
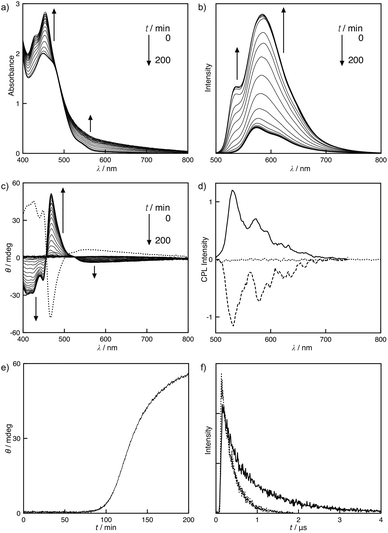 |
| | Fig. 3 Time dependent (a) UV-vis absorption and (b) emission spectra of S-1 in toluene at 25 °C. (c) CD spectral changes of S-1 as a function of time (solid line) and the corresponding spectrum of R-1 after the assembly (dotted line) in toluene at 25 °C. (d) CPL spectra of S-1 before (dotted line) and after (solid line) the assembly and R-1 after the assembly (dashed line) in toluene at 25 °C. (e) Plot of change in ellipticity θ at 469 nm versus time for a solution of S-1 in toluene at 25 °C. (f) Time-resolved emission decay curve of S-1 in toluene at 25 °C before (dotted line) and after (solid line) the assembly. [S-1] = [R-1] = 0.50 mmol L−1. λex = 444 nm. | |
Unusual AIEE8 was observed through the assembly. The emission strongly increased upon the formation of the assembly in toluene (Fig. 3b). The quantum yields of S-1 were also time dependent due to the slow assembly. Immediate cooling of the solution of S-1 to 25 °C resulted in an intense emission with a quantum yield of 0.023. After stirring for 24 h, the emission became more efficient with a quantum yield of 0.093. 3MMLCT emission band was not found in a toluene solution of S-1 after the assembly. The emission lifetime measurements of S-1 in toluene provided more detailed insight into the self-assembly. A rapidly cooled solution of S-1 exhibited a single-exponential emission decay function with a lifetime τ of 0.28 μs. When the solution was aged for 24 h, a double-exponential function was observed with lifetimes τ1 and τ2 of 0.25 and 1.1 μs, respectively (Fig. 3f). Accordingly, S-1 exists in two photochemically non-equivalent states: the lifetime τ1 corresponded to the monomeric form, and the long-lived species arose from the self-assembled aggregates. A longer emission lifetime τ2 of 1.1 μs reveals that the AIEE is due to the reduction of molecular motion through the self-assembly of S-1. The molecular motion is partially restricted in the stacked organization, which suppresses the non-radiative decay channels, thus increasing a portion of the radiative decay process.9
Circularly polarized luminescence (CPL) measurements were performed to understand the selective emission of right- and left-handed circularly polarized light from the molecules10 and chiral aggregates.4a,7c,11,12 No appreciable CPL was observed either from the monomeric state of the molecule or from solutions subjected to analysis immediately after cooling to 25 °C. Interestingly, the helical assembly of S-1 and R-1 exhibited mirror image CPL spectra at the fluorescence wavelength corresponding to the molecular aggregates with a high dissymmetry factor (glum) of 0.01 (Fig. 3d). These findings clearly suggest that the helical assemblies can act as efficient sources of circularly polarized light. Moreover, the CPL of S- and R-1 is regulated via assembling and disassembling of S-1 and R-1, respectively. The observed luminescence dissymmetric factor is significantly larger than the typical glum values exhibited by chiral organic molecules.10
Polymorphism and memory effect
An unusual polymorphism and the structural memory effect13 were observed in toluene. S-1 generated two types of luminescent solids, A and B, obtained by slow evaporation of its chloroform solution and toluene solution, respectively. The 1H NMR spectra of these solids dissolved in chloroform-d were almost identical to one another (Fig. S13†). The emission spectra of these solids were inconsistent with each other. Solid A displayed two emission bands at 564 and 733 nm corresponding to the 3MLCT and 3MMLCT bands of the non-helical structures in a chloroform solution of S-1, respectively (Fig. 4a). Solid B generated an emission band at 583 nm, as observed in the helical assemblies in toluene. The weak 3MMLCT emission was observed at approximately 750 nm. The quantum yields of solids A and B were 0.003 and 0.025, respectively (Fig. S12†). Accordingly, the assembled structures found in the chloroform and toluene solutions were maintained, even in the solid state (Fig. 2b and 3b). When solid A was dissolved in toluene at room temperature, two emission bands at 574 and 732 nm unexpectedly corresponded to those of the non-helical structures of the assemblies (Fig. 4b dotted line), and the CD was silent (Fig. S9†). These findings reveal that the non-helical structures are trapped in metastable states in toluene. After warming the solution to 50 °C, the emission band at 732 nm disappeared, indicating that a monomeric form of S-1 was dominant (Fig. 4b, dashed line). Helical structures developed while the solution was maintained at room temperature for a certain period, displaying the intense emission bands at 580 nm and the strong Cotton effect (Fig. 3a–c, 4b, solid line). Although the helical structures were thermodynamically more stable than the non-helical structures in toluene, a sizable energetic barrier existed between the non-helical and helical forms, the interconversion of which was relatively slow with a half-life of 66 h at 25 °C (Fig. S10†).
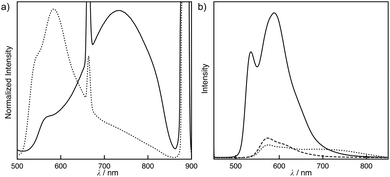 |
| | Fig. 4 (a) Emission spectra of solid A (solid line) and solid B (dotted line). (b) Emission spectra at various temperatures of solid A dissolved in toluene. The temperatures are 25 °C before heating (dotted line), 50 °C (dashed line), and 25 °C after heating (solid line). [S-1] = 0.50 mmol L−1. λex = 444 nm. | |
Microscopic study
The morphologies of the assemblies of S-1 were observed using transmission electron microscopy (TEM) and atomic force microscopy (AFM). The TEM image of the assembly showed nanofibers with an average width of 5–8 nm (Fig. 5a). The length of the molecule is ca. 1.8 nm, which indicates that the observed fibers are bundles of the assemblies. The AFM images provided more detailed insights into the assembled structures. When the toluene solution of S-1 was spin-coated on mica, networked fibers with a uniform height of 2 nm were observed (Fig. 5b and S14†). The height well fit to the length of S-1, suggesting that S-1 forms a one-dimensional stacked assembly. However, helical fibers were observed when the toluene solution was spin-coated on HOPG (Fig. 5c, d, and S15†). The pitch of the helix is ca. 30 nm. Interestingly, a bundle of the helices was observed (Fig. 5d).
 |
| | Fig. 5 (a) TEM image of S-1. (b) AFM image of S-1 on mica. (c), (d) AFM images of S-1 on HOPG. The sizes of the AFM images are (b) 5 μm × 5 μm, and (c), (d) 500 nm × 500 nm. The samples were prepared from the toluene solution of S-1 after the assembly. | |
Conclusions
We demonstrated the unique self-assembling behaviors and optical properties of novel platinum(II) phenylbipyridine complexes possessing bis(phenylisoxazolyl)phenylacetylene ligands. S-1 formed stacked assemblies in chloroform and in toluene, but the assembled structures were dependent on the solvent properties. In chloroform, S-1 stacked in a non-helical fashion, whereas S-1 formed helical assemblies in toluene. The MMLCT absorption and emission bands reveal that the assemblies are directed via the Pt–Pt, π–π stacking, and dipole–dipole interactions. The assembling and disassembling processes in toluene are slow on a human time scale, leading to the structure memory of the non-helical assemblies. The assemblies of S-1, formed in toluene after being aged, display AIEE and AICPL. These emission properties provide possible applications of S-1 as luminescent materials in the solid state or condensed phase.
Experimental section
5,5′-(5-Ethynyl-1,3-phenylene)bis(3-(4-((S)-3,7-dimethyloctyloxy)phenyl)isoxazole) (S-4)
To a solution of 1,3,5-triethynylbenzene 2 (750 mg, 5.00 mmol) and triethylamine (7.0 ml, 50 mmol) in CH2Cl2 (50 ml) was added chlorooxime S-3 (3.22 g, 10.3 mmol) in CH2Cl2 (10 ml). After being stirred at room temperature for 1 day under an argon atmosphere, the reaction mixture was concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexane) to give S-4 (696 mg, 20%) as a white solid. M.p. 98–99 °C; 1H NMR (300 MHz, CDCl3): δ 8.24 (t, J = 3.0 Hz, 1H), 7.98 (d, J = 3.0 Hz, 2H), 7.79 (d, J = 9.7 Hz, 4H), 6.99 (d, J = 9.7 Hz, 4H), 6.89 (s, 2H), 4.05 (t, J = 7.7 Hz, 4H), 3.24 (s, 1H), 1.90–1.81 (m, 4H), 1.75–1.47 (m, 8H), 1.42–1.10 (m, 8H), 0.96 (d, J = 7.0 Hz, 6H), and 0.88 (d, J = 7.3 Hz, 6H) ppm; 13C NMR (75 MHz, CDCl3): δ 168.3, 163.0, 161.0, 130.6, 128.9, 128.4, 124.3, 123.1, 121.1, 115.1, 98.8, 82.0, 79.6, 66.7, 39.5, 37.5, 36.3, 30.1, 28.2, 24.9, 22.9, and 19.9 ppm; IR (KBr): ν 2952, 2925, 2867, 2095, 1612, 1562, 1488, 1462, 1438, 1385, 1294, 1252, 1176, 1115, 949, 877, 836, 791, and 649 cm−1; HRMS (ESI+) calcd for C46H57N2O4m/z 701.4316 [M + H]+, found m/z 701.4313; Anal. calcd for C46H56N2O4: C 78.82, H 8.05, N 4.00, found C 78.76, H 8.02, N 4.02%.
5,5′-(5-Ethynyl-1,3-phenylene)bis(3-(4-((R)-3,7-dimethyloctyloxy)phenyl)isoxazole) (R-4)
To a solution of 1,3,5-triethynylbenzene 2 (403 mg, 2.68 mmol) and triethylamine (3.6 ml, 26 mmol) in CH2Cl2 (10 ml) was added chlorooxime R-3 (1.67 g, 5.37 mmol) in CH2Cl2 (10 ml). After being stirred at room temperature for 1 day under an argon atmosphere, the reaction mixture was concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexane) to give R-4 (300 mg, 16%) as a white solid. M.p. 98–99 °C; 1H NMR (300 MHz, CDCl3): δ 8.25 (t, J = 3.0 Hz, 1H), 8.00 (d, J = 3.0 Hz, 2H), 7.81 (d, J = 9.7 Hz, 4H), 7.01 (d, J = 9.7 Hz, 4H), 6.91 (s, 2H), 4.06 (m, 4H), 3.24 (s, 1H), 1.90–1.81 (m, 4H), 1.75–1.47 (m, 8H), 1.42–1.10 (m, 8H), 0.96 (d, J = 7.0 Hz, 6H), and 0.88 (d, J = 7.3 Hz, 6H) ppm; 13C NMR (75 MHz, CDCl3): δ 168.3, 163.1, 161.0130.6, 128.9, 128.4, 124.3, 123.1, 121.1, 115.2, 98.8, 82.0, 79.6, 66.7, 39.5, 37.5, 36.3, 30.1, 28.2, 24.9, 22.8, and 19.9 ppm; IR (KBr): ν 2952, 2925, 2095, 1611, 1564, 1526, 1488, 1460, 1439, 1385, 1295, 1251, 1176, 949, 876, 836, 790, 756, 730, and 649 cm−1; HRMS (ESI+) calcd for C46H57N2O4m/z 701.4321 [M + H]+, found m/z 701.4313; Anal. calcd for C46H56N2O4: C 78.82, H 8.05, N 4.00, found C 78.81, H 8.09, N 4.02%.
(6-Phenyl-2,2′-bipyridine){5,5′-(5-ethynyl-1,3-phenylene)bis(3-(4-((S)-3,7-dimethyloctyloxy)phenyl)isoxazole)}platinum (S-1)
To a solution of (6-phenyl-2,2′-bipyridine)chloroplatinum 5 (113 mg, 244 μmol) and ligand S-4 (171 mg, 244 μmol) in CH2Cl2 (5 ml) and DMF (5 ml) was added triethylamine (0.5 ml, 3.6 mmol). The mixture was degassed by bubbling argon for 30 min. CuI (8 mg, 42 μmol) was added to the reaction mixture and the resulting mixture was stirred at room temperature for 1 day under an argon atmosphere in the dark. The reaction mixture was extracted with CH2Cl2 and the organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude product was further purified by column chromatography on silica gel (EtOAc/hexane, CHCl3/MeOH) to give S-1 (216 mg, 79%) as an orange solid. M.p. 221–222 °C; 1H NMR (300 MHz, CDCl3): δ 9.21 (d, J = 6.0 Hz, 1H), 8.04 (s, 1H), 8.04 (s, 2H), 8.04–7.95 (m, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.81 (d, J = 9.7 Hz, 4H), 7.82–7.76 (m, 2H), 7.57 (d, J = 6.7 Hz, 1H), 7.55–7.52 (m, 1H), 7.49 (d, J = 3.3 Hz, 1H), 7.29 (t, J = 6.7 Hz, 1H), 7.20 (d, J = 10 Hz, 1H), 7.05 (t, J = 6.7 Hz, 1H), 6.99 (d, J = 10 Hz, 4H), 6.86 (s, 2H), 4.05 (t, J = 3.3 Hz, 4H), 1.93–1.78 (m, 4H), 1.75–1.48 (m, 8H), 1.43–1.09 (m, 8H), 0.96 (d, J = 10 Hz, 6H), and 0.88 (d, J = 6.7 Hz, 12H) ppm; 13C NMR (75 MHz, CDCl3): δ 169.6, 165.7, 162.9, 160.9, 158.0, 154.6, 152.2, 146.9, 142.1, 139.1, 139.0, 138.6, 131.8, 130.6, 128.4, 128.1, 127.9, 124.8, 124.1, 122.7, 121.5, 119.5, 118.7, 117.8, 115.1, 104.8, 98.15, 66.7, 39.5, 37.5, 36.4, 30.1, 28.21, 24.9, 22.8, and 19.9 ppm; IR (KBr): ν 3207, 2954, 2927, 2870, 1613, 1564, 1528, 1462, 1442, 1385, 1295, 1252, 1177, 1115, 1018, 950, 879, 837, 790, and 764 cm−1; HRMS (ESI+) calcd for C62H66N4O4NaPt m/z 1148.4630 [M + Na]+, found m/z 1148.4630; Anal. calcd for C62H66N4O4Pt·(H2O)1.3: C 64.77, H 6.01, N 4.87, found C 64.59, H 5.97, N 4.71%.
(6-Phenyl-2,2′-bipyridine){5,5′-(5-ethynyl-1,3-phenylene)bis(3-(4-((R)-3,7-dimethyloctyloxy)phenyl)isoxazole)}platinum (R-1)
To a solution of (6-phenyl-2,2′-bipyridine)chloroplatinum 5 (107 mg, 232 μmol) and isoxazolylbenzene R-4 (300 mg, 428 μmol) in CH2Cl2 (10 ml) was added triethylamine (0.5 ml, 3.6 mmol). The mixture was degassed by bubbling argon for 30 min. CuI (10 mg, 53 μmol) was added to the reaction mixture and the resulting mixture was stirred at room temperature for 1 day under an argon atmosphere in the dark. The reaction mixture was extracted with CH2Cl2 and the organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude product was further purified by column chromatography on silica gel (EtOAc/hexane, CHCl3/MeOH) to give R-1 (195 mg, 75%) as an orange solid. M.p. 221–222 °C; 1H NMR (300 MHz, CDCl3): δ 9.15 (d, J = 6.0 Hz, 1H), 8.03 (s, 1H), 8.03 (s, 2H), 8.04–7.95 (m, 2H), 7.81 (d, J = 9.7 Hz, 4H), 7.82–7.72 (m, 2H), 7.58–7.52 (m, 2H), 7.48 (d, J = 3.3 Hz, 1H), 7.30 (d, J = 6.7 Hz, 1H), 7.20 (t, J = 10 Hz, 1H), 7.05 (t, J = 6.7 Hz, 1H), 6.99 (d, J = 10 Hz, 4H), 6.86 (s, 2H), 4.05 (t, J = 3.3 Hz, 4H), 1.93–1.78 (m, 4H), 1.75–1.48 (m, 8H), 1.43–1.09 (m, 8H), 0.96 (d, J = 10 Hz, 6H), and 0.88 (d, J = 6.7 Hz, 12H) ppm; 13C NMR (75 MHz, CDCl3): δ 169.6, 165.7, 162.9, 160.9, 158.0, 154.7, 151.9, 147.0, 142.1, 139.1, 139.0, 138.6, 131.8, 130.5, 128.4, 128.1, 127.9, 124.7, 124.1, 122.9, 121.5, 119.5, 118.6, 117.9, 115.1, 104.9, 98.24, 66.7, 39.5, 37.5, 36.4, 30.1, 28.21, 24.9, 22.8, and 19.9 ppm; IR (KBr): ν 3737, 3567, 2954, 2927, 2870, 1717, 1613, 1563, 1528, 1509, 1459, 1385, 1295, 1177, 1115, 1018, 950, 837, 790, and 765 cm−1; HRMS (ESI+) calcd for C62H66N4O4NaPt m/z 1148.4630 [M + Na]+, found m/z 1148.4624; Anal. calcd for C62H66N4O4Pt·(H2O)1.3: C 64.77, H 6.01, N 4.87, found C 64.61, H 5.78, N 4.69%.
Acknowledgements
This work was supported by Grant-in-Aids for Scientific Research (B) (no. 24350060), Challenging Exploratory Research (no. 23655105), and Grant-in Aids for Young Scientists (B) (no. 26810051) of JSPS, as well as Grant-in-Aids for Scientific Research on Innovative Areas, “Stimuli-responsive Chemical Species for Creation of Functional Molecules” and “New Polymeric Materials Based on Element-Blocks” (no. 25109529, 25102532), and Platform for Dynamic Approaches to Living System of MEXT.
Notes and references
-
(a) S. I. Stupp and L. C. Palmer, Chem. Mater., 2014, 26, 507 CrossRef CAS;
(b) H. Li, J. Choi and T. Nakanishi, Langmuir, 2013, 29, 5394 CrossRef CAS PubMed;
(c) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed;
(d) P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009 CrossRef CAS PubMed;
(e) L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596 CrossRef CAS PubMed;
(f) T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38 CrossRef CAS PubMed;
(g) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS PubMed.
-
(a) Y. Chen, G. Cheng, K. Li, D. P. Shelar, W. Lu and C.-M. Che, Chem. Sci., 2014, 5, 1348 RSC;
(b) J.-J. Zhang, W. Lu, R. W.-Y. Sun and C.-M. Che, Angew. Chem., Int. Ed., 2012, 51, 4882 CrossRef CAS PubMed;
(c) W. Lu, K. T. Chan, S.-X. Wu, Y. Chen and C.-M. Che, Chem. Sci., 2012, 3, 752 RSC;
(d) H. O. Lintang, K. Kinbara, K. Tanaka, T. Yamashita and T. Aida, Angew. Chem., Int. Ed., 2010, 49, 4241 CrossRef CAS PubMed;
(e) A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179 CrossRef CAS PubMed.
- M. J. Mayoral, C. Rest, V. Stepanenko, J. Schellheimer, R. Q. Albuquerque and G. Fernández, J. Am. Chem. Soc., 2013, 135, 2148 CrossRef CAS PubMed.
-
(a) X.-P. Zhang, V. Y. Chang, J. Liu, X.-L. Yang, W. Huang, Y. Li, C.-H. Li, G. Muller and X.-Z. You, Inorg. Chem., 2015, 54, 143 CrossRef CAS PubMed;
(b) M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952 CrossRef CAS PubMed;
(c) R. Sekiya, Y. Tsutsui, W. Choi, T. Sakurai, S. Seki, Y. Bando and H. Maeda, Chem. Commun., 2014, 50, 10615 RSC;
(d) S. Y.-L. Leung and V. W.-W. Yam, Chem. Sci., 2013, 4, 4228 RSC;
(e) K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424 CrossRef CAS PubMed;
(f) C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136 CrossRef CAS PubMed;
(g) W. Lu, Y. Chen, V. A. L. Roy, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 7621 CrossRef CAS PubMed;
(h) M.-Y. Yuen, V. A. L. Roy, W. Lu, S. C. F. Kui, G. S. M. Tong, M.-H. So, S. S.-Y. Chui, M. Muccini, J. Q. Ning, S. J. Xu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9895 CrossRef CAS PubMed.
-
(a) N. Lanigan and X. Wang, Chem. Commun., 2013, 49, 8133 RSC;
(b) H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC;
(c) I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834 CrossRef CAS PubMed.
-
(a) A. Amar, H. Meghezzi, J. Boixel, H. Le Bozec, V. Guerchais, D. Jacquemin and A. Boucekkine, J. Phys. Chem. A, 2014, 118, 6278 CrossRef CAS PubMed;
(b) M. Mauro, A. Aliprandi, C. Cebrián, D. Wang, C. Kübel and L. De Cola, Chem. Commun., 2014, 50, 7269 RSC;
(c) Y.-J. Tian, E. W. Meijer and F. Wang, Chem. Commun., 2013, 49, 9197 RSC;
(d) A. Gross, T. Moriuchi and T. Hirao, Chem. Commun., 2013, 49, 1163 RSC;
(e) T. Moriuchi, Y. Sakamoto, S. Noguchi, T. Fujiwara, S. Akine, T. Nabeshima and T. Hirao, Dalton Trans., 2012, 41, 8524 RSC;
(f) C. A. Strassert, C.-H. Chien, M. D. G. Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946 CrossRef CAS PubMed.
-
(a) T. Haino, Y. Ueda, T. Hirao, T. Ikeda and M. Tanaka, Chem. Lett., 2014, 43, 414 CrossRef CAS;
(b) T. Haino, Y. Hirai, T. Ikeda and H. Saito, Org. Biomol. Chem., 2013, 11, 4164 RSC;
(c) T. Ikeda, T. Masuda, T. Hirao, J. Yuasa, H. Tsumatori, T. Kawai and T. Haino, Chem. Commun., 2012, 48, 6025 RSC;
(d) M. Tanaka, T. Ikeda, J. Mack, N. Kobayashi and T. Haino, J. Org. Chem., 2011, 76, 5082 CrossRef CAS PubMed;
(e) T. Haino and H. Saito, Aust. J. Chem., 2010, 63, 640 CrossRef CAS;
(f) T. Haino and H. Saito, Synth. Met., 2009, 159, 821 CrossRef CAS PubMed;
(g) T. Haino, M. Tanaka and Y. Fukazawa, Chem. Commun., 2008, 468 RSC;
(h) M. Tanaka, T. Haino, K. Ideta, K. Kubo, A. Mori and Y. Fukazawa, Tetrahedron, 2007, 63, 652 CrossRef CAS PubMed;
(i) T. Haino, M. Tanaka, K. Ideta, K. Kubo, A. Mori and Y. Fukazawa, Tetrahedron Lett., 2004, 45, 2277 CrossRef CAS PubMed.
-
(a) J. Seo, J. W. Chung, J. E. Kwon and S. Y. Park, Chem. Sci., 2014, 5, 4845 RSC;
(b) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC;
(c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC;
(d) Z. Zhao, S. Chen, X. Shen, F. Mahtab, Y. Yu, P. Lu, J. W. Y. Lam, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 686 RSC;
(e) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- The AIEE might be due to the restricted rotation of the C(sp2)–C(sp) bonds in the aggregated structures. See the following reference: S. K. Albert, H. V. P. Thelu, M. Golla, N. Krishnan, S. Chaudhary and R. Varghese, Angew. Chem., Int. Ed., 2014, 53, 8352–8357 CrossRef CAS PubMed.
-
(a) C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson, G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Réau and J. Crassous, Chem. Sci., 2014, 5, 1915 RSC;
(b) Y. Morisaki, M. Gon, T. Sasamori, N. Tokitoh and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 3350 CrossRef CAS PubMed;
(c) H. Oyama, K. Nakano, T. Harada, R. Kuroda, M. Naito, K. Nobusawa and K. Nozaki, Org. Lett., 2013, 15, 2104 CrossRef CAS PubMed;
(d) H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266 CrossRef CAS PubMed;
(e) J. E. Field, G. Muller, J. P. Riehl and D. Venkataraman, J. Am. Chem. Soc., 2003, 125, 11808 CrossRef CAS PubMed;
(f) E. Peeters, M. P. T. Christiaans, R. A. J. Janssen, H. F. M. Schoo, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1997, 119, 9909 CrossRef CAS;
(g) J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1 CrossRef CAS.
-
(a) H. Maeda, W. Hane, Y. Bando, Y. Terashima, Y. Haketa, H. Shibaguchi, T. Kawai, M. Naito, K. Takaishi, M. Uchiyama and A. Muranaka, Chem. – Eur. J., 2013, 19, 16263 CrossRef CAS PubMed;
(b) J. Liu, H. Su, L. Meng, Y. Zhao, C. Deng, J. C. Y. Ng, P. Lu, M. Faisal, J. W. Y. Lam, X. Huang, H. Wu, K. S. Wong and B. Z. Tang, Chem. Sci., 2012, 3, 2737 RSC;
(c) A. Gopal, M. Hifsudheen, S. Furumi, M. Takeuchi and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 10505 CrossRef CAS PubMed;
(d) T. Kaseyama, S. Furumi, X. Zhang, K. Tanaka and M. Takeuchi, Angew. Chem., Int. Ed., 2011, 50, 3684 CrossRef CAS PubMed;
(e) F. C. Spano, S. C. J. Meskers, E. Hennebicq and D. Beljonne, J. Am. Chem. Soc., 2007, 129, 7044 CrossRef CAS PubMed;
(f) A. Satrijo, S. C. J. Meskers and T. M. Swager, J. Am. Chem. Soc., 2006, 128, 9030 CrossRef CAS PubMed;
(g) K. E. S. Phillips, T. J. Katz, S. Jockusch, A. J. Lovinger and N. J. Turro, J. Am. Chem. Soc., 2001, 123, 11899 CrossRef CAS PubMed.
-
(a) J. Kumar, T. Nakashima, H. Tsumatori and T. Kawai, J. Phys. Chem. Lett., 2014, 5, 316 CrossRef CAS;
(b) J. Kumar, T. Nakashima, H. Tsumatori, M. Mori, M. Naito and T. Kawai, Chem. – Eur. J., 2013, 19, 14090 CrossRef CAS PubMed;
(c) H. Tsumatori, T. Nakashima and T. Kawai, Org. Lett., 2010, 12, 2362 CrossRef CAS PubMed;
(d) H. Tsumatori, T. Nakashima, J. Yuasa and T. Kawai, Synth. Met., 2009, 159, 952 CrossRef CAS PubMed.
-
(a) J.-S. Zhao, Y.-B. Ruan, R. Zhou and Y.-B. Jiang, Chem. Sci., 2011, 2, 937 RSC;
(b) F. Helmich, C. C. Lee, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 16753 CrossRef CAS PubMed;
(c) A. Mammana, A. D'Urso, R. Lauceri and R. Purrello, J. Am. Chem. Soc., 2007, 129, 8062 CrossRef CAS PubMed;
(d) M. Ziegler, A. V. Davis, D. W. Johnson and K. N. Raymond, Angew. Chem., Int. Ed., 2003, 42, 665 CrossRef CAS PubMed;
(e) L. J. Prins, F. De Jong, P. Timmerman and D. N. Reinhoudt, Nature, 2000, 408, 181 CrossRef CAS PubMed;
(f) A. Sugasaki, M. Ikeda, M. Takeuchi, A. Robertson and S. Shinkai, J. Chem. Soc., Perkin Trans. 1, 1999, 3259 RSC;
(g) Y. Furusho, T. Kimura, Y. Mizuno and T. Aida, J. Am. Chem. Soc., 1997, 119, 5267 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Results of TD-DFT calculation, spectroscopic data, photographs, AFM images, and copies of 1H and 13C NMR spectra for the reported compounds. See DOI: 10.1039/c5dt01284h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a