
Synthesis of magnesium ZIF-8 from Mg(BH4)2†
S.
Horike
*ab,
K.
Kadota
a,
T.
Itakura
c,
M.
Inukai
d and
S.
Kitagawa
ad
aDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: horike@sbchem.kyoto-u.ac.jp
bJapan Science and Technology Agency, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cDENSO Corporation, 1-1 Showa-cho, Kariya, Aichi 448-8661, Japan
dInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan
First published on 1st April 2015
Abstract
Porous Mg(2-methyl imidazolate)2 (Mg-ZIF-8) was synthesised from Mg(BH4)2 as a precursor under an Ar atmosphere. It possesses an uncommon tetrahedral Mg2+–N coordination geometry that is stabilised by the formation of a framework, and it exhibits a Brunauer–Emmett–Teller surface area greater than 1800 m2 g−1.
Porous coordination polymers (PCPs) or metal–organic frameworks (MOFs) that consist of metal ions and bridging ligands have been widely investigated for their possible applications in gas storage, separation and catalysis.1–3 Some properties of these materials are superior to those of conventional porous solids, and numerous attempts to adapt them to industrial applications have been reported.4 In view of their applications, greater chemical and thermal stabilities are demanded, and several examples of stable MOFs with stable and inert coordination geometries have been reported.5,6
On the other hand, the construction of MOFs with uncommon coordination geometries is particularly challenging. The stabilization of such geometries by lattice formation has recently been investigated. For example, a tetrahedral Ni2+–O coordination geometry was successfully incorporated into a Zn2+ MOF by post-synthetic ion metathesis.7 In this case, the maximum doping ratio of the tetrahedral Ni2+–O centre in the framework was 25%, and the doping of Ni2+ beyond 25% led to a structural collapse. The stabilisation of uncommon coordination geometries in MOFs is fundamentally important in the area of coordination chemistry and for understanding catalytic reactions. Among a large variety of coordination modes, a tetrahedral Mg2+–4X (X = N, O, and so on) coordination geometry is a rare example. The tetrahedral geometry in this case has X–Mg2+–X angles in the range of 105°–115° (ideal tetrahedral angle is 109.5°). According to the Cambridge Crystallographic Data Centre database, no crystal structure of a coordination polymer and an MOF having tetrahedral Mg2+–N and Mg2+–O coordination geometries is known. Only four crystal structures of discrete molecules with the tetrahedral Mg2+–N coordination geometry are known, and they all require bulky ligands.8–10 The literature contains five reports of the tetrahedral Mg2+–O coordination geometry, although many reports on distorted four-coordinate Mg2+ compounds have been published. For these reasons, in this study, we aimed at stabilising the tetrahedral Mg2+–N coordination geometry in an MOF and characterise its structural stability and porosity.
Moreover, to construct an MOF with the tetrahedral Mg2+–N coordination geometry, we employed a well-known cubic structure. [Zn(2 mim)2] (Fig. 1, Zn-ZIF-8, 2 mimH = 2-methyl imidazole) has a tetrahedral Zn2+–N coordination geometry and sodalite topology in its 3D extended structure.11,12 The N–Zn2+–N angle in the structure is 109.3°, and it exhibits a nondistorted, ideal tetrahedral coordination geometry. Zn-ZIF-8 has been widely studied in applications involving gas storage and separation because of its high porosity, high chemical stability and easy preparation. Co-ZIF-8 and Cd-ZIF-8 have been the isostructures of Zn-ZIF-8.12–14 Isostructural zeolitic boron frameworks that contain tetrahedral Li–N and B–N have also been reported.15
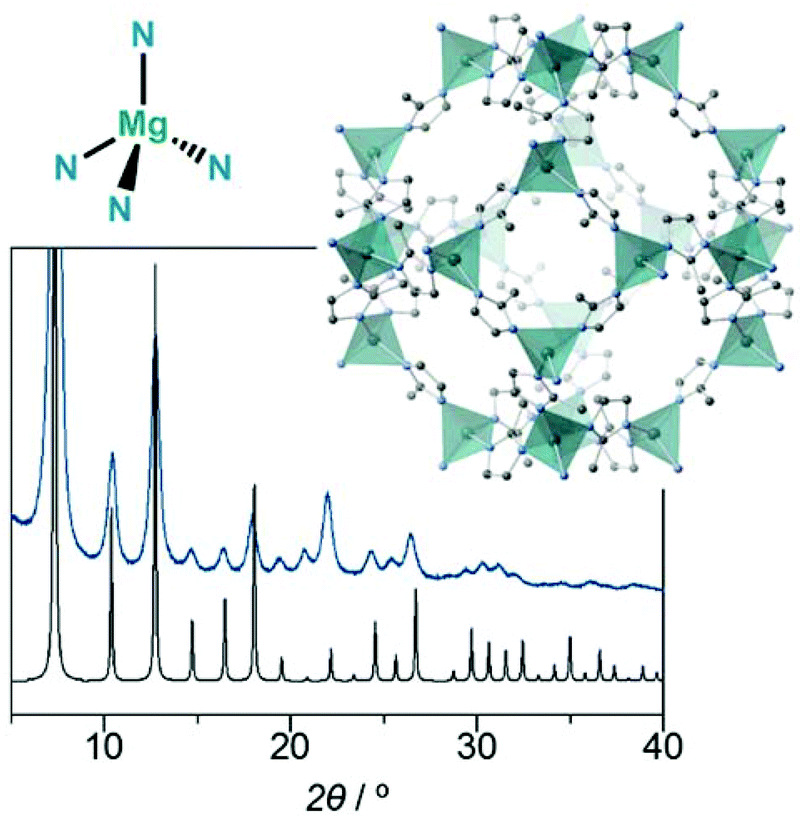 | ||
| Fig. 1 Powder X-ray diffraction pattern of Mg-ZIF-8 (blue) under an Ar atmosphere and the pattern of Zn-ZIF-8 simulated from its single-crystal structure (black). The crystal structure of Zn-ZIF-8 and the tetrahedral Mg2+−N coordination geometry are also shown. | ||
We first attempted to synthesise the Mg2+-based isostructure of Zn-ZIF-8 from anhydrous MgCl2 under an Ar atmosphere. Several different combinations of synthesis conditions always provided a discrete complex (MgCl2(2 mimH)3).16 Chloride ions coordinate to Mg2+ centres to prevent the formation of a tetrahedral Mg2+–N core. To construct an MOF with an uncommon coordination geometry, we need to consider the reaction equilibrium because coordinating counter anions and solvents easily interrupt the formation of such geometry. On the basis of this idea, we used magnesium borohydride, Mg(BH4)2, as a starting reagent. In general, metal hydrides are reactive and are widely used as reductants in organic syntheses. Three tetrahedral Mg2+–N discrete complexes among the four reports described above are Mg(NH2BH3)4-based structures;9,10 this suggests that the tetrahedral Mg2+–N core preferentially forms in the presence of a hydride ligand. In the case of MOF construction, the reaction of Mg(BH4)2 and 2 mimH is expected to have H2 gas as a by-product of MOF construction, thereby promoting the formation of an extended structure. To the best of our knowledge, the literature contains only one report about the synthesis of an MOF from a metal hydride.17 In that report, BH4− anions from Mg(BH4)2 coordinate to Mg2+ to form a nonporous framework. Our synthesis was performed as follows: 10 mL of an acetonitrile solution of 2 mimH (82.1 mg, 100 mM) was slowly added dropwise to 10 mL of an acetonitrile solution of Mg(BH4)2 (27.0 mg, 50 mM) at 298 K inside an Ar-filled glove box. Consequently, a white precipitate was generated when half of the ligand solution was added. The suspension was transferred to a hydrothermal bomb with a Teflon vial; the bomb was tightly sealed and heated at 373 K for 2 days. The white precipitate was filtered, and the powder was washed several times with acetonitrile and dried overnight at room temperature inside the glove box. We used two other concentrations for the reaction (2 mimH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mg(BH4)2 = 50:25, 150:75 (mM)), but they resulted in poorly crystalline compounds. We also used tetrahydrofuran as a synthesis solvent; this also resulted in the precipitation of poorly crystalline powders. The reactivity of Mg(BH4)2 precludes the use of protic solvents, and the optimisation of synthesis conditions is important to obtain a pure, highly crystalline compound.
:Mg(BH4)2 = 50:25, 150:75 (mM)), but they resulted in poorly crystalline compounds. We also used tetrahydrofuran as a synthesis solvent; this also resulted in the precipitation of poorly crystalline powders. The reactivity of Mg(BH4)2 precludes the use of protic solvents, and the optimisation of synthesis conditions is important to obtain a pure, highly crystalline compound.
Fig. 1 shows the powder X-ray diffraction (PXRD) pattern of the synthesised powder product; this pattern was obtained with the product under an Ar atmosphere. A simulated PXRD pattern of Zn-ZIF-8 is also shown. All diffraction peaks of the product in the 2θ range of 5°–35° match those of Zn-ZIF-8; this suggests that the product has the same topology as Zn-ZIF-8. Hereafter, we denote the product as Mg-ZIF-8. The broad peaks are because of the adsorption of X-ray by argon atmosphere, and probably the small particle size of the product, though it is impossible to observe them by using a scanning electron microscope. We determined the space group and cell parameters of the product on the basis of the PXRD data, i.e. the product crystallised in the same space group (I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m) as Zn-ZIF-8, with a lattice parameter of a = 17.28(5) Å. This space group suggests that Mg-ZIF-8 possesses a nondistorted tetrahedral Mg2+–N coordination geometry. The unit-cell parameter of Zn-ZIF-8 at 298 K is a = 17.01 Å, as calculated from the PXRD data of synthesised degased Zn-ZIF-8, which indicates that the volume of the unit cell for Mg-ZIF-8 (5159.8 Å3) is 5% larger than that of Zn-ZIF-8 (4921.7 Å3). We attempted to obtain the single-crystal structure of Mg-ZIF-8; however, we were unsuccessful. To determine the composition of Mg-ZIF-8, we degraded the dried sample and obtained its 1H-NMR spectrum. The spectrum exhibits only peaks of the 2 mimH ligand, and no decomposition or contamination was observed. Thus, we propose the reaction scheme as follows:
3m) as Zn-ZIF-8, with a lattice parameter of a = 17.28(5) Å. This space group suggests that Mg-ZIF-8 possesses a nondistorted tetrahedral Mg2+–N coordination geometry. The unit-cell parameter of Zn-ZIF-8 at 298 K is a = 17.01 Å, as calculated from the PXRD data of synthesised degased Zn-ZIF-8, which indicates that the volume of the unit cell for Mg-ZIF-8 (5159.8 Å3) is 5% larger than that of Zn-ZIF-8 (4921.7 Å3). We attempted to obtain the single-crystal structure of Mg-ZIF-8; however, we were unsuccessful. To determine the composition of Mg-ZIF-8, we degraded the dried sample and obtained its 1H-NMR spectrum. The spectrum exhibits only peaks of the 2 mimH ligand, and no decomposition or contamination was observed. Thus, we propose the reaction scheme as follows:
| Mg(BH4)2 + 2(2 mimH) → [Mg(2 mim)2] + 2BH3 + 2H2 |
This reaction produces only gases as by-products, which is the key to obtaining phase-pure Mg-ZIF-8. Indeed, we monitored the bubbling of gases during the reaction of Mg(BH4)2 and ligand solutions. The final yield of Mg-ZIF-8 was 81% based on Mg2+ ions used, which is sufficiently high for MOF synthesis. In contrast to Zn-ZIF-8, Mg-ZIF-8 is quite sensitive to humidity in air. The results of our PXRD study indicate that Mg-ZIF-8 immediately decomposes when exposed to air. We obtained the PXRD patterns for Mg-ZIF-8 after the exposure for 10 min, and the pattern is identical to that of pure 2 mimH ligand. The 2 mim ligands coordinating to Mg2+ were exchanged for H2O in air, and the released 2 mimH ligands recrystallised to form a bulk phase. This moisture sensitivity indicates that the chemical stability of Mg-ZIF-8 is not high. Because of the high air-reactivity of Mg-ZIF-8, we could not analyse it by thermogravimetric analysis. Differential scanning calorimetry was feasible under an Ar atmosphere, and the resulting trace did not show any peak in the temperature range of 298 K–473 K, suggesting that no phase transition or decomposition occurs in this temperature region. Thus, the thermal stability of Mg-ZIF-8 is sufficient to evaluate gas adsorption properties.
On the basis of the previously discussed results, we activated the powder sample of Mg-ZIF-8 at 353 K under vacuum for 12 h to remove all adsorbed solvents. The PXRD pattern of the activated powder under an Ar atmosphere is identical to that of the as-prepared sample. Fig. 2 shows the N2 and CO2 adsorption and desorption isotherms for Mg-ZIF-8 and Zn-ZIF-8. None of the sorption isotherms exhibit hysteresis, and all exhibit Type-I profiles; this adsorption behaviour is typical of microporous solids (i.e. those with pore diameters less than 2 nm).18 The amount of N2 adsorbed at 77 K by Mg-ZIF-8 was 589 mL g−1 at 100 kPa, which is higher than that adsorbed by Zn-ZIF-8 (410 mL g−1). The Brunauer–Emmett–Teller (BET) surface area of Mg-ZIF-8 was 1806 m2 g−1, whereas that of Zn-ZIF-8 was 1660 m2 g−1. The amount of CO2 adsorbed at 195 K by Mg-ZIF-8 was higher than that adsorbed by Zn-ZIF-8, reaching 322 mL g−1 at 99 kPa. The formula weights of Zn-ZIF-8 and Mg-ZIF-8 are 229.60 and 188.53 g mol−1, respectively, and the higher BET surface area of Mg-ZIF-8 is because of its lower molecular weight and larger cell volume than Zn-ZIF-8, i.e. the guest-accessible pore spaces of Zn-ZIF-8 and Mg-ZIF-8 are similar. The N2 and CO2 adsorption isotherms for Zn-ZIF-8 show a plateau above 30 kPa, whereas those of Mg-ZIF-8 show a gradual increase of gas uptake even at a pressure above 30 kPa. The PXRD pattern of the Mg-ZIF-8 sample under an Ar atmosphere after these gas-adsorption measurements was the same as that obtained before the sample was subjected to isotherm measurements, indicating that the original porous structure of Mg-ZIF-8 remained intact after gas adsorptions. At this stage we do not have direct evidence about the presence of hydride species inside the product, and further characterization such as solid state 1H NMR would be helpful.
 | ||
| Fig. 2 Adsorption (solid circles) and desorption (open circles) of Mg-ZIF-8 (blue) and Zn-ZIF-8 (black) for (a) N2 at 77 K and (b) CO2 at 195 K. | ||
In this study, the formation of the framework of Mg-ZIF-8 was demonstrated to significantly stabilise the tetrahedral Mg2+–N coordination geometry. We estimated the energies of the lattice formations of Zn-ZIF-8 and Mg-ZIF-8 using ab initio calculations, as implemented in the DMol code.19 Total energy calculations were performed for a periodic cell at the DFT/PBE level.20 Compared with the coordination bond of Zn2+–N in Zn-ZIF-8, the energy of the coordination bond of Mg2+–N in Mg-ZIF-8 is 138 kJ mol−1 lower. This instability is thermodynamically substantial, and the ability of such a small lattice formation energy to preserve the highly porous framework is surprising. The charge analysis results revealed that more electrons transfer from 2 mim to Zn2+via coordination bond formation in Zn-ZIF-8, indicating that the Zn2+–N bond exhibits a greater degree of covalent character than the Mg2+–N bond. However, in the case of Mg-ZIF-8, the charge on Mg2+ is more positive; thus, the nature of the Mg2+–N bond is more ionic. These calculation results suggest that the nature of the metal–N bonding in these compounds fundamentally differs.
Conclusions
We synthesised an Mg2+-ion-based zeolitic imidazolate framework, Mg-ZIF-8, with an uncommon tetrahedral Mg2+–N coordination geometry. The synthesis was made feasible by using Mg(BH4)2 as a starting reagent, and the resulting framework possesses permanent porosity, with a BET surface area greater than 1800 m2 g−1. This study demonstrates the potential of synthesising unique MOFs from various metal hydrides. Our stabilisation and characterisation of the uncommon coordination geometry of Mg2+ inside an MOF contributes to the fundamental advancement of coordination chemistry and to the understanding of catalytic reactions.Acknowledgements
This work was supported by a Grant-in-Aid for Young Scientists (A) and a Grant-in-Aid for Scientific Research on the Innovative Areas: ‘Fusion Materials’ from the Ministry of Education, Culture, Sports, Science and Technology, Japan and the PRESTO program of the Japan Science and Technology Agency (JST).Notes and references
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- C. K. Brozek and M. Dincă, Chem. Sci., 2012, 3, 2110 RSC.
- T. Y. Her, C. C. Chang and L. K. Liu, Inorg. Chem., 1992, 31, 2291–2294 CrossRef CAS.
- H. Wu, W. Zhou, F. E. Pinkerton, M. S. Meyer, Q. Yao, S. Gadipelli, T. J. Udovic, T. Yildirim and J. J. Rush, Chem. Commun., 2011, 47, 4102–4104 RSC.
- Y. S. Chua, W. Li, G. T. Wu, Z. T. Xiong and P. Chen, Chem. Mater., 2012, 24, 3574–3581 CrossRef CAS.
- X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- Y. Q. Tian, S. Y. Yao, D. Gu, K. H. Cui, D. W. Guo, G. Zhang, Z. X. Chen and D. Y. Zhao, Chem. – Eur. J., 2010, 16, 1137–1141 CrossRef CAS PubMed.
- J. Zhang, T. Wu, C. Zhou, S. Chen, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2009, 48, 2542–2545 CrossRef CAS PubMed.
- F. L. Phillips, F. M. Shreeve and A. C. Skapski, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 687–692 CrossRef.
- M. J. Ingleson, J. P. Barrio, J. Bacsa, A. Steiner, G. R. Darling, J. T. Jones, Y. Z. Khimyak and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2009, 48, 2012–2016 CrossRef CAS PubMed.
- K. S. W. Sing, Pure Appl. Chem., 1982, 54 Search PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, PXRD, and DSC. See DOI: 10.1039/c5dt01183c |
| This journal is © The Royal Society of Chemistry 2015 |