
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinuclear complexes of Ni(I) from 4-terphenyldithiophenol†
Felix
Koch
,
Hartmut
Schubert
,
Peter
Sirsch
and
Andreas
Berkefeld
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: andreas.berkefeld@anorg.uni-tuebingen.de; Tel: +49 7071 29 76213
First published on 1st July 2015
Abstract
Binuclear complexes of Ni(I) have been prepared from a 4-terphenyldithiophenol ligand. Steric effects were found to determine the formation of coordination isomeric structures that differ in the nature of metal-to-ligand bonding. Coordination of spatially demanding phosphine ligands PR3, R = C6H6, C6H11, at nickel sites results in a butterfly shaped thiolate-bridged Ni2(μ-S)2 motif. For smaller PMe3, the central π-system of the 4-terphenyl backbone adopts a bis-allyl like μ-syn-η3:η3-C6H4 structure due to significant d-π* Ni(I)-to-ligand charge transfer. Delocalisation indices δ(Ni–Ni) derived from DFT calculations provide a metric to assess the strength of electronic coupling of the Ni sites based on solid state structural data, and indicated less strong electronic coupling for the bis-allyl like structure with δ(Ni–Ni) = 0.225 as compared to 0.548 for the Ni2(μ-S)2 structural motif. A qualitative reactivity study toward CNCH3 as an auxiliary ligand has provided the first insight into the chemical properties of the bimetallic complexes presented.
Introduction
The class of binuclear complexes of Ni(I) features a remarkable structural diversity, and the ligand design plays a pivotal role in this regard. One can differentiate between structural motifs in which electronic coupling of the Ni(I) ions either occurs through bonding to (i) bridging conjugated π-systems in both syn- and antarafacial fashion,1–8 (ii) bridging amido,9 phosphido,10 thiolato,11,12 sulfido,13 halogenido,14–17 and hydrido18 ligands in the form of Ni2(μ-X)2 cores, (iii) bridging diphosphine,19,20 and biphenyldiyl15,21,22 ligands, or in the form of unsupported Ni–Ni bonds.23–25 Antiferromagnetic coupling through a ligand and direct Ni–Ni bonding results in diamagnetic behaviour in most instances, albeit triplet ground states have been proposed for the Ni2(μ-Br)2 core bound to the neutral form of a redox active 1,8-naphthyridine diimine ligand.16Based on solution reactivity and VT EPR studies on solid samples dinickel biradicals coexist in thermal equilibria with diamagnetic ground states. In the biradical state the Ni–Ni interaction is non-bonding and dissociation is a favourable and facile process.2,5,9 The reversible formation of reactive Ni(I) species appears to represent one of the two major modes of reactivity displayed by binuclear Ni(I) complexes. An early example of binuclear reactivity is the tetramerisation of ethyne to cyclooctatetraene as put forward by Wilke.26 Despite the exact mechanism of this process remaining unresolved, dinickel(I) complexes have been reported to catalyse reductive C–C bond formation in a cooperative fashion,15,21,22 to act as precursors in bimetallic catalytic group transfer reactions to form carbodiimides and isocyanates,17,27 and to activate secondary silanes for the catalytic hydrosilation of unsaturated substrates.28
The capability of thiolate ligands to bridge a pair of metals through bis-μ-thiolate coordination aids the formation of binuclear and higher nuclearity complexes,29–33 including well characterised examples for Ni(I) of type I in Scheme 1.10–12 In the context of a reactivity study of thiolate complexes of Ni(I), Tatsumi and co-workers reported the structure of a binuclear Ni(I) complex with bridging arene and thiolate groups, see II in Scheme 1.11
 | ||
| Scheme 1 General structural motifs of dinuclear Ni(I) complexes with L = neutral ligand, e.g. tert-phosphine, NHC, and R = aryl, tert-alkyl, H. | ||
We have set out to develop the coordination chemistry of binucleating terphenyldithiophenol ligands which combine the properties of metal–sulphur34,35 and labile metal–arene36 bonds. The use of a dianionic ligand framework eliminates the necessity for incorporating secondary anionic ligands that might bridge the nickel ions, which may provide structural flexibility to the bimetallic core. Herein we report on the synthesis and properties of mono- and binuclear complexes of nickel with such a type of ligand.
Results and discussion
Synthesis and structure of mono- and binuclear complexes 1–5
The preparation of the nickel complexes followed two different protocols. The first utilises the comproportionation of Ni(II) precursors 1 and 2 with Ni(COD)2, COD being 1,5-cyclooctadiene. Alternatively, reacting 4-terphenyldithiophenol with 2 equiv. of (PPh3)2NiN(SiMe3)2 as a source of Ni(I) affords bimetallic 5 directly, eliminating bis(trimethylsily)amine and PPh3.11Ni(II) precursor complexes 1 or 2 have been prepared by salt metathesis, with a reaction of the dipotassium salt of dithiophenol with Ni(II) chloride precursors in toluene solution at ambient temperature as shown in Scheme 2. The compounds have been isolated as purple solids in yields of 88 and 60% with λmax(thf) for 1 at 511 (ε/dm3 mol−1 cm−1 1128) and 2 at 550 (3545) nm. Note that 2 is a kinetic reaction product and was found to gradually convert into an unknown red precipitate, most likely an oligomeric composition. Substituting PCy3 for PPh3 affords a mixture of ill-defined products.
 | ||
| Scheme 2 Structure and characteristic δC (11.7 T) NMR chemical shifts of Ni(II) precursor complexes 1 and 2 (R′ = 2,4,6-(H3C)3C6H2, R = tBu). | ||
Single crystal XRD structural analysis verified square planar coordination geometries at the trans-dithiolatonickel(II) centres with average Ni–S bond lengths of 2.249(4) and 2.241(2) Å for 1 and 2 respectively which compare well with the Ni–S bond lengths reported for structurally related complexes.10–12 The molecular structure of 2 is depicted in Fig. 1. While two PMe3 donors complete the ligand shell of Ni(II) in 1, the use of the more bulky PCy3 (ΔΘTolman = 52°) ligand in 2 allows for binding just one phosphine ligand, with a Ni–P bond distance of 2.353(2) Å, and enforces η2-coordination of the central aryl ring with an averaged Ni–C distance of 2.285(4) Å.
 | ||
| Fig. 1 ORTEP representation and selected structural parameters of the molecular structure of 2 (thermal ellipsoids at the 50% probability level). Given Ni–C, and –S distances are averaged, H- and lattice solvent atoms are not shown for clarity. | ||
Also in the case of 1 the nickel ion resides above one of the C2H2 edges of the central arene ring, but the average Ni–C distance of 2.614(2) Å is significantly longer, as expected. The molecular structure of 1 is shown in Fig. S9, ESI.† Low-temperature 13C{1H} and 1H NMR data are consistent with η2-arene bonding to Ni(II) in 2. At 193 K in d2-CH2Cl2 solution, characteristic resonances of a Cs symmetric structure were observed at 108.4, broadened due to unresolved 2JC–P coupling, and 8.21 as well as 127.4 and 7.44 ppm which account for the Ni-bound and unbound, yet slowly exchanging C2H2 edges of the central aryl ring as defined in Scheme 2. At 298 K pseudo C2v symmetric spectra result for both complexes with an N-line pattern at 0.77 ppm, |2+4|JH–P = 7.8 Hz, for trans-(Me3P)2Ni(II) along with resonances of the nuclei at the central arene ring at 7.56 and 123.9 ppm for 1, and at 7.76 and 118.6 ppm in the case of 2.
Binuclear complexes 3–5 have been isolated in the form of distinct coordination isomers as depicted in Scheme 3 in yields of 51% (3, brown), 75% (4, yellow), and 73% (5, green) with λmax (thf)/nm 3, 520 (ε/dm3 mol−1 cm−1 2044), 4, 352 (3005), and 5, 450 (7821). Additional weak absorption bands were detected for each of the dinickel complexes at wavelengths of 940–990 nm (ε/dm3 mol−1 cm−1 200–500).
 | ||
| Scheme 3 Synthesis, structure, and characteristic δC (11.7 T) NMR chemical shifts of binuclear complexes 3–5 (R′ = 2,4,6-(H3C)3C6H2, R = tBu). | ||
Single crystal XRD structure analysis verified the binding modes of the dinickel cores to the dithiophenolate ligand as exemplified for 3 and 5 in Fig. 2 and 3. A distinct structural feature of 3 is the boat shaped conformation of the central π-system which is present in both the solid state and solution. The variation of bond lengths and deformation from planarity indicate the formation of a syn-μ-η3:η3 bonding motif, which likely results from a significant overlap of the occupied d-orbitals at both nickel ions with a π*-orbital of the arene π-system.36–38 Short distances of 1.949(4), 2.035(4), and 2.149(4) Å between Ni and o-, i-, and m-C atoms, and a long Ni–Ni separation of 2.659(1) Å support this description. The observed κ1-S–Ni bond lengths of 2.205(1) and 2.193(3) Å compare well with those in the literature.11 Such tight Ni2-arene bonding is likely a consequence of a strong charge donation from the thiolate and phosphine ligands to nickel. A structurally similar conformation was reported for the central π-system of a 4-terphenyldiphosphine ligand that stabilises a bridging μ-η1:η1-o,o′-biphenyldiyl dinickel fragment.15
 | ||
| Fig. 2 Left: ORTEP representation and selected structural parameters of 3 (thermal ellipsoids at the 50% probability level; H- and lattice solvent atoms omitted for clarity). Right: schematic representation of the overall exchange process of the Ni(I)2 fragment (as a visual aid, asterisks highlight identical C atoms). | ||
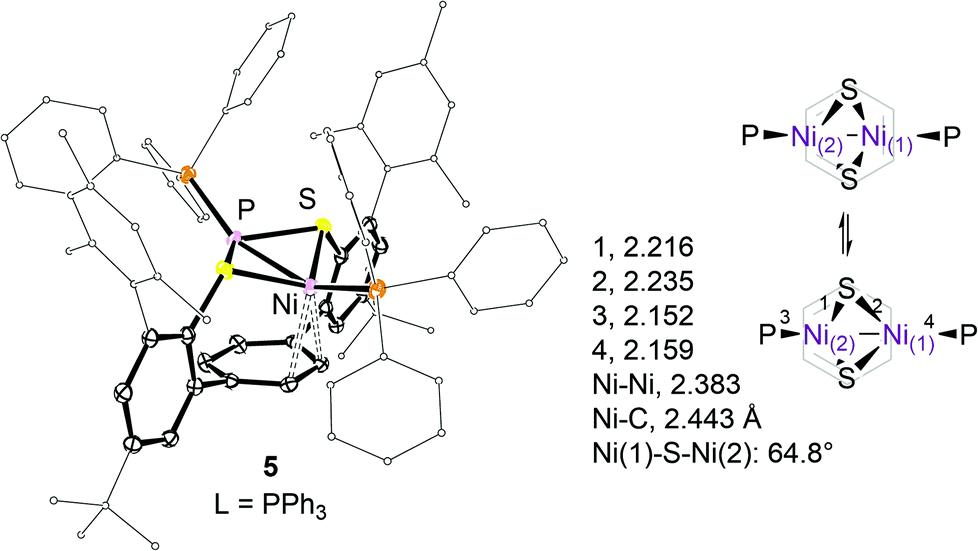 | ||
| Fig. 3 ORTEP representation and selected structural parameters of 5 (thermal ellipsoids at the 50% probability level; H- and lattice solvent atoms omitted for clarity), and schematic representation of a seesaw-like motion of the Ni2(μ-S)2 fragment. | ||
The geometry at each Ni ion may be best described as a distorted square planar defined by the sulphur, phosphorus, and the peripheral C-atoms of the η3-allyl moiety. The shortest distance between hydrogen atoms at the PMe3 ligands is 2.5 Å.
The dinickel core of 3 is dynamic. For an 11.7 T magnetic field, the resonance at δH 5.41 ppm of protons 8/9-H at the central π-system was observed to coalesce at 203 K. Further cooling to 178 K gave rise to broad 1H and 13C NMR resonances at 4.97 (ν1/2 ∼ 26 Hz) and 5.59 (ν1/2 ∼ 23 Hz) as well as 109 (i-C, ν1/2 ∼ 42 Hz), 74.2 (o-C, ν1/2 ∼ 60 Hz), and 98.5 (m-C, ν1/2 ∼ 55 Hz) ppm. Exchange cross peaks between protons 8/9-H are well established in 1H–1H NOESY data at 178 K. Apparent exchange of 8-H for 9-H requires the Ni(I) ions to formally change sites across the syn-μ-η3:η3-C6H4 system as schematised in Fig. 2. Whether this process evolves through an intermediate structure analogous to that of 4 and 5 is unknown.
Steric interactions between the mesityl and phosphine substituents in 4 and 5 impair a close approach of both nickel ions to the arene system. Instead, the P(1)–Ni(1)–Ni(2)–P(2) moiety is tilted upward relative to the aryl ring plane which allows one nickel ion, Ni(1), to weakly interact with the undistorted π-system as indicated in Fig. 3.
The observed average Ni(1)–C distances of 2.569(6) and 2.443(1)39 Å in 4 and 5 respectively are significantly longer than those found in 2 (2.285(4) Å). The formation of a syn-endo-Ni2(μ-S)2 structure29 compensates for the lack of metal–arene interactions at Ni(2). As a result, slightly shorter average μ-S–Ni(2) and Ni(2)–P bond distances of 2.213(1) (Δd = 0.02) and 2.148(1) (Δd = 0.009) Å as compared to the respective bonds to Ni(1) have been found. Acute Ni(1)–S–Ni(2) angles of 65° within the Ni2(μ-S)2 entity are accompanied by a short Ni–Ni separation of 2.383(1) Å.10,11,29
Exchange averaged singlet 1H NMR resonances of the C2H2 moieties of the central aryl rings in 4 and 5 have been observed at 8.41 (ν1/2 ∼ 95 Hz) and 7.72 (ν1/2 ∼ 33 Hz) ppm at 168 K. Whether or not this exchange involves preliminary dissociation of one phosphine ligand is unclear albeit no spectral differences have been observed in the presence of excess (≤5%) phosphine. As for 3, the apparent exchange of the C2H2 sites requires an overall seesaw-like motion as schematised in Fig. 3.
Computational study
A comparison of Ni–Ni and Ni–C(arene) bond lengths calculated from single crystal X-ray diffraction data suggests a higher contribution of direct metal–metal (M–M) bonding to the coupling of the nickel ions in the case of the syn-endo-Ni2(μ-S)2 structural motif in 4 and 5 than in 3. To gain deeper insight into the nature and actual strength of Ni–Ni bonding in these structurally different complexes, we carried out a topological analysis of their electron density distribution, ρ(r), as derived from the DFT calculations of 3 and 5, using the “quantum theory of atoms in molecules” (QTAIM) approach.40,41 DFT calculations were performed using Gaussian42 at the B3LYP/6-311G(d,p) level43–49 with dispersion corrections, including Becke–Johnson damping,50,51 and substituting the tBu groups on the ligand for CH3 in the theoretical model systems. The optimised geometries were in excellent agreement with the experimental counterparts, and the Ni–Ni distances differed by 2 and 6.1 pm for 3 and 5, respectively. For computational details and the full geometries, see the ESI.†Although numerous experimental and theoretical electron density studies on M–M bonding in dinuclear complexes have been reported, a fundamental understanding of this interaction remains elusive.52,53 In both 3 and 5, a stable bond path54 between the two nickel ions could be identified which implies direct M–M interactions in both systems. This observation is noteworthy, as the presence of bridging ligands usually results in the loss of M–M bond paths, in particular for metal atoms linked by formal single bonds, as concluded by Farrugia and Macchi in a recent review.53 The topological parameters of ρ(r) at the Ni–Ni bond critical points (BCPs)55 in 3 and 5 are listed in Table 1, and the extended topological data and the molecular graphs of 3 and 5 are provided in the ESI.† In both systems, the value for ρ(rb) is relatively small, a feature commonly observed for M–M bonds which has been attributed to the diffuse nature of this interaction.53,56 The slightly negative values for the total energy density, H(rb), imply that the potential energy density at these BCPs is somewhat larger than the kinetic energy density which is indicative of covalent bonding, despite the positive value for the Laplacian of ρ(rb), ∇2ρ(rb).56,57 A better metric than ρ(rb) to assess the actual strength of metal–metal bonding is provided by the delocalization index, δ(A–B), which measures the number of electron pairs shared between two atoms A and B, irrespective of the existing bond paths.53,56,58 For 3 and 5, the values of δ(Ni–Ni) are equal to 0.225 and 0.548, respectively. These smaller, fractional bond orders indicate what has been termed as a partial covalent character for formally M–M single bonds, in the sense that not a whole pair of electrons is involved in direct interactions between the atoms.53 To summarise, the bonding between the Ni atoms in both 3 and 5 can be described as a combination of through-ligand and direct M–M bonding. The latter contribution was clearly identified through the existence of Ni–Ni bond paths. The large increase in the Ni–Ni separation by 27.6 pm [DFT: 31.7 pm] in 3 compared to 5 also reflects a significant reduction of Ni–Ni bond strength: the respective value of δ(Ni–Ni) in 5 is more than 2.4 times higher than the one in 3, which can therefore be classified as a bimetallic complex exhibiting only very weak Ni–Ni interactions.
Solution properties of 3–5
VT 1H NMR data of 3 and 5 revealed a marked dependence of chemical shifts and line-widths of specific proton resonances with temperature and solvent polarity. In the case of 3, substituting the solvent d8-thf with d6-C6H6 at 299 K resulted in a ΔδH of +1 ppm for protons 8/9-H at the central π-system. As shown in Fig. 4, variation of the temperature of a d8-thf solution of 3 results in a reversible, non-linear variation of δH between 5.39 at 198 K and 11 ppm at 405 K, ΔδH(8/9-H) = 5.61 ppm.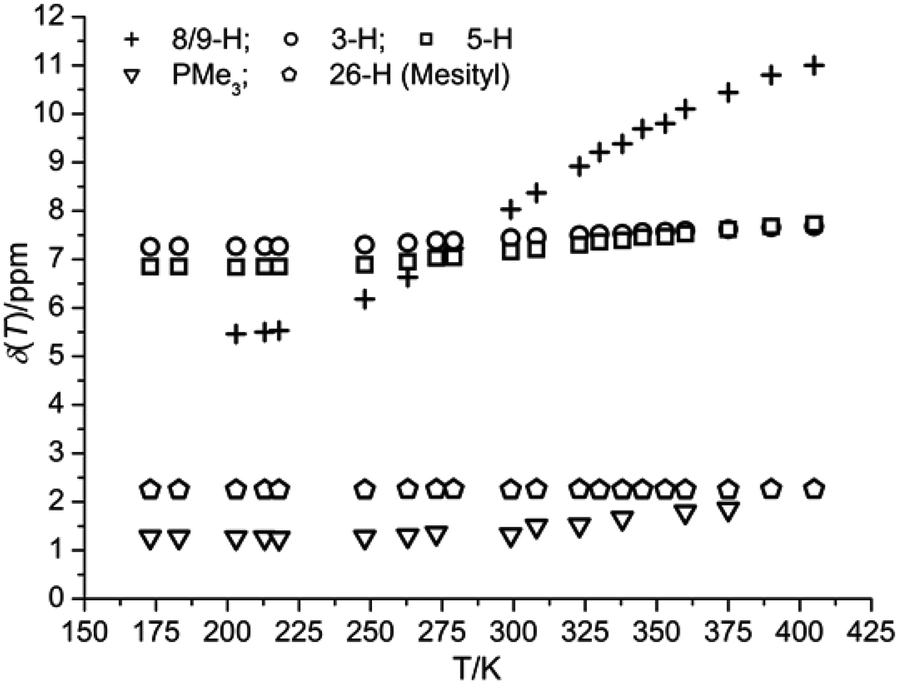 | ||
| Fig. 4 Temperature dependence of δH observed for 3 (500 MHz, 21 mM in d8-thf). | ||
In comparison, the observed chemical shift changes for the protons at the flanking phenyl rings, ΔδH(3-H) = 0.42, and 0.88 (5-H) ppm, and for the PMe3 ligands (0.58 ppm) are smaller by about one order of magnitude but vary in the same non-linear fashion. The largest shift difference in the latter series accounts for the protons most distant from the nickel ions (5-H; for numbering cf. to Scheme 3) whereas resonances of the mesityl (26-H) and tBu substituents are unaffected.
A similarly marked dependence emerged for 5 in d8-thf with ΔδH(8,9-H) = 4.94 ppm and δH(8,9-H, 299 K) = 10.12 ppm (12.01 ppm in d6-C6H6 and d8-toluene) whereas it was found to be small in the case of 4, with ΔδH(8,9-H) = 0.51 ppm and δH(8,9-H, 299 K) = 8.69 ppm. Plots of δHvs. temperature for selected types of protons for complexes 4 and 5 are provided in Fig. S7 and S8, ESI.† The respective values of δH and the variation in temperature have been observed for both raw products and single as well as double recrystallized samples of 3–5 that were obtained from different batches. This indicates that putative Ni(I) impurities most likely are not the reason for this solution behaviour.
Whereas the 1H and 13C NMR chemical shifts determined at low temperatures account for the respective structural features observed in the solid state structures of 3–5, the continuous shift of δH to lower fields at higher temperatures, especially δH(8/9-H) being progressively greater than 11 ppm, suggests thermal population of a paramagnetic biradical (S = 1) state2,9 which would add contact and dipolar shift contributions to the observed chemical shift.28,59,60 An independent parameter to probe for variable net electron spin moments is the trend of the temperature dependence of the 1H spin–lattice relaxation time, T1, which is expected to be positive for protons in diamagnetic environments. Values of T1 for 8/9-H of 3–5 were found to decrease markedly with increasing temperatures, e.g. T1(8/9-H) for 3 in d8-thf decreased from 1300 ms at 223 K to 110 ms at 323 K; cf. to Fig. S3–S6, ESI.† Surprisingly, invariable effective magnetic moments of 0.9 B.M. for 3 and 1.0 B.M. for 5 in both toluene and thf solutions were determined by Evan's method61,62 over the same temperature range at a field of 11.7 T. This finding challenges the hypothesis of the thermal equilibrium population of a paramagnetic spin state. Heating a solution of 5 in d8-toluene to 373 K for 3 h was found to slightly increase the effective magnetic moment by ∼0.1 B.M. but did not have any effect on the temperature dependence of δH.
Chemical properties of dinickel cores
Compounds 3–5 displayed appreciable thermal stability in VT NMR spectroscopic studies of their toluene and thf solutions. While heating a solution of 3 in d6-benzene to 343 K for 2 h resulted in low conversion, ∼3%, back into 1 no changes were observed in d8-thf under identical conditions. When dissolved in d2-CH2Cl2 under ambient conditions gradual deterioration to yet unknown products occurs in all cases. To gain a more detailed insight into the chemical properties of these bimetallic systems the reactivities of 3–5 were studied qualitatively toward methyl isocyanide (CNCH3 = L′) by VT NMR spectroscopy.
1H and 31P NMR spectroscopic monitoring showed that 5 (δP 68 ppm) reacts with L′ in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry at T ≤ 213 K to produce a single product 5*2L′ with characteristic resonances δH(Ni–L′) 2.10 and δP 22.51 ppm as described in Scheme 4. Coordination of both PPh3 and L′ to both Ni sites is supported by 1H–1H COSY and 1H–31P HSQC data which established the presence of scalar JH and JP coupling of the methyl protons of L′ with the o- and m-1H and 31P nuclei of coordinate PPh3. Albeit we cannot assign a molecular structure based on the NMR data available, 5*2L′ must be a bimetallic complex. Warming the sample to 263 K in the NMR probe resulted in the progressive formation of (Ph3P)2(L′)2Ni(0) at the expense of the resonances of 5*2L′, with characteristic 1H and 31P NMR resonances at 2.85 ppm (N-line pattern, L′) and δP 33.2 ppm. The identity of this Ni(0) product has been established unequivocally through independent synthesis from Ni(COD)2, 2 equiv. of PPh3 and L′ in an NMR tube reaction under otherwise identical conditions.
:2 stoichiometry at T ≤ 213 K to produce a single product 5*2L′ with characteristic resonances δH(Ni–L′) 2.10 and δP 22.51 ppm as described in Scheme 4. Coordination of both PPh3 and L′ to both Ni sites is supported by 1H–1H COSY and 1H–31P HSQC data which established the presence of scalar JH and JP coupling of the methyl protons of L′ with the o- and m-1H and 31P nuclei of coordinate PPh3. Albeit we cannot assign a molecular structure based on the NMR data available, 5*2L′ must be a bimetallic complex. Warming the sample to 263 K in the NMR probe resulted in the progressive formation of (Ph3P)2(L′)2Ni(0) at the expense of the resonances of 5*2L′, with characteristic 1H and 31P NMR resonances at 2.85 ppm (N-line pattern, L′) and δP 33.2 ppm. The identity of this Ni(0) product has been established unequivocally through independent synthesis from Ni(COD)2, 2 equiv. of PPh3 and L′ in an NMR tube reaction under otherwise identical conditions.
 | ||
| Scheme 4 Reactivity of binuclear 5 toward CNCH3 (L′) monitored in situ by VT NMR spectroscopy in d8-thf (δNucleus in ppm at 11.7 T). | ||
IR spectra obtained with a dip probe at 193 K showed the appearance of an intense band at 2051 cm−1, along with a weak band at 2152 cm−1, for 5*2L′ whose intensity reached a maximum after the addition of 2 equiv. of L′. As expected, both bands disappeared upon warming the solution to room temperature due to the disproportionation of 5*2L′, and were replaced by broad bands at 2109 and 2060 cm−1. These bands compare well with the literature reported values for complexes of the type (Ph3P)n(RNC)4−nNi(0), with n = 1–3, and R = tBu, Cy, and PhCH2.63
In clear contrast, addition of 1 equiv. of L′ to 3 at 213 K afforded an asymmetric compound which we assign as 6 shown in Scheme 5. The complete assignment of 1H and 31P NMR data of putative 6 is given in the ESI.† The PMe3 ligands in 6 are non-equivalent but appear to coordinate to the same nickel site, with 2JP–P = |54| Hz.64 The magnitude of 3JP–P of the putative isomeric structure 6iso may be expected to be significantly smaller if detectable at all.20 Selective 31P decoupling of 1H NMR data aided in the assignment of the two PMe3 ligands. Both phosphine ligands showed 1H–1H NOE contacts to L′ (δH 3.32 ppm), which would not have been possible if PMe3 and L′ were coordinating in a trans-fashion to the same nickel ion as in 6iso. Ni-arene d-π* back bonding appears to be insignificant, judging from the chemical shifts of the four distinct resonances of the central ring protons, δH being 7.74, 6.53, 6.47, and 6.18 ppm at 193 K. The central π-system of the 4-terphenyl backbone may act as a σ-donor to complete the ligand shell at the Ni–L′ site as indicated in Scheme 5. As observed for complex 2, the shift of one of the above 1H resonances to a lower field may reflect dative bonding of the π-system to nickel which binds another strong σ-donor ligand such as PCy3 and L′ in the trans-position.
 | ||
| Scheme 5 Reactivity of 3 toward CNCH3 (L′) monitored by in situ VT NMR spectroscopy (L = PMe3, R′ = 2,4,6-(H3C)3C6H2, R = tBu). Dashed line accounts for the capability of L′ to μ-bonding. | ||
The structure of 6 is dynamic in solution. Exchange spectra show that the PMe3 ligands slowly exchange Ni sites with L′ at 213 K, see Fig. S1 and S2 in the ESI.† The known capability of L′ to coordinate in a μ-fashion19,65 likely triggers this exchange process as indicated in Scheme 5.
Monitoring the addition of L′ to 3 at 193 K in thf by IR spectroscopy showed the gradual appearance of a broad band at 2064 cm−1, along with a significantly weaker broad band centred at 2124 cm−1. Bands at wavenumbers greater than 2000 cm−1 have been reported as characteristic for terminal rather than bridging coordination of isocyanide ligands in binuclear complexes of low-valent nickel.19,65 Compared to a reference sample with no added 3, L′ coordination results in a shift of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching mode by 100 cm−1 to lower energy, indicating a significant d-π* charge transfer from Ni to L′. At 234 K, 1H NMR resonances of the central arene ring protons are subject to exchange broadening and resonances of 1 began to gradually grow into the spectra. The addition of two equivalents of L′ to 3 resulted in complete conversion of 6 into 1. IR spectra obtained at the same temperatures showed a broad band centred at 1855 cm−1 that irreversibly formed at the expense of the bands at 2124 and 2064 cm−1 that had originally been observed upon L′ addition at 193 K.
N stretching mode by 100 cm−1 to lower energy, indicating a significant d-π* charge transfer from Ni to L′. At 234 K, 1H NMR resonances of the central arene ring protons are subject to exchange broadening and resonances of 1 began to gradually grow into the spectra. The addition of two equivalents of L′ to 3 resulted in complete conversion of 6 into 1. IR spectra obtained at the same temperatures showed a broad band centred at 1855 cm−1 that irreversibly formed at the expense of the bands at 2124 and 2064 cm−1 that had originally been observed upon L′ addition at 193 K.
Conclusions
Mono- and binuclear complexes of Ni(I) have been prepared from a 4-terphenyldithiophenol ligand. Steric interactions between phosphine and substituents at the 4-terphenyl backbone result in the formation of coordination isomeric structures with thiolate bonding either in a bridging or terminal fashion. In the latter case, significant d-π* charge transfer causes the 4-terphenyl ligand backbone to coordinate to the Ni ions in a syn-μ-η3:η3 fashion. Electronic coupling of metal centres within the Ni2(μ-S)2 core displayed in 4 and 5 involves bonding interactions via the bridging thiolates and also direct M–M bonding with calculated δ5(Ni–Ni) = 0.548, whereas a delocalisation index of 0.225 indicates significantly weaker coupling of the Ni(I) ions in 3. The origin of the chemical shift dependence on temperature observed for 3–5 is currently not understood but most likely reflects changes of the electronic properties of the bimetallic core. Whereas metal–thiolate bonding couples protons 3/5-H to the Ni ions, 8/9-H at the central π-system bind directly to the latter. Albeit plausible this hypothesis necessitates further studies.Coordination of a π-acceptor ligand such as methyl isocyanide to 3 and 5 subjects the bimetallic fragments to disproportionation. Interestingly, the nature of the phosphine ligand determines the character of intermediately formed species and the temperature at which Ni(0) species are extruded. The binuclear Ni2(μ-S)2 structure in 5 persists upon binding of 2 equiv. of isocyanide at low temperatures. In the case of 3, apparent PMe3 migration to the same nickel site is triggered by isocyanide bonding. This structural reorganisation reaction is remarkably facile even at low temperature which may be a consequence of the weak electronic coupling of the Ni ions, as also reflected by the calculated delocalisation index from solid state structural data.
The magnitude of metal–metal electronic coupling may be taken as a parameter which determines the electronic flexibility and thus the reactivity of bimetallic structures.66 The bimetallic complexes of nickel described herein provide a complementary set of model compounds suitable for studies on the structural effects on the reactivity of binuclear systems toward electrophilic reactants.
Experimental
General considerations
All reactions were carried out under a dry argon atmosphere using standard Schlenk or glove box techniques (MBraun, MB 150-GI). All solvents were purified and dried prior to use. Dichloromethane and hexane were dried over Grubbs columns of a MBraun solvent purification system. Benzene, diethyl ether, pentane, tetrahydrofuran, and toluene were pre-dried over activated 3 Å molecular sieves and distilled from sodium benzophenone ketyl or potassium metal under argon. Methanol was dried over activated neutral alumina. d6-Benzene, d8-toluene, and d8-thf were dried over and distilled from the NaK alloy whereas d-CHCl3 and d2-CH2Cl2 were dried over and vacuum transferred from 3 Å molecular sieves. All solvents were stored over 3 Å molecular sieves under argon. Molecular sieves and neutral alumina were activated by heating under dynamic vacuum (10−3 mbar) at 250 °C for 24–48 hours. UV-Vis spectra were collected on a PerkinElmer Lambda 35 spectrophotometer. The range from 200 to 1100 nm was scanned at a speed of 480 nm per minute, using 1 cm quartz cuvettes sealed with Teflon stoppers. Combustion analyses were performed on an Elementar Vario MICRO instrument. NMR data were recorded on Bruker Avance II 400 and DRX 250 instruments. VT NMR spectra were collected on a Bruker AVII+500 spectrometer. δ Values are given in ppm, J values in Hz. 1H and 13C{1H} NMR chemical shifts are referenced to the residual proton and naturally abundant carbon resonances of the solvents: 7.16/128.06 (d6-C6H6), 3.58/67.21 (d8-thf), 5.32/53.84 (d2-CH2Cl2), and 7.26/77.16 (d-CHCl3) ppm. 31P NMR chemical shifts are referenced to an external standard sample of 85% H3PO4 set to 0 ppm. VT solution IR spectra were obtained with a Mettler Toledo ReactIR 15 system equipped with a Sicomp dip probe at a spectral range from 2600 to 650 cm−1. Ligand preparation is described in the ESI.† The compounds (Me3P)2NiCl2,67 Ni(COD)2,68 (Ph3P)2NiN(SiMe3)2,69 (pyridine)4NiCl2,70 and methyl isocyanide71 were prepared following the procedures adapted from the literature. Caution: Methyl isocyanide is toxic and has a very unpleasant odour. All manipulations with this reagent should be carried out in a fume hood. X-ray data were collected on a Bruker Smart APEX II diffractometer with graphite-monochromated Mo Kα radiation. The programs used were Bruker's APEX2 v2011.8-0, including SADABS for absorption correction and SAINT for structure solution,72 the WinGX suite of programs version 2013.3,73 SHELXS for structure solution, SHELXL for structure refinement,74,75 and PLATON.76 Crystals were, unless otherwise noted, coated in a perfluorinated polyether oil and mounted on a 100 μm MiTeGen MicroMounts™ loop that was placed on the goniometer head under a stream of dry nitrogen at 100 K.Preparation and characterization of 1
Ligand (500 mg, 0.776 mmol), benzyl potassium (210 mg, 1.550 mmol), and toluene (40 mL) were combined in a Schlenk flask. The resulting mixture was stirred for 0.5 hour (h) at room temperature (r.t.) to form a clear yellow solution. After this time, (Me3P)2NiCl2 (220 mg, 0.780 mmol) was added and stirring was continued for an additional 2 h at r.t. The solution gradually turned dark purple and a white solid separated. The solid was filtered off, the solvent removed under vacuum, and the residual dark purple solid repeatedly (3×) washed with 10 mL portions of methanol, and dried under dynamic vacuum to produce a purple solid 1 (576 mg, 88%). Bulk crystallisation by slow diffusion of methanol layered on top of a concentrated solution of 1 in benzene produced single crystals also suitable for XRD analysis.δ H (400 MHz; d6-C6H6; 298 K) 7.76 (2 H, d, J3−5 2.4, 4, 3-, 18-H), 7.56 (4 H, s, 8-, 9-, 11-, 12-H), 7.05 (2 H, d, J5−3 2.6, 4, 5-, 16-H), 6.95 (4 H, s, 21-, 23-, 32-, 34-H), 2.29 (12 H, s, 25-, 27-, 36-, 38-H), 2.17 (6 H, s, 26-, 37-H), 1.33 (18 H, s, 4-, and 17-tBu), 0.77 (18 H, N-line pattern |2+4|JH–P 7.83, P(CH3)3).
δ
C (100 MHz; d6-C6H6; 298 K) 144.22, 143.41, 141.83 (t, ![[J with combining macron]](https://www.rsc.org/images/entities/i_char_004a_0304.gif) C–P 2.1, C-1, -14), 141.63 (C-19, -30), 140.75, 140.12, 136.10 (C-20, -24, -31, -35), 135.65 (C-22, -33), 128.36 (C-21, -23, -32, -34), 126.34 (C-5, -16), 123.85 (C-8, -9, -11, -12), 122.77 (C-3, -18), 34.19 (C-28, -40), 31.63 (C-29, -39), 21.19 (C-26, -37), 20.97 (C-25, -27, -36, -38), 12.47 (N-line pattern |1+3|JC–P 27.7, P(CH3)3).
C–P 2.1, C-1, -14), 141.63 (C-19, -30), 140.75, 140.12, 136.10 (C-20, -24, -31, -35), 135.65 (C-22, -33), 128.36 (C-21, -23, -32, -34), 126.34 (C-5, -16), 123.85 (C-8, -9, -11, -12), 122.77 (C-3, -18), 34.19 (C-28, -40), 31.63 (C-29, -39), 21.19 (C-26, -37), 20.97 (C-25, -27, -36, -38), 12.47 (N-line pattern |1+3|JC–P 27.7, P(CH3)3).
δ P (162 MHz, d6-C6H6, 298 K) −27.98 (Ni-P(CH3)3).
Elemental analysis found: C, 68.26; H, 8.35; S, 6.73. Calc. for C50H66P2S2·2CH3OH: C, 68.18; H, 8.16; S, 7.00%.
UV-Vis: λmax (thf) nm 257 (ε/dm3 mol−1 cm−1 67671), 280 (52736), 351 (13220) and 511 (1128).
Crystal data: C50H66P2S2·2CH3OH, M = 915.88, monoclinic, a = 13.5105(2), b = 12.1898(2), c = 29.9385(5) Å, U = 4928.25(14) Å3, T = 100(2) K, space group P121/c, Z = 4, 27715 reflections measured, 10728 unique (Rint = 0.0223) which were used in all calculations. The final wR(F2) was 0.1114 (all data).
Preparation and characterization of 3
1 (200 mg, 0.234 mmol), Ni(COD)2 (65 mg, 0.234 mmol), and thf (25 mL) were combined in a Schlenk flask. The resulting mixture was stirred for 2 h at r.t. to form a brown solution. The solvent was removed under vacuum, and the residual brown solid repeatedly washed with 5 ml portions of pentane (3×), and dried under dynamic vacuum to produce a brown solid 3 (110 mg, 51%). Slow evaporation of a concentrated solution of 3 in pentane produced single crystals suitable for XRD analysis. Further purification was carried out by cooling a saturated solution of 3 (200 mg) in diethyl ether/pentane (1:1) to 238 K to yield brown crystals of 3 (110 mg, 55%). Prolonged evacuation of solid 3 must be avoided since concomitant removal of volatile PMe3 leads to gradual sample degradation.
δ H (500 MHz, d8-thf, 178 K) 7.29 (2 H, d, J3−5 1.7, 4, 3-, 18-H), 6.87 (2 H, d, J5−3 1.7, 4, 5-, 16-H), 6.83 (4 H, s, 21-, 23-, 32-, 34-H), 5.59 (2 H, br s), 4.97 (2 H, br s), 2.28 (6 H, s, 26-, 37-H), 1.95 (12 H, s, 25-, 27-, 36-, 38-H), 1.33 (18 H, s, 4-, and 17-tBu), 1.29 and 1.28 (s, P(CH3)3).
δ C (126 MHz, d8-thf, 178 K) 152.42 (C-1, -14), 144.05, 140.80, 139.03 (C-19, -30), 136.00, 135.34, 134.81, 127.53 (C-21, -23, -32, -34), 125.75 (C-5, -16), 122.21 (C-3, -18), 109.43, 98.51, 74.23, 34.12 (C-28, -40), 31.11 (C-29, -39), 20.5 (C-25, -27, -36, and -38), 20.5 (C-26, -37), 15.72 (JC−7−P 26.5, P(CH3)3).
δ P (202 MHz, d8-thf, 178 K) −13.66.
Elemental analysis found: C, 66.08; H, 7.45; S, 6.83. Calc. for C50H66S2: C, 65.96; H, 7.31; S, 7.04%.
UV-Vis: λmax (thf)/nm 266 (ε/dm3 mol−1 cm−1 25089), 328 (20496), 373 (15074), 520 (2044) and 943 (310).
Crystal data: C50H66Ni2P2S2·C5H12, M = 982.65, monoclinic, a = 15.8803(8), b = 20.1160(10), c = 17.6539(9) Å, U = 5371.9(5) Å3, T = 100(2) K, space group P121/n, Z = 4, 75839 reflections measured, 15591 unique (Rint = 0.0321) which were used in all calculations. The final wR(F2) was 0.1389 (all data).
Preparation and characterization of 2
Ligand (500 mg, 0.776 mmol), benzyl potassium (210 mg, 1.550 mmol), and toluene (40 mL) were combined in a Schlenk flask. The resulting mixture was stirred for 30 minutes at r.t. to form a clear yellow solution. After (pyridine)4NiCl2 (360 mg, 0.776 mmol) and tricyclohexylphosphine (210 mg, 0.776 mmol) were added, stirring was continued for an additional 2 h at r.t. The solution turned brown and a white solid separated. After filtration, the solvent was removed under vacuum, the residual brown solid repeatedly (3×) washed with 10 mL portions of methanol, and dried under dynamic vacuum to produce a dark purple solid 2 (456 mg, 60%). Crystallisation by slow diffusion of pentane vapour into a concentrated solution of 2 in thf produced single crystals suitable for XRD analysis.δ H (500 MHz; d2-CH2Cl2; 273 K) 7.76 (4 H, s, 8-, 9-, 11-, 12-H), 7.27 (2 H, d, J3−5 2.3, 4, 3-, 18-H), 6.78 (4 H, s, 21-, 23-, 32-, 34-H), 6.73 (2 H, d, J5−3 2.3, 4, 5-, 16-H), 2.19 (6 H, s, 26-, 37-H), 1.91 (12 H, s, 25-, 27-, 36-, 38-H), 1.27 (18 H, s, 4-, and 17-tBu), 2.04–0.75 (m, P(C6H11)3).
δ C (126 MHz; d2-CH2Cl2; 273 K) 146.25 (C-4, -17), 141.72 (C-6, -15), 141.02 (C-19, -30), 140.71 (C-2, -13), 139.60 (C-7, -10), 137.93 (C-1, -14), 136.07 (C-20, -24, -31, -35), 135.88 (C-22, -33), 128.08 (C-21, -23, -32, -34), 127.59 (C-5, -16), 121.49 (C-3, -18), 118.62 (C-8, -9, -11, -12), 34.56 (C-28, -40), 33.53 (d, P(C6H11)3), 31.42 (C-29, -39), 30.11 (d, P(C6H11)3), 27.19 (d, P(C6H11)3), 26.82 (d, P(C6H11)3), 21.15 (C-26, -37), 20.88 (C-25, -27, -36, -38).
δ P (162 MHz, d2-CH2Cl2, 273 K) 15.42.
Elemental analysis found: C, 74.32; H, 8.44; S, 6.27. Calc. for C62H81S2: C, 75.98; H, 8.33; S, 6.54%.
UV-Vis: λmax (thf)/nm 253 (ε/dm3 mol−1 cm−1 28630), 269 (27354), 283 (24012), 298 (21892), 453 (4545) and 550 (3545).
Crystal data: C62H81NiPS2·C4H8O, M = 1052.17, monoclinic, a = 9.7947(5), b = 31.5469(16), c = 19.7427(11) Å, U = 6084.0(6) Å3, T = 100(2) K, space group P121/n, Z = 4, 81481 reflections measured, 12065 unique (Rint = 0.0491) which were used in all calculations. The final wR(F2) was 0.2111 (all data).
Preparation and characterization of 4
Compound 2 (100 mg, 0.102 mmol), Ni(COD)2 (28 mg, 0.102 mmol), tricyclohexylphosphine (29 mg, 0.103 mmol), and thf (25 mL) were combined in a Schlenk flask. The resulting mixture was stirred for 2 h at r.t. to form a yellow solution. After removing the solvent under dynamic vacuum, the residual yellow solid was repeatedly (3×) washed with 5 mL portions of pentane, and dried under dynamic vacuum to produce solid yellow 4 (80 mg, 75%). Bulk crystallisation by slow diffusion of pentane layered on top of a concentrated thf solution produced single crystals suitable for XRD analysis.δ H (500 MHz, d8-thf, 253 K) 8.59 (4 H, br s, 8-, 9-, 11-, 12-H), 7.35 (2 H, d, J3−5 2.3, 4, 3-, 18-H), 6.81 (2 H, d, J5−3 2.2, 4, 5-, 16-H), 6.74 (4 H, s, 21-, 23-, 32-, 34-H), 2.23 (6 H, s, 26-, 37-H), 1.94 (12 H, s, 25-, 27-, 36-, 38-H), 1.30 (18 H, s, 4-, and 17-tBu), 2.05–0.72 (P(C6H11)3).
δ C (126 MHz, d8-thf, 253 K) 164.86 (C-2, -13), 150.45 (C-6, -15), 146.71 (C-4, -17), 143.19 (C-7, -10), 141.81 (C-19, -30), 139.52 (C-1, -14), 136.32 (C-20, -24, -31, -35), 135.47 (C-22, -33), 127.75 (C-21, -23, -32, -34), 126.30 (C-5, -16), 124.95 (C-8, -9, -11, -12), 121.56 (C-3, -18), 35.92 (PCy3), 34.88 (C-28, -40), 31.47 (C-29, -39), 30.63 (PCy3), 28.10 (PCy3), 27.20 (PCy3), 23.08 (C-25, -27, -36, -38), 21.12 (C-26, -37).
δ P (202 MHz, d8-thf, 253 K): 35 (ν1/2 ∼ 5000 Hz).
Elemental analysis found: C, 72.28; H, 8.74; S, 4.60. Calc. for C80H114S2: C, 72.84; H, 8.71; S, 4.86%.
UV-Vis: λmax (thf)/nm 220 (ε/dm3 mol−1 cm−1 15789), 248 (25725), 311 (6257), 352 (3005), 789 (233) and 972 (227).
Crystal data: C80H114Ni2P2S2, M = 1319.19, triclinic, a = 11.1421(4), b = 12.9165(5), c = 26.8959(10) Å, T = 130(2) K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 51911 reflections measured, 17549 unique (Rint = 0.0224) which were used in all calculations. The final wR(F2) was 0.1013 (all data).
, Z = 2, 51911 reflections measured, 17549 unique (Rint = 0.0224) which were used in all calculations. The final wR(F2) was 0.1013 (all data).
Preparation and characterization of 5
To a yellow solution of (Ph3P)2NiN(SiMe3)2 (463 mg, 0.622 mmol) in thf (50 mL) was added a solution of the ligand (200 mg, 0.311 mmol) in thf (5 mL) dropwise, and the resulting mixture stirred for 2 h at r.t. The solution gradually turned dark green. After removing the solvent under dynamic vacuum, the residual green solid was repeatedly (3×) washed with 10 mL portions of pentane, and dried under dynamic vacuum to produce solid 5 (215 mg). The raw product contains additional PPh3, typically ≤5%, as the only impurity. NMR spectroscopic properties of raw products are identical to samples recovered after one and two recrystallization steps. Recrystallization was performed by slow vapour diffusion of pentane, or hexane, into a concentrated solution of 5 in thf and produced green single crystals which were also suitable for XRD analysis (155 mg 39%). Other than the co-crystallized hydrocarbon, the material contains varying amounts of trapped thf, rendering elemental analysis difficult.δ H (500 MHz, d6-C6H6, 299 K) 12.01 (4 H, br s, 8-, 9-, 11-, 12-H), 7.99 (2 H, d, J3−5 1.6, 4, 3-, 18-H), 7.86 (2 H, d, J5−3 1.8, 4, 5-, 16-H), 7.48 (m, PPh3), 7.02 (m, PPh3), 6.84 (m, PPh3), 6.78 (4 H, s, 21-, 23-, 32-, 34-H), 2.28 (6 H, s, 26-, 37-H), 1.68 (12 H, s, 25-, 27-, 36-, 38-H), 1.22 (18 H, s, 4-, and 17-tBu).
δ C (126 MHz, d6-C6H6, 299 K) 164.71, 152.34, 142.79, 136.41, 136.37, 135.04, 133.70, 129.49, 128.02, 126.01, 115.33, 33.20, 32.25, 21.27, 20.73.
δ H (500 MHz, d8-thf, 298 K) 10.12 (4 H, br s, 8-, 9-, 11-, 12-H), 7.53 (2 H, d, J3−5 2.2, 4, 3-, 18-H), 7.36 (2 H, d, J5−3 2.3, 4, 5-, 16-H), 7.29 (m, PPh3), 7.17 (m, PPh3), 6.55 (4 H, s, 21-, 23-, 32-, 34-H), 2.26 (6 H, s, 26-, 37-H), 1.36 (18 H, s, 4-, and 17-tBu), 1.26 (12 H, s, 25-, 27-, 36-, 38-H).
δ C (126 MHz, d8-thf, 298 K) 159.11, 150.80 (C-4, -17), 147.85, 142.56 (C-19, -30), 137.45, 137.09 (PPh3), 136.05, 135.47 (PPh3), 130.58 (C-8, -9, -11, -12), 130.09 (PPh3), 129.24, 128.80 (PPh3), 128.23 (C-21, -23, -32, -34), 127.73 (C-5, -16), 125.60 (C-1, -14), 124.93 (C-3, -18), 34.57 (C-28, -40), 32.39 (C-29, -39), 21.60 (C-25, -27, -36, -38), 21.10 (C-26, -37).
δ P (202 MHz, d8-thf, 213 K) 68 (ν1/2 ∼ 800 Hz).
Elemental analysis found: C, 73.84; H, 6.22; S, 4.87. Calc. for C80H78S2: C, 74.90; H, 6.13; S, 5.00%.
UV-Vis: λmax (thf)/nm 315 (ε/dm3 mol−1 cm−1 13205), 380 (17718), 450 (7821), 725 (576) and 990 (527).
Crystal data: C80H78Ni2P2S2, M = 1282.9, triclinic, a = 11.9127(3), b = 21.1860(5), c = 28.2328(7) Å, T = 100(2) K, space group P, Z = 4, 275458 reflections measured, 30564 unique (Rint = 0.0235) which were used in all calculations. The final wR(F2) was 0.0936 (all data).
Computational details
DFT calculations of 3 and 5 were performed using the Gaussian 09 program suite,42 using the B3LYP density functional,43–45 along with the implemented 6-311G(d,p) basis set46–49 and dispersion corrections, including Becke–Johnson damping.50,51 For the theoretical model systems, the tBu substituents on the bridging 4-terphenyldithiophenolate ligand were replaced by methyl groups. All geometry optimizations were initially carried out without imposing any symmetry constraints. The geometry of 3, however, converged close to C2 symmetry and was subsequently optimised within this symmetry. All geometrical parameters were in excellent agreement with their experimental counterparts. The optimized structures were confirmed as true minima on the respective potential energy surface by calculating analytical frequencies. Modes with imaginary frequencies were absent for both 3 and 5. The topology of the electron density was analysed using the software package Aimall.41 Plots were generated using Aimall and Chemcraft.77Acknowledgements
This work was funded by the Fonds der Chemischen Industrie, FCI, in the form of a Liebig scholarship to A.B. which includes a doctoral scholarship to F.K. We thank Prof. Rainer Winter and Stefan Scheerer, University of Konstanz, for helpful discussions and Dr Klaus Eichele for assistance with and discussion of NMR data. Umicore AG & Co. KG, Hanau, Germany is acknowledged for the generous donation of (tBu3P)2Pd.Notes and references
- K. T. Horak, A. Velian, M. W. Day and T. Agapie, Chem. Commun., 2014, 50, 4427–4429 RSC.
- J. Wu, A. Nova, D. Balcells, G. W. Brudvig, W. Dai, L. M. Guard, N. Hazari, P.-H. Lin, R. Pokhrel and M. K. Takase, Chem. – Eur. J., 2014, 20, 5327–5337 CrossRef CAS PubMed.
- R. Beck and S. A. Johnson, Organometallics, 2013, 32, 2944–2951 CrossRef CAS.
- S. T. Chao, N. C. Lara, S. Lin, M. W. Day and T. Agapie, Angew. Chem., Int. Ed., 2011, 50, 7529–7532 CrossRef CAS PubMed.
- C. Jones, C. Schulten, L. Fohlmeister, A. Stasch, K. S. Murray, B. Moubaraki, S. Kohl, M. Z. Ertem, L. Gagliardi and C. J. Cramer, Chem. – Eur. J., 2011, 17, 1294–1303 CrossRef CAS PubMed.
- A. L. Keen and S. A. Johnson, J. Am. Chem. Soc., 2006, 128, 1806–1807 CrossRef CAS PubMed.
- G. Bai, P. Wei and D. W. Stephan, Organometallics, 2005, 24, 5901–5908 CrossRef CAS.
- J. J. Schneider, D. Wolf, U. Denninger, R. Goddard and C. Krüger, J. Organomet. Chem., 1999, 579, 139–146 CrossRef CAS.
- D. Adhikari, S. Mossin, F. Basuli, B. R. Dible, M. Chipara, H. Fan, J. C. Huffman, K. Meyer and D. J. Mindiola, Inorg. Chem., 2008, 47, 10479–10490 CrossRef CAS PubMed.
- R. Beck, M. Shoshani, J. Krasinkiewicz, J. A. Hatnean and S. A. Johnson, Dalton Trans., 2013, 42, 1461–1475 RSC.
- M. Ito, T. Matsumoto and K. Tatsumi, Inorg. Chem., 2009, 48, 2215–2223 CrossRef CAS PubMed.
- A. Mondragón, M. Flores-Alamo, P. R. Martínez-Alanis, G. Aullón, V. M. Ugalde-Saldívar and I. Castillo, Inorg. Chem., 2015, 54, 619–627 CrossRef PubMed.
- D. A. Vicic and W. D. Jones, J. Am. Chem. Soc., 1997, 119, 10855–10856 CrossRef CAS.
- B. R. Dible, M. S. Sigman and A. M. Arif, Inorg. Chem., 2005, 44, 3774–3776 CrossRef CAS PubMed.
- A. Velian, S. Lin, A. J. M. Miller, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2010, 132, 6296–6297 CrossRef CAS PubMed.
- Y.-Y. Zhou, D. R. Hartline, T. J. Steiman, P. E. Fanwick and C. Uyeda, Inorg. Chem., 2014, 53, 11770–11777 CrossRef CAS PubMed.
- C. A. Laskowski and G. L. Hillhouse, Organometallics, 2009, 28, 6114–6120 CrossRef CAS.
- K. Jonas and G. Wilke, Angew. Chem., Int. Ed. Engl., 1970, 9, 312–313 CrossRef CAS.
- G. M. Ferrence, E. Simón-Manso, B. K. Breedlove, L. Meeuwenberg and C. P. Kubiak, Inorg. Chem., 2004, 43, 1071–1081 CrossRef CAS PubMed.
- A. Miedaner and D. L. DuBois, Inorg. Chem., 1988, 27, 2479–2484 CrossRef CAS.
- J. J. Eisch, A. M. Piotrowski, K. I. Han, C. Kruger and Y. H. Tsay, Organometallics, 1985, 4, 224–231 CrossRef CAS.
- R. Beck and S. A. Johnson, Chem. Commun., 2011, 47, 9233–9235 RSC.
- U. Denninger, J. J. Schneider, G. Wilke, R. Goddard and C. Krüger, Inorg. Chim. Acta, 1993, 213, 129–140 CrossRef CAS.
- J. J. Schneider, U. Denninger, J. Hagen, C. Krüger, D. Bläser and R. Boese, Chem. Ber., 1997, 130, 1433–1440 CrossRef CAS.
- O. Jarchow, H. Schulz and R. Nast, Angew. Chem., Int. Ed. Engl., 1970, 9, 71 CrossRef CAS.
- G. Wilke, Angew. Chem., Int. Ed. Engl., 1988, 27, 185–206 CrossRef.
- C. A. Laskowski and G. L. Hillhouse, Chem. Sci., 2011, 2, 321–325 RSC.
- T. J. Steiman and C. Uyeda, J. Am. Chem. Soc., 2015, 137, 6104–6110 CrossRef CAS PubMed.
- P. J. Blower and J. R. Dilworth, Coord. Chem. Rev., 1987, 76, 121–185 CrossRef CAS.
- D. W. Stephan and T. Timothy Nadasdi, Coord. Chem. Rev., 1996, 147, 147–208 CrossRef CAS.
- E. Bouwman and J. Reedijk, Coord. Chem. Rev., 2005, 249, 1555–1581 CrossRef CAS.
- P. Kilian, F. R. Knight and J. D. Woollins, Coord. Chem. Rev., 2011, 255, 1387–1413 CrossRef CAS.
- J. C. Bayón, C. Claver and A. M. Masdeu-Bultó, Coord. Chem. Rev., 1999, 193–195, 73–145 CrossRef.
- E. I. Solomon, S. I. Gorelsky and A. Dey, J. Comput. Chem., 2006, 27, 1415–1428 CrossRef CAS PubMed.
- D. Sellmann and J. Sutter, Acc. Chem. Res., 1997, 30, 460–469 CrossRef CAS.
- H. Wadepohl, Angew. Chem., Int. Ed. Engl., 1992, 31, 247–262 CrossRef.
- T. Murahashi, K. Takase, M.-a. Oka and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 14908–14911 CrossRef CAS PubMed.
- A. Falceto, D. Casanova, P. Alemany and S. Alvarez, Chem. – Eur. J., 2014, 20, 14674–14689 CrossRef CAS PubMed.
- The longer Ni–C distance of 256.5(2) pm found for the second crystallographically independent formula unit results from packing effects between the C6H5 rings of the respective PPh3 ligands. See the ESI† for details.
- R. F. Bader, Atoms in molecules : a quantum theory, Clarendon Pr., Oxford, 1990 Search PubMed.
- T. A. Keith, AIMALL, TK Gristmill Software, Overland Park KS, USA, 2014, aim.tkgristmill.com Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Solid State, 1988, 37, 785–789 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033–1036 CrossRef CAS.
- K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062–1065 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- L. J. Farrugia, C. Evans, H. M. Senn, M. M. Hänninen and R. Sillanpää, Organometallics, 2012, 31, 2559–2570 CrossRef CAS.
- L. J. Farrugia and P. Macchi, Struct. Bonding, 2012, 146, 127–158 CrossRef CAS.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- The BCP represents the point of minimum electron density, ρ(r), along the bond path between two atoms, in which ρ(r) is locally greater than in any direction away from the path.
- P. Macchi and A. Sironi, Coord. Chem. Rev., 2003, 238–239, 383–412 CrossRef CAS.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed. Engl., 1984, 23, 627–628 CrossRef.
- X. Fradera, M. A. Austen and R. F. W. Bader, J. Phys. Chem. A, 1998, 103, 304–314 CrossRef.
- S. Pfirrmann, C. Limberg, C. Herwig, C. Knispel, B. Braun, E. Bill and R. Stösser, J. Am. Chem. Soc., 2010, 132, 13684–13691 CrossRef CAS PubMed.
- R. Beck, M. Shoshani and S. A. Johnson, Angew. Chem., Int. Ed., 2012, 51, 11753–11756 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- C. Piguet, J. Chem. Educ., 1997, 74, 815 CrossRef CAS.
- E. Wenschuh, D. Wilhelm, H. Hartung and U. Baumeister, Z. Anorg. Allg. Chem., 1994, 620, 2048–2052 CrossRef CAS.
- D. J. Elliot, D. G. Holah, A. N. Hughes, H. A. Mirza and E. Zawada, J. Chem. Soc., Chem. Commun., 1990, 32–33 RSC.
- D. L. DeLaet, P. E. Fanwick and C. P. Kubiak, Organometallics, 1986, 5, 1807–1811 CrossRef CAS.
- P. J. Hay, J. C. Thibeault and R. Hoffmann, J. Am. Chem. Soc., 1975, 97, 4884–4899 CrossRef CAS.
- O. Dahl, Acta Chem. Scand., 1969, 23, 2342–2354 CrossRef CAS.
- D. J. Krysan and P. B. Mackenzie, J. Org. Chem., 1990, 55, 4229–4230 CrossRef CAS.
- D. C. Bradley, M. B. Hursthouse, R. J. Smallwood and A. J. Welch, J. Chem. Soc., Chem. Commun., 1972, 872–873 RSC.
- S. M. Nelson and T. M. Shepherd, J. Chem. Soc., 1965, 3276–3284 RSC.
- R. E. Schuster, J. E. Scott and J. Casanova Jr., Org. Synth., 1966, 46, 75–77 CrossRef CAS.
- APEX2, Bruker AXS Inc., v 2011.8–0.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- G. M. S. C. B. Hübschle and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Speck, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef PubMed.
- G. A. Zhurko, CHEMCRAFT, chemcraft.com.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational, experimental, and NMR spectroscopic details. CCDC 1042645 (1), 1042646 (2), 1042647 (3), 1042648 (4), and 1042649 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01178g |
| This journal is © The Royal Society of Chemistry 2015 |