
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrothermal synthesis of Group 13 metal trifluoride complexes with neutral N-donor ligands†‡
Rajiv
Bhalla
a,
William
Levason
b,
Sajinder K.
Luthra
c,
Graeme
McRobbie
c,
Francesco M.
Monzittu
b,
Jazmyn
Palmer
b,
Gillian
Reid
*b,
George
Sanderson
b and
Wenjian
Zhang
b
aCentre for Advanced Imaging, University of Queensland, Brisbane, Queensland 4072, Australia
bSchool of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
cGE Healthcare UK, White Lion Road, Amersham HP7 9LL, UK
First published on 20th April 2015
Abstract
The reactions of the hydrated Group 13 fluorides, MF3·3H2O (M = Al, Ga or In) with 2,2′:6′,2′′-terpyridyl, 2,2′-bipyridyl or 1,10-phenanthroline under hydrothermal conditions (180 °C/15 h) produced high yields of the complexes [MF3(terpy)]·3H2O, [MF3(bipy)(OH2)]·2H2O and [MF3(phen)(OH2)]. X-Ray crystal structures of [M′F3(terpy)]·3H2O (M′ = Al or Ga), [M′F3(bipy)(OH2)]·2H2O and [GaF3(phen)(OH2)] show that all of them contain distorted octahedral geometries at the metal with mer-trifluoride coordination. Extensive H-bonding (F⋯H–OH) links the molecules. The complexes have been further characterised by microanalysis, IR, 1H, 19F{1H} and 27Al NMR spectroscopy. In contrast, reactions of the trifluorides with the acyclic triamine, N,N,N′,N′,N′′-pentamethyldiethylenetriamine, under similar hydrothermal conditions results in cleavage of the triamine and ring-closure to form the 1,1,4-trimethylpiperazinium cation, [⊂Me2N(CH2)2NMe(CH2)2]+, with fluorometallate anions, and confirmed by X-ray analysis of [⊂Me2N(CH2)2NMe(CH2)2]2[Al2F8(OH2)2]·2H2O. The strongly H-bonded [GaF3(terpy)]·3H2O was also obtained by Cl/F exchange from [GaCl3(terpy)] and [NBu4]F or [K(2,2,2-crypt)]F. Crystallisation of a mixture of [NH4][PF6] and [GaF3(terpy)]·3H2O from aqueous solution produced the edge-bridged cationic complex, [{Ga(terpy)F}2(μ-F)2][PF6]2. The synthesis of the more sterically bulky [GaCl3(tBu3-terpy)] (tBu3-terpy = 4,4′4′′-tris-tBu-2,2′:6′,2′′-terpyridyl) and the crystal structure of [GaCl2(tBu3-terpy)][GaCl4], which contains a trigonal bipyramidal cation, are also reported.
Introduction
The anhydrous Group 13 metal fluorides MF3 (M = Al, Ga or In) are inert polymeric solids and generally unreactive towards neutral ligands. This has hindered attempts to explore their coordination chemistry, hence very few adducts have been described.1 The hydrates, MF3·3H2O, are more reactive, but have very poor solubility in organic solvents or water.1 Two structural forms of AlF3·3H2O are known, the α-form with discrete [AlF3(OH2)3] molecules, and the β-form which is a polymer, [{AlF2(OH2)2(μ-F)}n]·nH2O.2 The structure of GaF3·3H2O is unclear, but InF3·3H2O is also a fluoride bridged polymer.2 Prolonged reflux of GaF3·3H2O with pyridine in thf afforded the mer octahedral complex [GaF3(py)3],3 whilst the reaction of GaF3·3H2O with 1,4,7-tris(2-amino-3,5-di-tert-butylbenzyl)-1,4,7-triazacyclononane (L) under reflux conditions in ethanol, followed by crystallisation from MeOH, afforded [GaF3(L)]·6MeOH, as an S6-symmetric MeOH hexamer.4 Complexes of aluminium, gallium and indium fluorides with (neutral) NH3 ligands have been formed from the reaction of AlN or InN with NH4F in supercritical ammonia at 400 °C, from NH4F or NH4HF2 and elemental Ga, and from [NH4]3[MF6] and gaseous NH3, or MF3·3H2O and liquid NH3.5–9 The structures are usually polymeric with F-bridges. Studies in aqueous solution of various ethylenediamine derivatives of Al and Ga10,11 and of the indium fluoride-2,2′-bipyridyl-H2O-HF system12 have also been reported. The X-ray crystal structures of [InF3(2,2′-bipy)(OH2)]·2H2O and [InF3(phen)(OH2)] obtained during the latter study have been described.12,13 The complexes [MF3(4,4′-bipy)] (M = Ga and In) were formed hydrothermally from the respective MF3·3H2O and 4,4′-bipyridyl in aqueous HF at 180 °C.14 Hydrothermal synthesis has also been used with other N-donor ligands such as the macrocyclic Me3-tacn and BzMe2-tacn (Me3-tacn = 1,4,7-trimethyl-1,4,7-triazacyclononane, BzMe2-tacn = 1,4,dimethyl-7-benzyl-1,4,7-triazacyclononane, L′) to form fac-[MF3(L′)]·xH2O for each of aluminium, gallium and indium.15 The compounds formed with Me3-tacn were found to crystallise as tetrahydrates, and extensive hydrogen bonding interactions between the water molecules of crystallisation and the coordinated fluorides were observed.An alternative method for the production of the metal-fluoro complexes is to first synthesise the metal chloride analogue and then perform a halide exchange reaction using reagents such as Me3SiF or Me3SnF. For example, [AlF2(py)4]Cl was successfully formed from the reaction of [AlCl3(py)n] (n = 1 to 3) with Me3SiF in pyridine.16
The use of aluminium15 and gallium17 fluoride complexes incorporating 18F as a radiolabel, has attracted much recent interest as diagnostic imaging agents for PET (positron emission tomography). Key to their potential clinical suitability is the ability to incorporate the short-lived 18F isotope (t1/2 = 110 min.) rapidly and cleanly in water; the resulting aluminium fluoride complexes are stable under physiological conditions,15 while the gallium fluoride radio-product is stable in phosphate buffered saline (PBS) solution.17 A fuller understanding of the coordination chemistry of these Group 13 fluorides is expected to contribute to advancing the design of the next generation of 18F imaging agents.
Here we report on the systematic study of the preparation, spectroscopic and structural features of several series of complexes of the three Group 13 trifluorides with bi- and poly-dentate N-donor ligands, to explore the suitability of hydrothermal synthesis for other neutral ligands. The work also investigates the effect of replacing amine with neutral N-heterocyclic ligands, and introducing the mer-trifluoride geometry, rather than the fac geometry present in the [MF3(R3-tacn)] systems.
Experimental
The MF3·3H2O (M = Al, Ga or In), GaCl3, 2,2′-bipyridyl, 1,10-phenanthroline, 2,2′:6′,2′′-terpyridyl, 4,4′,4′′-tris-t-butyl,-2,2′:6′,2′′-terpyridyl (tBu3-terpy), and N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDTA), were obtained from Sigma-Aldrich or Alfa-Aesar and used as supplied. Solvents were dried by distillation prior to use, CH2Cl2 and CH3CN from CaH2, hexane and toluene from sodium, THF from sodium benzophenone ketyl, methanol from magnesium/diiodine. All preparations of chloro-complexes were performed under an atmosphere of dry N2 using Schlenk techniques and spectroscopic samples were prepared in a dry, N2-purged glove box. Hydrothermal preparations were conducted in a 23 mL Teflon reactor vessel placed in a Parr stainless steel bomb. 1H NMR spectra were recorded in CD3OD on a Bruker AVII 400 spectrometer and referenced to the residual proton resonance. 19F{1H} NMR spectra were recorded in CD3OD on Bruker AVII 400 and DPX400 spectrometers, with CFCl3 as external reference. 27Al, 71Ga and 115In NMR spectra were recorded in CH3OH/CD3OD on a Bruker AVII 400 NMR spectrometer and were referenced to [Al(OH2)6]3+, [Ga(OH2)6]3+ and [In(OH2)6]3+, respectively, in H2O/D2O at pH = 1. Infra-red spectra were recorded as Nujol mulls between CsI plates on a Perkin Elmer Spectrum100 spectrometer over the range 4000–200 cm−1. Microanalyses were undertaken by London Metropolitan University.[AlF3(terpy)]·3H2O
AlF3·3H2O (0.100 g, 0.72 mmol) was suspended in freshly distilled water (7 mL) and terpy (0.169 g, 0.72 mmol) was then added. The suspension was transferred into a Teflon container and loaded into a stainless steel high pressure vessel (Parr) and heated to 180 °C for 15 h. The vessel was then allowed to cool. A pale yellow solution had formed, a small aliquot of which was retained to grow crystals. For the remaining reaction mixture the solvent was removed in vacuo, yielding a pale orange solid. Yield: 0.18 g, 70%. IR (Nujol, ν/cm−1): 3370 (ν O–H), 1640 (δ H–O–H) 656, 631, 620 (Al–F). 1H NMR (298 K): δ = 8.90 (d, JHH 8.0 Hz, [2H], ArH,) 8.76 (d, JHH 8.0 Hz, [2H], ArH), 8.54 (d, JHH 8.0 Hz, [2H], ArH), 8.39 (t, JHH 8.0 Hz, [2H], ArH), 8.28 (t, JHH 8.0 Hz, [H], ArH), 7.83 (t, JHH 6.0 Hz, [2H], ArH), 4.87 (s, H2O). 19F{1H} NMR (298 K): δ = −162.0 (d, 2JFF 23 Hz, [2F]), −177.3 (t, 2JFF 23 Hz, [F]). 27Al NMR (298 K): δ = 16.7 (br). Microanalyses on several batches, which were pure by spectroscopic analysis (including the single crystals), consistently gave H and N content as expected, but very variable (low) C content for this complex. Slow evaporation of the reaction solvent gave crystals suitable for X-ray diffraction.[GaF3(terpy)]·3H2O
Method 1: Method as for [AlF3(terpy)]·3H2O using GaF3·3H2O (0.200 g, 1.11 mmol) and terpy (0.259 g, 1.11 mmol). Orange solid. Yield 0.366 g, 80%. Required for C15H17F3GaN3O3 (414.0): C, 43.5; H, 4.1; N, 10.2. Found: C, 43.3; H, 4.0; N, 10.3%. IR (Nujol, ν/cm−1): 3407 (ν O–H), 1644 (δ H–O–H), 548, 504, 490(sh) (Ga–F). 1H NMR (298 K): δ = 9.04 (d, JHH 8.0 Hz, [2H], ArH,) 8.80 (d, JHH 8.0 Hz, [2H], ArH), 8.78 (d, JHH 8.0 Hz, [2H], ArH), 8.64 (t, JHH 8.0 Hz, [H], ArH), 8.49 (t, JHH 8.0 Hz, [2H], ArH), 8.03 (t, JHH 6.0 Hz, [2H], ArH), 4.87 (s, H2O). 19F{1H} NMR (298 K): δ = −156.3 (s, [2F]), −189.7 (s, [F]); (183 K) δ = −154.5 (s, [2F]), −188.9 (s, [F]). Slow evaporation of the reaction solvent gave crystals suitable for X-ray diffraction.Method 2: A suspension of [GaCl3(terpy)] (0.06 g, 0.15 mmol) in anhydrous MeCN (5 mL) was treated with 0.45 mL (0.45 mmol) of a 1.0 M solution of [NBu4]F in thf. Addition of the fluoride source resulted in the dissolution of the chloride precursor and the formation of a pale yellow solution. The mixture was stirred at room temperature for 1 h and then the volatiles were removed in vacuo to yield a yellow gum. This was dissolved in a minimum volume of CH2Cl2 (ca. 2 mL) and layered with hexane. A pale yellow precipitate formed overnight. Yield 0.042 g, 67%. Spectroscopic data matched that observed for Method 1 and recrystallisation from CH2Cl2/hexane yielded small crystals whose unit cell dimensions matched those of the crystals obtained via Method 1.
Method 3: [GaCl3(terpy)] (0.020 g, 0.050 mmol) was suspended in anhydrous MeCN (5 mL). A solution of [K(2.2.2-crypt)]F (0.067 g, 0.150 mmol) in 3 mL anhydrous MeCN was added dropwise to the chloride precursor. Addition of the fluoride source resulted in the dissolution of the chloride precursor and the formation of a colourless solution. The mixture was stirred at room temperature for 1 h, then the volatiles were removed in vacuo to give a white solid containing both the expected fluoride complex and the [K(2.2.2-crypt)]Cl by-product. Spectroscopic data for the former matched that observed from Method 1. No further purification was undertaken.
[{Ga(terpy)F}2(μ-F)2][PF6]2·4H2O
[GaF3(terpy)]·3H2O (0.013 g, 0.025 mmol) was dissolved in a mixture CH3CN/H2O (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 1.5 mL) and added to [NH4][PF6] (0.004 g, 0.025 mmol) in H2O (1.5 mL). The reaction solution was left to evaporate slowly at room temperature, resulting in the formation of orange crystals suitable for single crystal X-ray diffraction. Required for C30H30F16Ga2N6O4P2: C, 34.5; H, 2.9; N, 8.1. Found: C, 34.4; H, 2.9; N, 8.2%. 19F{1H} NMR (CD3OD, 183 K): δ = −75.1 (d, [PF6]−), −156.4 (s, [2F]), −189.8 (s, [F]).
:3, 1.5 mL) and added to [NH4][PF6] (0.004 g, 0.025 mmol) in H2O (1.5 mL). The reaction solution was left to evaporate slowly at room temperature, resulting in the formation of orange crystals suitable for single crystal X-ray diffraction. Required for C30H30F16Ga2N6O4P2: C, 34.5; H, 2.9; N, 8.1. Found: C, 34.4; H, 2.9; N, 8.2%. 19F{1H} NMR (CD3OD, 183 K): δ = −75.1 (d, [PF6]−), −156.4 (s, [2F]), −189.8 (s, [F]).
[InF3(terpy)]·3H2O
Method as for [AlF3(terpy]·3H2O using InF3·3H2O (0.100 g, 0.44 mmol) and terpy (0.1032 g, 0.44 mmol). Pale orange solid. Yield: 0.10 g, 49%. Required for C15H17F3InN3O3 (459.1): C, 39.2; H, 3.7; N, 9.2. Found: C, 39.4; H, 3.9; N, 9.3%. IR (Nujol, ν/cm−1): 3490, 3400(ν O–H), 1654 (δ H–O–H), 476, 427, 406 (In–F). 1H NMR (298 K): δ = 8.68 (m, [2H], ArH), 8.61 (d, JHH 8.0 Hz, [2H], ArH), 8.39 (d, JHH 8.0 Hz, [2H], ArH), 8.07–7.97 (m, [3H], ArH), 7.47 (m, [2H], ArH), 4.87 (s, H2O). 19F{1H} NMR (298 K): δ = −157.8 (br s [2F]), −181.6 (br s [F]); (183 K): −157.5 (br s [2F]), −178.5 (br s [F]).[AlF3(bipy)(OH2)]·2H2O
Method as for [AlF3(terpy)]·3H2O using AlF3·3H2O (0.200 g, 1.45 mmol) and bipy (0.226 g, 1.45 mmol). The resulting pale pink solid was washed with acetonitrile and filtered to leave a white solid which was dried in vacuo. Yield: 0.20 g, 47%. Required for C10H14AlF3N2O3 (294.2): C, 40.8; H, 4.8; N, 9.5. Found: C, 40.6; H, 4.6; N, 9.7%. IR (Nujol, ν/cm−1): 3533, 3422 (ν O–H), 1689 br (δ H–O–H), 610, 590(sh) (Al–F). 1H NMR (CD3OD, 298 K): δ = 9.04 (d, JHH 4.0 Hz, [1H], ArH), 8.76 (d, JHH 4.4 Hz, [1H], ArH), 8.59 (d, JHH 7.6 Hz, [1H], ArH), 8.44 (d, JHH 8.1 Hz, [1H], ArH), 8.33 (t, JHH 7.7 Hz, [1H], ArH), 8.14 (t, JHH 7.5 Hz, [1H], ArH), 7.81–7.85 (m, [1H], ArH), 7.63 (t, JHH 5.9 Hz, [1H], ArH), 4.75 (s, H2O). 19F{1H} NMR (298 K): δ = −172.8 (br s); (183 K): −162.3 (s, [F]), −176.4 (s, [2F]). 27Al NMR (298 K): δ = 8.3 (br s, w1/2 = 400 Hz), resonance lost on cooling below 233 K. Slow evaporation of the reaction solvent gave crystals suitable for X-ray diffraction.[GaF3(bipy)(OH2)]·2H2O
Method as for [AlF3(terpy)]·3H2O using GaF3·3H2O (0.200 g, 1.11 mmol) and bipy (0.158 g, 1.11 mmol). Pale pink solid. Yield 0.314 g, 84%. Required for C10H14F3GaN2O3 (337.0): C, 35.7; H, 4.1; N, 8.3. Found: C, 35.6; H, 4.1; N, 8.2%. IR (Nujol, ν/cm−1): 3500, 3380 (ν O–H), 1660 br (δ H–O–H), 527, 473 (Ga–F). 1H NMR (CD3OD, 298 K): δ = 9.07 (br, [2H], ArH), 8.66 (d, JHH 8 Hz, [2H], ArH), 8.37 (t, JHH ∼ 8 Hz, [2H], ArH), 7.89 (s, [2H], ArH); (183 K): 9.09 (s, [2H]), 8.81 (br, s [2H], ArH), 8.47 (s, [2H]), 7.97 (s, [2H]), 4.75 (s, H2O). 19F{1H} NMR (298 K): δ = −176.2 (br s); (183 K): −160.9 (s, [F]), −181.5 (s, [2F]). Slow evaporation of the reaction solvent gave crystals suitable for X-ray diffraction.[InF3(bipy)(OH2)]·2H2O
Method as for [AlF3(terpy)]·3H2O using InF3·3H2O (0.200 g, 0.89 mmol) and bipy (0.138 g, 0.89 mmol), yielding a pale yellow solid. Yield: 0.17 g, 50%. Required for C10H14F3InN2O3 (382.1): C, 31.4; H, 3.7; N, 7.3. Found: C, 31.6; H, 3.6; N, 7.4%. IR (Nujol, ν/cm−1): 3422 br (ν O–H), 1655 br (δ H–O–H), 442, 428, 404 (In–F). 1H NMR (CD3OD, 298 K): δ = 8.78 (br s, [2H], ArH), 8.68 (br s, [2H], ArH), 8.45 (br s, [2H], ArH), 7.96 (br s, [2H], ArH), 4.75 (s, H2O); (183 K): 8.99 (br, s), 8.95 (br s,), 8.66 (s), 8.52 (vbr), 8.05–7.99 (m). 19F{1H} NMR (CD3OD, 298 K): not observed; (183 K): δ = −152.9 (s, [F]), −178.1 (s, [2F]).[AlF3(phen)(OH2)]
Method as for [AlF3(terpy)(OH2)]·2H2O, but using AlF3·3H2O (0.200 g, 1.45 mmol) and phen (0.261 g, 1.45 mmol). White solid. Yield: 0.254 g, 62%. Required for C12H10AlF3N2O (282.2): C, 51.1; H, 3.6; N, 9.9. Found: C, 50.9; H, 3.4; N, 10.0%. IR (Nujol, ν/cm−1): 3200br (ν O–H), 1670 (δ H–O–H), 637, 608 (Al–F). 1H NMR (298 K): δ = 9.30 (d, JHH 5.1 Hz, [1H], ArH), 9.17 (d, JHH 3.7 Hz, [1H], ArH), 8.88 (d, JHH 7.6 Hz, [1H], ArH), 8.66 (d, JHH 8.8 Hz, [1H], ArH), 8.25 (s, [1H], ArH), 8.14 (m, [1H], ArH), 8.09 (s, [1H], ArH), 7.93 (m, JHH 4.2 Hz, [1H], ArH), 4.75 (s, H2O). 19F{1H} NMR (298 K): δ = −157.3 (br s [F]), −174.8 (s, [2F]); (183 K): δ = −162.5 (br s [F]), −177.3 (s, [2F]);. 27Al NMR (298 K): δ = 8.8 (br s, w1/2 450 Hz).[GaF3(phen)(OH2)]
Method as for [AlF3(terpy)]·3H2O, using GaF3·3H2O (0.200 g, 1.11 mmol) and phen (0.200 g, 1.11 mmol). Pale red solid. Yield 0.315 g, 79%. Required for C12H10F3GaN2O (324.9): C, 44.4; H, 3.1; N, 8.6. Found: C, 44.4; H, 3.1; N, 8.5%. IR (Nujol, ν/cm−1): 3200 br (ν O–H), 1680 (δ H–O–H), 560, 543, 511 (Ga–F). 1H NMR (298 K): δ = 9.34 (d, JHH 4 Hz, [2H], ArH), 8.89 (d, JHH 12 Hz, [2H], ArH), 8.26 (s, [2H], ArH), 8.15 (br, [2H], ArH) 4.87 (s, H2O); (183 K): 9.34 (s), 9.02 (d), 8.94 (d), 8.33–8.21 (m). 19F{1H} NMR (298 K): δ = −152 (br s [F]), −176 (br, [2F]); (183 K): δ = −151.0 (br s [F]), −184.0 (s, [2F]). Slow evaporation of the reaction solvent gave crystals suitable for X-ray diffraction.[InF3(phen)(OH2)]
Method as for [AlF3(terpy)]·3H2O, but using InF3·3H2O (0.200 g, 0.89 mmol) and phen (0.160 g, 0.89 mmol). Pale orange solid. Yield: 0.16 g, 49%. Required for C12H10F3InN2O (370.0): C, 39.0; H, 2.7; N, 7.6. Found: C, 39.2; H, 2.8; N, 7.7%. IR (Nujol, ν/cm−1): 3360 br, 3177 (ν O–H), 1660 w (δ H–O–H), 446, 422 br, 405(sh) (In–F). 1H NMR (298 K): δ = 9.30 (br s, [1H], ArH), 9.02–8.96 (br [3H], ArH), 8.29–8.07 (br, m, [4H], ArH), 4.87 (s, H2O); (183 K): 9.28 (s), 8.98 (s), 8.76 (s), 8.33–8.22 (m), 7.95 (m). 19F{1H} NMR (298 K): not observed; (183 K): δ = −157 (br, [F]), −199 (br, [2F]).[GaCl3(terpy)]
A solution of terpy (0.258 g, 1.11 mmol) in anhydrous CH2Cl2 (5 mL) was added dropwise to a solution of GaCl3 (0.195 g, 1.11 mmol) in 5 mL anhydrous CH2Cl2. A white precipitate formed upon addition of the ligand. The mixture was stirred at room temperature for 1 h. The product was isolated by filtration, washed with hexane and dried in vacuo. White solid. Yield: 0.402 g, 88%. Required for C15H11Cl3GaN3: C, 44.0; H, 2.7; N, 10.3. Found: C, 43.5; H, 2.6; N, 10.1%. IR (Nujol, ν/cm−1): 267 s, 254 br, (Ga–Cl). 1H NMR (CD3CN, 298 K): δ = 9.06–9.04 (m, [2H], ArH), 8.75–8.73 (m, [3H], ArH), 8.67–8.65 (m, [2H], ArH), 8.53–8.49 (m, [2H], ArH), 8.08–8.05 (m, [2H], ArH).[GaCl3(tBu3-terpy)]
A solution of tBu3-terpy (0.111 g, 0.278 mmol) in anhydrous CH2Cl2 (5 mL) was added dropwise to a solution of GaCl3 (0.025 g, 0.139 mmol) in anhydrous CH2Cl2 (5 mL). Addition of the ligand resulted in the formation of a white precipitate. The mixture was stirred at room temperature for 1 h. The solid was isolated by filtration, washed with a further 10 mL CH2Cl2 and dried in vacuo. White solid. Yield 0.062 g, 77%. Required for C27H35Cl3GaN3·CH2Cl2 (662.60): C, 50.7; H, 5.6; N, 6.3. Found: C, 50.4; H, 5.6; N, 6.5%. 1H NMR (CD3CN, 298 K): δ = 1.42 (s, [18H], tBu), 1.44 (s, [9H], tBu), 7.47–7.46 (br m, [2H], ArH), 8.50 (br s, [2H], ArH), 8.61–8.60 (br m, [2H], ArH), 8.77 (br s, [2H], ArH).The same reaction performed in a 1:1 stoichiometric ratio gave a spectroscopically identical product. However, crystals formed from the filtrate gave the ionic by-product [GaCl2(tBu3-terpy)][GaCl4]·CH2Cl2.
[⊂Me2N(CH2)2NMe(CH2)2]2[Al2F8(OH2)2]·2H2O
Method as for [AlF3(bipy)(OH2)]·2H2O, but using AlF3·3H2O (0.200 g, 1.45 mmol) and PMDTA (0.3 mL, 1.45 mmol). A brown gum was obtained after removing the solvent. This was washed with MeCN, producing a white powder. Yield: 0.44 g, 58%. Required for C14H42Al2F8N4O4 (536.4): C, 31.4; H, 7.8; N, 10.4. Found: C, 31.3; H, 7.8; N, 10.4%. IR (Nujol, ν/cm−1): 3370 br (ν O–H), 1673 (δ H–O–H), 616 br, 569 br, (Al–F). 1H NMR (CD3CN, 298 K): δ = 4.87 (s, H2O), 3.35 (t, JHH 6.0 Hz, [4H], CH2), 3.07 (s, [6H], Me), 2.67 (br s, [4H], CH2CH2), 2.32 (s, [3H], Me). 19F{1H} NMR (CD3CN, 298 K): δ = −194.6 (6 lines 1JAlF = 38 Hz). 27Al NMR (CH3CN, 298 K): δ = 48.7 (s). Slow evaporation of the reaction solvent gave crystals suitable for X-ray diffraction.The corresponding gallium and indium fluoride reactions were conducted similarly.
[⊂Me2N(CH2)2NMe(CH2)2]2[Ga2F8(OH2)2]·2H2O
Required for C14H42F8Ga2N4O4 (619.9): C, 27.0; H, 6.8; N, 9.0. Found: C, 26.7; H, 6.4; N, 9.8%. 1H NMR (CD3CN, 298 K): δ = 4.87 (s, H2O), 3.35 (t, JHH 6.0 Hz, [4H], CH2), 3.07 (s, [6H], Me), 2.67 (br s, [4H], CH2CH2), 2.32 (s, [3H], Me). 13C{1H} NMR (CD3CN): δ = 44.18 (s), 44.84 (s), 48.04 (s), 61.59 (s). IR (Nujol, ν/cm−1): 3420 br (ν O–H), 1673 (δ H–O–H), 518 br, 477 br, (Ga–F).A small number of crystals obtained from the Ga and In reaction mixtures were found to be [⊂Me2N(CH2)2NMe(CH2)2]Cl (see text and ESI‡).
X-Ray experimental
Details of the crystallographic data collection and refinement parameters are given in Table 1. Crystals suitable for single crystal X-ray analysis were obtained as described above. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 μm focus) with the crystal held at 100 K (N2 cryostream). Structure solution and refinements were performed with either SHELX(S/L)97 or SHELX(S/L)201318 and were straightforward, except where detailed below. H atoms bonded to C were placed in calculated positions using the default C–H distance and refined using a riding model. In the case of the [AlF3(bipy)(OH2)]·2H2O structure, the H-atoms on the co-crystallised water molecules were not located in the difference map. While not included in the refinement, the H-atoms are inferred from the H-bonding distances of F⋯O and O⋯O and are thus included in the formulae. The H-atoms on both the coordinated and co-crystallised water molecules could not be located in the structure of [⊂Me2N(CH2)2NMe(CH2)2]2[Al2F8(OH2)2]·2H2O. While not included in the refinement, the H-atoms are similarly inferred and included in the formulae. CCDC numbers 1053047–1053048 and 1053152–1053158.| Compound | [AlF3(terpy)]·3H2O | [GaF3(terpy)]·3H2O | [AlF3(bipy)(OH2)]·2H2O | [GaF3(bipy)(OH2)]·2H2O |
|---|---|---|---|---|
| Formula | C15H17AlF3N3O3 | C15H17F3GaN3O3 | C10H14AlF3N2O3 | C10H14F3GaN2O3 |
| M | 371.30 | 414.04 | 294.20 | 336.95 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group (no.) | P21/c (14) | P21/c (14) | P21/n (14) | P21/n (14) |
| a/Å | 9.4806(9) | 10.323(5) | 8.976(5) | 9.056(3) |
| b/Å | 25.104(2) | 9.708(5) | 7.300(5) | 7.371(2) |
| c/Å | 7.0846(6) | 16.131(5) | 19.062(5) | 18.904(6) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 104.425(7) | 100.484(5) | 93.331(5) | 93.462(6) |
| γ/° | 90 | 90 | 90 | 90 |
| U/Å3 | 1633.0(3) | 1589.6(12) | 1246.9(11) | 1259.6(7) |
| Z | 4 | 4 | 4 | 4 |
| μ(Mo-Kα)/mm–1 | 0.177 | 1.785 | 0.207 | 2.228 |
| F(000) | 768 | 840 | 608 | 680 |
| Total number reflections | 6803 | 9623 | 8257 | 11425 |
| R int | 0.062 | 0.048 | 0.078 | 0.154 |
| Unique reflections | 2163 | 3646 | 2848 | 2883 |
| No. of parameters, restraints | 294,0 | 250, 0 | 188, 0 | 196, 8 |
| R 1, wR2 [I > 2σ(I)]b | 0.071, 0.133 | 0.037, 0.084 | 0.060, 0.144 | 0.048, 0.130 |
| R 1, wR2 (all data) | 0.146, 0.172 | 0.056, 0.089 | 0.104, 0.168 | 0.051, 0.132 |
| Compound | [GaF3(phen)(OH2)] | [GaCl2(tBu3-terpy)]-[GaCl4]·CH2Cl2 | [⊂Me2N(CH2)2NMe(CH2)2]2[Al2F8(OH2)2]·2H2O | [{Ga(terpy)F}2(μ-F)2][PF6]2·4H2O |
|---|---|---|---|---|
| a Common items: T = 100 K; wavelength (Mo-Kα) = 0.71073 Å; θ(max) = 27.5°. b R 1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑wFo2]1/2. | ||||
| Formula | C12H10F3GaN2O | C14H18.5Cl4GaN1.5 | C14H42Al2F8N4O4 | C30H30F16Ga2N6O4P2 |
| M | 324.94 | 419.32 | 538.4 | 1043.98 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group (no.) | P21/n (14) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2) (2) |
C2/c (15) |
P (2) |
| a/Å | 8.3906(5) | 11.674(3) | 24.054(10) | 9.774(2) |
| b/Å | 7.1940(5) | 12.642(4) | 7.558(3) | 10.460(2) |
| c/Å | 19.0124(13) | 13.756(5) | 16.389(7) | 10.646(2) |
| α/° | 90 | 89.137(17) | 90 | 78.064(12) |
| β/° | 100.631(3) | 78.682(16) | 123.374(4) | 62.736(8) |
| γ/° | 90 | 65.276(15) | 90 | 85.448(14) |
| U/Å3 | 1127.93(13) | 1803.1(11) | 2488.3(17) | 946.5(3) |
| Z | 4 | 4 | 6 | 1 |
| μ(Mo-Kα)/mm–1 | 2.472 | 2.111 | 0.214 | 1.633 |
| F(000) | 648 | 848 | 1200 | 520 |
| Total number reflections | 12632 |
16476 |
5785 | 17446 |
| R int | 0.051 | 0.141 | 0.035 | 0.079 |
| Unique reflections | 2581 | 7870 | 2822 | 3925 |
| No. of parameters, restraints | 178, 2 | 379, 0 | 170, 6 | 271, 0 |
| R 1, wR2 [I > 2σ(I)]b | 0.025, 0.064 | 0.086, 0.192 | 0.078, 0.212 | 0.043, 0.109 |
| R 1, wR2 (all data) | 0.028, 0.066 | 0.125, 0.211 | 0.098, 0.231 | 0.049, 0.111 |
Results and discussion
The unreactive and poorly soluble nature of the MF3·3H2O makes reaction with neutral ligands in organic solvents difficult or impossible. We therefore used the hydrothermal approach (180 °C/15 h) and found this gave high yields of [MF3(terpy)]·3H2O (below). The same approach was then extended to reactions with N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDTA), and to the diimines, 2,2′-bipyridyl and 1,10-phenanthroline.[MF3(terpy)]·3H2O (M = Al, Ga or In)
The reaction of the MF3·3H2O with terpy in a 1:1 molar ratio in water at 180 °C/15 h, followed by removal of the solvent in vacuo at room temperature, gave high yields of the [MF3(terpy)]·3H2O as pale orange solids. Slow evaporation of a small portion of the mother liquor at ambient temperatures gave crystals of [MF3(terpy)]·3H2O (M = Al or Ga). For M = Al, the structure shows (Fig. 1) a distorted octahedral geometry about the aluminium, the distortions being largely due to the rigid terpy ligand which results in N–Al–N angles significantly less than 90°/180°, whereas the F–Al–F and (cis) F–Al–N angles are close to those expected for an octahedron.
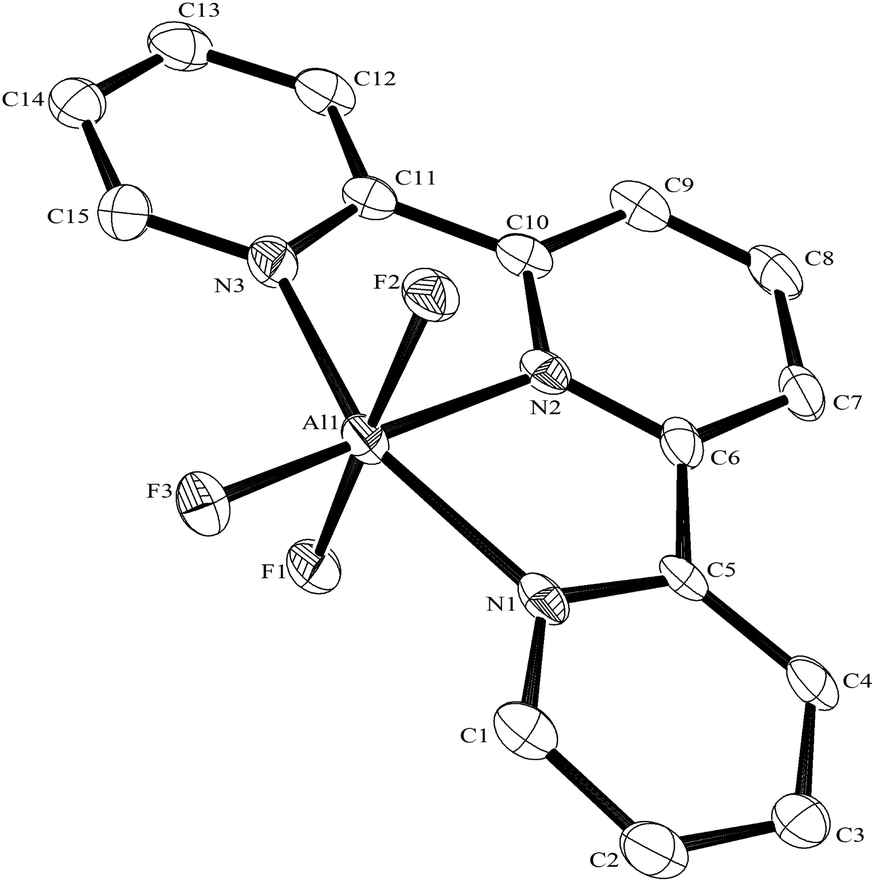 | ||
| Fig. 1 The structure of the Al species in [AlF3(terpy)]·3H2O with ellipsoids drawn at the 50% probability level. Hydrogen atoms on terpy are omitted for clarity. Selected bond lengths (Å) and angles (°) are Al1–F1 = 1.761(2), Al1–F2 = 1.762(2), Al1–F3 = 1.737(2), Al1–N1 = 2.057(3), Al1–N2 = 2.010(3), Al1–N3 = 2.059(3), F1–Al1–F3 = 92.55(12), F2–Al1–F3 = 91.08(11), F1–Al1–F2 = 176.34(13), N1–Al1–N2 = 77.58(13), N2–Al1–N3 = 78.17(13), N1–Al1–N3 = 155.73(13). | ||
There is extensive H-bonding between the fluoride ligands and the lattice water molecules (Fig. 2), as well as π-stacking of the aromatic rings (Fig. 3).
 | ||
| Fig. 2 The H-bonding network (red) in the crystals of [AlF3(terpy)]·3H2O. | ||
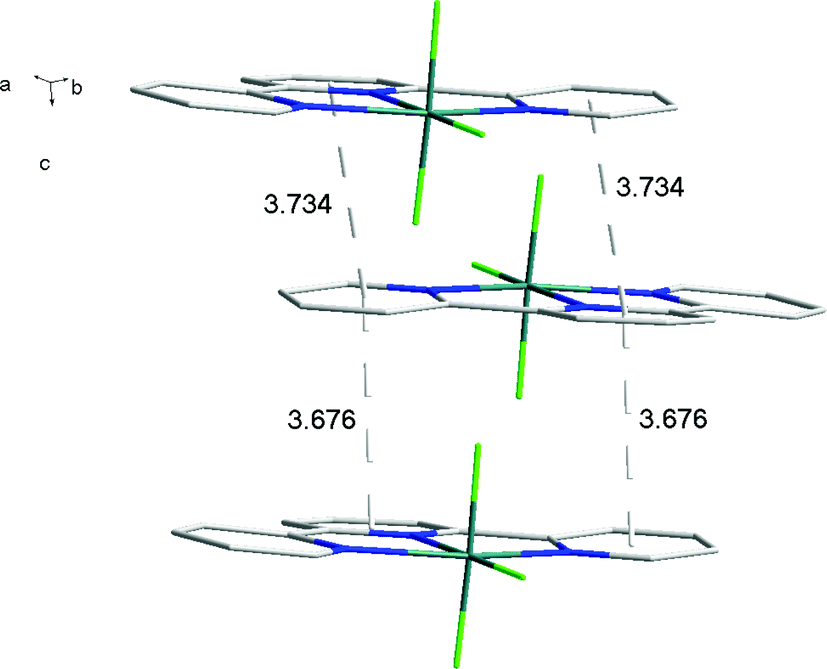 | ||
| Fig. 3 π-stacking in the structure of [AlF3(terpy)]·3H2O with lattice water omitted for clarity. | ||
The Al–F bond lengths are very similar to those observed in fac-[AlF3(Me3-tacn)]·4H2O,15 but the Al–N bonds are shorter by ∼0.05 Å, suggesting the macrocyclic ring may limit the close approach of the N atoms to the metal centre. The molecular structure of [GaF3(terpy)]·3H2O is very similar to that of the Al(III) complex (Fig. 4) with the Ga–F bonds ca. 0.1 Å longer than the corresponding Al–F, whilst the Ga–N and Al–N are little different.
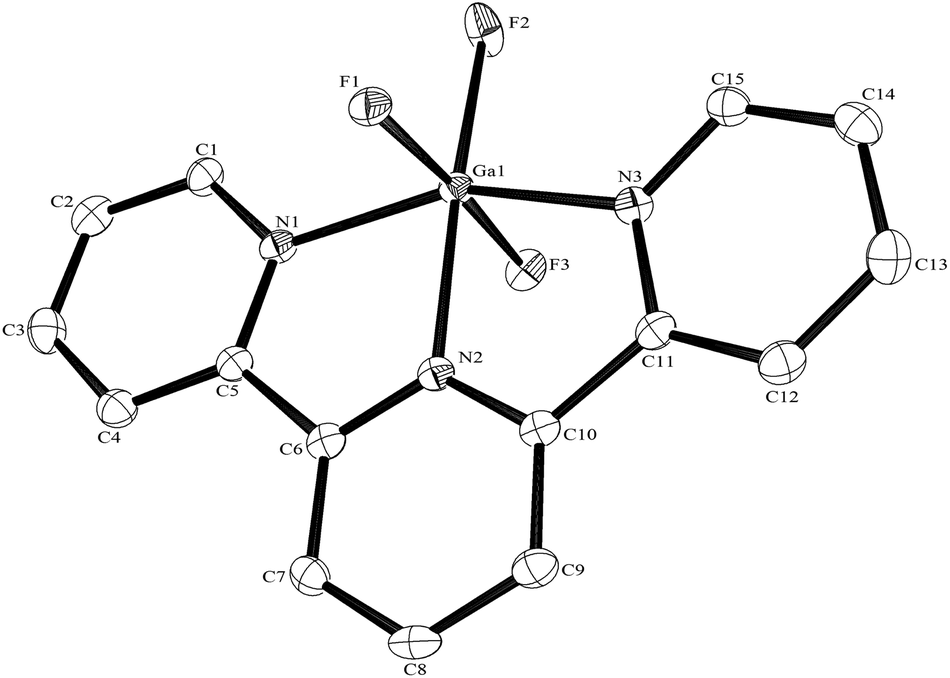 | ||
| Fig. 4 The structure of the Ga species in [GaF3(terpy)]·3H2O with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°):Ga1–F1 = 1.8781(17), Ga1–F2 = 1.8780(16), Ga1–F3 = 1.8895(17), Ga1–N1 = 2.082(2), Ga1–N2 = 2.045(2), Ga1–N3 = 2.093(2), F1–Ga1–F2 = 92.12(7), F2–Ga1–F3 = 90.60(7), F1–Ga1–F3 = 176.29(7), N1–Ga1–N2 = 77.21(8), N2–Ga1–N3 = 77.26(9), N1–Ga1–N3 = 154.28(8). | ||
As found in the aluminium complex, the structure of [GaF3(terpy)]·3H2O shows extensive H-bonding as shown in Fig. 5 as well as π-stacking interactions of 3.57 and 3.62 Å (see ESI Fig. S1‡), although the arrangements differ in detail. The Ga–F distances are very similar to those found in fac-[GaF3(Me3-tacn)]·4H2O.15 The Ga–N distances are also not significantly different to those found in the [GaX3(terpy)] (X = Cl or Br),19 although the extensive H-bonding in the fluoride complex is absent in the structures of the heavier halides. The single bond covalent radii of Al(III) and Ga(III) are quoted in standard texts as nearly identical (∼1.25 Å), although the ionic radius of Ga3+ is ∼0.07 Å larger than that of Al3+.20 The limited number of structurally characterised complexes of the trifluorides limits detailed comparisons, but it seems that the metal-donor bond length may be very sensitive to the electronegativity of the donor atom, with little difference between Al–L and Ga–L with heavier donor ligands and halides,21 but significant differences in the M–F bond lengths. The effects of solvent molecules and hydrogen bonding also complicates the interpretation of small differences in metal-donor bond lengths,19,22 and these may be a significant factor in the extensively hydrogen bonded metal fluoride complexes.
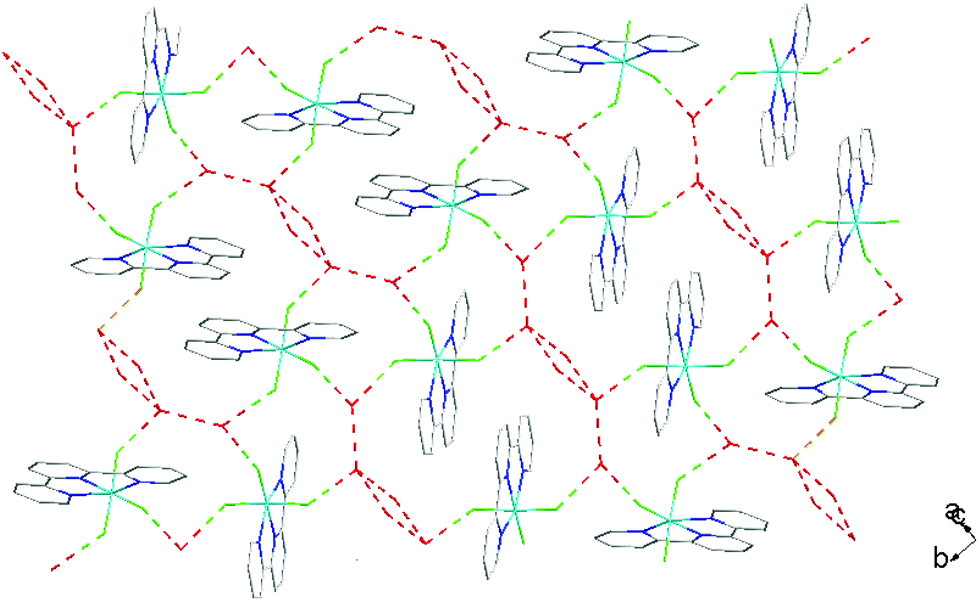 | ||
| Fig. 5 View showing the H-bonding network (red) in [GaF3(terpy)]·3H2O. | ||
Interpretation of the spectroscopic properties (Experimental section) of the three terpy complexes is straightforward. The IR spectra show strong, very broad features due to the ν(OH) and δ(HOH) modes of the water and three ν(MF) stretches, as expected for a mer-trifluoride (theory 2A1 + B1). The 1H NMR spectra in CD3OD exhibit sharp multiplet resonances for the terpy protons and a broad signal for the water, whilst the 19F{1H} NMR spectra show two resonances in a 2:1 ratio due to FtransF and FtransN respectively. The 19F{1H} resonances for the [AlF3(terpy)] show doublet and triplet 2JFF couplings of 23 Hz, but for the gallium and indium complexes only broad singlets are observed over the temperature range 298–183 K.23 The aluminium complex also exhibited a 27Al NMR resonance at δ = 16.7 as a broad singlet with no resolved 1JAlF coupling, which is in the range expected for six-coordinate aluminium.24 Neither the gallium nor indium complex exhibited a metal nucleus resonance, probably due to fast quadrupolar relaxation. The multinuclear NMR data show that the molecular structures of these complexes are maintained in MeOH solution.
The synthesis of the [GaF3(terpy)] by halide exchange was also explored. The [GaCl3(terpy)] precursor was made in high yield by reaction of GaCl3 with terpy in anhydrous CH2Cl2 solution, and its composition confirmed by microanalysis and spectroscopically. Complete fluorination of [GaCl3(terpy)] in MeCN solution was achieved at room temperature with [nBu4N]F in thf or [K(2.2.2-crypt)]F in MeCN; the [GaF3(terpy)] produced was spectroscopically identical to samples produced by the hydrothermal route, and crystals obtained using the [nBu4N]F route had the same unit cell. Fluorination with [K(2.2.2-crypt)]F was also successful at 80 °C in MeCN, confirming the gallium species is thermally stable under these conditions. The [GaCl3(terpy)] was relatively poorly soluble in organic solvents and in an attempt to increase the solubility, the corresponding complex of 4,4′,4′′-tris-t-butyl,-2,2′:6′,2′′-terpyridyl, [GaCl3(tBu3-terpy)], was prepared. Its properties were very similar to the terpy complex (Experimental section), but it was in fact rather less soluble in common organic solvents. However, crystals grown from the filtrate from one synthesis were found to be [GaCl2(tBu3-terpy)][GaCl4]·CH2Cl2, containing a five-coordinate cation. This minor by-product probably results from the presence of a small excess of GaCl3 in the synthesis, which extracts a chloride from the neutral species to form the stable [GaCl4]− anion. The structure of the cation is shown in Fig. 6. The geometry is a distorted trigonal bipyramid with apical N, distorted by the steric constraints of the tBu3-terpy (N3–Ga1–N1 = 155.1(2)°), as also found in the neutral [GaCl3(terpy)] complex above. Comparisons of the bond lengths between [GaCl2(tBu3-terpy)]+ and [GaCl3(terpy)],19 as expected, show shorter bonds in the five-coordinate cation (Ga–Cl = 2.169(2), 2.195(2), Ga–N = 1.995(5)–2.092(5) Å) compared to the six-coordinate neutral complex (Ga–Cl = 2.2511(5) −2.4118(6), Ga–N = 2.0412(5)–2.1024(15) Å).
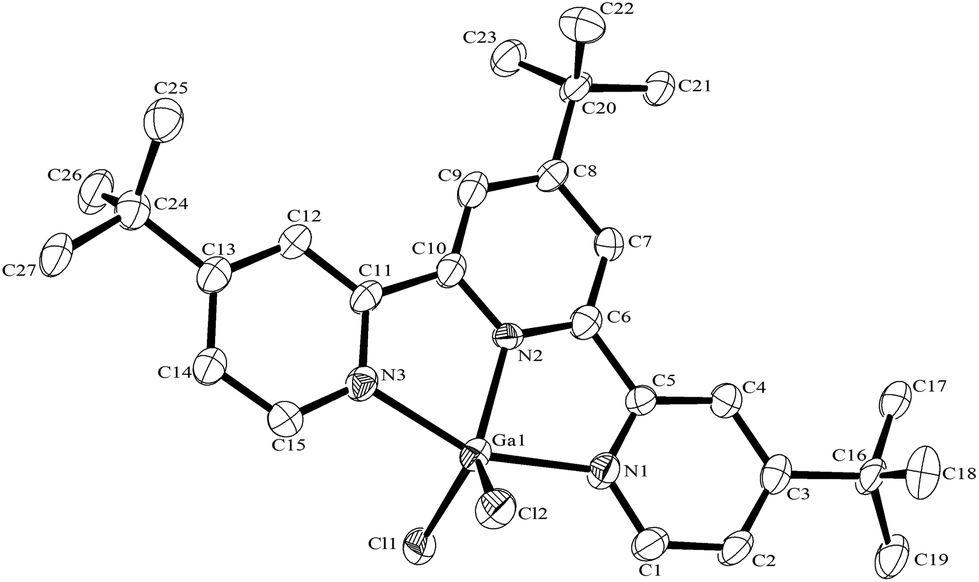 | ||
| Fig. 6 The structure of the cation in [GaCl2(tBu3-terpy)][GaCl4] with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Ga1–N1 = 2.092(5), Ga1–N2 = 1.995(5), Ga1–N3 = 2.066(5), Ga1–Cl1 = 2.195(2), Ga1–Cl2 = 2.169(2), N2–Ga1–N3 = 77.9(2), N1–Ga1–N2 = 77.3(2), N1–Ga1–N3 = 155.1(2), N2–Ga1–Cl2 = 125.68(18), N3–Ga1–Cl2 = 97.76(17), N1–Ga1–Cl2 = 95.78(17), N2–Ga1–Cl1 = 120.20(18), N3–Ga1–Cl1 = 96.80(16), N1–Ga1–Cl1 = 96.56(18), Cl1–Ga1–Cl2 = 114.10(8). | ||
[{Ga(terpy)F}2(μ-F)2][PF6]2·4H2O
The fac-[GaF3(BzMe2-tacn)] has been shown to function as a neutral ‘metalloligand’ through the coordinated fluorides towards alkali metal and ammonium cations in aqueous solution, leading to supramolecular arrays with Ga–F–M linkages,25 whilst combination of Gd3+ and fac-[GaF3(Me3-tacn)] leads to ‘[Gd3Ga2]’ cores that are of interest as molecular magnets.26 A key feature of these systems is that the three facial Ga–F bonds remain intact throughout. To explore if the meridional trifluoride arrangement present in [GaF3(terpy)] could function in a similar manner, a H2O-MeCN solution containing [GaF3(terpy)] and [NH4][PF6] was allowed to evaporate slowly. Orange crystals formed and an X-ray structure analysis on these showed that rather than forming an ammonium-metalloligand complex, the dimer, [{Ga(terpy)F}2(μ-F)2][PF6]2·4H2O had formed.The centrosymmetric cation (Fig. 7) contains six-coordinate gallium, severely distorted from regular octahedral by the steric constraints of the terpy ligand (N1–Ga1–N3 = 153.85(10)°), and the fluoride bridges are asymmetric (Ga1–F1 = 1.889(2), Ga1–F1a = 2.003(2) Å). There is extensive hydrogen bonding linking the lattice water molecules, the cations and the [PF6]− anions (Fig. 8). The complex, formed by dissociation of one fluoride from each gallium centre, followed by dimerisation, dissolves in CD3OD with decomposition and formation of a white precipitate. The 19F{1H} NMR spectrum of the supernatant shows only [GaF3(terpy)] and [PF6]− as significant species.
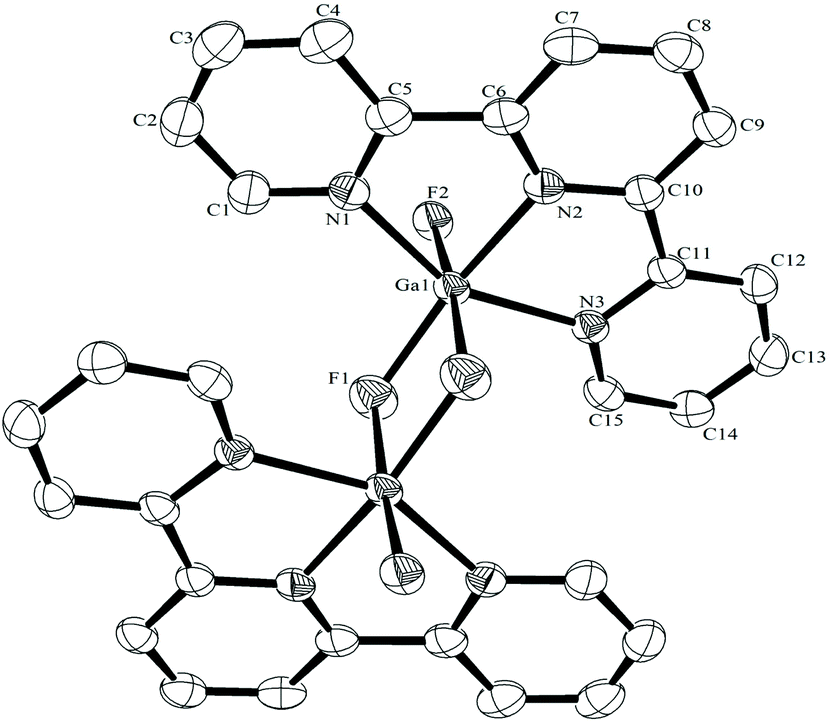 | ||
| Fig. 7 The structure of the centrosymmetric cation in [{Ga(terpy)F}2(μ-F)2][PF6]2·4H2O with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Ga1–F2 = 1.8564(17), Ga1–F1 = 1.889(2), Ga1–F1a = 2.003(2), Ga1–N2 = 2.026(2), Ga1–N1 = 2.095(3), Ga1–N3 2.110(2), F2–Ga1–F1 = 98.39(8), F2–Ga1–F1a = 174.58(7), F1–Ga1–F1a = 76.19(9), F2–Ga1–N2 = 97.02(8), F1–Ga1–N2 = 88.40(9), F2–Ga1–N1 = 88.84(9), F1a–Ga–N1 = 102.47(9), F1–Ga1–N1 = 92.37(9), N2–Ga1–N1 = 77.26(10), F2–Ga1–N3 = 88.19(8), F1a–Ga1–N3 = 103.67(9), F1–Ga1–N3 = 93.03(9), N2–Ga1–N3 = 77.34(9), N1–Ga1–N3 = 153.85(10), Ga1–F1–Ga1a = 103.81(9). | ||
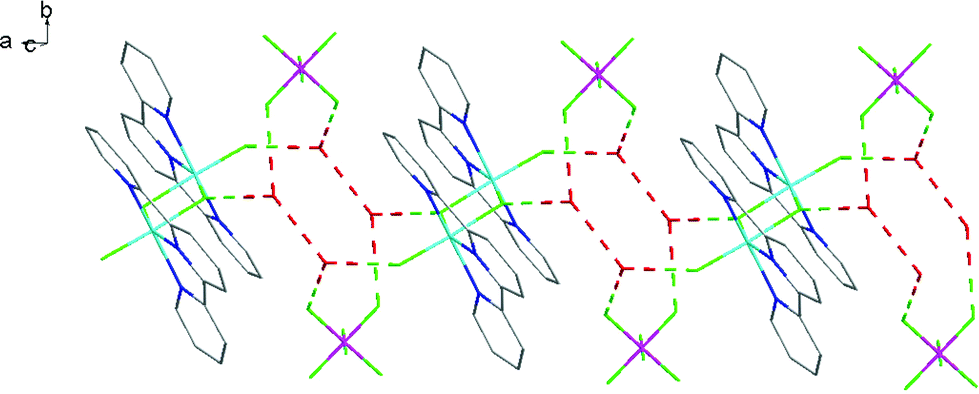 | ||
| Fig. 8 View showing the H-bonding network (red) in [{Ga(terpy)F}2(μ-F)2][PF6]2·4H2O. | ||
[MF3(diimine)(OH2)]·nH2O
The hydrothermal method also proved a suitable route to obtain [MF3(bipy)(OH2)]·2H2O and [MF3(phen)(OH2)] in good yields. The structures of [InF3(phen)(OH2)] and [InF3(bipy)(OH2)]·2H2O obtained from samples made from aqueous HF solution have been reported previously.13 The [MF3(bipy)(OH2)]·2H2O (M = Al or Ga) are monoclinic, P21/n, with six-coordinate metal centres and a mer-fluoride arrangement, which means the neutral bipy ligand is trans to OH2/F (Fig. 9 and 10).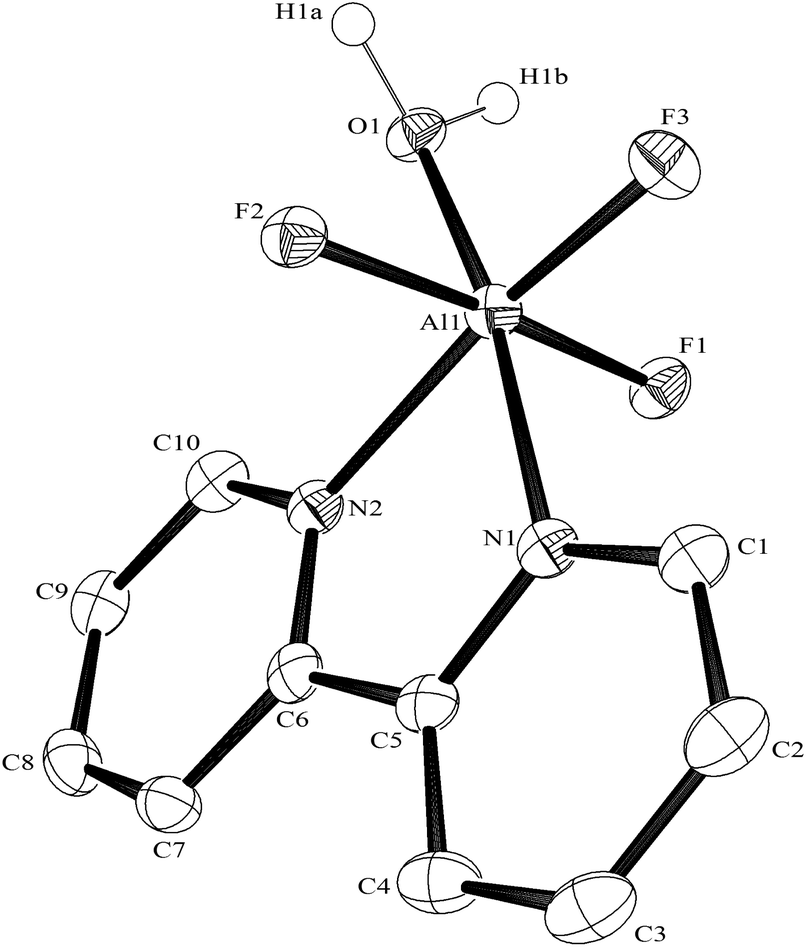 | ||
| Fig. 9 The structure of the Al complex in [AlF3(bipy)(OH2)]·2H2O with ellipsoids drawn at the 50% probability level. Hydrogen atoms (except those on the coordinated water) are omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–F1 = 1.786(2), Al1–F2 = 1.781(2), Al1–F3 = 1.743(2), Al1–O1 = 1.900(2), Al1–N1 = 2.050(3), Al1–N2 = 2.072(3), F2–Al1–F3 = 93.35(11), F1–Al1–F3 = 92.22(11), F3–Al1–O1 = 97.24(10), F2–Al1–O1 = 88.30(11), F1–Al1–O1 = 89.16(11), F3–Al1–N1 = 92.19(10), F2–Al1–N1 = 90.08(10), F1–Al1–N1 = 91.55(10), F2–Al1–N2 = 86.50(11), F1–Al1–N2 = 88.32(11), O1–Al1–N2 = 92.39(11), N1–Al1–N2 = 78.18(11). | ||
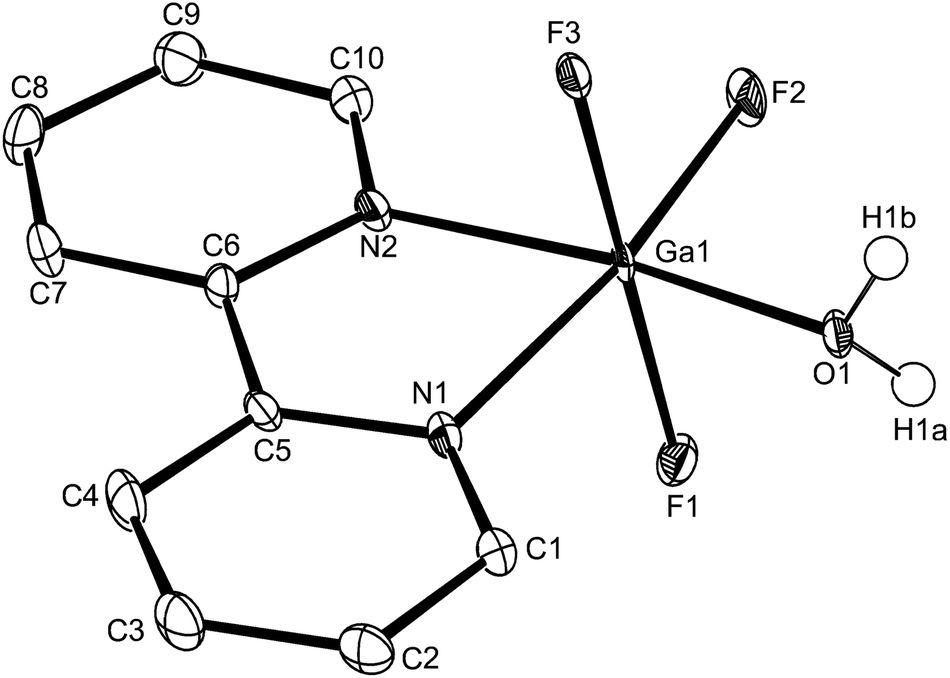 | ||
| Fig. 10 The structure of Ga species in [GaF3(bipy)(OH2)]·2H2O with ellipsoids drawn at the 50% probability level. Hydrogen atoms (except those on the coordinated water) are omitted for clarity. Selected bond lengths (Å) and angles (°): Ga1–F1 = 1.8892(15), Ga1–F2 = 1.8442(17), Ga1–F3 = 1.8857(14), Ga1–O1 = 1.962(2), Ga1–N1 = 2.093(2), Ga1–N2 = 2.064(3), F2–Ga1–F3 = 92.41(7), F1–Ga1–F2 = 90.74(7), F1–Ga1–F3 = 175.68(7), F2–Ga1–O1 = 96.70(8), F3–Ga1–O1 = 87.76(7), F1–Ga1–O1 = 88.94(7), F2–Ga1–N2 = 92.28(9), F3–Ga1–N2 = 90.64(8), F1–Ga1–N2 = 92.19(8), F3–Ga1–N1 = 87.52(7), F1–Ga1–N1 = 89.86(7), O1–Ga1–N1 = 92.67(8), N2–Ga1–N1 = 78.35(9). | ||
For [InF3(bipy)(OH2)]·2H2O,13 In–FtransF = 2.065(2), 2.070(2), In–FtransN = 2.002(3), In–NtransF = 2.260(4), In–NtransO = 2.243(4), In–O = 2.130(4) Å. Comparing the bond lengths in the three compounds reveals some interesting trends. The M–N distances are only slightly different (≤0.02 Å) between Al and Ga, whereas the M–F and M–O distances increase by between 0.06–0.1 Å; as noted with the terpy compounds above, the more electronegative donors have the greatest differences. The corresponding bond lengths all increase in the indium complex as expected, but here the differences with donor type are less clear, with all showing an increase of ∼0.15–0.2 Å over the gallium analogue. In all of the complexes H-bonding to the lattice water and π-stacking of the aromatic rings is also evident (Fig. 11–13 and S2‡).
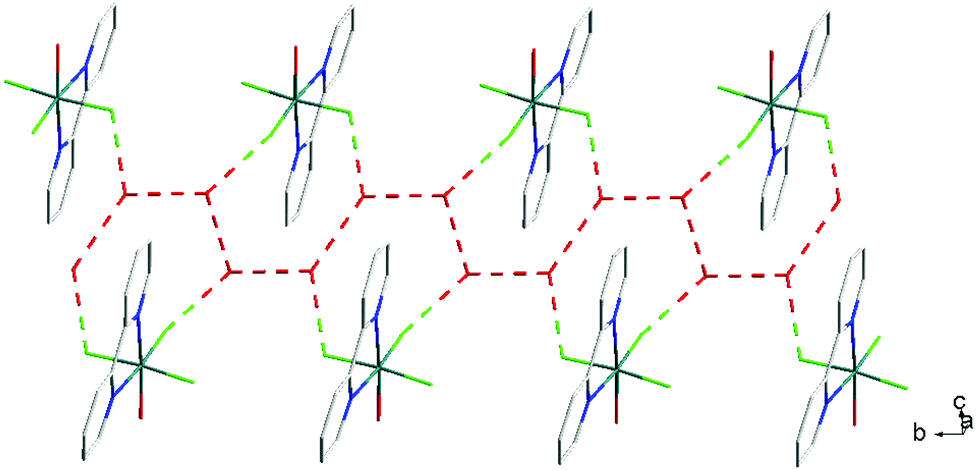 | ||
| Fig. 11 View showing the H-bonding network (red) involving the F− ligands and lattice H2O present in the structure of [AlF3(bipy)(OH2)]·2H2O. | ||
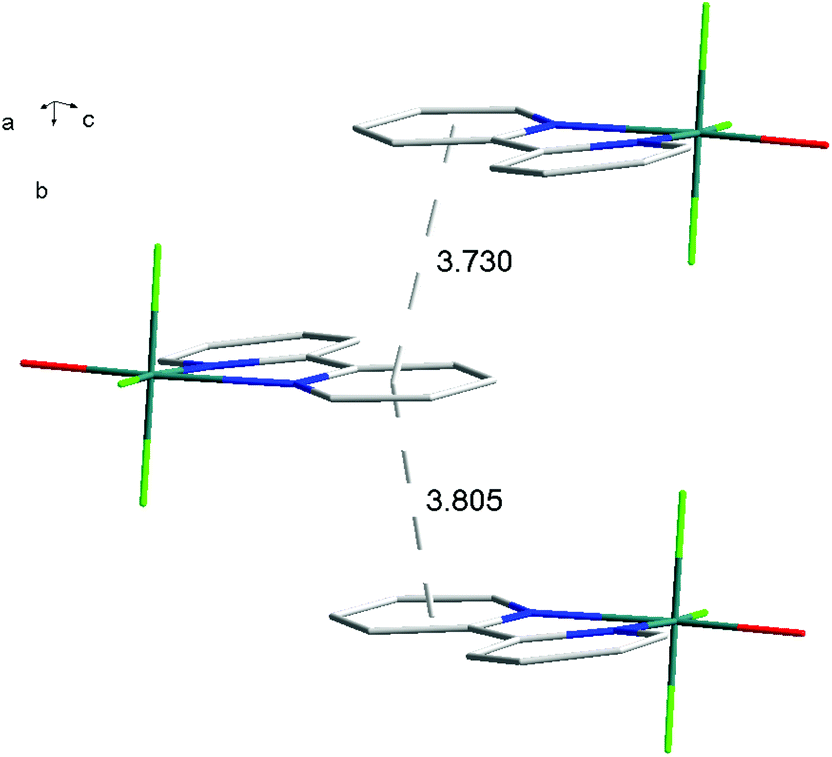 | ||
| Fig. 12 View showing the π-stacking (lattice H2O omitted for clarity) present in the structure of [AlF3(bipy)(OH2)]·2H2O. | ||
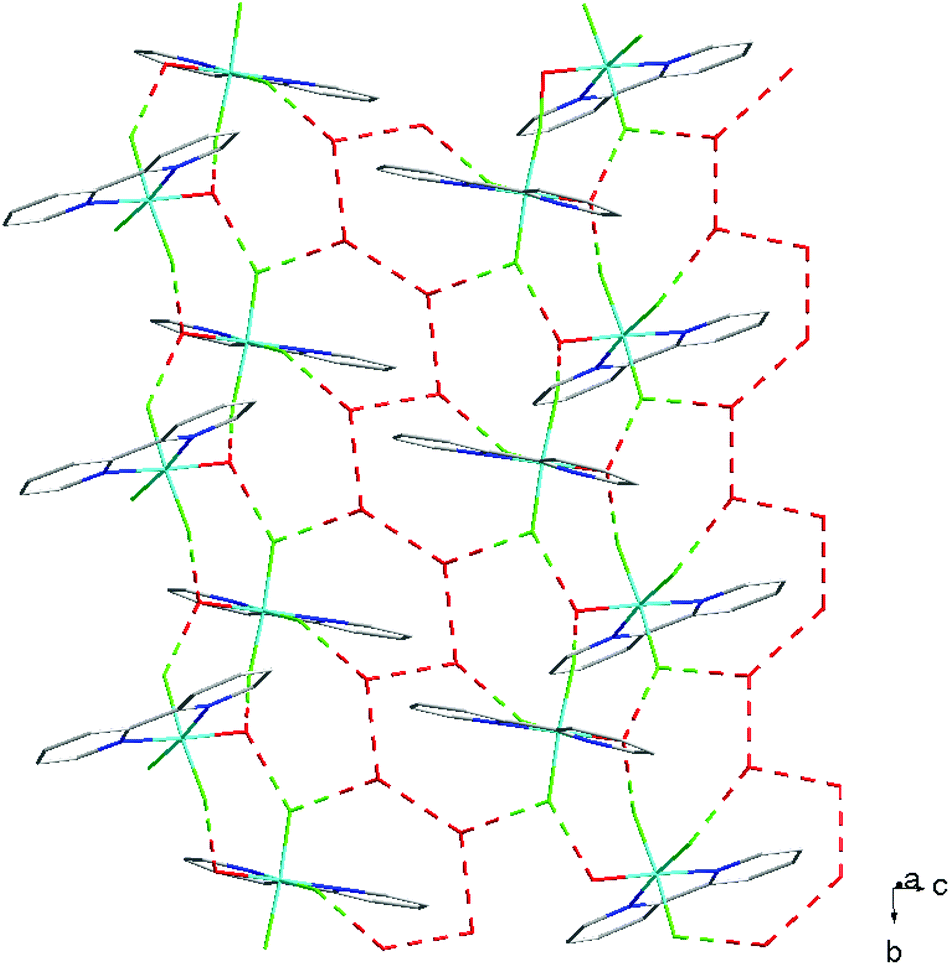 | ||
| Fig. 13 View showing the H-bonding network (red) present in the structure of [GaF3(bipy)(OH2)]·2H2O. | ||
Overall, the structural data reinforce earlier conclusions that trends in the bond lengths in comparable Al and Ga complexes often differ from those predicted on the basis of simple Lewis acidity in the gas phase;22,27–29 other factors, including the presence or absence of lattice solvent and hydrogen bonding also need to be considered.13,19,21,22 The effects are less noticeable at indium, where corresponding bonds are typically ∼0.2 Å longer than for Ga, reflecting the increased radius of the metal centre.
Using the same hydrothermal route with 1,10-phenanthroline in place of 2,2′-bipyridyl, gave [MF3(phen)(OH2)], notably without lattice water (in contrast to the terpy and bipy complexes above). The structure of crystals of [GaF3(phen)(OH2)] is shown in Fig. 14, and the [InF3(phen)(OH2)] is isomorphous.13 Corresponding bond lengths are again ∼0.15–0.20 Å longer for the indium complex. The [GaF3(phen)(OH2)] also shows π-stacking of the aromatic rings (Fig. S3‡) and intermolecular F⋯H hydrogen bonding, in this case involving the coordinated water molecules (Fig. 15).
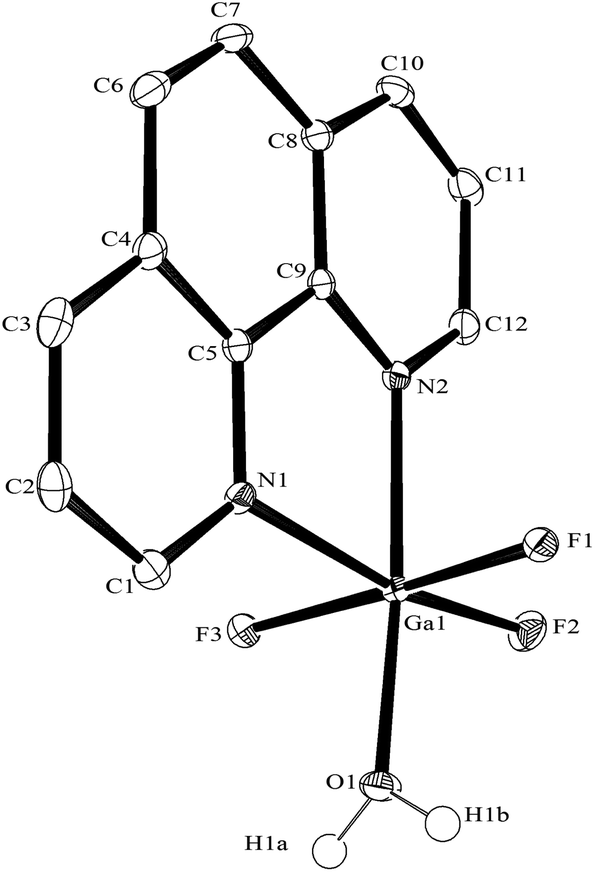 | ||
| Fig. 14 The structure of [GaF3(phen)(OH2)] with ellipsoids drawn at the 50% probability level. Hydrogen atoms (except those on the coordinated water) are omitted for clarity. Selected bond lengths (Å) and angles (°): Ga1–F1 = 1.8990(11), Ga1–F2 = 1.8242(12), Ga1–F3 = 1.8790(11), Ga1–N1 = 2.1250(16), Ga1–N2 = 2.0897(16), Ga1–O1 = 1.9594(14), F2–Ga1–F3 = 95.13(5), F1–Ga1–F2 = 93.69(5), F2–Ga1–O1 = 98.11(6), F3–Ga1–O1 = 89.79(5), F1–Ga1–O1 = 86.92(5), F2–Ga1–N2 = 92.02(6), F3–Ga1–N2 = 93.11(5), F1–Ga1–N2 = 88.65(5), F3–Ga1–N1 = 87.18(6), F1–Ga1–N1 = 84.47(6), O1–Ga1–N1 = 91.54(6), N2–Ga1–N1 = 78.20(6). | ||
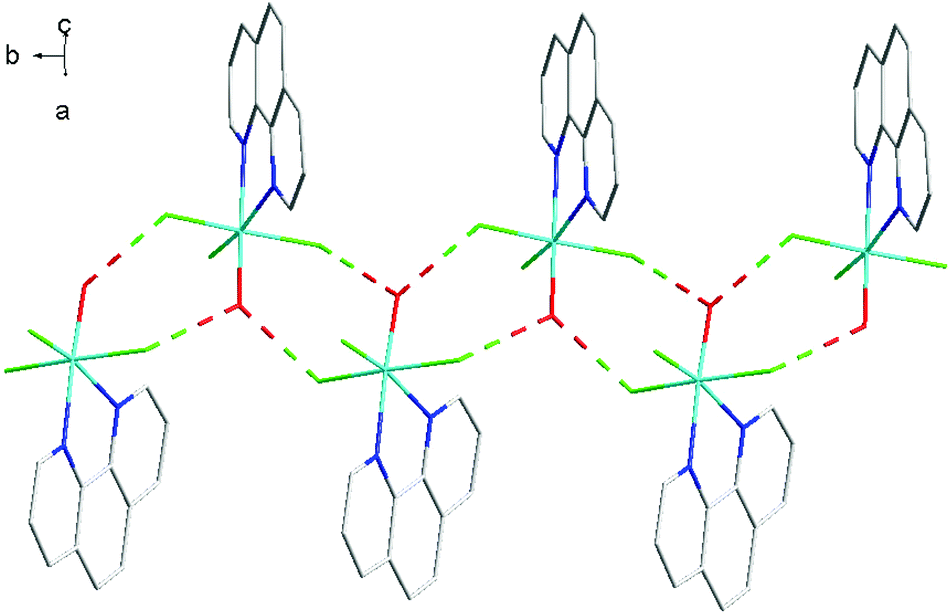 | ||
| Fig. 15 View showing the H-bonding network (red) present in the structure of [GaF3(phen)(OH2)]. | ||
The IR spectra of the [MF3(phen)(OH2)] complexes show quite weak features due to ν(OH) and δ(HOH), whereas in the hydrated [MF3(bipy)(OH2)]·2H2O the corresponding features are much stronger and show several overlapping bands. The ν(MF) fall in energy as expected Al > Ga > In, but for the corresponding Al and Ga complexes (less clearly in the In complexes), those in [MF3(bipy)(OH2)]·2H2O occur at lower frequency than in [MF3(phen)(OH2)], which is probably due to the extensive hydrogen bonding in the former.
The multinuclear NMR data show interesting trends as the metal is changed. The complexes were insoluble or very poorly soluble in chlorocarbons and CD3CN, and therefore spectra were obtained from freshly prepared solutions in CD3OD. The solutions decompose slowly forming F− or HF2−, qualitatively the indium complexes seem less stable in solution than those of the lighter metals. At 183 K, all six complexes show two resonances with relative intensities 1:2 in the 19F{1H} NMR spectra, consistent with the solid state structures. On warming, the fluorine resonances of the indium complexes are lost, and those of the gallium complexes and of [AlF3(bipy)(OH2)] broaden and then coalesce; only the 19F{1H} NMR spectrum of [AlF3(phen)(OH2)] shows two fluorine resonances at room temperature. The 1H NMR spectra of the aluminium complexes show inequivalent pyridyl rings, as expected from the static structures, but those of the gallium and indium show broad resonances even at low temperatures. The temperature dependence of the spectra is consistent with reversible ligand dissociation, possibly of the water, to give a fluxional five-coordinate intermediate, the dissociation increasing with decreased Lewis acid strength Al > Ga > In.
Reactions of MF3·3H2O with PMDTA
PMDTA, Me2N(CH2)2NMe(CH2)2NMe2, is a flexible, aliphatic acyclic triamine analogue of Me3-tacn and terpy, and its reactions with the hydrated Group 13 fluorides were explored to provide a third series of complexes for comparison. In contrast to the reactions with the other two N3-donor ligands, the hydrothermal route using PMDTA resulted in cleavage of the triamine and the formation of the 1,1,4-trimethylpiperazinium cation, [⊂Me2N(CH2)2NMe(CH2)2]+. After removing all volatiles from the reaction mixtures and washing the residue with MeCN, the 1H and 13C{1H} NMR spectra show the cyclic cation to be the only organic species in the bulk products. For the aluminium reaction, crystals were obtained, showing the solid contained [⊂Me2N(CH2)2NMe(CH2)2]2[Al2F8(OH2)2]·2H2O. The anion (Fig. 16) has been obtained previously as the [NMe4]+ salt,32 and breaks up in solution to form [AlF4]−, which was identified by a combination of 27Al (δ = 48.7 (s)) and 19F{1H} NMR data (δ = –194.6, 6 lines, 1JAlF = 38 Hz).32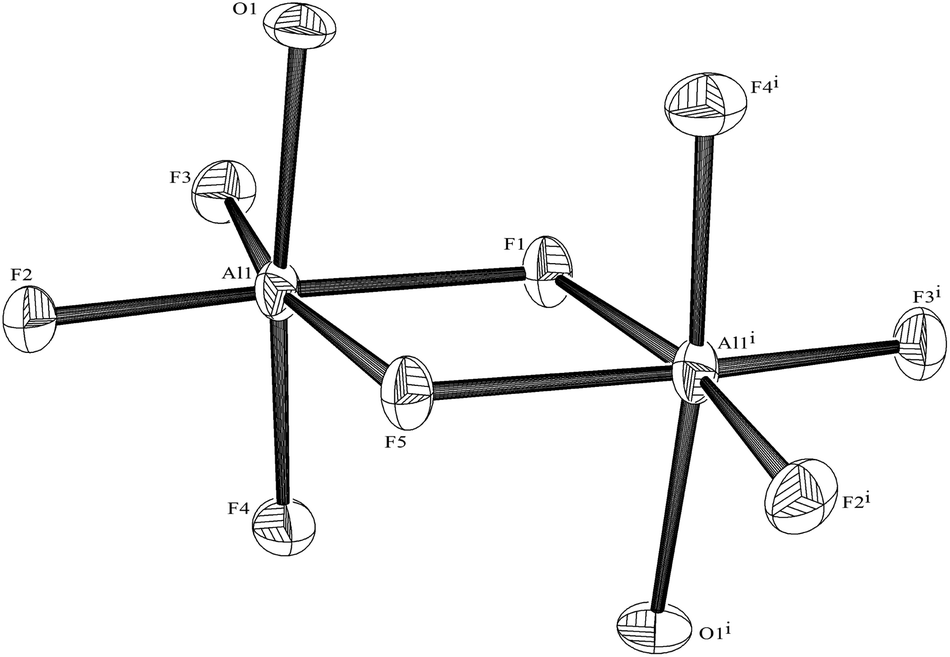 | ||
| Fig. 16 The structure of the [Al2F8(OH2)2]2− anion with ellipsoids drawn at the 50% probability level and with (O–H) hydrogens omitted. Selected bond lengths (Å) and angles (°): Al1–F2 = 1.748(2), Al1– F3 = 1.749(2), Al1–F4 = 1.778(2), Al1–O1 = 1.895(2), Al1–F5 = 1.8968(19), Al1–F1 = 1.9048(19), F2–Al1–F3 = 99.65(11), F2–Al1–F4 = 93.28(10), F3–Al1–F4 = 93.83(10), F2–Al1–O1 = 90.62(10), F3–Al1–O1 = 90.65(10), F2–Al1–F5 = 92.34(10), F4–Al1–F5 = 88.95(8), O1–Al1–F5 = 85.68(8), F3–Al1–F1 = 91.94(10), F4–Al1–F1 = 88.61(8), O1–Al1–F1 = 86.52(8), F5–Al1–F1 = 75.93(10), Al1–F5–Al1i = 104.38(13), Al1–F1–Al1i = 103.77(13). | ||
In the cases of the gallium and indium reactions, a few crystals of the same cation were obtained as the chloride salt, from traces of chloride in the reaction. The structure of [⊂Me2N(CH2)2NMe(CH2)2]Cl has been reported previously31 and the crystals obtained in this study were identical, and hence are not discussed further (see ESI‡). The data on the bulk product from the gallium reaction fitted the constitution [⊂Me2N(CH2)2NMe(CH2)2]2 [Ga2F8(OH2)2]·2H2O, analogous to the aluminium complex, although in the absence of X-ray structural data, the anion present cannot be confirmed. The relative instability of the fluoro-metallate anions in solution and the sensitivity of the 19F chemical shifts to solvent,30,32 make identification of the anions uncertain without structural data. The reactions of the MF3·3H2O with PMDTA were also attempted in refluxing methanol solution, since it was reasoned that the milder conditions (compared to the hydrothermal preparations) might have prevented cleavage of the PMDTA. No reaction occurred in the case of indium fluoride, whilst with AlF3·3H2O, 1H and 19F{1H} NMR spectra of the crude product showed protonated PMDTA and [AlF4]− as the only significant species. The contrast between the instability of PMDTA and the robust Me3-tacn which has similar groups in these Group 13 fluoride reactions may be due to the ring structure of the latter preventing close approach of an amine function polarised by coordination to the metal, to the next CH2NMe unit, which is presumably the first stage in C–N bond fission and formation of the small ring.
Conclusions
Hydrothermal synthesis using MF3·3H2O as the metal source has been shown to be a convenient method to form a range of complexes of the otherwise rather intractable Group 13 trifluorides with nitrogen heterocycles in high yield. X-Ray crystallographic studies show all of the new complexes contain a mer arrangement of fluorides, contrasting with the fac geometry present in the triaza-macrocyclic complexes15 reported previously. Extensive H-bonding and π-stacking networks are present in the complexes of all three imines with the three metal ions, although the details differ. These studies significantly extend the known coordination chemistry of the Group 13 trifluorides. The relatively high stability of the trifluoride complexes contrasts with the moisture sensitivity of complexes of the Group 13 elements with heavier halides. However, this work has also shown that unlike [GaF3(BzMe2-tacn)]·4H2O, which is stable towards alkali metal or ammonium salts in aqueous MeCN, forming heterometallic (or mixed gallium-ammonium) complexes, competitive ions such as [NH4]+ and [PF6]−, [GaF3(terpy)]·3H2O reacts, with loss of a fluoride ligand, to form the dinuclear product, [{Ga(terpy)F}2(μ-F)2][PF6]2·4H2O.Future work will aim to establish whether the hydrothermal approach is also suitable for oxygen donor ligands and whether soft donor ligands such as thioethers or phosphines can form complexes with the Group 13 fluorides.
Acknowledgements
We thank EPSRC and GE Healthcare for support (EP/I501673/1, EP/L505651/1 and EP/1033394/1).References
- (a) S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460 RSC; (b) Chemistry of aluminium, gallium, indium and thallium, ed. A. J. Downs, Blackie, London, 1st edn, 1993 Search PubMed.
- (a) E. Kemnitz, U. Gross, St. Ruediger, G. Scholz, D. Heideman, S. I. Troyanov, I. V. Morosov and M.-H. Lemee-Cailleau, Solid State Sci., 2006, 8, 1443 CrossRef CAS; (b) P. Bukovec and V. Kaucic, Inorg. Nucl. Chem. Lett., 1978, 14, 79 CrossRef CAS.
- S. Petricek, A. Demsar, P. Bukovec, L. Golic and J. V. Brencic, Acta Chim. Slov., 1997, 44, 317 CAS.
- F. N. Penkert, T. Weyhermuller and K. Wieghardt, Chem. Commun., 1998, 557 CAS.
- D. R. Ketchum, G. L. Schimek, W. T. Pennington and J. W. Kolis, Inorg. Chim. Acta, 1999, 294, 200 CrossRef CAS.
- (a) M. Roos and G. Meyer, Z. Anorg. Allg. Chem., 1999, 625, 1129 CrossRef CASM. Roos and G. Meyer, Z. Anorg. Allg. Chem., 1999, 625, 1843 CrossRef CAS.
- S. A. Polyshchuk, S. P. Kozerenko and Y. V. Gagarinsky, J. Less-Common Met., 1972, 27, 45 CrossRef.
- S. P. Kozerenko, S. A. Polyshchuk and N. I. Sigula, Russ. J. Inorg. Chem., 1972, 17, 954 Search PubMed.
- M. Roos, J. Wittrock, G. Meyer, S. Fritz and J. Straehle, Z. Anorg. Allg. Chem., 2000, 626, 1179 CrossRef CAS.
- S. P. Petrosyants and E. R. Buslaeva, Russ. J. Inorg. Chem., 1989, 34, 932 Search PubMed.
- S. A. Polyshchuk and G. F. Krysenko, Koord. Khim., 1977, 3, 816 Search PubMed.
- M. A. Malyarik, S. P. Petrosyants, A. B. Ilyukhin and Yu. A. Buslaev, Russ. J. Inorg. Chem., 1991, 36, 2816 CAS.
- (a) A. B. Ilyukhin and M. A. Malyarik, Koord. Khim., 1999, 44, 1511 CAS; (b) A. B. Ilyukhin and M. A. Malyarik, Krystallographiya, 1994, 39, 439 CAS.
- S. P. Petrosyants and A. B. Ilyukhin, Russ. J. Inorg. Chem., 2010, 55, 30 CrossRef CAS.
- R. Bhalla, C. Darby, W. Levason, S. K. Luthra, G. McRobbie, G. Reid, G. W. Sanderson and W. Zhang, Chem. Sci., 2014, 5, 381 RSC.
- (a) A. Dimitrov, D. Heidemann, K. I. Khallow and E. Kemnitz, Inorg. Chem., 2012, 51, 11612 CrossRef CAS PubMed; (b) A. Dimitrov, D. Heidemann and E. Kemnitz, Inorg. Chem., 2006, 45, 10807 CrossRef CAS PubMed.
- (a) P. Laverman, W. McBride, R. Sharkey, A. Eek, L. Joosten, W. Oyen, D. Goldenberg and O. Boerman, J. Nucl. Med., 2010, 51, 454 CrossRef CAS PubMed; (b) W. McBride, C. D'Souza, R. Sharkey, H. Karacay, E. Rossi, C. Chang and D. Goldenberg, Bioconjugate Chem., 2010, 21, 1331 CrossRef CAS PubMed; (c) W. McBride, R. Sharkey, H. Karacay, C. D'Souza, E. Rosso, P. Laverman, C. Chang, O. Boerman and D. Goldenberg, J. Nucl. Med. Chem., 2009, 50, 991 CrossRef CAS PubMed; (d) W. McBride, C. D'Souza, H. Karacay, R. Sharkey and D. Goldenberg, Bioconjugate Chem., 2012, 23, 538 CrossRef CAS PubMed; (e) D. Shetty, S. Y. Choi, J. M. Jeong, J. Y. Lee, L. Hoigebazar, Y.-S. Lee, D. S. Lee, J. K. Chung, M. C. Lee and Y. K. Chung, Chem. Commun., 2011, 47, 9732 RSC; (f) R. Bhalla, W. Levason, S. K. Luthra, G. McRobbie, G. Sanderson and G. Reid, Chem. – Eur. J., 2015, 21, 4688 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- I. V. Kazakov, M. Bodensteiner and A. Y. Timoshkin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2014, 70, 312 CAS.
- (a) M. J. Taylor and P. J. Brothers, in The chemistry of aluminium, gallium, indium and thallium, ed. and A. J. Downs, Blackie, London, 1993, p. 111 Search PubMed; (b) B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- (a) J. Burt, W. Levason, M. E. Light and G. Reid, Dalton Trans., 2014, 43, 14600 RSC; (b) K. George, M. Jura, W. Levason, M. E. Light and G. Reid, Dalton Trans., 2014, 43, 3637 RSC.
- A. Y. Timoshkin, M. Bodensteiner, T. N. Sevastianova, A. S. Lisovenko, E. I. Davydova, M. Scheer, C. Grassi and A. V. Butlak, Inorg. Chem., 2012, 51, 11602 CrossRef CAS PubMed.
- 27Al [I = 5/2, 100% abundance, Ξ = 26.08 MHz, Q = 0.149 × 10–28 m2, RC = 1170], 69Ga [I = 3/2, 60.4% abundance, Ξ = 24.07 MHz, Q = 0.178 × 10–28 m2, RC = 237] 71Ga [I = 3/2, 39.6% abundance, Ξ = 30.58 MHz, Q = 0.112 × 10–28 m2, RC = 322] 115In [I = 9/2, 95.7% abundance, Ξ = 22.04 MHz, Q = 1.16 × 10–28 m2, RC = 1920].
- J. Mason, Multinuclear NMR, Plenum, New York, 1987 Search PubMed.
- R. Bhalla, W. Levason, S. J. Luthra, G. McRobbie, G. Reid, G. Sanderson and W. Zhang, Chem. Commun., 2014, 50, 12673 RSC.
- K. S. Pedersen, G. Lorusso, J. J. Morales, T. Weyhermuller, S. Piligkos, S. K. Singh, D. Larsen, M. Schau-Magnussen, G. Rajaraman, M. Evanelisti and J. Bendix, Angew. Chem., Int. Ed., 2014, 53, 2394 CrossRef CAS PubMed.
- F. Bessac and G. Frenking, Inorg. Chem., 2006, 45, 6956 CrossRef CAS PubMed.
- J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65 CrossRef CAS , and refs therein.
- A. V. Timoshkin, A. V. Suvorov, H. F. Bettinger and H. F. Schaefer III, J. Am. Chem. Soc., 1999, 121, 5687 CrossRef CAS.
- (a) N. Herron, D. L. Thorn, R. L. Harlow and F. Davidson, J. Am. Chem. Soc., 1993, 115, 3028 CrossRef CAS; (b) N. Herron, R. L. Harlow and D. L. Thorn, Inorg. Chem., 1993, 32, 2985 CrossRef CAS.
- W. Massa and K. Dehnicke, Z. Anorg. Allg. Chem., 2009, 635, 287 CrossRef CAS.
- S. P. Petrosyants and A. B. Ilyukhin, Russ. J. Inorg. Chem., 2011, 56, 1250 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Professor Ken Wade, FRS, in recognition of his many significant contributions to inorganic chemistry. |
| ‡ Electronic supplementary information (ESI) available: X-ray structure of [⊂Me2N(CH2)2NMe(CH2)2]Cl. CCDC 1053047–1053048 and 1053152-1053158. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01120e |
| This journal is © The Royal Society of Chemistry 2015 |