
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The importance of second shell effects in the simulation of hydrated Sr2+ hydroxide complexes†
Eszter
Makkos
a,
Andrew
Kerridge
*ab and
Nikolas
Kaltsoyannis
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: a.kerridge@lancaster.ac.uk; n.kaltsoyannis@ucl.ac.uk
bDepartment of Chemistry, Lancaster University, Bailrigg, Lancaster LA1 4YP, UK
First published on 22nd May 2015
Abstract
Density functional theory at the meta-GGA level is employed to study the microsolvation of Sr2+ hydroxides, in order to establish likely candidate species for the interaction of nuclear fission-generated strontium with corroded Magnox fuel cladding in high pH spent nuclear fuel storage ponds. A combination of the COSMO continuum solvation model and one or two shells of explicit water molecules is employed. Inclusion of only a single explicit solvation shell is unsatisfactory; open regions are present in the strontium coordination shell which would not exist in real aqueous complexes, and many optimised structures possess unavoidable energetic instabilities. Incorporation of a second shell of explicit waters, however, yields energetically minimal structures without open regions in the first strontium coordination shell. The most stable systems with one, two or three hydroxide ions are all 6-coordinated with a distorted trigonal antiprismatic geometry, whereas systems with four OH− ions have a most stable coordination number of five. Transformation, via a proton transfer mechanism, from one coordination mode to another (e.g. from a system with two hydroxides bound directly to the strontium to one in which a hydroxide ion migrates into the second coordination shell) is found to be energetically facile. It is concluded that the most likely strontium-hydroxide complexes to be found in high pH aqueous solutions are mono- and dihydroxides, and that these coexist.
Introduction
The spent uranium fuel rods and associated cladding from civil nuclear reactors are held in storage ponds at reactor sites and reprocessing facilities prior to reprocessing or final disposal. The corrosion behaviour of the fuel rods and any corrosion products generated depends on several factors. One is the type of cladding material used, which can vary across the world; in the first generation of British civil nuclear reactors a magnesium-aluminium based alloy, known as Magnox, was used.1 Other important factors are the storage period and condition of the ponds. Since the ponds are filled with water to act as a radioactivity shield and as a cooling medium, the cladding may corrode over time and in the case of Magnox, form the mineral brucite (Mg(OH)2).2 Magnox corrosion and resultant brucite sludge formation is a significant problem for a number of UK “legacy” facilities including fuel storage ponds in which Magnox fuel elements have been stored for decades and wet silos used for storing stripped Magnox casings. Besides the corrosion products, the sludge and liquor contain leached actinides (238U, 239Pu and 241Am) and their fission products (mostly 137Cs and 90Sr) in significant concentrations which can form aquo and hydroxide complexes.2 Brucite has a hydroxide-terminated (0001) surface which is highly reactive, and it has been shown experimentally that it can absorb some of the above mentioned ions.3 Moreover, its sorption capacity increases at higher pH4 and the base molality in the ponds is generally high. The composition of the waste changes during storage, and the monitoring and investigation of the exact conditions is difficult because of the high radiological hazard.5–8 This situation creates a challenging problem for the removal and disposal of material in order to decommission facilities.In order to investigate the interactions between brucite surfaces and the solvated ions, it is essential to have a detailed understanding of the microsolvation of those ions, such as Sr2+, for the identification of possible candidate species for surface adsorption. The structures of Sr2+ hydrates are both experimentally and theoretically well studied. According to HPMS9 and TCID10 measurements the most stable hydrate coordination is 6 in the gas phase. Whilst early theoretical models11 predicted a higher coordination number of 8, more recent higher quality calculations12,13 also find a coordination number of 6, in agreement with the experimental results. X-ray diffraction (XRD) studies in aqueous solution showed 8 as the most stable coordination,14,15 but Albright indicated that a coordination number of 6 or 8 is equally possible in the case of the Sr2+ ion.14 Aqueous phase large angle X-ray scattering (LAXS) and extended X-ray adsorption fine structure spectroscopy (EXAFS) measurements reported coordination numbers over a wide range: 8,16 7.317 and up to 10.3.18 There are several factors which can vary between different studies, such as the concentration of the ion, the counter ion used, the temperature, and most of all, the chosen model compound for interpreting the measured results. All of these can cause deviation in the calculated coordination number. For instance, Persson et al.19 found 8.1 water molecules in the first shell with an average Sr–O distance of 2.63 Å at room temperature, choosing the crystal structure of the Sr hydroxide octahydrate as their model system. D'Angelo and coworkers18 analysed the data based on molecular dynamics simulations and included multielectron excitations in their model to obtain 10.3 as an average coordination number. Early theoretical work by Spohr and coworkers yielded 9.8 as the average coordination from molecular dynamics,20 although later computational works narrowed down the range of coordination numbers; Harris et al.21 predicted a coordination number of 7.5 from ab initio molecular dynamics calculations, and Kerridge et al.13 found a maximum coordination number of 7 from quantum chemical calculations in aqueous media.
Sr2+ hydroxide can be crystallised in several hydrate forms: Sr(OH)2, Sr(OH)2·H2O and Sr(OH)2·8H2O. It acts as a moderate base in aqueous solution and is well soluble in water at room temperature (8 g l−1 at 20 °C), although its solubility depends on its crystal structure and decreases at higher pH.22,23 Studying the formation of aqueous species in solution is challenging due to the low concentration of the solute and the fact that the structure is dominated by the solvent.11 To our knowledge, there are no experimental data regarding Sr hydroxide structure in aqueous solution in the literature, although solid state structural analysis can reveal important information to aid understanding the solvation. XRD studies have shown that anhydrous Sr(OH)2 has a polyhedral coordination in which the Sr2+ ion is surrounded by seven OH− ions with an average Sr–O distance of 2.60 Å.24 Strontium hydroxide monohydrate is relatively unstable, partially decomposing to the anhydrous hydroxide by prolonged evacuation at room temperature.25 According to single crystal Raman measurements, the monohydrate form demonstrates hydroxide coordination in a trigonal prismatic structure around the Sr2+ ion, bicapped by the oxygen atoms of the water molecules.26 IR and Raman spectra reveal that in such formations, coordinating waters are strongly hydrogen bonded but, by contrast, the hydroxide ions do not act as hydrogen-bond acceptors.27 Sr(OH)2·8H2O has an entirely different structure: based on XRD19,28 and neutron diffraction studies,29 oxygen atoms from eight water molecules coordinate the Sr2+ ion in a tetragonal antiprismatic conformation, with each water engaged in at least in three hydrogen bonds. The hydroxide ions are not coordinated directly to the Sr2+ centre, but instead form chains of acceptor and donor bonds, with each hydroxide oxygen involved in four hydrogen bonds with neighbouring waters.
Only gas phase theoretical studies exist concerning the hydrolysis of Sr2+. Felmy et al. introduced a hydroxide ion into hydrate complexes by removal of a proton from Sr(H2O)62+ and Sr(H2O)82+.11 Interestingly, they found that the addition of water molecules to the system dissociates the hydroxide ion from the central ion, i.e. in Sr(H2O)7(OH)+ the OH− ion is not directly connected to the Sr2+; instead, it has three hydrogen bonds to water molecules in the primary solvation shell. This result suggests only weak hydrolysis of aqueous Sr2+, although the authors note that the delocalisation of the hydroxide ion may be overestimated in the gas phase compared with aqueous phase calculations due to the lack of a background potential and/or competitive explicit second shell water molecules.
Kerridge et al. published a more detailed investigation of the hydroxide complexes of Sr2+ in 2011,30 in which they studied a series of Sr(H2O)8−n(OH)n(2−n) complexes up to n = 4. From analysis of the coordination modes they deduced that systems with more than 8 − n water molecules are not stable. Quantum Theory of Atoms in Molecules (QTAIM) calculations revealed that hydroxide ions have a weakening effect on the Sr interactions with water oxygens (Ow), with hydrogen bonding between water molecules becoming energetically more favourable and waters consequently beginning to occupy the second solvation shell instead of directly coordinating the Sr2+.
Modelling solvation or ligand exchange in solvated complexes requires different methods than for reactions involving strong chemical bonds, because these complexes are generally formed by weak and labile bonds where the effects of the solvent–solute interactions cannot be neglected.31 Thus, in addition to the selection of an appropriate model chemistry, several other factors such as the solvent model and the inclusion of explicit first and even second solvation shells can significantly affect the results.32,33
In the present work we have investigated the effects of bulk solvent and large explicit water clusters on different Sr2+ hydroxide complexes in order to find a suitable and robust solvation model. The aim is to understand the dependence of the coordination on the number of hydroxide ions in the model and to rationalise the previously reported hydroxyl group migration into the second solvation shell. A reliable model of solvation is necessary in our broader goal, namely to identify possible candidate species with which to investigate the interaction of solvated ions with brucite surfaces.
Computational details
The model chemistry was chosen to be the same as in the previous work of Kerridge et al. since this was tested for small gas-phase Sr2+ hydrates and exhibited excellent agreement with experimental results.13 The present calculations were therefore carried out with version 6.5 of the Turbomole code34 using resolution-of-the-identity density functional theory (RI-DFT).35 The TPSS exchange–correlation functional,36 which employs the meta-generalised gradient approximation, and the def2-TZVP basis sets of polarised triple-ζ quality37–39 was used for all atoms along with the associated Sr effective core potential (ECP), which replaces the electrons occupying the core 1s–3d orbitals. Calculations were carried out with the m4 integration grid and the following tight convergence criteria: SCF energy: 10−9, structural energy: 10−6 and energy gradient: 10−3.The effects of the bulk solvent (water) were taken into account via the COSMO continuum solvent model40 with the default Turbomole 6.5 parameters, i.e. a relative permittivity of εr = ∞ and molecular cavities constructed of spheres of radius 2.223 Å for Sr, 1.720 Å for O, 1.300 Å for H. Test calculations on structures with small energy differences between them (those in columns 2 and 3 of Fig. 3) using εr = 80 (the dielectric constant of water at 20 °C) revealed extremely small changes in geometry (average εr = ∞/80 difference in r(Sr–O) and r(O–H) in hydroxide groups and neighbouring water molecules = 0.0001 Å, average change in the H–O–H angle in those water molecules = 0.004°) and energy (a maximum number of 0.6 kJ mol−1 difference in relative Gibbs energy; see the ESI† for more details).
Grimme-type41 DFT-D3 dispersion corrected single point energies were calculated in the case of the systems with two complete solvation shells (see Fig. 3).
Zero-point vibrational frequencies and thermochemical enthalpic and entropic contributions at 298.15 K were obtained via numerical frequency analysis in the aqueous media. A frequency scaling factor of 1 was used throughout, since according to test calculations with the TPSS functional and a basis set of similar quality to that employed here (6-311G(d,p)) a scaling factor of 0.9999 was found to be appropriate in order to minimise the RMS error of the zero-point vibrational energies (ZPVEs).42 Results were visualised with the MOLDRAW chemical graphical software43–45 and Cartesian coordinates of all calculated structures, along with their SCF energies, are provided in the ESI.†
Results and discussion
In this section we present the results of quantum chemical calculations in which a combination of the COSMO implicit solvent model and explicit solvent molecules was used to study the aqueous solvation of strontium mono-, di-, tri-, and tetrahydroxides. The following labelling scheme will be employed to classify the studied systems:[Sr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :a/b:α/β](2−n) :a/b:α/β](2−n) |
:5/17:1/1] is a neutral, 6-coordinated Sr2+ monohydroxide with five water molecules and one OH− ion in the first shell, and 17 waters and one OH− ion in the second.
Sr2+ hydroxide complexes with a first solvation shell
As discussed above, previous gas phase studies have identified the maximum coordination number of Sr2+ hydroxide complexes to be seven, with a tendency to decrease with increasing number of OH− ions in the system.30 Therefore, we kept the total number of molecules in the system at seven and studied structures with one to four hydroxyl groups while the number of explicit waters was reduced concurrently from six to three. A continuum solvent model was employed in all cases.We attempted to map all possible coordinations while keeping the number of waters and OH− ions constant in the system. However, despite repeated efforts, we were unable to stabilise complexes with certain coordination numbers. Hence the following complexes are absent from our study: [Sr:4/2:1/0]+, [Sr:5/1:1/1]+, [Sr:6/0:1/0]+, [Sr:5/0:2/0] and [Sr:3/0:4/0]2−. Attempts to optimise these structures resulted in imaginary frequencies which proved impossible to eliminate. For all other complexes, optimised structures together with their relative SCF and Gibbs energies (ΔESCF/ΔG) are summarised in Fig. 1. Relative energies were calculated with reference to the energy of the most stable complex for a given number of hydroxide ions and water molecules.
 | ||
| Fig. 1 Ball and stick images of optimised strontium hydroxides with a 1st solvation shell, along with their relative SCF and Gibbs free energies (ΔESCF/ΔG). (magenta – Sr2+ ion, red – O atoms in water molecules, blue – O atoms in hydroxide ions, white – H atoms). All energies are given in kJ mol−1. | ||
When we compare the relative stabilities obtained from SCF energies to the order based on Gibbs free energies, we find qualitative differences, demonstrating that thermochemical effects are crucial to the understanding of strontium hydroxide speciation in aqueous environments. Considering the ΔG results, the most stable coordination number is five for the di- and trihydroxide complexes ([Sr:3/2:2/0] and [Sr:2/2:3/0]−), while it is four for the tetrahydroxides ([Sr:0/3:4/0]2−). These results are in partial agreement with the previous gas phase study30 in which the maximum coordination number of seven decreases to four with an increasing number of hydroxide ions. Furthermore, the reduction in total coordination number from five to four found here is consistent with the previous observation from QTAIM analysis that increasing the number of hydroxide ions destabilises higher coordinations.
Inspection of Fig. 1 reveals that for most species, large parts of the first coordination sphere are open, an unphysical situation which would not be possible in real solvated systems. This, and the problems we encountered with energetic instability in several structures, led us to conclude that the combination of an implicit bulk solvent and explicit first coordination shell does not contain enough explicit coordination to accurately model the solvation of Sr2+ hydroxides. To overcome this problem, we moved on to study the effect of using additional explicit waters in our solvation model.
Sr2+ hydroxide complexes with first and second solvation shells
When deciding on the total number of water molecules required to complete two solvation shells, we took into account the possibility that the hydroxide ions can in principle migrate to the second shell, in which case it would be possible to form an eight coordinated hydrate complex.19 Therefore we chose the maximum number of water molecules to be 24 − n, (where n is the number of hydroxide ions in the system) considering that only two second shell waters are able to coordinate each first shell water.As a starting point, we used the 24 water molecule cluster (W24) previously optimised by Ludwig and Weinhold.46 These authors found that a tetrakaidecahedral cluster composed of two hexagons and twelve pentagons was energetically most stable (see Fig. 2). For modelling the solvation of Sr2+ hydroxides, we modified this cluster by placing a Sr2+ ion at its centre and replacing between one and four waters with hydroxide ions.
 | ||
| Fig. 2 Optimised structure of the W24 water cluster from ref. 46 viewed from the side (a), and the top (b). | ||
To verify the suitability of the chosen water cluster for this particular problem, we first studied simple Sr2+ hydrates. We found 6- and 7-coordinated aqua complexes to be energetically very similar (see ESI†). These results are in a good agreement with the previously published theoretical data in which the preferred coordination was seven13,21 and are at the lower end of the experimental range.17–19,21 We also considered the extent to which our model produces structures in which the second solvation shell of water molecules is complete. Defining a precise number of second shell waters, or measuring the completeness of the shell, is not necessarily straightforward, as the water molecules beyond the well-defined first shell are generally weakly bound and can move easily from one shell to the other.30 However, we can take the average number of hydrogen bonds per second shell water molecule as a measure of the completeness of the second shell, and find this to be 3.47 over all of our structures (shown in Fig. 3 and discussed below). This average lies toward the top of the range of values previously determined for bulk water;47–53 between 2.247 and 3.7353 depending upon the definition of hydrogen bonding and the method of investigation. Therefore we concluded that the W24 water cluster provides an appropriate starting point from which to model two complete solvation shells.
 | ||
| Fig. 3 Optimised structures with a first solvation shell (ball and stick) surrounded by an explicit second solvation shell (represented as tubes), along with their relative SCF, dispersion corrected SCF, and free energies (ΔESCF (ΔED3)/ΔG).‡ (magenta – Sr2+ ion, red – O atoms in water molecules, blue – O atoms in hydroxide ions, white – H atoms). All energies are given in kJ mol−1. | ||
We also attempted to create complexes with a second solvation shell by systematically increasing the number of water molecules around the previously optimised ‘single shell’ complexes. We found however, that these structures were always energetically unfavourable when compared with those based on the W24 cluster; presumably the high symmetry of the W24 cluster and the closed hydrogen bond network stabilises the systems obtained from this approach. Therefore only the W24 cluster based results are presented here.
The optimised structures of the Sr2+ hydroxides are summarised in Fig. 3, and are compared with the most stable structures for a given number of OH− ions and waters in the system. By contrast to the structures reported in the previous section, here we found no difficulties in optimising the geometries. Furthermore, examination of the structures in Fig. 3 reveals that they all have a largely closed hydrogen bond network, without the open spaces of the structures presented in Fig. 1. Comparing the average number of hydrogen bonds per molecule in the system confirms this statement, since the average number of hydrogen bonds is higher in the systems with two complete solvation shells (3.39 for water molecules and 3.27 for hydroxide groups), than for the structures with one solvation shell (only 2.33 for water molecules and 2.26 for hydroxides). These differences support our previous assertion that a second explicit solvation shell is required to accurately describe hydroxide speciation.
We find a coordination number of six to be preferred in the mono-, di-, and trihydroxides, whereas the tetrahydroxide is most stable with a coordination number of five. Therefore the inclusion of two explicit solvation shells results in an increase in the coordination number of the most stable complexes when compared with the structures determined with only a single shell.
The six coordinated complexes have a slightly distorted trigonal antiprismatic structure and in all cases only molecules from the two hexagons (visible on the top and bottom of the W24 cluster in Fig. 2a) are coordinated to the Sr2+ ion. On complex formation, the original planes of the hexagons distort and the cluster compresses as shown in the example in Fig. 4. Those molecules not directly coordinating the Sr2+ ion occupy the second and form a partially occupied third, solvation shell, but since the third shell water molecules cannot clearly be identified based on their distances from the Sr ion, they are treated as forming part of the second shell.
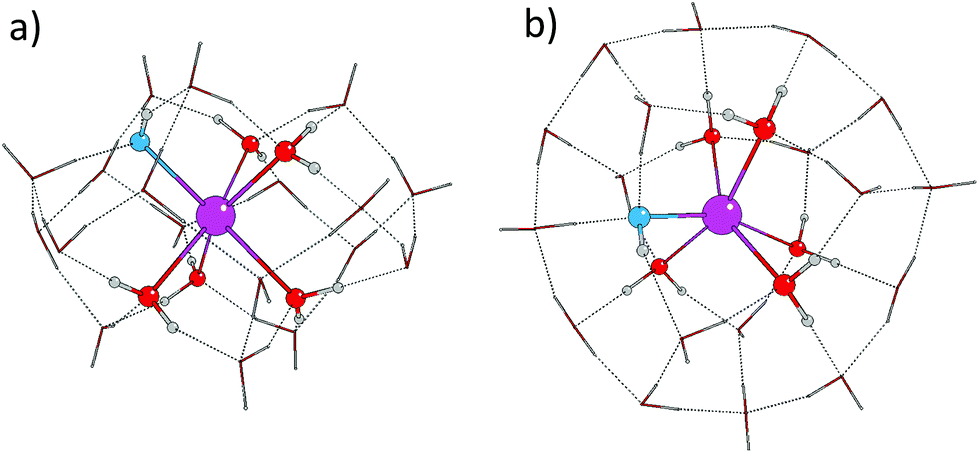 | ||
| Fig. 4 Ball and stick figure of the optimised geometry of the [Sr:5/18:1/0]+ monohydroxide with two complete solvation shells, based on the W24 water cluster, viewed from the side (a), and the top (b). The second shell waters are represented by tubes. (magenta – Sr2+ ion, red – O atoms in water molecules, blue – O atoms in hydroxide ions, white – H atoms). | ||
The average Sr–O distances for the Sr2+ hydroxides and hydrates with a complete second shell are summarised in Table 1. The average Sr–Ow distance in the first shell of the hydrate complexes varies from 2.60016 to 2.643 Å18 in previous experimental studies, in very good agreement with our calculated bond length (2.616 Å). With more hydroxide ions in the system, the Sr–Ow distances of the first shell water molecules increase; it was previously noted that higher hydroxide coordination weakens these bonds.30 It might be expected that the increasing Sr–Ow distances would be accompanied by a reduction of the partial charge on the metal, and the average Mulliken Sr charge does indeed decrease from +1.151 to +1.089, +1.024 and +1.019 in the mono through tetra hydroxo systems. The Sr–OOH distances in the first and second shells also increase, presumably for the same reason. Conversely, the Sr–Ow distances to the second shell waters decrease, as the weakening of the Sr first shell water bonds leads to a stronger bonding between the first and second shells. However, the calculated average bond length for the hydrate complex (4.464 Å) is still some way from the experimentally detected 4.940 Å. It is even less than the Sr–Ow distance measured in the solid crystal structure (4.760 Å). This may well be the effect of overestimated hydrogen bond strength in the whole system, due to the incomplete description of the environment by the COSMO.
| Sr−OOH | Sr−Ow | Sr−Ow (exp) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1st | 2nd | 1st | 2nd | 1st | 2nd | ||||||||
| Average | SD | Average | SD | Average | SD | Average | SD | Ref. 15 | Ref. 16 | Ref. 18 | Ref. 15 | Ref. 19 | |
| Hydrate | — | — | — | — | 2.616 | (0.037) | 4.464 | (0.333) | 2.640 | 2.600 | 2.643 | 4.940 | 4.760 |
| Mono | 2.455 | (0.008) | 3.818 | (0.009) | 2.621 | (0.020) | 4.409 | (0.324) | — | — | — | — | — |
| Di | 2.476 | (0.027) | 3.874 | (0.101) | 2.634 | (0.027) | 4.401 | (0.342) | — | — | — | — | — |
| Tri | 2.481 | (0.028) | 4.049 | — | 2.668 | (0.027) | 4.394 | (0.376) | — | — | — | — | — |
| Tetra | 2.482 | (0.018) | — | — | 2.708 | — | 4.397 | (0.410) | — | — | — | — | — |
If we compare the geometries of the different type of hydroxides in a given column of Fig. 3, we find that structures which have fewer OH− ions coordinated to the Sr2+ can be derived directly from those with more coordinated hydroxides via a proton transfer from a second shell water molecule to a first shell hydroxyl group. In addition, the energies of the structures in a given column are close to one another. For example, the Gibbs free energies of the systems with three hydroxide ions and 21 water molecules span less than 3 kJ mol−1: a room temperature Boltzmann distribution suggests that these structures may coexist in the following proportions: 19%, 26% and 55% for mono-, tri- and dihydroxides respectively. Such small energy differences can be rationalised by the fact that OH− ions are stronger hydrogen bond acceptors and weaker hydrogen bond donors than water molecules.26,29 Therefore, a system which contains a direct Sr2+–OH− interaction with the coordinated OH− ion involved in two strong hydrogen bonds as a donor and one weak hydrogen bond as an acceptor (e.g. [Sr:4/18:2/0]) is competitively close in energy to a system with a direct Sr2+–H2O interaction and an uncoordinated OH− ion which forms 3 strong hydrogen bonds with its oxygen atom while its hydrogen atom is directed away from the structure (e.g. [Sr:5/17:1/1]).
A tendency for one hydroxide ion to migrate from the first to the second solvation shell was reported previously for gas phase Sr2+ hydroxide complexes with one complete solvation shell11,30 but, to the best of our knowledge, proton transfer between shells has not been previously studied. We therefore sought the transition state for the Sr2+ dihydroxide ([Sr:4/18:2/0]) → Sr2+ monohydroxide ([Sr:5/17:1/1]) reaction. The results are summarised in Table 2 and the energy profile plotted in Fig. 5, from which we can see that the Gibbs free energy barrier for this reaction is very small (3.0 kJ mol−1). Thus, the coordination can easily interchange between di- and monohydroxide in this system, suggesting that our explicit two shell model well reflects the dynamical nature of the solutions, i.e. the frequent exchange of solvent molecules and anions between the ion solvation shells and the bulk solvent.54,55
 | ||
| Fig. 5 Free energy profile (ΔG) of proton transfer between the most stable dihydroxide ([Sr:4/18:2/0]) and monohydroxide ([Sr:5/17:1/1]) complexes from Fig. 3. TS = transition state. | ||
| (ESCF) (a.u.) | ΔEscf (kJ mol−1) | (SCF + Gcorr) (a.u.) | ΔG (kJ mol−1) | |
|---|---|---|---|---|
| Di [Sr:4/18:2/0] |
−1865.326957 | 0.0 | −1864.85131 | 0.0 |
| TS | −1865.326349 | 1.6 | −1864.850185 | 3.0 |
| Mono [Sr:5/17:1/1] |
−1865.328093 | −3.0 | −1864.852268 | −2.5 |
The relative stability of Sr2+ hydroxide complexes in the presence of two explicit solvation shells
To study the effect of increasing the number of hydroxide ions present in the system, we have explored the energetics of two reactions. In the first of these, we have compared the free energies of the different Sr2+ hydroxide species with the most stable Sr2+ hydrate complex plus the solvated hydroxide ions (eqn (1)). These reaction free energies are presented in Table 3 and in Fig. 6.| Sr(H2O)242+ + n(OH)(H2O)23− = Sr(OH)n(H2O)24−n(2−n) + n(H2O)24 | (1) |
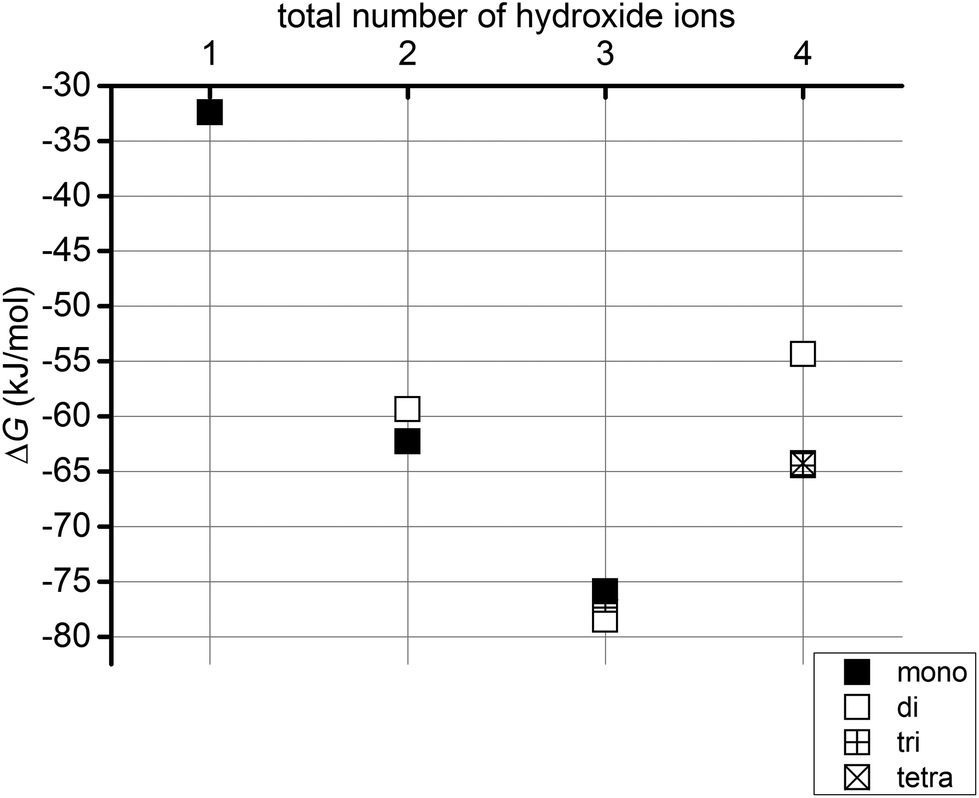 | ||
| Fig. 6 Gibbs free energies (ΔG, kJ mol−1) for eqn (1) for systems with 1 to 4 hydroxide ions. | ||
| Type of hydroxide | Total number of hydroxide ions | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||
| ΔESCF | ΔG | ΔESCF | ΔG | ΔESCF | ΔG | ΔESCF | ΔG | |
| a In order to probe the effects of basis set size on reaction energies, we carried out single point calculations for eqn (1) with different basis sets (def2-SVP and def2-QZVP in addition to our chosen def2-TZVP) and compare the SCF energies in Table S2 of the ESI. It can be seen that, while the reaction energies obtained with polarised double-ζ (SVP) and polarised triple-ζ basis sets (TZVP) vary significantly, for a given total number of hydroxides there is an essentially constant energy shift between the polarised triple-ζ and the larger quadruple-ζ quality basis set (QZVP) data. This suggests that TZVP is sufficient to obtain reliable relative energies. | ||||||||
| Mono | −36.3 | −32.5 | −60.9 | −62.3 | −74.5 | −75.6 | — | — |
| Di | — | — | −57.9 | −59.4 | −77.3 | −78.5 | −49.2 | −54.4 |
| Tri | — | — | — | — | −72.2 | −76.7 | −60.1 | −64.5 |
| Tetra | — | — | — | — | — | — | −57.6 | −64.3 |
We also wished to establish whether, for a structure with a given number of first or second shell hydroxides, it is energetically preferable for an additional hydroxyl group to enter the bulk solution or to become part of the strontium's primary or secondary coordination shell. We therefore calculated the energies of the reactions described by eqn (2), and these are collected in Table 4 and Fig. 7.
| Sr(OH)n−1(H2O)24−(n−1)(2−(n−1)) + (OH)(H2O)23− = Sr(OH)n(H2O)24−n(2−n) + (H2O)24 | (2) |
 | ||
| Fig. 7 Gibbs free energies (ΔG, kJ mol−1) for eqn (2) for systems with 1 to 4 hydroxide ions. | ||
| Type of hydroxide | Total number of hydroxide ions | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||
| ΔESCF | ΔG | ΔESCF | ΔG | ΔESCF | ΔG | ΔESCF | ΔG | |
| Mono | −36.3 | −32.5 | −24.6 | −29.8 | −13.6 | −13.3 | — | — |
| Di | — | — | −21.6 | −26.9 | −16.4 | −16.2 | 28.1 | 24.1 |
| Tri | — | — | — | — | −11.3 | −14.4 | 17.2 | 14.0 |
| Tetra | — | — | — | — | — | — | 19.6 | 14.2 |
Fig. 6 shows that in all cases it is energetically favourable for hydroxides to coordinate the Sr2+ ion than to all exist as hydrated OH−. If we introduce one hydroxide ion into the system, the formation of a monohydroxide lowers the energy by c. 32 kJ mol−1. On increasing the number of hydroxide ions up to three, the reaction energy becomes increasingly negative, before becoming less so for four hydroxides. Fig. 7 reveals that the successive addition of one more hydroxide ion into the Sr coordination sphere has a less and less stabilising effect, to the point that the reaction energy associated with the introduction of a fourth OH− ion into one of the Sr solvation shells is positive, i.e. the OH− prefers to exist as a hydrated ion. Thus, although a system with four hydroxides in the Sr coordination sphere is more stable than when all hydroxides exist as hydrated ions (Fig. 6), such a complex is significantly less stable than one in which three hydroxides coordinate the Sr2+ ion, with the fourth existing as a hydrated ion (Fig. 7). This allows us to predict that dianionic tetrahydroxide species will not exist. In agreement with this conclusion, Sr(OH)2 is known to be a moderately strong base without amphoteric properties,22 and as such it should not form negatively charged hydroxide complexes even in the presence of strong(er) bases.§
Fig. 7 indicates that monoanionic species containing three hydroxides are stable within the confines of our model. For these systems, however, the most stable structure ([Sr:4/17:2/1]−) has two hydroxides in the primary coordination shell with the third in the second shell. It is therefore entirely possible that monoanionic species are also unstable in solution, as the second shell hydroxide in [Sr:4/17:2/1]− may well prefer to migrate out of the Sr coordination environment altogether. Unfortunately, we cannot test this hypothesis within the limits of our present model, as to include an explicit third solvation would be computationally prohibitive.
Conclusions
We have carried out DFT quantum chemical calculations to model the aqueous solvation of Sr2+ mono-, di-, tri- and tetrahydroxides, using a combination of the COSMO continuum solvent model and one or two explicit solvation shells. We have shown that including a second explicit solvation shell is essential for the accurate modelling of these systems.With only a single explicit solvation shell, the coordination number of the most stable mono-, di-, and trihydroxides is five, decreasing to four for the tetrahydroxides. In all cases we find open regions in the Sr coordination shell which would not exist in real aqueous complexes, and we often found our optimised structures to possess unavoidable energetic instabilities. Including a second shell of explicit waters, however, resulted in energetically minimal structures without open regions in the first Sr coordination shell. The energetically-preferred coordination numbers obtained from the two-shell calculations increased with respect to those found in with the single shell model. For systems with one, two or three hydroxide ions, we found the most stable complexes to all be 6-coordinated with a distorted trigonal antiprismatic geometry, whereas systems with four OH− ions have a most stable coordination number of five.
Comparison of the SCF and Gibbs free energies of the systems with two explicit solvation shells shows that there is only a small energy difference between the different types of hydroxide (c. 3 kJ mol−1). Transformation from one coordination mode to another can easily occur via a proton transfer mechanism; e.g. the barrier in the Sr2+ dihydroxide ([Sr:4/18:2/0]) → Sr2+ monohydroxide ([Sr:5/17:1/1]) reaction is only 3.0 kJ mol−1.
We have evaluated the relative stabilities of the mono-, di-, tri- and tetrahydroxide species by calculating the energies of two reactions (defined by eqn (1) and (2)). We found that in all cases the Sr2+ hydroxide complexes are more stable than a Sr2+ hydrate plus hydrated OH− ions. However, the addition of more hydroxide ions has a systematically weaker stabilising effect, terminating at the point when adding a fourth OH− ion to the trihydroxide species is significantly energetically unfavourable. Furthermore, the most stable trihydroxide structure has only two hydroxides in the first coordination shell of the Sr2+ ion, and we suggest that the third hydroxide ligand may migrate away from the Sr coordination environment altogether if our model contained a significantly larger number of explicit water molecules.
We conclude that the most likely Sr-hydroxide complexes to be found in high pH aqueous solutions are mono- and dihydroxides, and that they coexist. These species are therefore the most likely candidates for adsorption onto brucite surfaces, and we are currently exploring these interactions.
Acknowledgements
We are grateful to the Nuclear Decommissioning Authority and UCL for financial support for a PhD studentship to EM. AK thanks the EPSRC for the award of a career acceleration fellowship (grant EP/J002208/1). We also thank UCL for computing resources via the Research Computing “Legion” cluster (Legion@UCL) and associated services, and also the “Iridis” facility of the e-Infrastructure South Consortium's Centre for Innovation. We are also grateful to Dr Jonathan Austin of the National Nuclear Laboratory for helpful discussions.References
- Sellafield Ltd., Sellafield Integrated Waste Strategy (Version 2 Report and Recommendations), GEN-1880A, 2007.
- C. R. Gregson, D. T. Goddard, M. J. Sarsfield and R. J. Taylor, J. Nucl. Mater., 2011, 412, 145 CrossRef CAS.
- J. D. Farr, M. P. Neu, R. K. Schulze and B. D. Honeyman, J. Alloys Compd., 2007, 444–445, 533 CrossRef CAS.
- G. R. Bochkarev and G. I. Pushkareva, J. Min. Sci., 2009, 45, 290 CrossRef.
- Toxilogical profile for Cesium, U.S. Department of Health and Human Services, Agency for Toxic Substances and Disease Registry, 2004.
- Toxilogical profile of Strontium, U.S. Department of Health and Human Services, Agency for Toxic Substances and Disease Registry, 2004.
- J. Peterson, M. MacDonell, L. Haroun, F. Monette, R. D. Hildebrand and A. Toboas, Radiological and Chemical Fact Sheets to Support Health Risk Analyses for Contaminated Areas, Argonne National Laboratory Environmental Science Division and U.S. Department of Energy, 2007 Search PubMed.
- Toxicological profile for Uranium, U.S. Department of Health and Human Services, Agency for Toxic Substances and Disease Registry, 2013.
- M. Peschke, A. T. Blades and P. Kebarle, J. Phys. Chem. A, 1998, 102, 9978 CrossRef CAS.
- D. R. Carl, B. K. Chatterjee and P. B. Armentrout, J. Chem. Phys., 2010, 132, 044303 CrossRef CAS PubMed.
- A. R. Felmy, D. A. Dixon, J. R. Rustad, M. J. Mason and L. M. Onishi, J. Chem. Thermodyn., 1998, 30, 1103 CrossRef CAS.
- E. D. Glendening and D. Feller, J. Phys. Chem., 1996, 100, 4790 CrossRef CAS.
- A. Kerridge and N. Kaltsoyannis, Chem. – Eur. J., 2011, 17, 5060 CrossRef CAS PubMed.
- J. N. Albright, J. Chem. Phys., 1972, 56, 3783 CrossRef CAS.
- R. Caminiti, A. Musinu, G. Paschina and G. Pinna, J. Appl. Crystallogr., 1982, 15, 482 CrossRef CAS.
- G. Moreau, L. Helm, J. Purans and A. E. Merbach, J. Phys. Chem. A, 2002, 106, 3034 CrossRef CAS.
- D. M. Pfund, J. G. Darab, J. L. Fulton and Y. Ma, J. Phys. Chem., 1994, 98, 13102 CrossRef CAS.
- P. D'Angelo, H.-F. Nolting and N. V. Pavel, Phys. Rev. A, 1996, 53, 798 CrossRef.
- I. Persson, M. Sandström, H. Yokoyama and M. Chaudhry, Z. Naturforsch., A: Phys. Sci., 1995, 50, 21 CrossRef CAS.
- E. Spohr, G. Pálinkás, K. Heinzinger, P. Bopp and M. M. Probst, J. Phys. Chem., 1988, 92, 6754 CrossRef CAS.
- D. J. Harris, J. P. Brodholt and D. M. Sherman, J. Phys. Chem. B, 2003, 107, 9056 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, in Chemistry of the Elements, Elsevier Ltd., 2nd edn, 1997, ch. 5, vol. 1, pp. 107–138 Search PubMed.
- P. Patnaik, in Handbook of Inorganic Chemicals, McGraw-Hill, New York, 2002, p. 886 Search PubMed.
- Von H. W. Grueninger and H. Bärnighausen, Z. Anorg. Allg. Chem., 1969, 368, 53 CrossRef.
- M. D. Judd and M. I. Pope, J. Therm. Anal., 1971, 3, 397 CrossRef CAS.
- H. D. Lutz, P. Kuske and J. Henning, J. Mol. Struct., 1988, 176, 149 CrossRef CAS.
- H. D. Lutz, W. Eckers, G. Schneider and H. Haeuseler, Spectrochim. Acta, Part A, 1981, 37, 561 CrossRef.
- H. G. Smith, Acta Crystallogr., 1953, 6, 604 CrossRef CAS.
- J. S. Ricci, R. C. Stevens, R. K. McMullan and W. T. Klooster, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 381 CAS.
- A. Kerridge and N. Kaltsoyannis, Dalton Trans., 2011, 40, 11258 RSC.
- V. Vallet, P. Macak, U. Wahlgren and I. Grenthe, Theor. Chem. Acc., 2006, 115, 145 CrossRef CAS.
- D. Rios, M. C. Michelini, A. F. Lucena, J. Marçalo, T. H. Bray and J. K. Gibson, Inorg. Chem., 2012, 51, 6603 CrossRef CAS PubMed.
- K. E. Gutowski and D. A. Dixon, J. Phys. Chem. A, 2006, 110, 8840 CrossRef CAS PubMed.
- TURBOMOLE V6.5 2013, a development of University of Karlsruhe and Forschungszentrum; Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- M. Kaupp, P. v. R. Schleyer, H. Stoll and H. Preuss, J. Chem. Phys., 1991, 94, 1360 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683 CrossRef CAS PubMed.
- P. Ugliengo, MOLDRAW: A Program to Display and Manipulate Molecular and Crystal Structures, Torino (2006) available on the web at: http:\\www.moldraw.unito.it.
- P. Ugliengo, G. Borzani and D. Viterbo, J. Appl. Crystallogr., 1988, 21, 75 CrossRef.
- P. Ugliengo, D. Viterbo and G. Chiari, Z. Kristallogr., 1993, 207, 9 CAS.
- R. Ludwig and F. Weinhold, J. Chem. Phys., 1999, 110, 508 CrossRef CAS.
- Ph. Wernet, D. Nordlund, U. Bergmann, M. Cavalleri, M. Odelius, H. Ogasawara, L. Å. Näslund, T. K. Hirsch, L. Ojamäe, P. Glatzel, L. G. M. Pettersson and A. Nilsson, Science, 2004, 304, 995 CrossRef CAS PubMed.
- S. Myneni, Y. Luo, L. Å. Näslund, M. Cavalleri, L. Ojamäe, H. Ogasawara, A. Pelmenschikov, Ph. Wernet, P. Väterlein, C. Heske, Z. Hussain, L. G. M. Pettersson and A. Nilsson, J. Phys.: Condens. Matter, 2002, 14, L213 CrossRef CAS.
- E. Schwegler, G. Galli and F. Gygi, Phys. Rev. Lett., 2000, 84, 2429 CrossRef CAS PubMed.
- J. D. Smith, C. D. Cappa, K. R. Wilson, B. M. Messer, R. C. Cohen and R. J. Saykally, Science, 2004, 306, 851 CrossRef CAS PubMed.
- H. S. Lee and M. E. Tuckerman, J. Chem. Phys., 2006, 125, 154507 CrossRef PubMed.
- A. K. Soper, F. Bruni and M. A. Ricci, J. Chem. Phys., 1997, 106, 247 CrossRef CAS.
- C. Nieto-Draghi, J. B. Avalos and B. Rousseau, J. Chem. Phys., 2003, 118, 7954 CrossRef CAS.
- H. Ohtaki and T. Radnai, Chem. Rev., 1993, 93, 1157 CrossRef CAS.
- J. Burgess, in Ions in Solution - Basic Principles of Chemical Interactions, Horwood Publishing Limited, Chichester, West Sussex, England, 1999, ch. 9, pp. 111–123 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt01110h |
| ‡ Inclusion of the DFT-D3 dispersion corrections does not result in any qualitative difference in the relative energies of the isomers of a given system. |
| § The pH of our model systems, even with two shells of explicit waters, is >14. Explicit inclusion of 1250 water molecules for every hydroxide ion would be required to lower the pH to 13. |
| This journal is © The Royal Society of Chemistry 2015 |